Regulation of Angiogenesis by Aminoacyl-tRNA Synthetases

Abstract
:1. Introduction

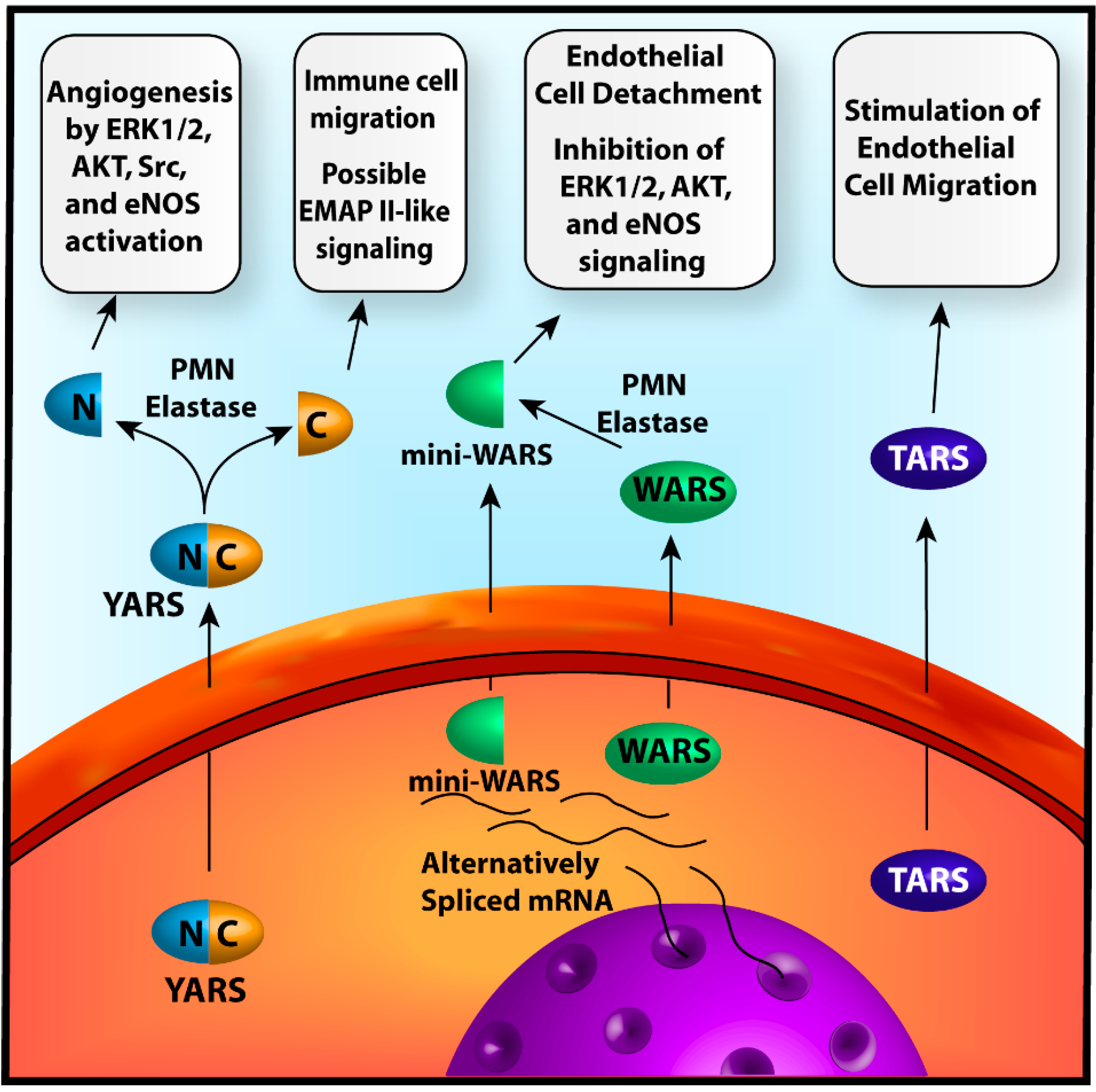{kind=link}
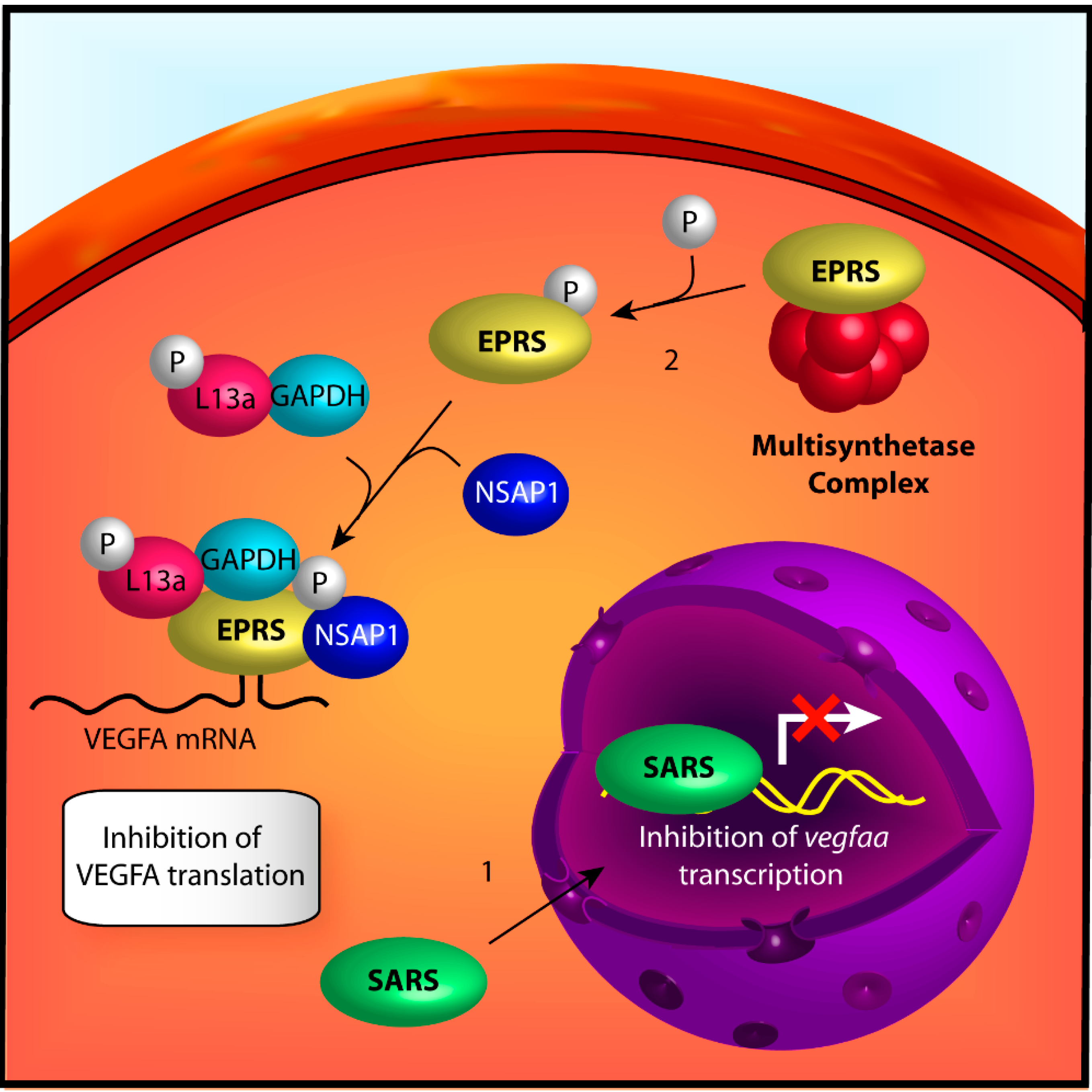{kind=link}
{kind=link}
| ARS or Associated Factor | Location of Angiogenic Function | Effect on Angiogenesis | Associated Domains | Details |
|---|---|---|---|---|
| Externally Acting ARS | ||||
| YARS | Extracellular | Pro | ELR motif, EMAP II-like [12,13] | Secreted and cleaved into N and C cytokine fragments; N-terminal ELR stimulates angiogenesis; C-terminus may possess EMAP II-like signaling [12,13,14,15] |
| WARS | Extracellular | Static | WHEP domain [16] | Secreted and WHEP-domain removed by secretion or alternative splicing; loss of WHEP-domain allows for interactions with E-cadherin [16,17,18,19] |
| TARS | Extracellular | Pro | Catalytic domain (possibly others) [20] | Secreted and stimulates vessel migration, patterning, and maturation; Based on borrelidin binding site, the mechanism is likely dependent, at least partially, on the catalytic domain [20,21] |
| Internally Acting ARS | ||||
| SARS | Nucleus | Static | NLS [22,23,24] | Directed to nucleus by NLS; Binds to vegfaa promoter and disrupts cMyc induction of vegfaa mRNA [22,23,24] |
| EPRS | Cytoplasm | Static | Three WHEP domains [25] | IFNγ stimulates the release of EPRS from the MSC and its association with the GAIT complex; complex binds to mRNA 3' elements and inhibits translation [25,26,27,28,29,30,31,32] |
| ARS Associated Factors | ||||
| AIMP1 | Extracellular | Pro (low conc.) Anti (high conc.) | EMAP II [33] | Released from MSC in response to apoptotic signals; secreted as full-length AIMP1 or EMAP II; stimulates angiogensis at low concentrations but induces EC apoptosis at high concentrations [33,34,35,36,37,38,39,40,41] |
2. Regulation of Angiogenesis by Aminoacyl-tRNA Synthetases
2.1. Angiogenic Signaling by Extracellular ARSs
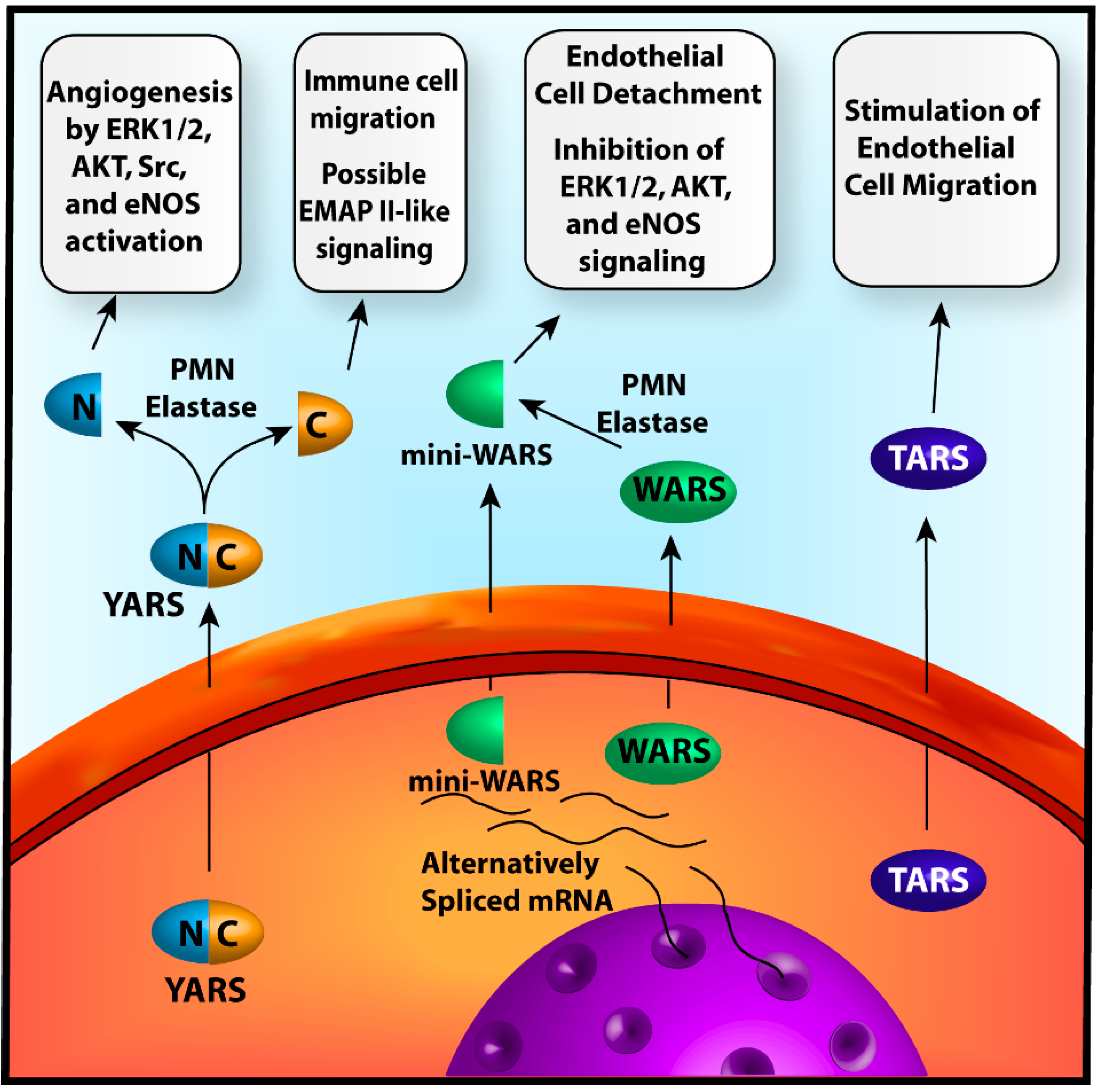
2.1.1. Tyrosyl-tRNA Synthetase
2.1.2. Tryptophanyl-tRNA Synthetase
2.1.3. Threonyl-tRNA Synthetase
2.2. Transcriptional and Translational Regulation of Angiogenesis by Aminoacyl-tRNA Synthetases (ARSs)
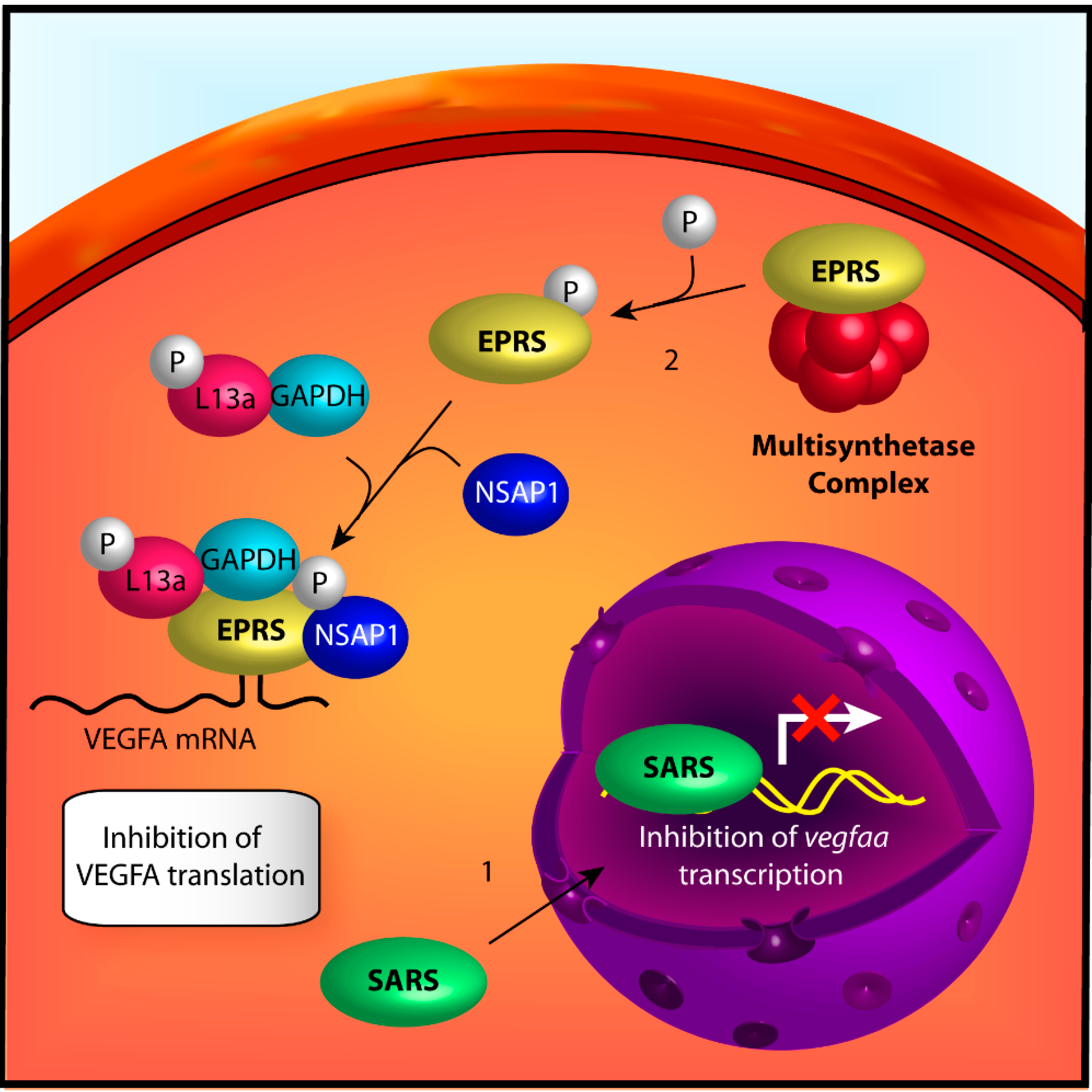
2.2.1. Seryl-tRNA Synthetase
2.2.2. Glutamyl-Prolyl-tRNA Synthetase
2.3. Regulation of Angiogenesis by ARS-Associated Proteins
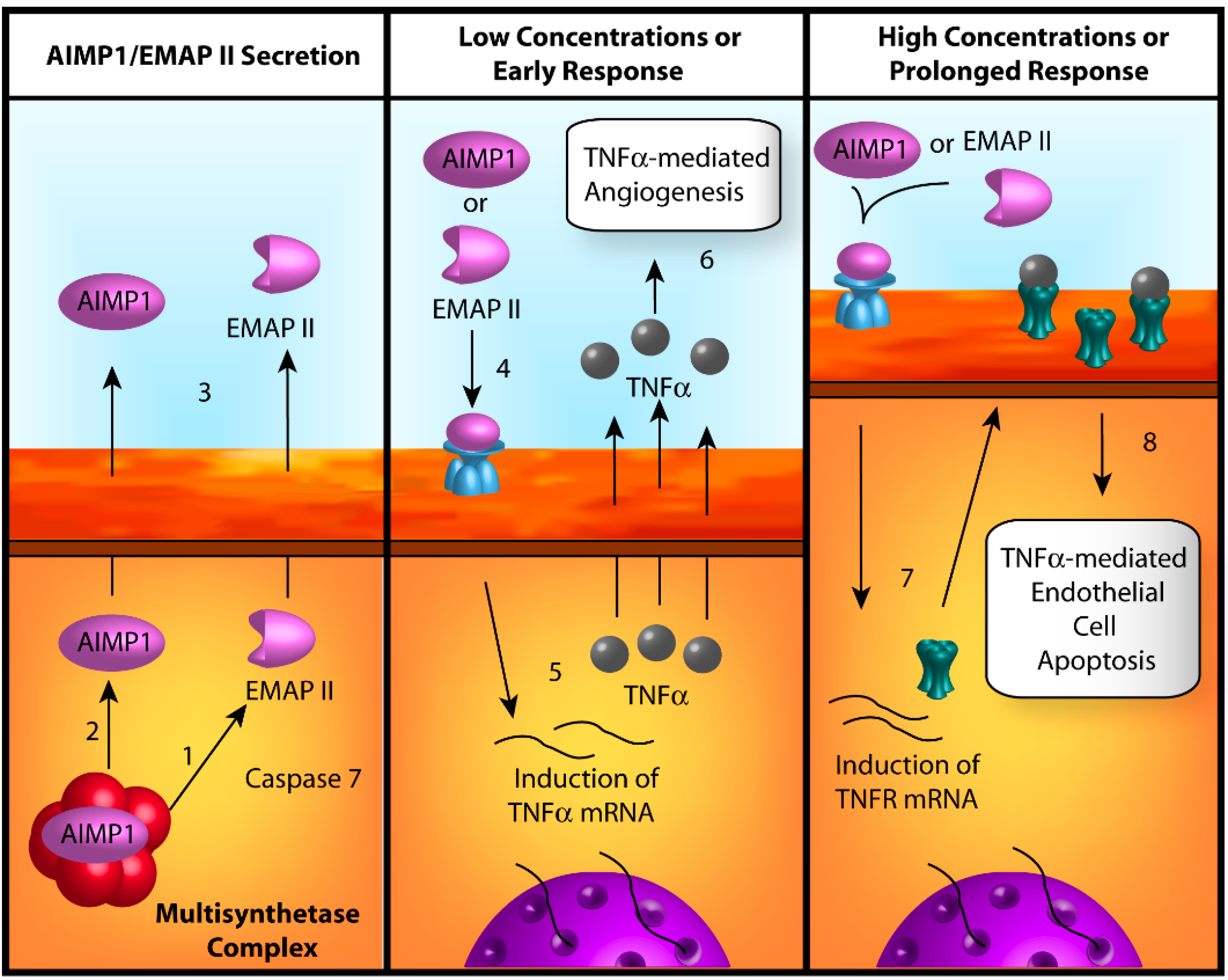
3. Conclusions
3.1. Aminoacyl-tRNA Synthetases as Nutrient Sensors
3.2. Mechanistic Separation of Angiogenesis-Associated ARSs
3.3. Associations of Angiogenesis-Related ARSs to Inflammation
3.4. The Role of Appended Domains in ARS-Mediated Angiogenesis
3.5. ARSs as Targets for Anti-Angiogenic Therapy
