KRAS and MAPK1 Gene Amplification in Type II Ovarian Carcinomas

Abstract
:1. Introduction
2. Results
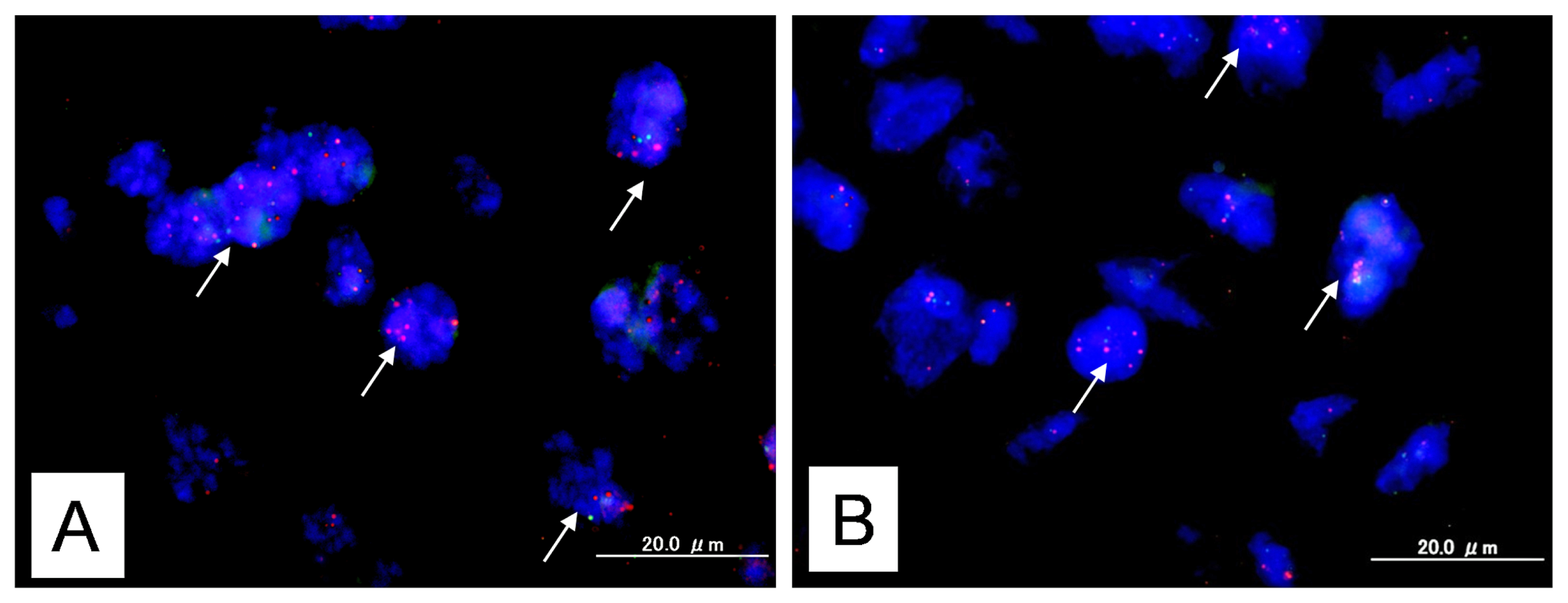
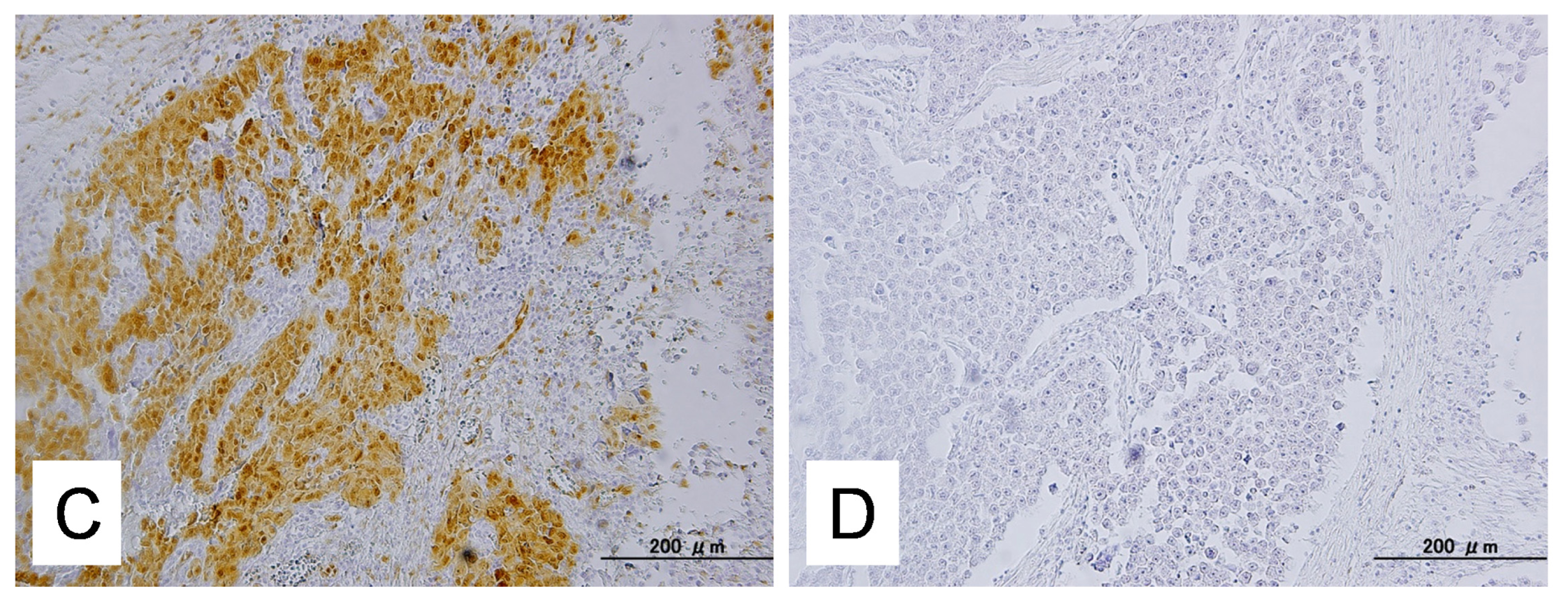
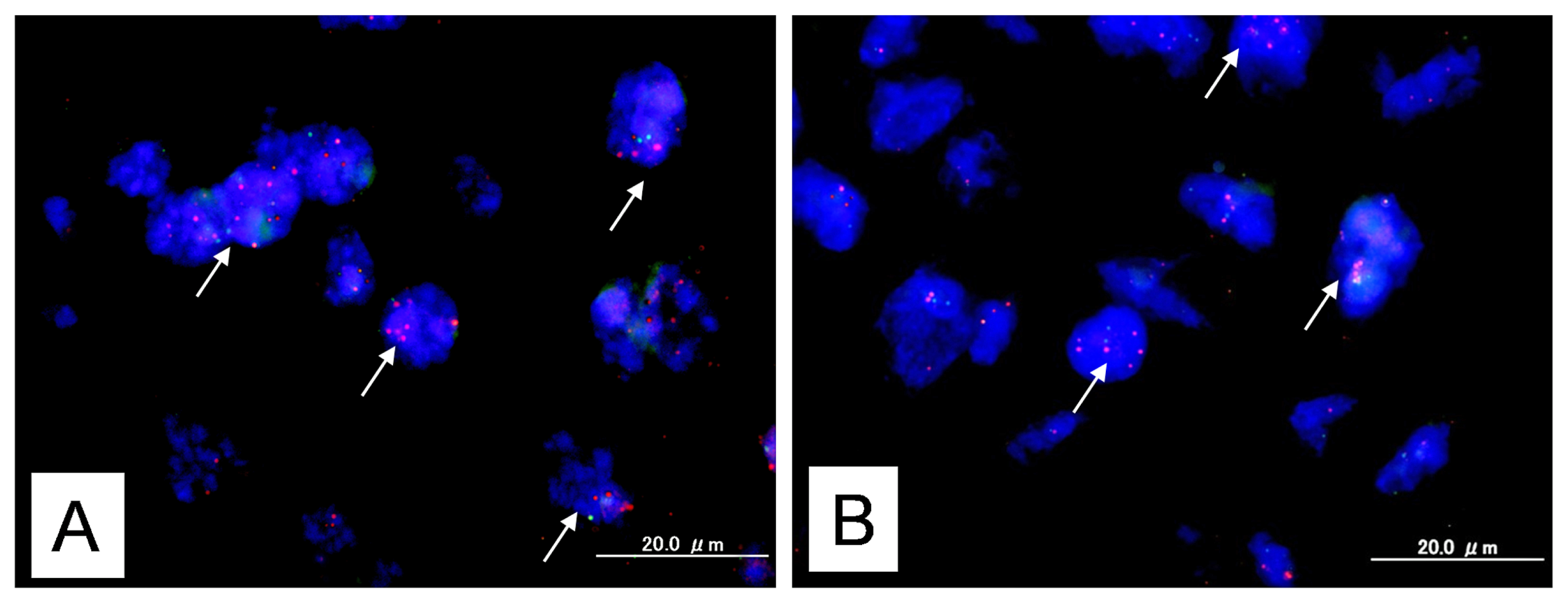
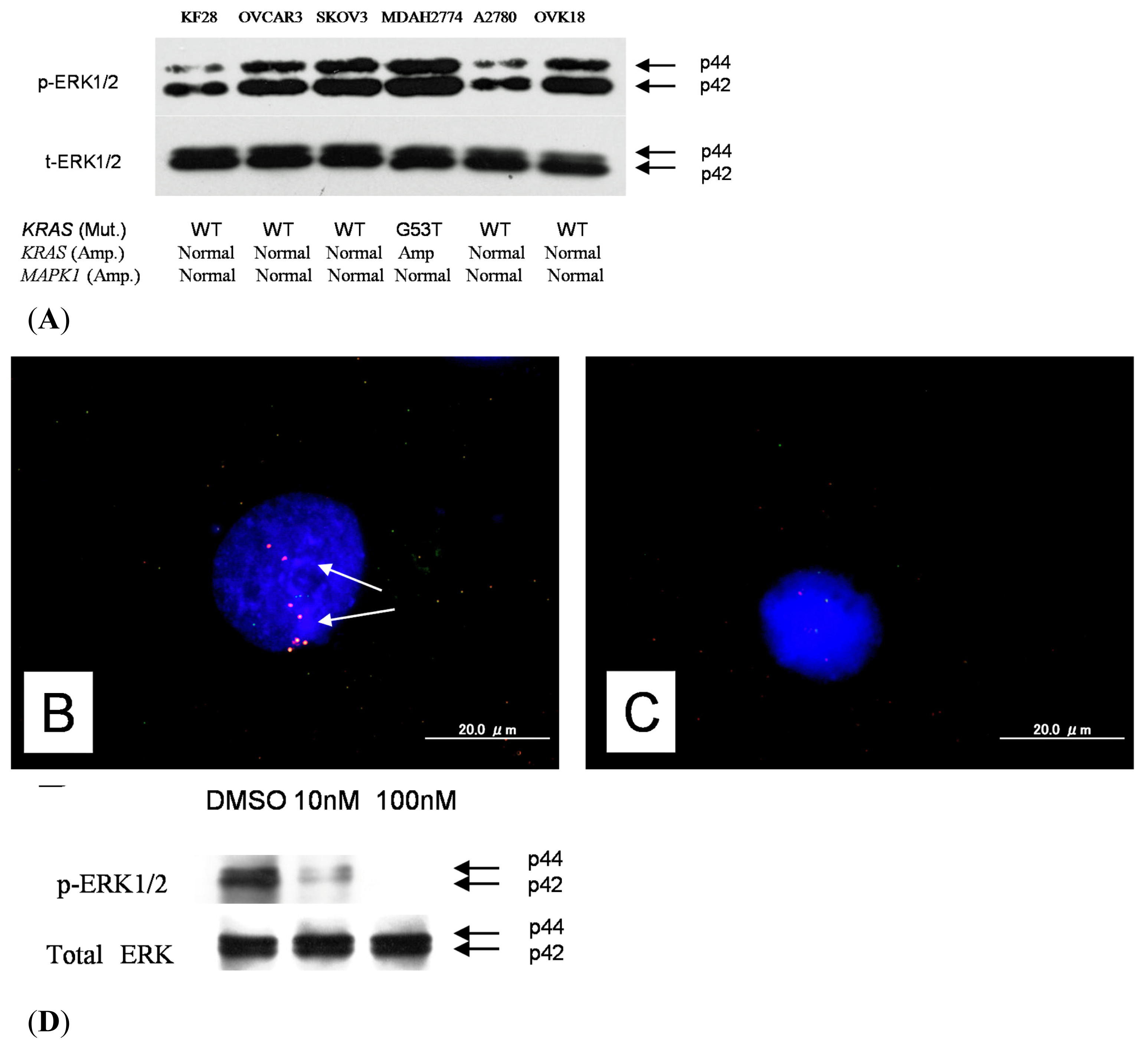
2.1. Identification of KRAS or MAPK1 Amplification and Correlation with p-ERK Immunostaining
2.2. Relationship between KRAS or MAPK1 Amplification or p-ERK1/2 Expression and Clinicopathological Factors
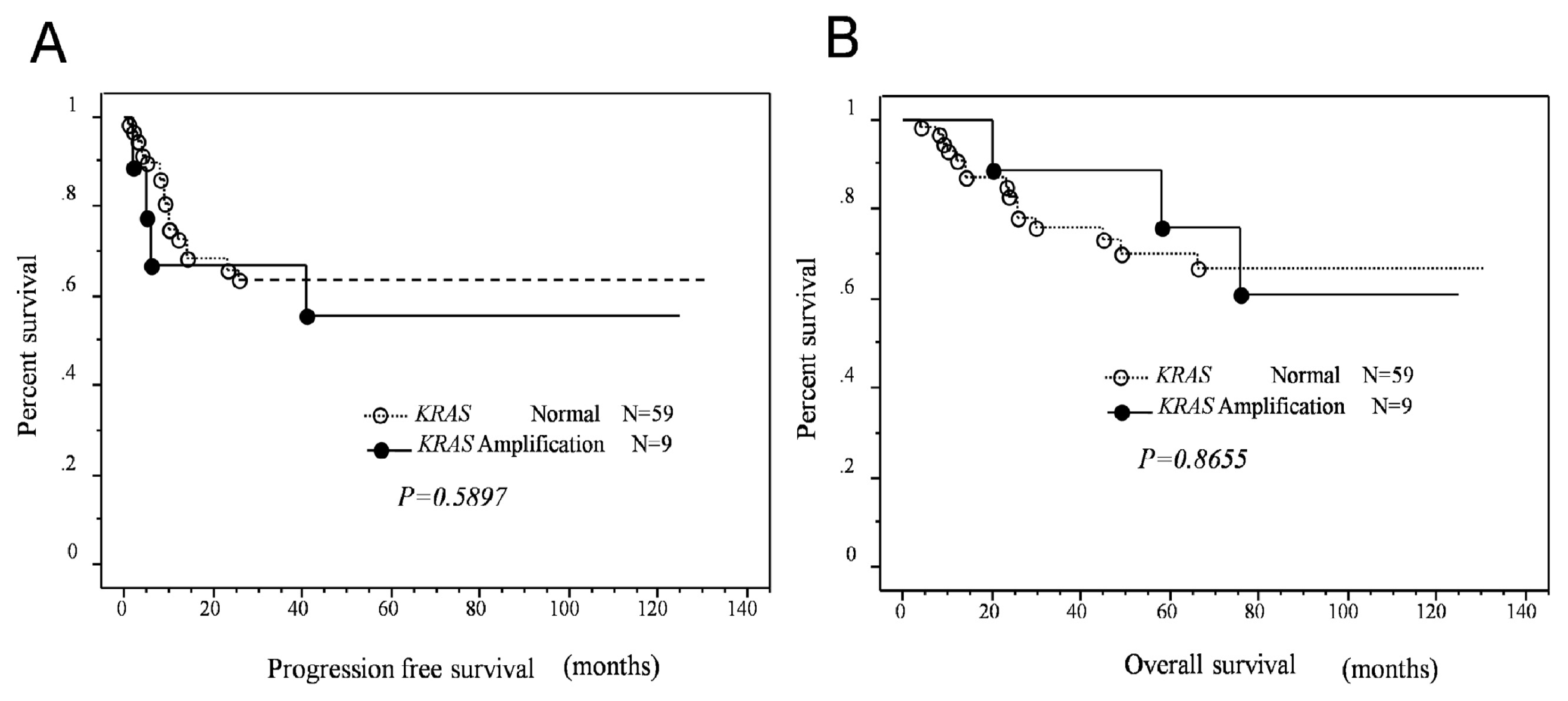
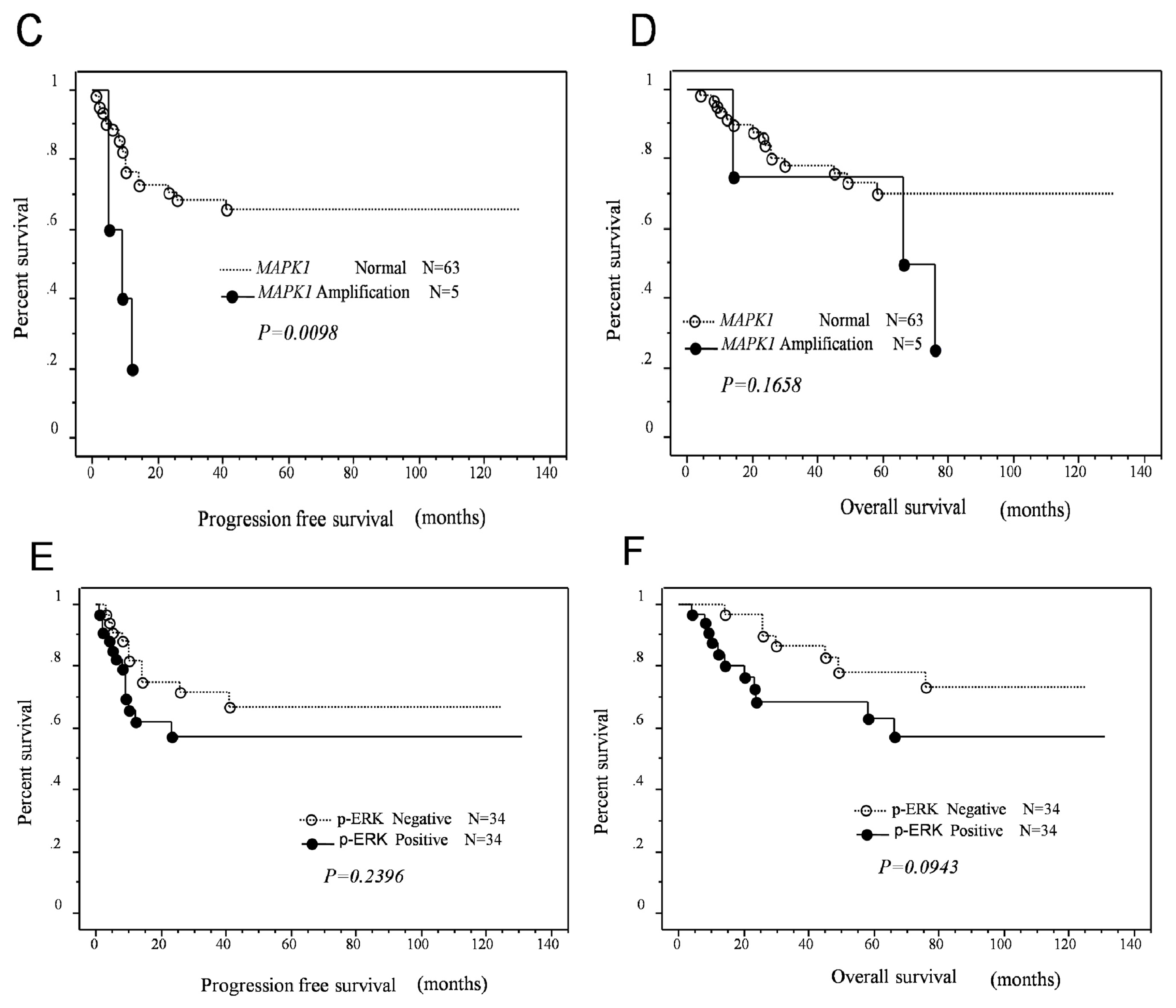
2.3. Effects of KRAS or MAPK1 Amplification and p-ERK1/2 Status on the Prognosis of Type II Ovarian Carcinomas
