CREG Promotes the Proliferation of Human Umbilical Vein Endothelial Cells through the ERK/Cyclin E Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
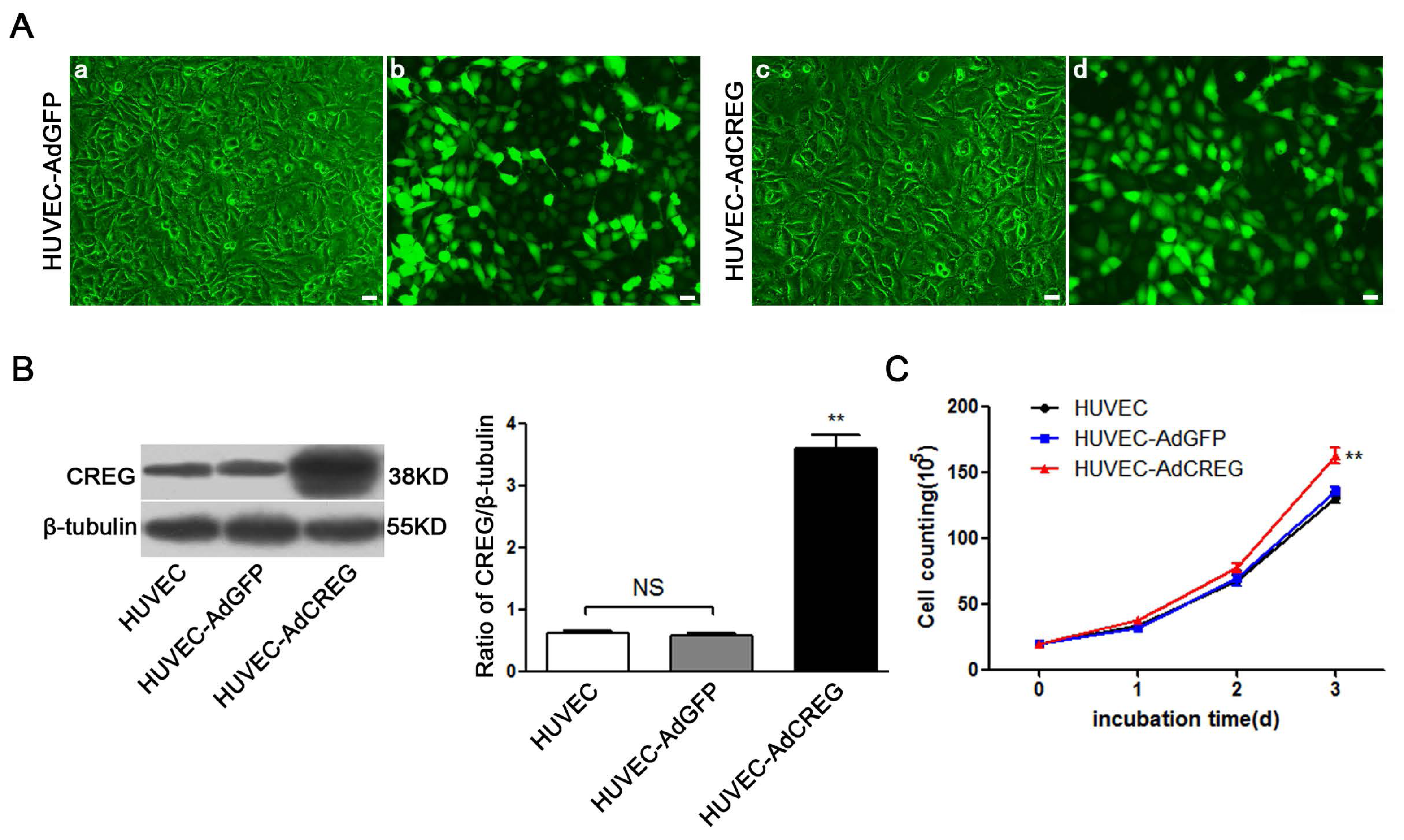
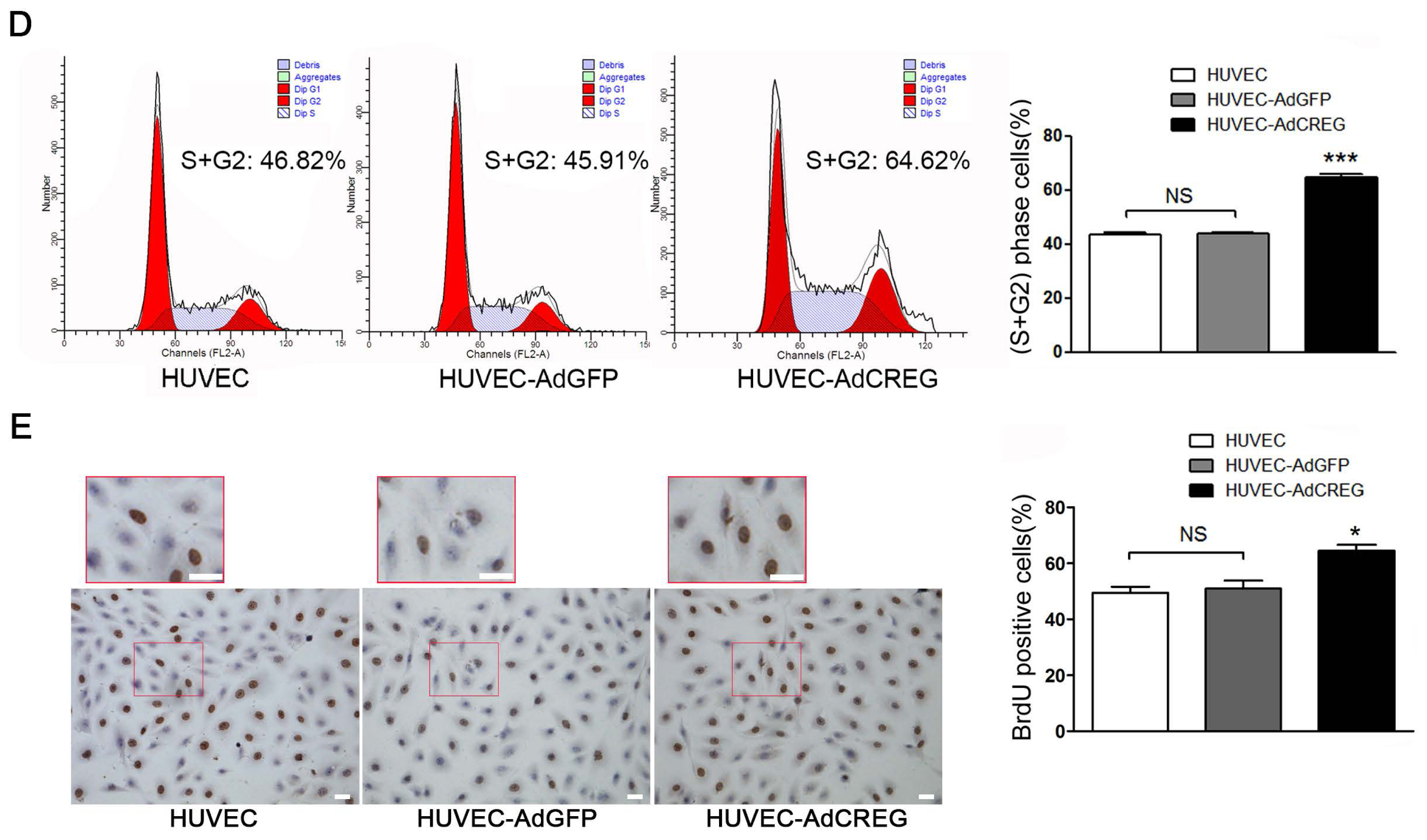
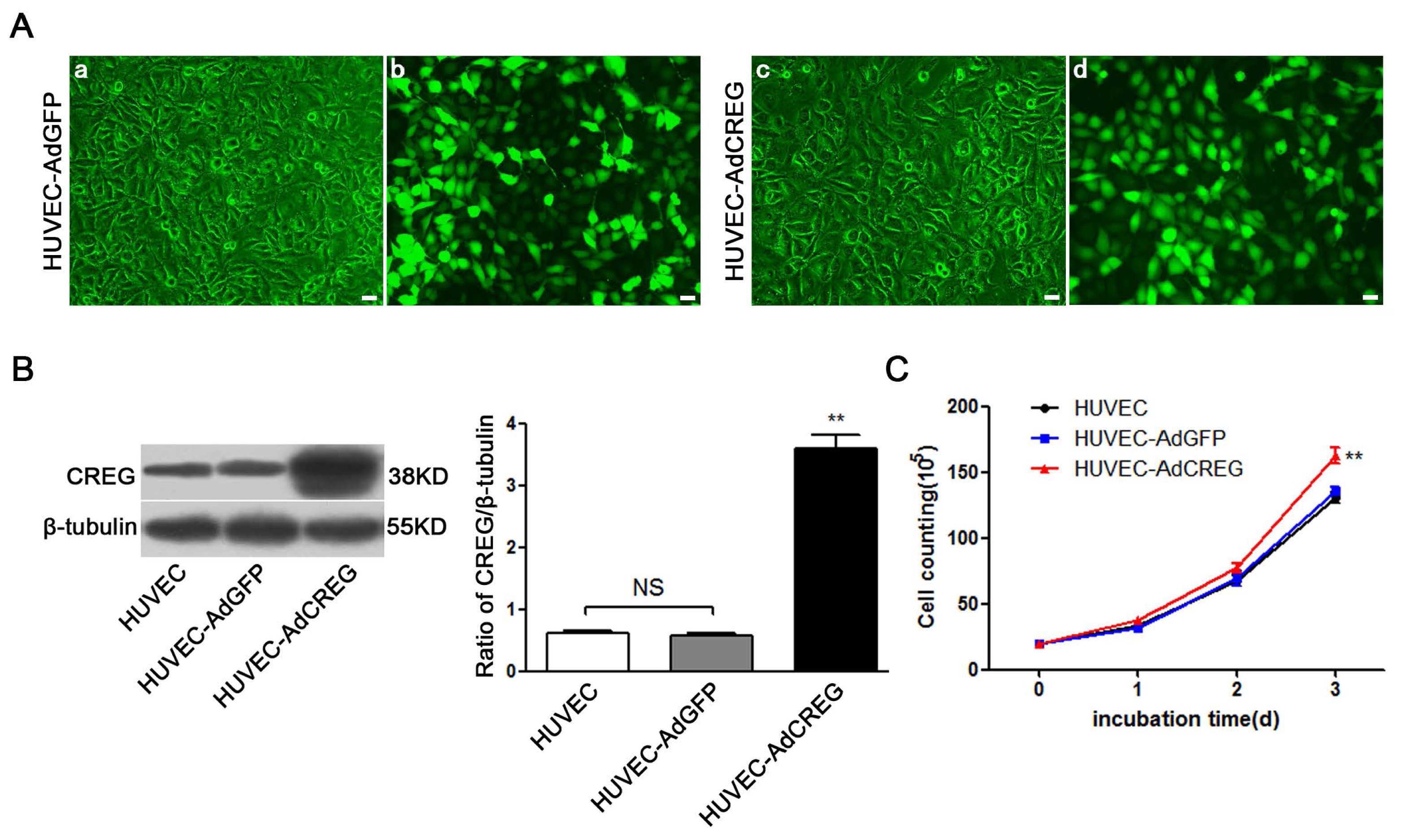
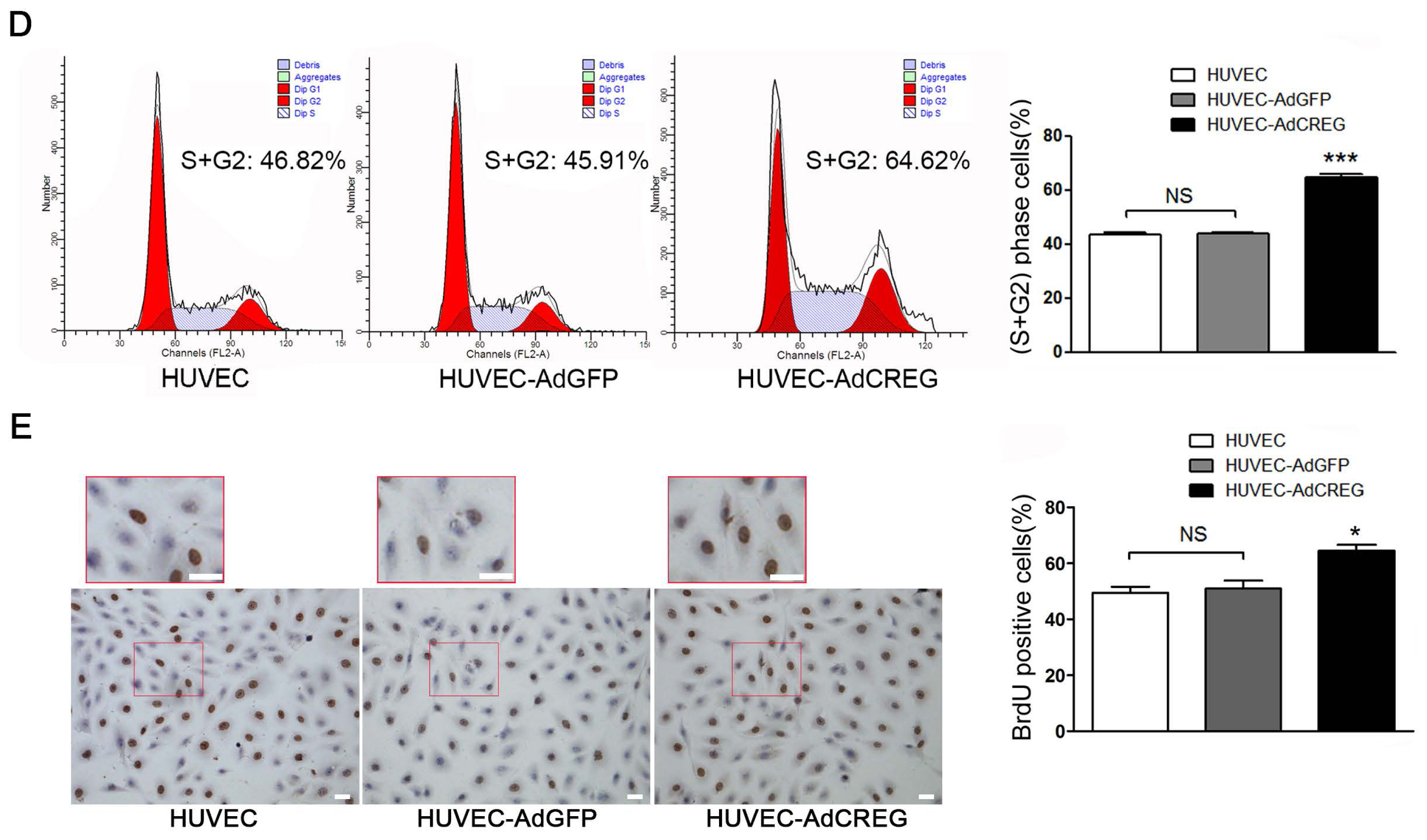
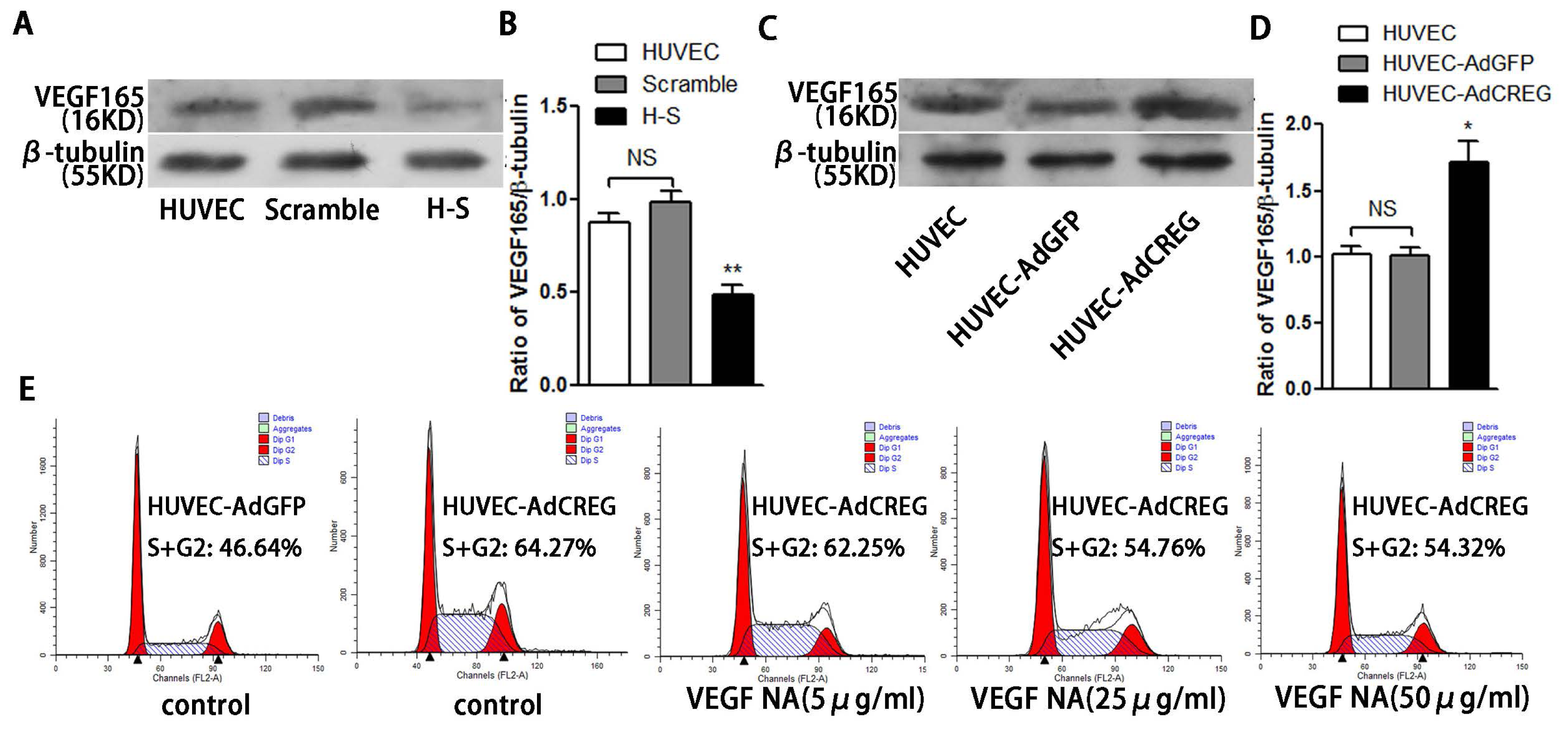
2.1. Effect of CREG Overexpression on Proliferation of HUVEC in Culture
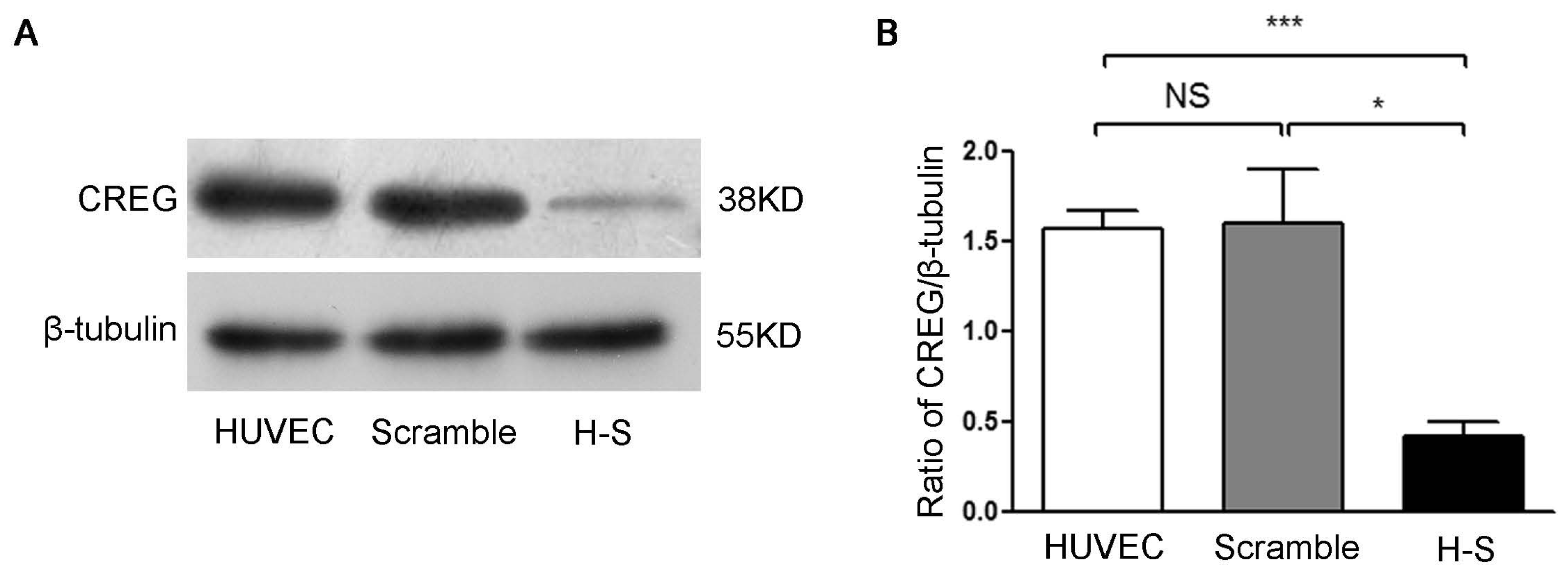
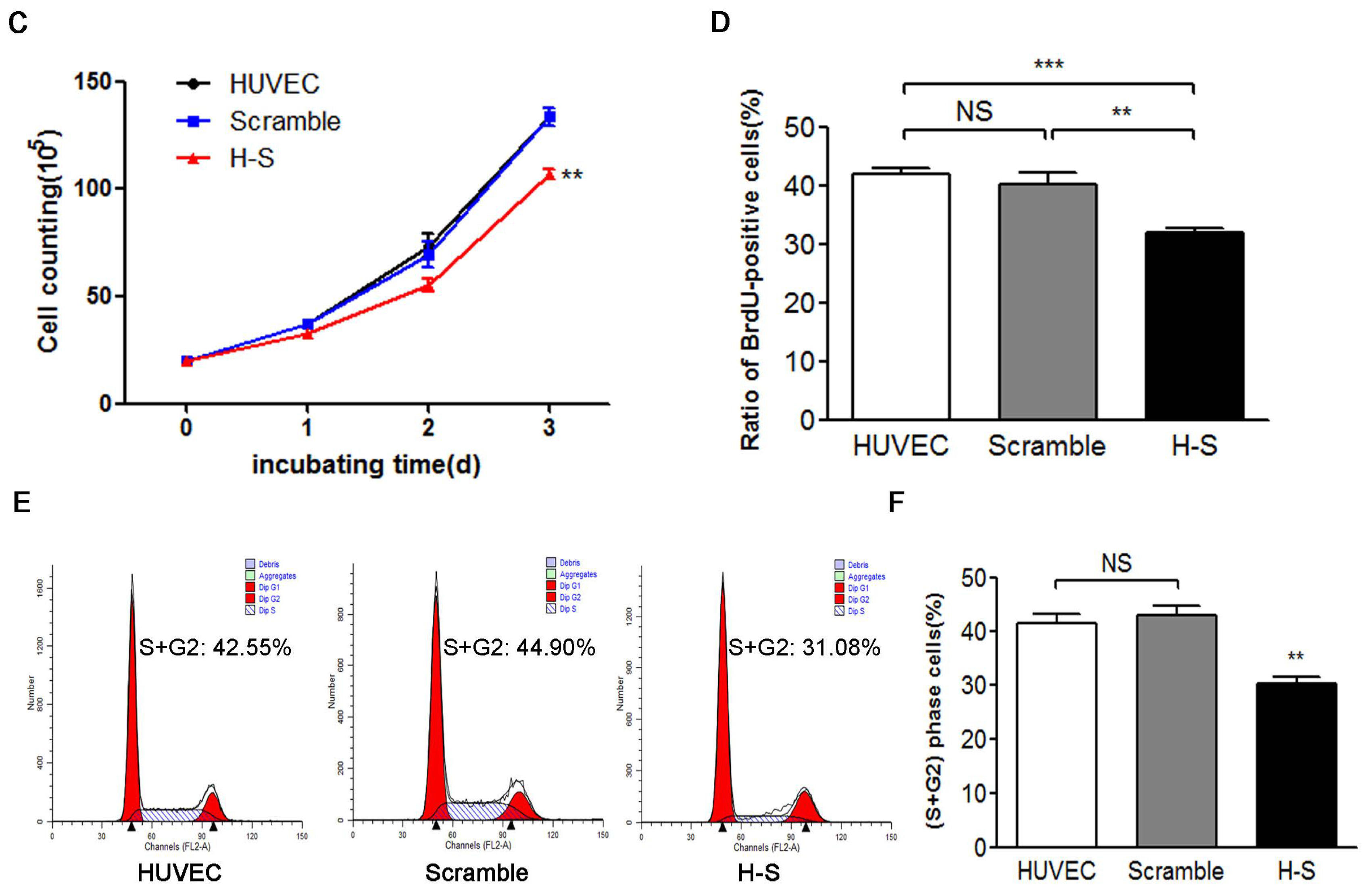
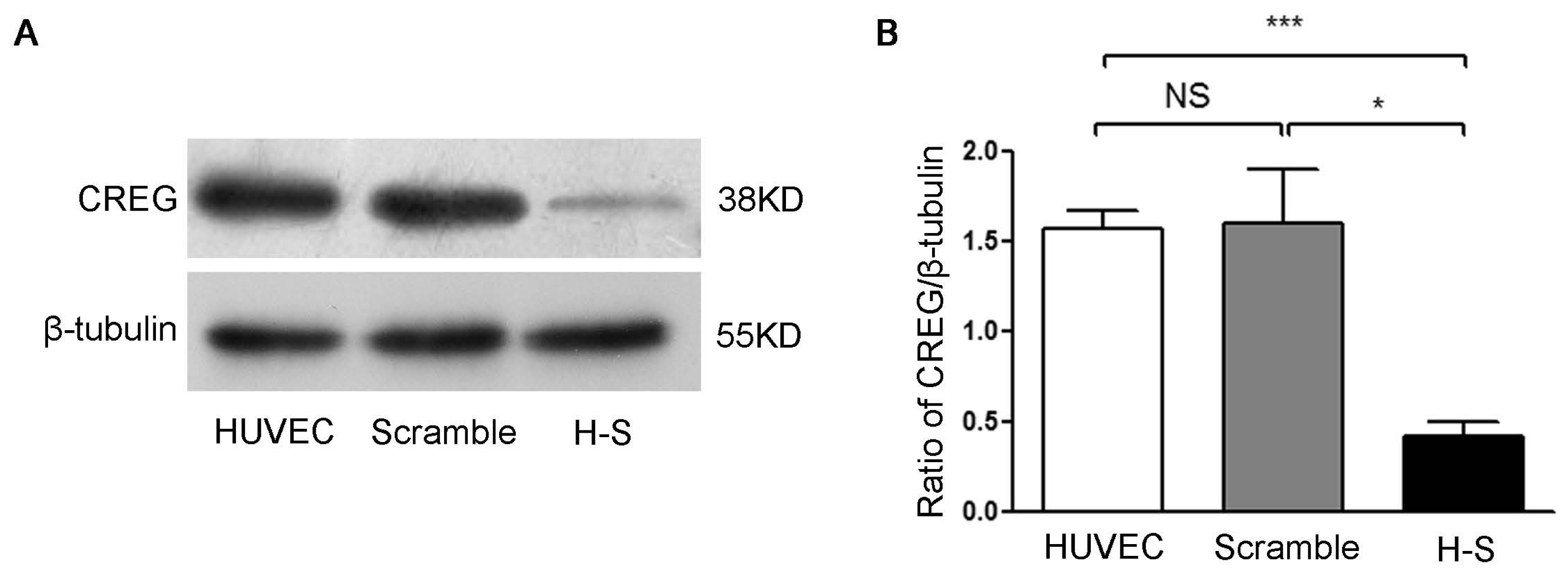
2.2. CREG Knocked down Exhibits an Inhibitory Effect on HUVEC Proliferation
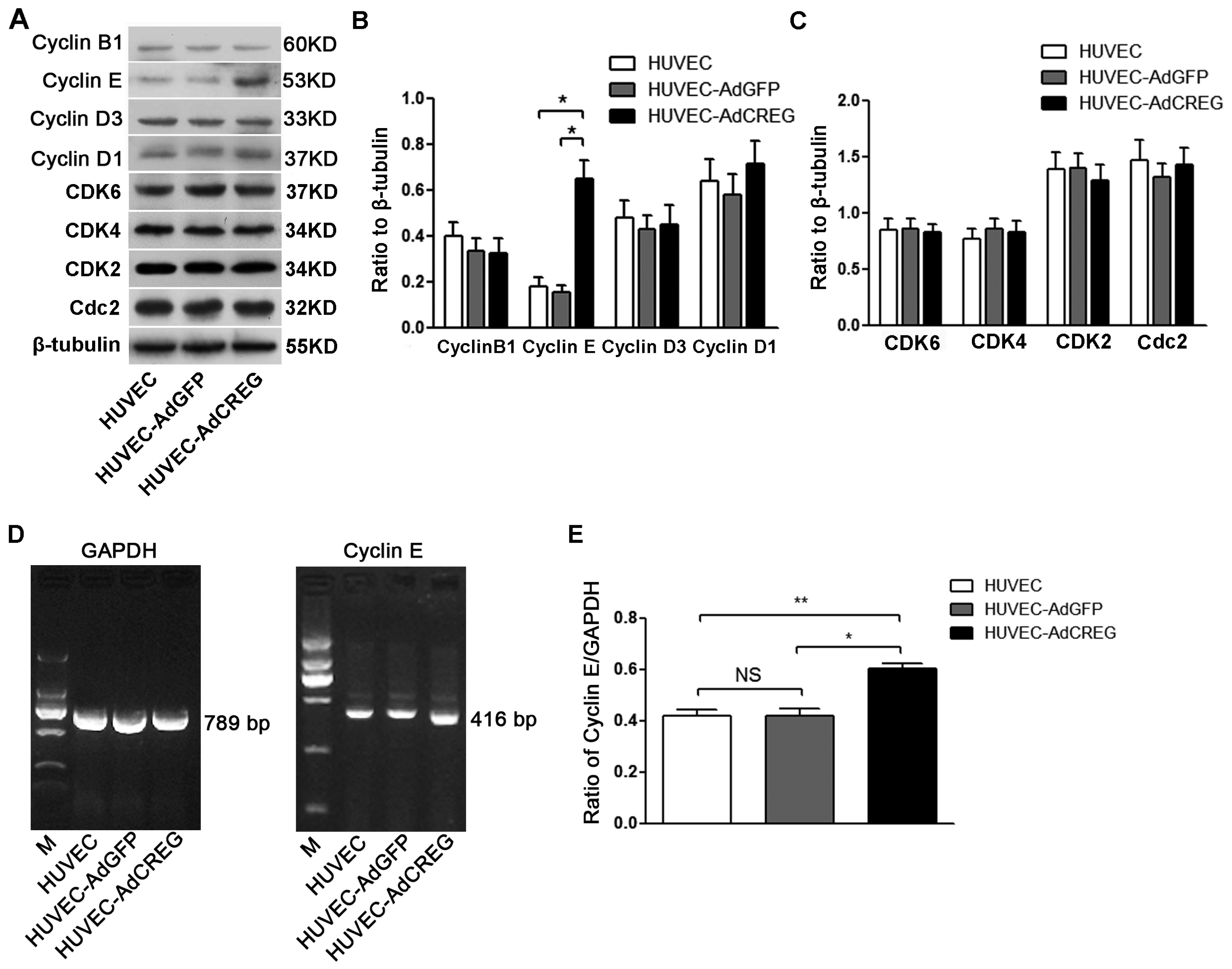
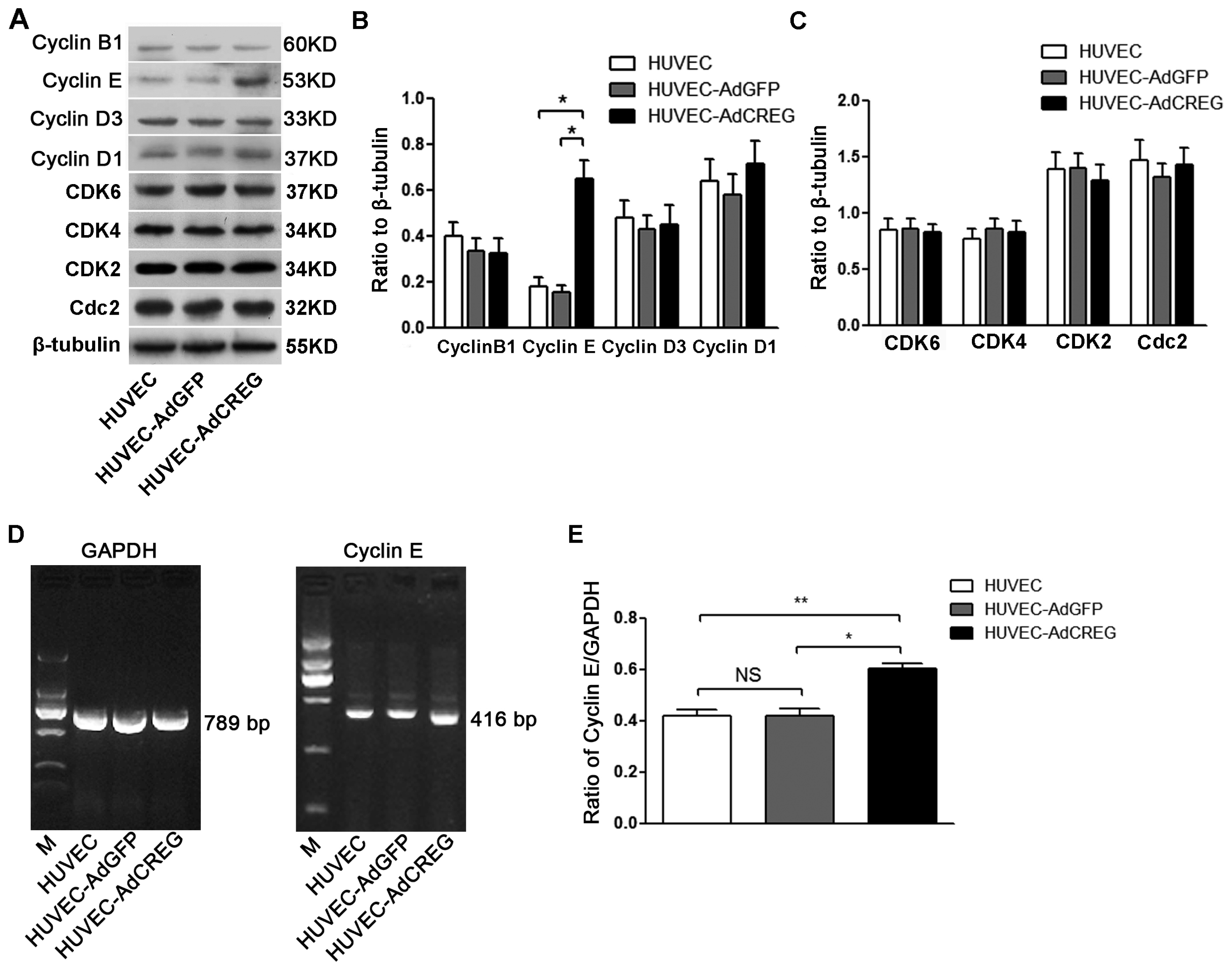
2.3. Overexpression of CREG Enhances the Expression of Cyclin E in HUVEC
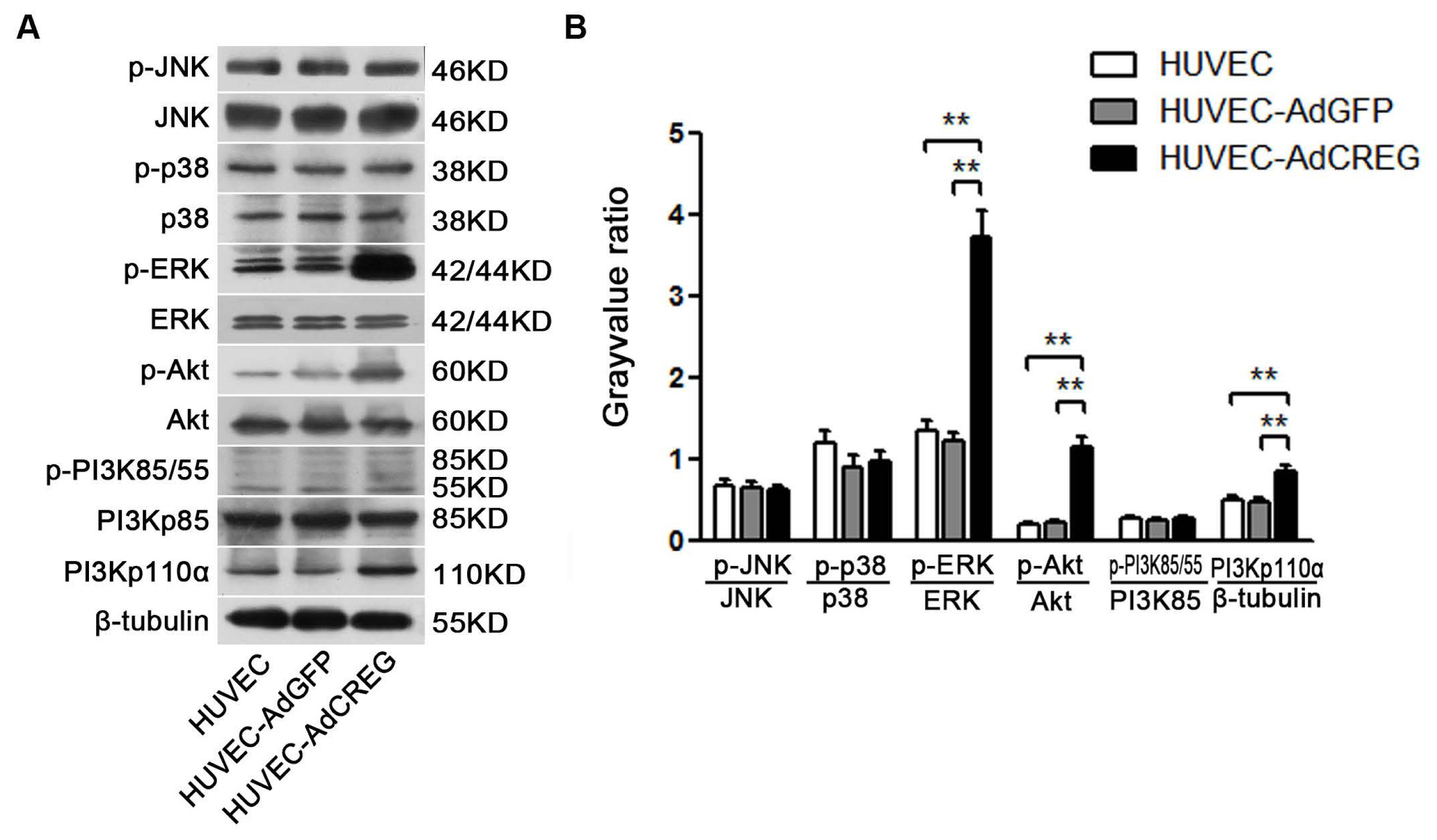
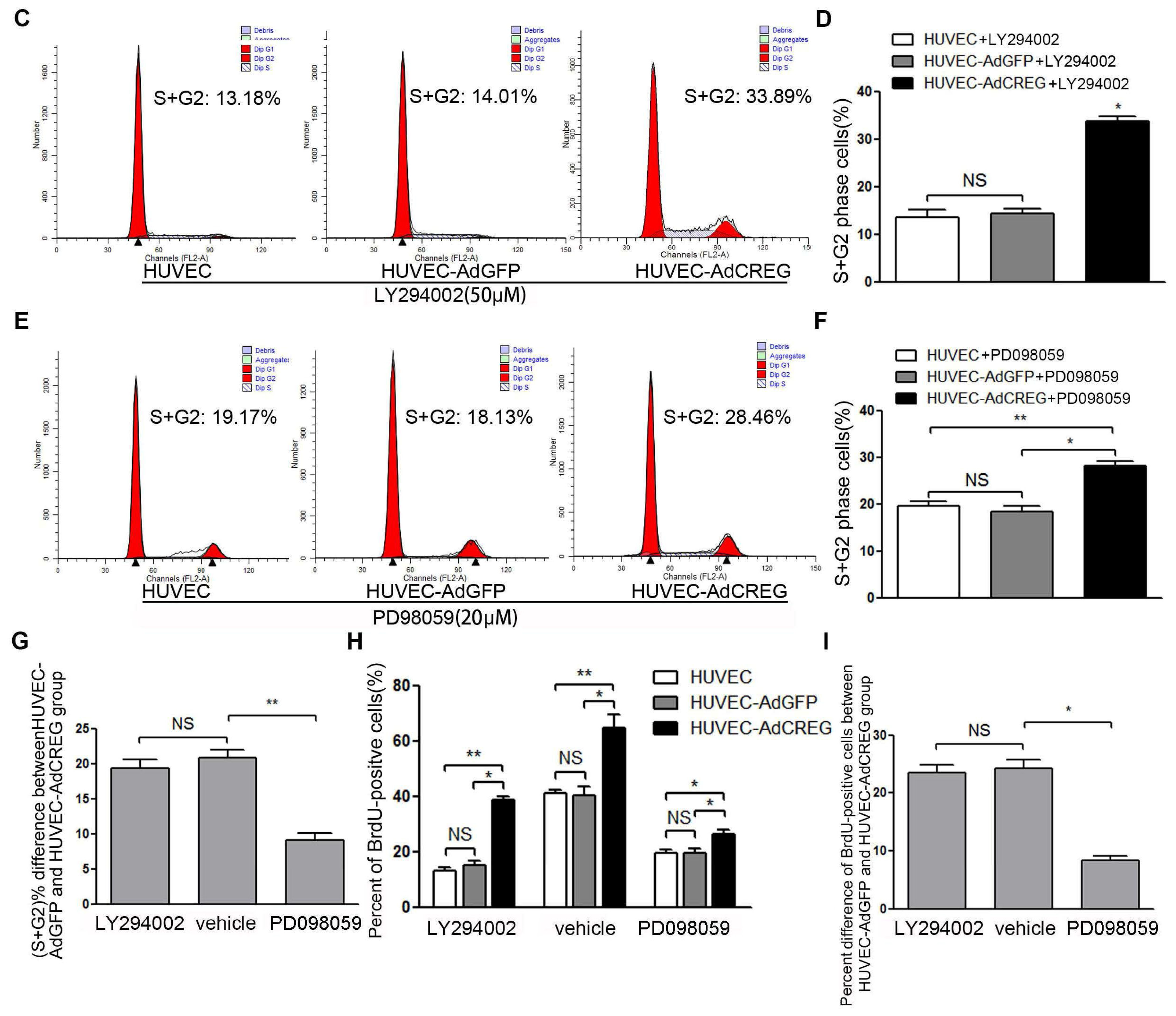
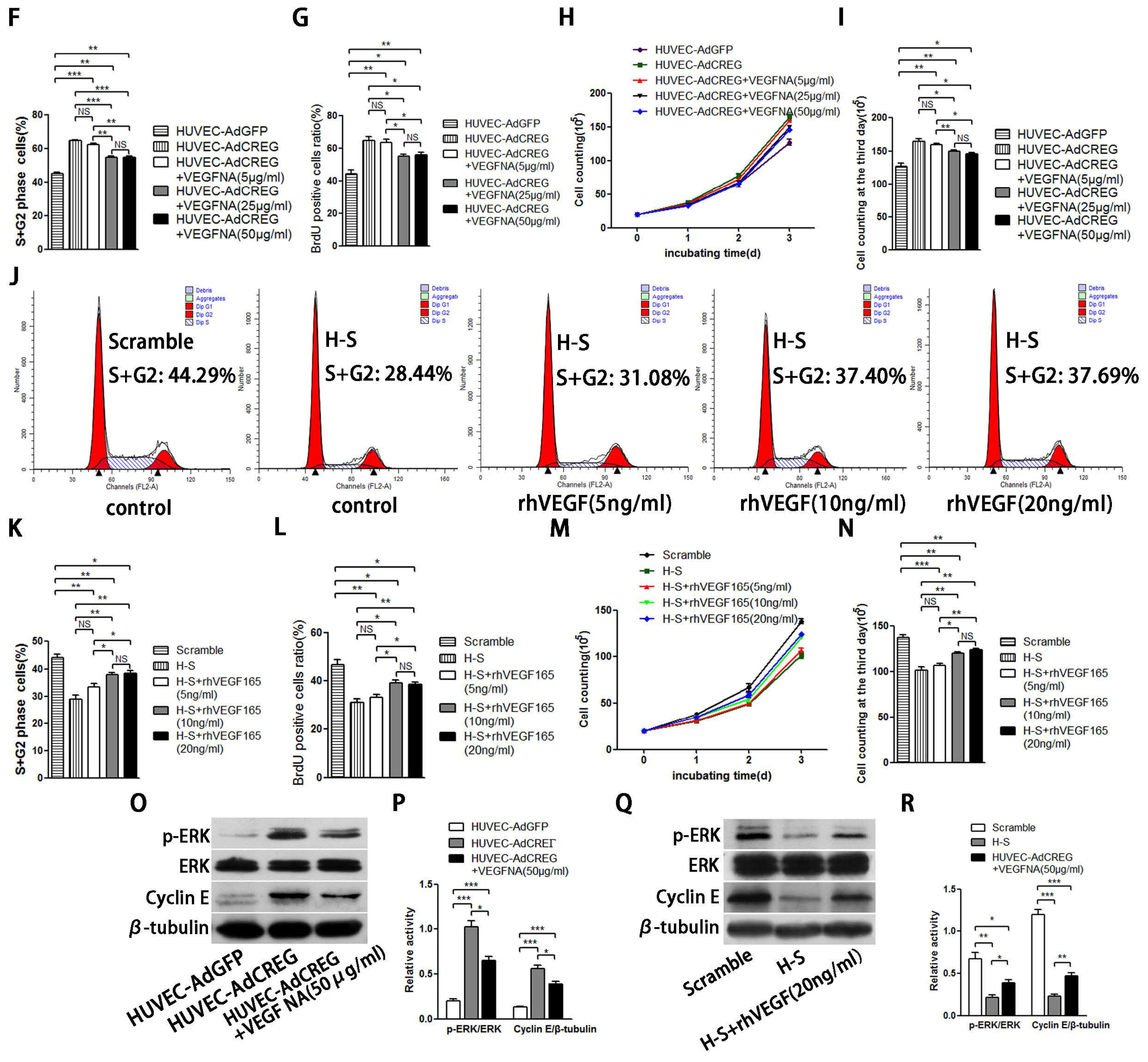
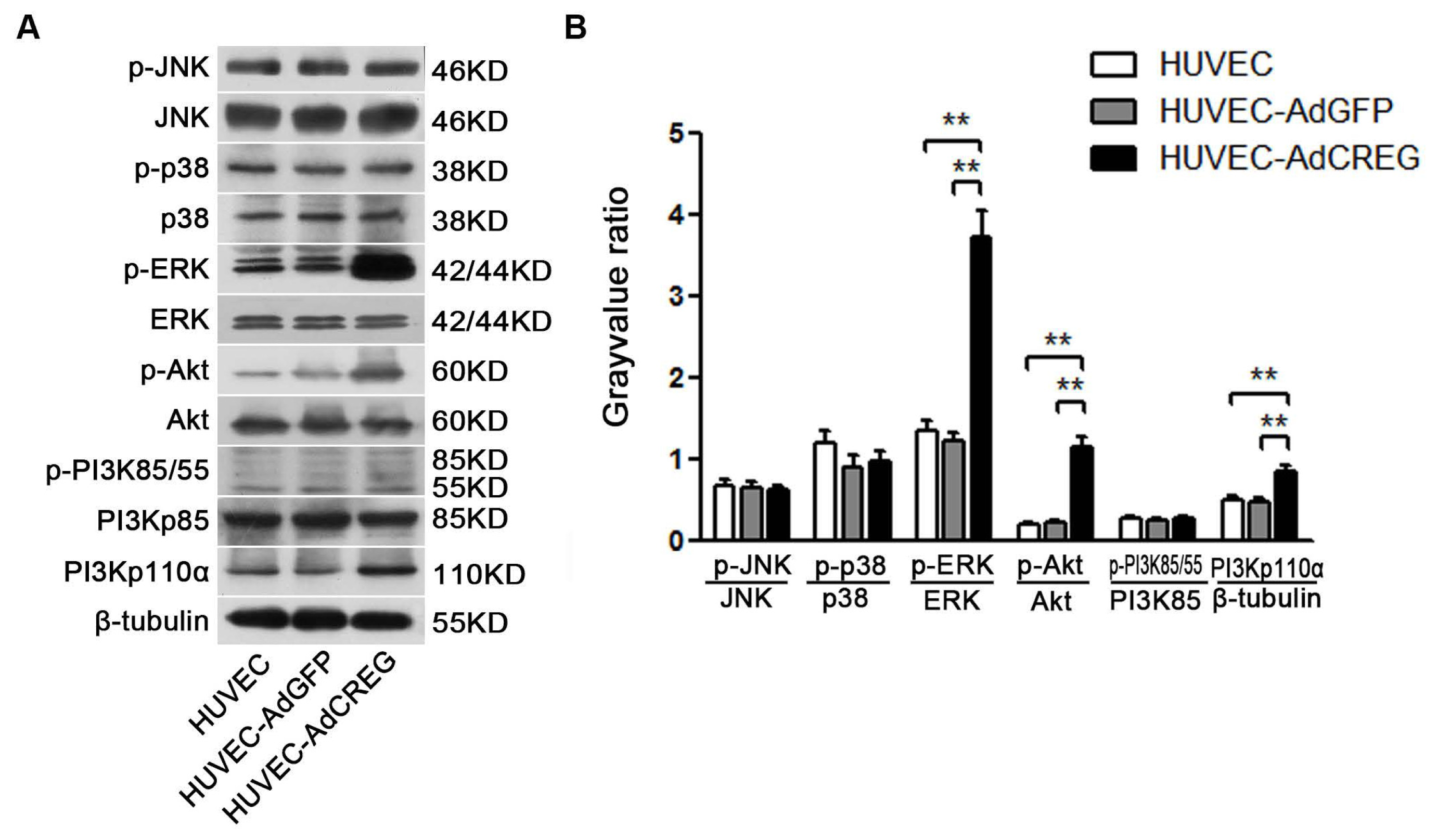
2.4. ERK Signaling Pathway Mediates the Proliferative Effect of CREG on HUVEC
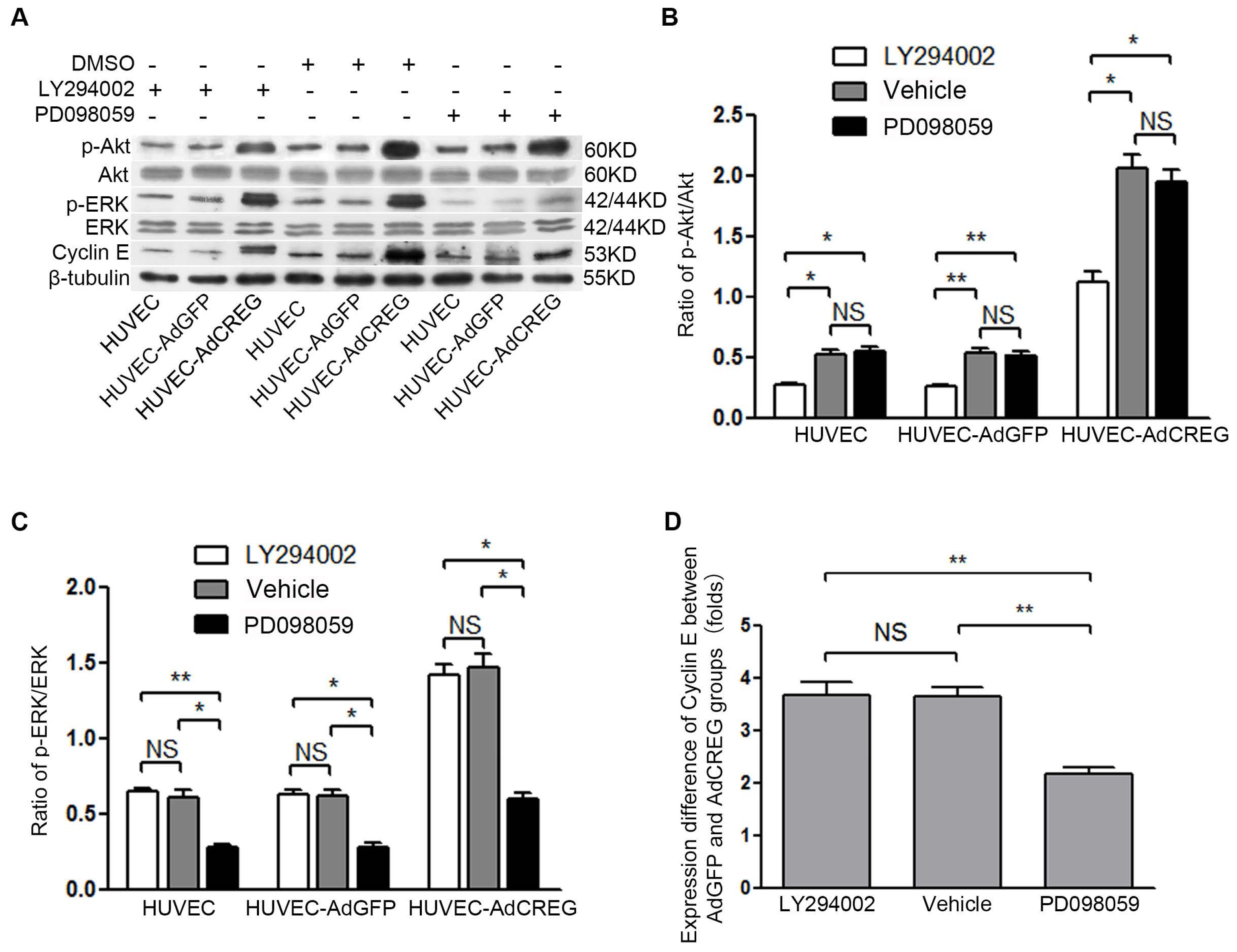
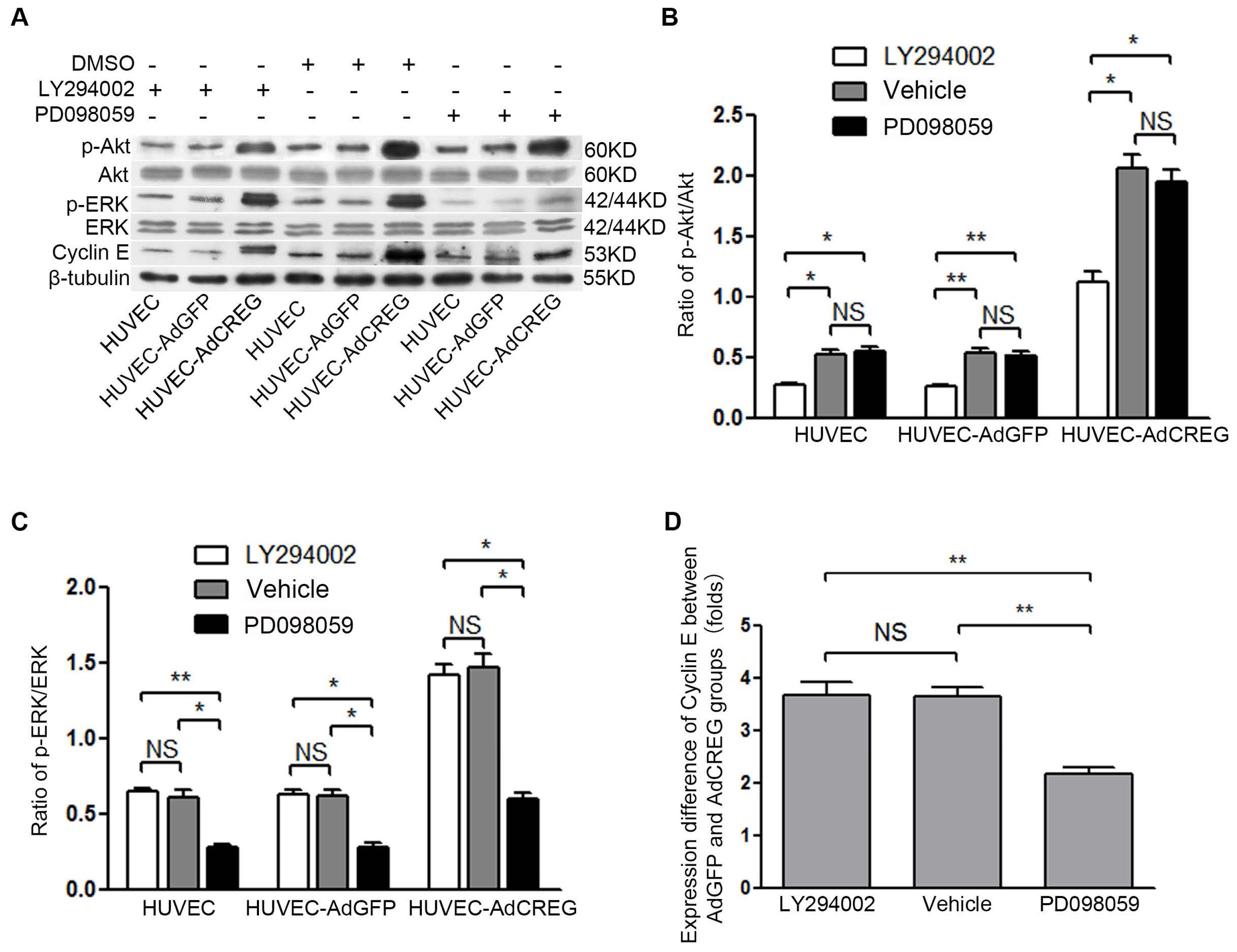