3.6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, G.Y.; Becam, A.M.; Slonimski, P.P.; Herbert, C.J. In vitro mutagenesis of the mitochondrial leucyl tRNA synthetase of Saccharomyces cerevisiae shows that the suppressor activity of the mutant proteins is related to the splicing function of the wild-type protein. Mol. Gen. Genet. 1996, 252, 667–675. [Google Scholar] [PubMed]
- Mohr, G.; Rennard, R.; Cherniack, A.D.; Stryker, J.; Lambowitz, A.M. Function of the Neurospora crassa mitochondrial tyrosyl-tRNA synthetase in RNA splicing. Role of the idiosyncratic N-terminal extension and different modes of interaction with different group I introns. J. Mol. Biol. 2001, 307, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Nechushtan, H.; Figov, N.; Razin, E. The function of lysyl-tRNA synthetase and Ap4A as signaling regulators of MITF activity in FcepsilonRI-activated mast cells. Immunity 2004, 20, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Putney, S.D.; Schimmel, P. An aminoacyl tRNA synthetase binds to a specific DNA sequence and regulates its gene transcription. Nature 1981, 291, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Howard, O.M.; Dong, H.F.; Yang, D.; Raben, N.; Nagaraju, K.; Rosen, A.; Casciola-Rosen, L.; Hartlein, M.; Kron, M.; Yang, D.; et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J. Exp. Med. 2002, 196, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.G.; Kim, E.Y.; Kim, T.; Park, H.; Park, H.S.; Choi, E.J.; Kim, S. Glutamine-dependent antiapoptotic interaction of human glutaminyl-tRNA synthetase with apoptosis signal-regulating kinase 1. J. Biol. Chem. 2001, 276, 6030–6036. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Sottile, J. Regulation of angiogenesis by extracellular matrix. Biochim. Biophys. Acta 2004, 1654, 13–22. [Google Scholar] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; van Oosterom, A.T.; de Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Kleeman, T.A.; Wei, D.; Simpson, K.L.; First, E.A. Human tyrosyl-tRNA synthetase shares amino acid sequence homology with a putative cytokine. J. Biol. Chem. 1997, 272, 14420–14425. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, K.; Schimmel, P. Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science 1999, 284, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, K.; Slike, B.M.; Hood, J.; Ewalt, K.L.; Cheresh, D.A.; Schimmel, P. Induction of angiogenesis by a fragment of human tyrosyl-tRNA synthetase. J. Biol. Chem. 2002, 277, 20124–20126. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Chapuli, R.; Quesada, A.R.; Medina, M.A. Angiogenesis and signal transduction in endothelial cells. Cell. Mol. Life Sci. 2004, 61, 2224–2243. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Kapoor, M.; Guo, M.; Belani, R.; Xu, X.; Kiosses, W.B.; Hanan, M.; Park, C.; Armour, E.; Do, M.H.; et al. Orthogonal use of a human tRNA synthetase active site to achieve multifunctionality. Nat. Struct. Mol. Biol. 2010, 17, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, K.; Slike, B.M.; Hood, J.; Otani, A.; Ewalt, K.L.; Friedlander, M.; Cheresh, D.A.; Schimmel, P. A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Reader, J.S.; Irani-Tehrani, M.; Ewalt, K.L.; Schwartz, M.A.; Schimmel, P. VE-cadherin links tRNA synthetase cytokine to anti-angiogenic function. J. Biol. Chem. 2005, 280, 2405–2408. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Reader, J.S.; Irani-Tehrani, M.; Ewalt, K.L.; Schwartz, M.A.; Schimmel, P. Biologically active fragment of a human tRNA synthetase inhibits fluid shear stress-activated responses of endothelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 14903–14907. [Google Scholar] [CrossRef] [PubMed]
- Ruan, B.; Bovee, M.L.; Sacher, M.; Stathopoulos, C.; Poralla, K.; Francklyn, C.S.; Soll, D. A unique hydrophobic cluster near the active site contributes to differences in borrelidin inhibition among threonyl-tRNA synthetases. J. Biol. Chem. 2005, 280, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.F.; Mirando, A.C.; Wilkinson, B.; Francklyn, C.S.; Lounsbury, K.M. Secreted threonyl-tRNA synthetase stimulates endothelial cell migration and angiogenesis. Sci. Rep. 2013, 3, 1317. [Google Scholar] [PubMed]
- Fukui, H.; Hanaoka, R.; Kawahara, A. Noncanonical activity of seryl-tRNA synthetase is involved in vascular development. Circ. Res. 2009, 104, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shi, Y.; Zhang, H.M.; Swindell, E.C.; Marshall, A.G.; Guo, M.; Kishi, S.; Yang, X.L. Unique domain appended to vertebrate tRNA synthetase is essential for vascular development. Nat. Commun. 2012, 3, 681. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xu, X.; Zhang, Q.; Fu, G.; Mo, Z.; Wang, G.S.; Kishi, S.; Yang, X.L. tRNA synthetase counteracts c-Myc to develop functional vasculature. eLife 2014, 3, e02349. [Google Scholar] [PubMed]
- Jia, J.; Arif, A.; Ray, P.S.; Fox, P.L. WHEP domains direct noncanonical function of glutamyl-Prolyl tRNA synthetase in translational control of gene expression. Mol. Cell 2008, 29, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, B.; Fox, P.L. Delayed translational silencing of ceruloplasmin transcript in γ interferon-activated U937 monocytic cells: Role of the 3' untranslated region. Mol. Cell Biol. 1999, 19, 6898–6905. [Google Scholar] [PubMed]
- Sampath, P.; Mazumder, B.; Seshadri, V.; Fox, P.L. Transcript-selective translational silencing by gamma interferon is directed by a novel structural element in the ceruloplasmin mRNA 3' untranslated region. Mol. Cell Biol. 2003, 23, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, B.; Sampath, P.; Seshadri, V.; Maitra, R.K.; DiCorleto, P.E.; Fox, P.L. Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell 2003, 115, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Sampath, P.; Mazumder, B.; Seshadri, V.; Gerber, C.A.; Chavatte, L.; Kinter, M.; Ting, S.M.; Dignam, J.D.; Kim, S.; Driscoll, D.M.; et al. Noncanonical function of glutamyl-prolyl-tRNA synthetase: Gene-specific silencing of translation. Cell 2004, 119, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Kapasi, P.; Chaudhuri, S.; Vyas, K.; Baus, D.; Komar, A.A.; Fox, P.L.; Merrick, W.C.; Mazumder, B. L13a blocks 48S assembly: Role of a general initiation factor in mRNA-specific translational control. Mol. Cell 2007, 25, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R.; Jia, J.; Arif, A.; Ray, P.S.; Fox, P.L. The GAIT system: A gatekeeper of inflammatory gene expression. Trends Biochem. Sci. 2009, 34, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.S.; Fox, P.L. A post-transcriptional pathway represses monocyte VEGF-A expression and angiogenic activity. EMBO J. 2007, 26, 3360–3372. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, S.; Agou, F.; Robinson, J.C.; Mirande, M. The p43 component of the mammalian multi-synthetase complex is likely to be the precursor of the endothelial monocyte-activating polypeptide II cytokine. J. Biol. Chem. 1997, 272, 32573–32579. [Google Scholar] [CrossRef] [PubMed]
- Shalak, V.; Kaminska, M.; Mitnacht-Kraus, R.; Vandenabeele, P.; Clauss, M.; Mirande, M. The EMAPII cytokine is released from the mammalian multisynthetase complex after cleavage of its p43/proEMAPII component. J. Biol. Chem. 2001, 276, 23769–23776. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Kang, Y.S.; Ahn, Y.H.; Lee, S.H.; Kim, K.R.; Kim, K.W.; Koh, G.Y.; Ko, Y.G.; Kim, S. Dose-dependent biphasic activity of tRNA synthetase-associating factor, p43, in angiogenesis. J. Biol. Chem. 2002, 277, 45243–45248. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.C.; Alexander, H.R.; Tang, G.; Wu, P.S.; Hewitt, S.M.; Turner, E.; Kruger, E.; Figg, W.D.; Grove, A.; Kohn, E.; et al. Endothelial monocyte activating polypeptide II induces endothelial cell apoptosis and may inhibit tumor angiogenesis. Microvasc. Res. 2000, 60, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.G.; Park, H.; Kim, T.; Lee, J.W.; Park, S.G.; Seol, W.; Kim, J.E.; Lee, W.H.; Kim, S.H.; Park, J.E.; et al. A cofactor of tRNA synthetase, p43, is secreted to up-regulate proinflammatory genes. J. Biol. Chem. 2001, 276, 23028–23033. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Park, M.C.; Kim, D.G.; Cho, K.; Park, Y.W.; Han, J.M.; Kim, S. Identification of CD23 as a functional receptor for the proinflammatory cytokine AIMP1/p43. J. Cell Sci. 2012, 125, 4620–4629. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.A.; Zheng, H.; Liu, J.; Corbett, S.; Schwarz, R.E. Endothelial-monocyte activating polypeptide II alters fibronectin based endothelial cell adhesion and matrix assembly via α5 β1 integrin. Exp. Cell Res. 2005, 311, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.; Schwarz, M.A.; Verma, V.; Cappiello, C.; Schwarz, R.E. Endothelial monocyte activating polypeptide II interferes with VEGF-induced proangiogenic signaling. Lab. Investig. 2009, 89, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Tandle, A.T.; Calvani, M.; Uranchimeg, B.; Zahavi, D.; Melillo, G.; Libutti, S.K. Endothelial monocyte activating polypeptide-II modulates endothelial cell responses by degrading hypoxia-inducible factor-1α through interaction with PSMA7, a component of the proteasome. Exp. Cell Res. 2009, 315, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, Y.; King, M.; Kiosses, W.B.; Ewalt, K.; Yang, X.; Schimmel, P.; Reader, J.S.; Tzima, E. The novel fragment of tyrosyl tRNA synthetase, mini-TyrRS, is secreted to induce an angiogenic response in endothelial cells. FASEB J. 2008, 22, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Patera, A.C.; Pong-Kennedy, A.; Deno, G.; Gonsiorek, W.; Manfra, D.J.; Vassileva, G.; Zeng, M.; Jackson, C.; Sullivan, L.; et al. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J. Biol. Chem. 2007, 282, 11658–11666. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Gohongi, T.; Kadambi, A.; Izumi, Y.; Ang, J.; Yun, C.O.; Buerk, D.G.; Huang, P.L.; Jain, R.K. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. USA 2001, 98, 2604–2609. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, G.; Dorizzi, M.; Spotorno, G.; Labouesse, B. Purification of tryptophan tRNA synthetase from beef pancreas. Bull. Soc. Chim. Biol. 1969, 51, 495–510. [Google Scholar] [PubMed]
- Beresten, S.F.; Zargarova, T.A.; Favorova, O.O.; Rubikaite, B.I.; Ryazanov, A.G.; Kisselev, L.L. Molecular and cellular studies of tryptophanyl-tRNA synthetase using monoclonal antibodies. Evaluation of a common antigenic determinant in eukaryotic, prokaryotic and archaebacterial enzymes which maps outside the catalytic domain. Eur. J. Biochem. 1989, 184, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Zhou, Q.; Otero, F.; Myers, C.A.; Bates, A.; Belani, R.; Liu, J.; Luo, J.K.; Tzima, E.; Zhang, D.E.; et al. Evidence for annexin II-S100A10 complex and plasmin in mobilization of cytokine activity of human TrpRS. J. Biol. Chem. 2008, 283, 2070–2077. [Google Scholar] [CrossRef] [PubMed]
- Renigunta, V.; Yuan, H.; Zuzarte, M.; Rinne, S.; Koch, A.; Wischmeyer, E.; Schlichthorl, G.; Gao, Y.; Karschin, A.; Jacob, R.; et al. The retention factor p11 confers an endoplasmic reticulum-localization signal to the potassium channel TASK-1. Traffic 2006, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Paetz, W.; Nass, G. Biochemical and immunological characterization of threonyl-tRNA synthetase of two borrelidin-resistant mutants of Escherichia coli K12. Eur. J. Biochem. 