2.4. Identification of KRAS Mutations
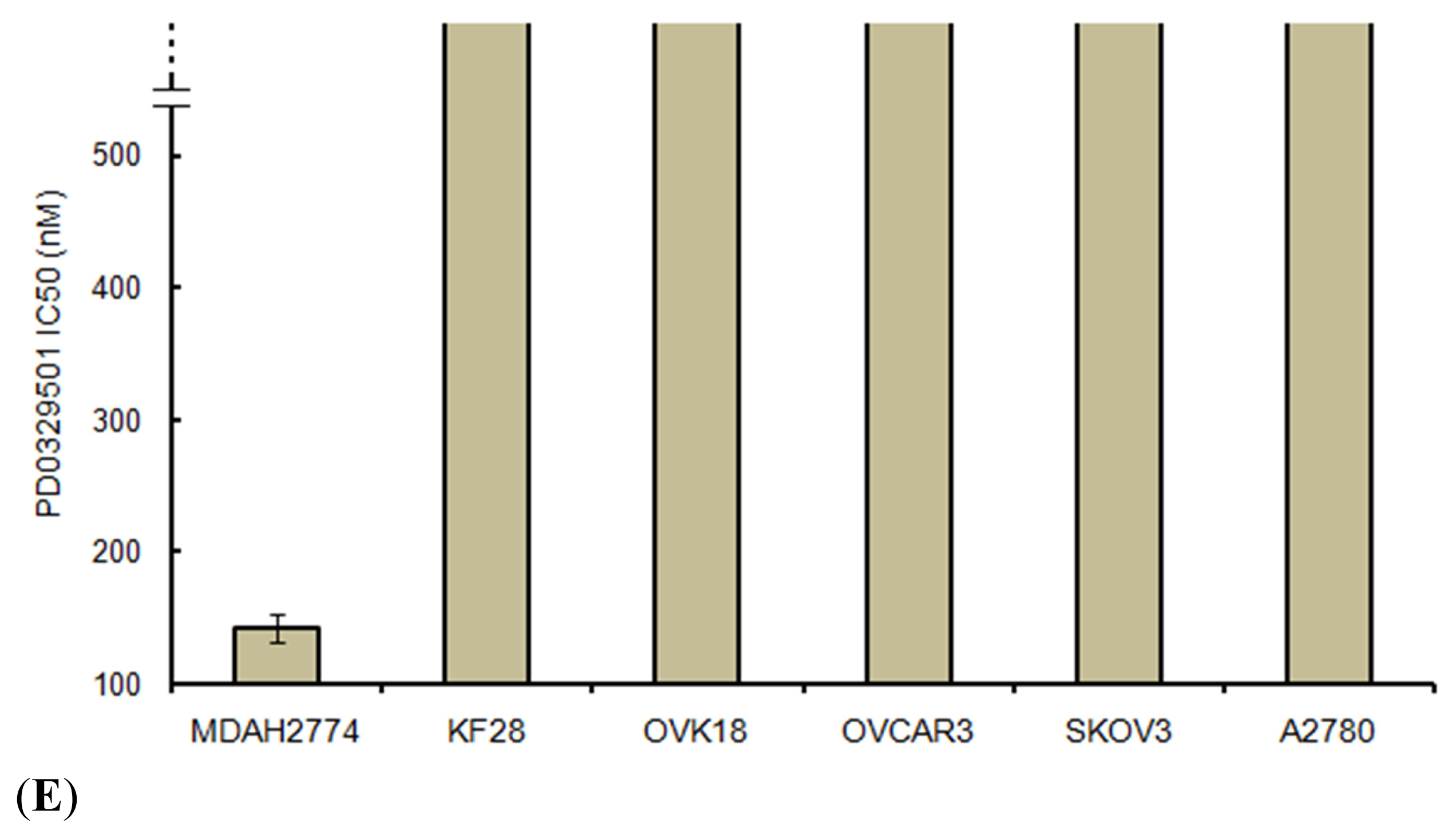
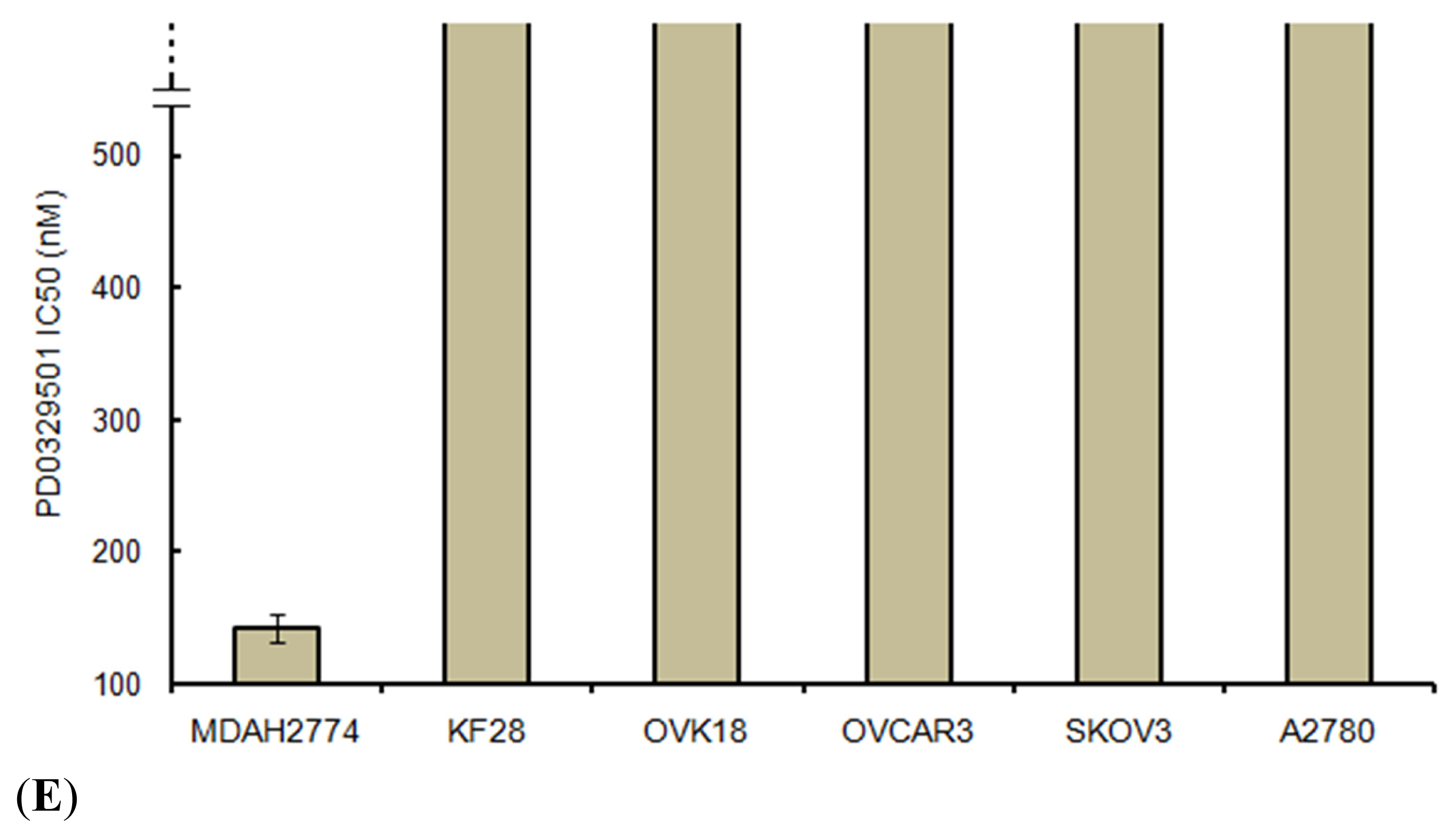
2.5. Effects of ERK1/2 Inactivation on High-Grade Serous Ovarian Carcinoma in Vitro
3. Discussion
4. Experimental Section
4.1. Tissue Samples
4.2. Fluorescence in Situ Hybridization
4.3. Mutational Analysis of KRAS
4.4. Immunohistochemistry
4.5. Western Blot Analysis
4.6. Cell Growth Assays
4.7. Statistical Methods for Clinical Correlations
5. Conclusions
Acknowledgement
Conflicts of Interest
References
- Wingo, P.A.; Tong, T.; Bolden, S. Cancer statistics 1995. CA Cancer J. Clin 1995, 45, 8–30. [Google Scholar]