2.5. ERK Activation Mediates CREG-Induced Cyclin E Expression
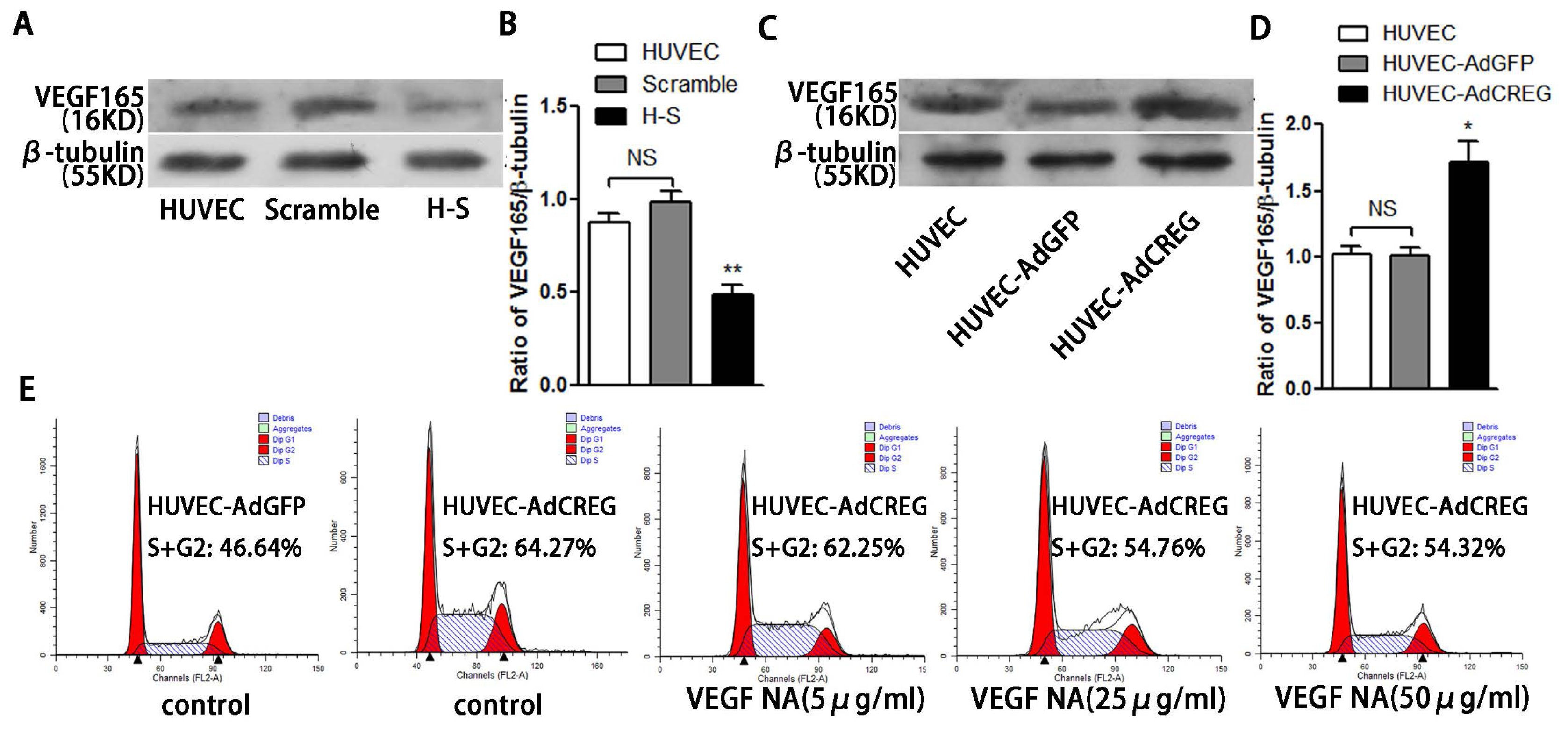
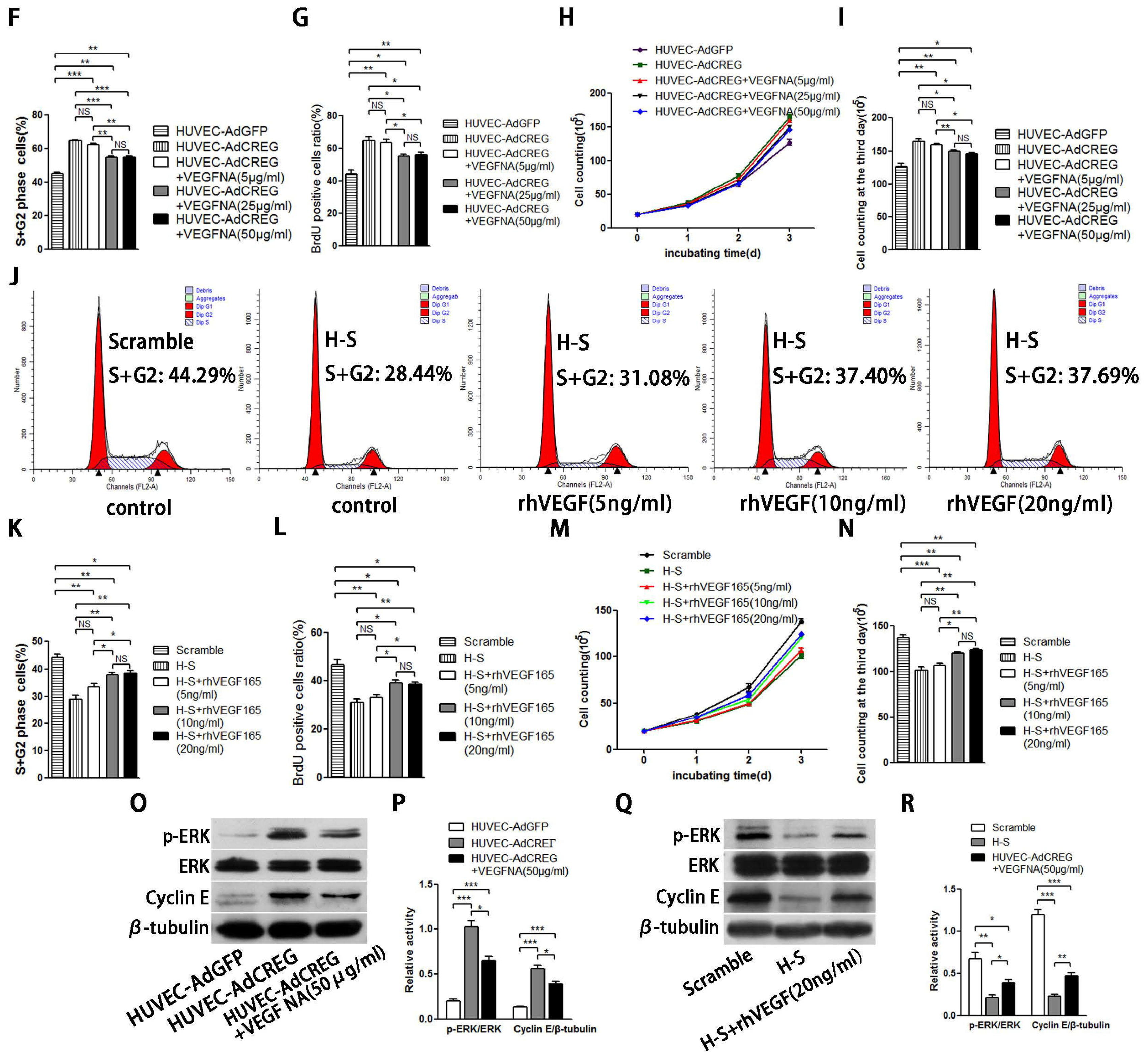
2.6. VEGF165 Partially Mediated the Role of CREG in Regulating HUVEC Proliferation through the Perk and Cyclin Pathway
3. Discussion
4. Material and Methods
4.1. Reagents
4.2. Culture of Primary HUVEC
4.4. Generation of CREG Knocked down Endothelial Cell Lines
4.5. FCM and 5-bromo-2′-deoxy-uridine (BrdU) Incorporation Assays
4.6. Cell Counting
4.7. Western Blot Analysis
4.8. RT-PCR
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Triggle, C.R.; Samuel, S.M.; Ravishankar, S.; Marei, I.; Arunachalam, G.; Ding, H. The endothelium: Influencing vascular smooth muscle in many ways. Can. J. Physiol. Pharmacol 2012, 90, 713–738. [Google Scholar]
- Otsuka, F.; Finn, A.V.; Yazdani, S.K.; Nakano, M.; Kolodgie, F.D.; Virmani, R. The importance of the endothelium in atherothrombosis and coronary stenting. Nat. Rev. Cardiol 2012, 9, 439–453. [Google Scholar]
- Veal, E.; Eisenstein, M.; Tseng, Z.H.; Gill, G. A cellular repressor of E1A-stimulated genes that inhibits activation by E2F. Mol. Cell. Biol 1998, 18, 5032–5041. [Google Scholar]