1973, 35, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Gantt, J.S.; Bennett, C.A.; Arfin, S.M. Increased levels of threonyl-tRNA synthetase in a borrelidin-resistant Chinese hamster ovary cell line. Proc. Natl. Acad. Sci. USA 1981, 78, 5367–5370. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Kageyama, R.; Naruse, N.; Tsukahara, N.; Funahashi, Y.; Kitoh, K.; Watanabe, Y. Borrelidin is an angiogenesis inhibitor; disruption of angiogenic capillary vessels in a rat aorta matrix culture model. J. Antibiot. 1997, 50, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Sundrud, M.S.; Koralov, S.B.; Feuerer, M.; Calado, D.P.; Kozhaya, A.E.; Rhule-Smith, A.; Lefebvre, R.E.; Unutmaz, D.; Mazitschek, R.; Waldner, H.; et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science 2009, 324, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Liu, D.; Towle, M.J.; Kageyama, R.; Tsukahara, N.; Wakabayashi, T.; Littlefield, B.A. Anti-angiogenesis effects of borrelidin are mediated through distinct pathways: threonyl-tRNA synthetase and caspases are independently involved in suppression of proliferation and induction of apoptosis in endothelial cells. J. Antibiot. 2003, 56, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Wellman, T.L.; Eckenstein, M.; Wong, C.; Rincon, M.; Ashikaga, T.; Mount, S.L.; Francklyn, C.S.; Lounsbury, K.M. Threonyl-tRNA synthetase overexpression correlates with angiogenic markers and progression of human ovarian cancer. BMC Cancer 2014, 14, 620. [Google Scholar] [CrossRef] [PubMed]
- Brunel, C.; Romby, P.; Moine, H.; Caillet, J.; Grunberg-Manago, M.; Springer, M.; Ehresmann, B.; Ehresmann, C. Translational regulation of the Escherichia coli threonyl-tRNA synthetase gene: Structural and functional importance of the thrS operator domains. Biochimie 1993, 75, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.W.; Herzog, W.; Santoro, M.M.; Mitchell, T.S.; Frantsve, J.; Jungblut, B.; Beis, D.; Scott, I.C.; D’Amico, L.A.; Ober, E.A.; et al. A transgene-assisted genetic screen identifies essential regulators of vascular development in vertebrate embryos. Dev. Biol. 2007, 307, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Herzog, W.; Muller, K.; Huisken, J.; Stainier, D.Y. Genetic evidence for a noncanonical function of seryl-tRNA synthetase in vascular development. Circ. Res. 2009, 104, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Amsterdam, A.; Nissen, R.M.; Sun, Z.; Swindell, E.C.; Farrington, S.; Hopkins, N. Identification of 315 genes essential for early zebrafish development. Proc. Natl. Acad. Sci. USA 2004, 101, 12792–12797. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. TLR signaling. Cell Death Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Polunovsky, V.A.; Wendt, C.H.; Ingbar, D.H.; Peterson, M.S.; Bitterman, P.B. Induction of endothelial cell apoptosis by TNF alpha: modulation by inhibitors of protein synthesis. Exp. Cell Res. 1994, 214, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, R.; Rens, J.A.; Schipper, D.; Eggermont, A.M.; ten Hagen, T.L. EMAP-II facilitates TNF-R1 apoptotic signalling in endothelial cells and induces TRADD mobilization. Apoptosis 2006, 11, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Segev, N.; Hay, N. Hijacking leucyl-tRNA synthetase for amino acid-dependent regulation of TORC1. Mol. Cell 2012, 46, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Hao, Z.; Tian, Q.; Zhang, Z.; Zhou, C.; Xie, W. Cocrystal structures of glycyl-tRNA synthetase in complex with tRNA suggest multiple conformational states in glycylation. J. Biol. Chem. 2014, 289, 20359–20369. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lee, E.H.; Park, M.; Kim, J.H.; Kim, J.; Kim, S.; Jeon, Y.H.; Hwang, K.Y. Conformational changes in human prolyl-tRNA synthetase upon binding of the substrates proline and ATP and the inhibitor halofuginone. Acta Crystallogr. Sect. D 2013, 69, 2136–2145. [Google Scholar] [CrossRef]
- Romagnani, P.; Lasagni, L.; Annunziato, F.; Serio, M.; Romagnani, S. CXC chemokines: The regulatory link between inflammation and angiogenesis. Trends Immunol. 2004, 25, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, L.F.; Kwan, H.H.; Kowalski, J.; Prionas, S.D.; Allison, A.C. Dual role of tumor necrosis factor-alpha in angiogenesis. Am. J. Pathol. 1992, 140, 539–544. [Google Scholar] [PubMed]
- Belperio, J.A.; Keane, M.P.; Arenberg, D.A.; Addison, C.L.; Ehlert, J.E.; Burdick, M.D.; Strieter, R.M. CXC chemokines in angiogenesis. J. Leukoc. Biol. 2000, 68, 1–8. [Google Scholar] [PubMed]
- Ohmori, Y.; Schreiber, R.D.; Hamilton, T.A. Synergy between interferon-γ and tumor necrosis factor-α in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor κB. J. Biol. Chem. 1997, 272, 14899–14907. [Google Scholar] [CrossRef] [PubMed]
- Labirua, A.; Lundberg, I.E. Interstitial lung disease and idiopathic inflammatory myopathies: Progress and pitfalls. Curr. Opin. Rheumatol. 2010, 22, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Suber, T.L.; Casciola-Rosen, L.; Rosen, A. Mechanisms of disease: Autoantigens as clues to the pathogenesis of myositis. Nat. Clin. Pract. Rheumatol. 2008, 4, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Bonello, S. The cross-talk between NF-kappaB and HIF-1: Further evidence for a significant liaison. Biochem. J. 2008, 412, e17–e19. [Google Scholar] [PubMed]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Grundtman, C.; Tham, E.; Ulfgren, A.K.; Lundberg, I.E. Vascular endothelial growth factor is highly expressed in muscle tissue of patients with polymyositis and patients with dermatomyositis. Arthritis Rheumatol. 2008, 58, 3224–3238. [Google Scholar] [CrossRef]