- Hoskins, W.J.; McGuire, W.P.; Brady, M.F.; Homesley, H.D.; Creasman, W.T.; Berman, M.; Ball, H.; Berek, J.S. The effect of diameter of largest residual disease on survival after primary cytoreductive surgery in patients with suboptimal residual epithelial ovarian carcinoma. Am. J. Obstet. Gynecol 1994, 170, 974–979, , discussion 979–980.. [Google Scholar]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J. Clin. Oncol 2003, 21, 3194–3200. [Google Scholar]
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N. Engl. J. Med 1996, 334, 1–6. [Google Scholar]
- Kurman, R.J.; Shih, le-M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—Shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615.[Green Version]
- Mizumoto, Y.; Kyo, S.; Mori, N.; Sakaguchi, J.; Ohno, S.; Maida, Y.; Hashimoto, M.; Takakura, M.; Inoue, M. Activation of ERK1/2 occurs independently of KRAS or BRAF status in endometrial cancer and is associated with favorable prognosis. Cancer Sci 2007, 98, 652–658. [Google Scholar]
- Nakayama, N.; Nakayama, K.; Yeasmin, S.; Ishibashi, M.; Katagiri, A.; Iida, K.; Fukumoto, M.; Miyazaki, K. KRAS or BRAF mutation status is a useful predictor of sensitivity to MEK inhibition in ovarian cancer. Br. J. Cancer 2008, 99, 2020–2028. [Google Scholar]
- Cho, K.R.; Shih, le-M. Ovarian cancer. Annu. Rev. Pathol. 2009, 4, 287–313. [Google Scholar]