- Veal, E.; Groisman, R.; Eisenstein, M.; Gill, G. The secreted glycoprotein CREG enhances differentiation of NTERA-2 human embryonal carcinoma cells. Oncogene 2000, 19, 2120–2128. [Google Scholar]
- Kunita, R.; Otomo, A.; Ikeda, J.E. Identification and characterization of novel members of the CREG family, putative secreted glycoproteins expressed specifically in brain. Genomics 2002, 80, 456–460. [Google Scholar]
- Schahs, P.; Weidinger, P.; Probst, O.C.; Svoboda, B.; Stadlmann, J.; Beug, H.; Waerner, T.; Mach, L. Cellular repressor of E1A-stimulated genes is a bona fide lysosomal protein which undergoes proteolytic maturation during its biosynthesis. Exp. Cell Res 2008, 314, 3036–3047. [Google Scholar]
- Di Bacco, A.; Gill, G. The secreted glycoprotein CREG inhibits cell growth dependent on the mannose-6-phosphate/insulin-like growth factor II receptor. Oncogene 2003, 22, 5436–5445. [Google Scholar]
- Xu, L.; Liu, J.M.; Chen, L.Y. CREG, a new regulator of ERK1/2 in cardiac hypertrophy. J. Hypertens 2004, 22, 1579–1587. [Google Scholar]