- Guo, M.; Schimmel, P.; Yang, X.L. Functional expansion of human tRNA synthetases achieved by structural inventions. FEBS Lett. 2010, 584, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Cahuzac, B.; Berthonneau, E.; Birlirakis, N.; Guittet, E.; Mirande, M. A recurrent RNA-binding domain is appended to eukaryotic aminoacyl-tRNA synthetases. EMBO J. 2000, 19, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Yang, X.L.; Schimmel, P. New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell. Biol. 2010, 11, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.S.; Fox, P.L. Origin and evolution of glutamyl-prolyl tRNA synthetase WHEP domains reveal evolutionary relationships within Holozoa. PLoS One 2014, 9, e98493. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, Y.; Wakabayashi, T.; Semba, T.; Sonoda, J.; Kitoh, K.; Yoshimatsu, K. Establishment of a quantitative mouse dorsal air sac model and its application to evaluate a new angiogenesis inhibitor. Oncol. Res. 1999, 11, 319–329. [Google Scholar] [PubMed]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crepin, T.; Zhou, H.; Zhang, Y.K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Hothersall, J.; Willis, C.L.; Simpson, T.J. Resistance to and synthesis of the antibiotic mupirocin. Nat. Rev. Microbiol. 2010, 8, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Van de Vijver, P.; Ostrowski, T.; Sproat, B.; Goebels, J.; Rutgeerts, O.; van Aerschot, A.; Waer, M.; Herdewijn, P. Aminoacyl-tRNA synthetase inhibitors as potent and synergistic immunosuppressants. J. Med. Chem. 2008, 51, 3020–3029. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.G.; Lee, J.Y.; Kwon, N.H.; Fang, P.; Zhang, Q.; Wang, J.; Young, N.L.; Guo, M.; Cho, H.Y.; Mushtaq, A.U.; et al. Chemical inhibition of prometastatic lysyl-tRNA synthetase-laminin receptor interaction. Nat. Chem. Biol. 2014, 10, 29–34. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirando, A.C.; Francklyn, C.S.; Lounsbury, K.M. Regulation of Angiogenesis by Aminoacyl-tRNA Synthetases. Int. J. Mol. Sci. 2014, 15, 23725-23748. https://doi.org/10.3390/ijms151223725
Mirando AC, Francklyn CS, Lounsbury KM. Regulation of Angiogenesis by Aminoacyl-tRNA Synthetases. International Journal of Molecular Sciences. 2014; 15(12):23725-23748. https://doi.org/10.3390/ijms151223725
Chicago/Turabian StyleMirando, Adam C., Christopher S. Francklyn, and Karen M. Lounsbury. 2014. "Regulation of Angiogenesis by Aminoacyl-tRNA Synthetases" International Journal of Molecular Sciences 15, no. 12: 23725-23748. https://doi.org/10.3390/ijms151223725
APA StyleMirando, A. C., Francklyn, C. S., & Lounsbury, K. M. (2014). Regulation of Angiogenesis by Aminoacyl-tRNA Synthetases. International Journal of Molecular Sciences, 15(12), 23725-23748. https://doi.org/10.3390/ijms151223725