- Nakayama, K.; Nakayama, N.; Kurman, R.J.; Cope, L.; Pohl, G.; Samuels, Y.; Velculescu, V.E.; Wang, T.L.; Shih, le-M. Sequence mutations and amplification of PIK3CA and AKT2 genes in purified ovarian serous neoplasms. Cancer Biol. Ther. 2006, 5, 779–785. [Google Scholar]
- Small, G.W.; Shi, Y.Y.; Higgins, L.S.; Orlowski, R.Z. Mitogen-activated protein kinase phosphatase-1 is a mediator of breast cancer chemoresistance. Cancer Res 2007, 67, 4459–4466. [Google Scholar]
- Vicent, S.; Garayoa, M.; López-Picazo, J.M.; Lozano, M.D.; Toledo, G.; Thunnissen, F.B.; Manzano, R.G.; Montuenga, L.M. Mitogen-activated protein kinase phosphatase-1 is overexpressed in non-small cell lung cancer and is an independent predictor of outcome in patients. Clin. Cancer Res 2004, 10, 3639–3649. [Google Scholar]
- Kelly, K.; Chu, Y. The Regulation of MAP Kinase Pathways by MAP Kinase Phosphatases. In Signaling Networks and Cell Cycle Control; Gutkind, J.S., Ed.; Humana Press: Totowa, NJ, USA, 2000; pp. 165–182. [Google Scholar]
- Tanaka, Y.; Terai, Y.; Tanabe, A.; Sasaki, H.; Sekijima, T.; Fujiwara, S.; Yamashita, Y.; Kanemura, M.; Ueda, M.; Sugita, M.; et al. Prognostic effect of epidermal growth factor receptor gene mutations and the aberrant phosphorylation of Akt and ERK in ovarian cancer. Cancer Biol. Ther 2011, 11, 50–57. [Google Scholar]