- Wang, N.; Han, Y.; Tao, J.; Huang, M.; You, Y.; Zhang, H.; Liu, S.; Zhang, X.; Yan, C. Overexpression of CREG attenuates atherosclerotic endothelium apoptosis via VEGF/PI3K/AKT pathway. Atherosclerosis 2011, 218, 543–551. [Google Scholar]
- Zhang, H.; Han, Y.; Tao, J.; Liu, S.; Yan, C.; Li, S. Cellular repressor of E1A-stimulated genes regulates vascular endothelial cell migration by the ILK/AKT/mTOR/VEGF(165) signaling pathway. Exp. Cell Res 2011, 317, 2904–2913. [Google Scholar]
- Kisielewska, J.; Philipova, R.; Huang, J.Y.; Whitaker, M. MAP kinase dependent cyclinE/cdk2 activity promotes DNA replication in early sea urchin embryos. Dev. Biol 2009, 334, 383–394. [Google Scholar]
- Wang, Y.; Shenouda, S.; Baranwal, S.; Rathinam, R.; Jain, P.; Bao, L.; Hazari, S.; Dash, S.; Alahari, S.K. Integrin subunits alpha5 and alpha6 regulate cell cycle by modulating the chk1 and Rb/E2F pathways to affect breast cancer metastasis. Mol. Cancer 2011, 10, 84. [Google Scholar]
- Morello, F.; Perino, A.; Hirsch, E. Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc. Res 2009, 82, 261–271. [Google Scholar]
- Ammit, A.J.; Panettieri, R.A., Jr. Invited review: The circle of life: Cell cycle regulation in airway smooth muscle. J. Appl. Physiol. 2001, 91, 1431–1437. [Google Scholar]
- Zhang, F.; Dong, L.; Ge, J. Effect of statins pretreatment on periprocedural myocardial infarction in patients undergoing percutaneous coronary intervention: A meta-analysis. Ann. Med 2010, 42, 171–177. [Google Scholar]
- Ishii, Y.; Langberg, J.; Rosborough, K.; Mikawa, T. Endothelial cell lineages of the heart. Cell Tissue Res 2009, 335, 67–73. [Google Scholar]
- Mood, G.R.; Bavry, A.A.; Roukoz, H.; Bhatt, D.L. Meta-analysis of the role of statin therapy in reducing myocardial infarction following elective percutaneous coronary intervention. Am. J. Cardiol 2007, 100, 919–923. [Google Scholar]
- Han, Y.; Deng, J.; Guo, L.; Yan, C.; Liang, M.; Kang, J.; Liu, H.; Graham, A.M.; Li, S. CREG promotes a mature smooth muscle cell phenotype and reduces neointimal formation in balloon-injured rat carotid artery. Cardiovasc. Res 2008, 78, 597–604. [Google Scholar]
- Han, Y.; Wu, G.; Deng, J.; Tao, J.; Guo, L.; Tian, X.; Kang, J.; Zhang, X.; Yan, C. Cellular repressor of E1A-stimulated genes inhibits human vascular smooth muscle cell apoptosis via blocking P38/JNK MAP kinase activation. J. Mol. Cell. Cardiol 2010, 48, 1225–1235. [Google Scholar]
- Peng, C.F.; Han, Y.L.; Deng, J.; Yan, C.H.; Kang, J.; Luan, B.; Li, J. Overexpression of cellular repressor of E1A-stimulated genes inhibits TNF-alpha-induced apoptosis via NF-kappaB in mesenchymal stem cells. Biochem. Biophys. Res. Commun 2011, 406, 601–607. [Google Scholar]
- Xu, L.; Wang, F.; Liu, H.; Xu, X.F.; Mo, W.H.; Xia, Y.J.; Wan, R.; Wang, X.P.; Guo, C.Y. Increased expression of cellular repressor of E1A-stimulated gene (CREG) in gastric cancer patients: A mechanism of proliferation and metastasis in cancer. Dig. Dis. Sci 2011, 56, 1645–1655. [Google Scholar]
- Won, K.A.; Reed, S.I. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J 1996, 15, 4182–4193. [Google Scholar]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev 1999, 13, 1501–1512. [Google Scholar]
- Carcagno, A.L.; Marazita, M.C.; Ogara, M.F.; Ceruti, J.M.; Sonzogni, S.V.; Scassa, M.E.; Giono, L.E.; Canepa, E.T. E2F1-mediated upregulation of p19INK4d determines its periodic expression during cell cycle and regulates cellular proliferation. PLoS One 2011, 6, e21938. [Google Scholar]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004, 18, 2699–2711. [Google Scholar]
- Myatt, S.S.; Lam, E.W. Promiscuous and lineage-specific roles of cell cycle regulators in haematopoiesis. Cell Div 2007, 2, 6. [Google Scholar]
- Taylor, S.M.; Nevis, K.R.; Park, H.L.; Rogers, G.C.; Rogers, S.L.; Cook, J.G.; Bautch, V.L. Angiogenic factor signaling regulates centrosome duplication in endothelial cells of developing blood vessels. Blood 2010, 116, 3108–3117. [Google Scholar]
- Deng, J.; Han, Y.; Sun, M.; Tao, J.; Yan, C.; Kang, J.; Li, S. Nanoporous CREG-eluting stent attenuates in-stent neointimal formation in porcine coronary arteries. PLoS One 2013, 8, e60735. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tao, J.; Yan, C.; Tian, X.; Liu, S.; Li, Y.; Zhang, J.; Sun, M.; Ma, X.; Han, Y. CREG Promotes the Proliferation of Human Umbilical Vein Endothelial Cells through the ERK/Cyclin E Signaling Pathway. Int. J. Mol. Sci. 2013, 14, 18437-18456. https://doi.org/10.3390/ijms140918437
Tao J, Yan C, Tian X, Liu S, Li Y, Zhang J, Sun M, Ma X, Han Y. CREG Promotes the Proliferation of Human Umbilical Vein Endothelial Cells through the ERK/Cyclin E Signaling Pathway. International Journal of Molecular Sciences. 2013; 14(9):18437-18456. https://doi.org/10.3390/ijms140918437
Chicago/Turabian StyleTao, Jie, Chenghui Yan, Xiaoxiang Tian, Shaowei Liu, Yang Li, Jian Zhang, Mingyu Sun, Xinliang Ma, and Yaling Han. 2013. "CREG Promotes the Proliferation of Human Umbilical Vein Endothelial Cells through the ERK/Cyclin E Signaling Pathway" International Journal of Molecular Sciences 14, no. 9: 18437-18456. https://doi.org/10.3390/ijms140918437
APA StyleTao, J., Yan, C., Tian, X., Liu, S., Li, Y., Zhang, J., Sun, M., Ma, X., & Han, Y. (2013). CREG Promotes the Proliferation of Human Umbilical Vein Endothelial Cells through the ERK/Cyclin E Signaling Pathway. International Journal of Molecular Sciences, 14(9), 18437-18456. https://doi.org/10.3390/ijms140918437