- Lanitis, E.; Dangaj, D.; Hagemann, I.S.; Song, D.G.; Best, A.; Sandaltzopoulos, R.; Coukos, G.; Powell, D.J., Jr. Primary human ovarian epithelial cancer cells broadly express HER2 at immunologically-detectable levels. PLoS One 2012, 7, e49829. [Google Scholar]
- Nakayama, K.; Nakayama, N.; Jinawath, N.; Salani, R.; Kurman, R.J.; Shih, le-M.; Wang, T.L. Amplicon profiles in ovarian serous carcinomas. Int. J. Cancer 2007, 120, 2613–2617. [Google Scholar]
- Scott, M.; Hall, P.A. Prognostic and predictive factors. Methods Mol. Med 2004, 97, 1–11. [Google Scholar]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res 2008, 68, 3077–3080. [Google Scholar]
- Nakayama, N.; Nakayama, K.; Shamima, Y.; Ishikawa, M.; Katagiri, A.; Iida, K.; Miyazaki, K. Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer. Cancer 2010, 116, 2621–2634. [Google Scholar]
- Iida, K.; Nakayama, K.; Rahman, M.T.; Rahman, M.; Ishikawa, M.; Katagiri, A.; Yeasmin, S.; Otsuki, Y.; Kobayashi, H.; Nakayama, S.; et al. EGFR gene amplification is related to adverse clinical outcomes in cervical squamous cell carcinoma; making the EGFR pathway a novel therapeutic target. Br. J. Cancer 2011, 105, 420–427. [Google Scholar]
- Wagner, P.L.; Stiedl, A.C.; Wilbertz, T.; Petersen, K.; Scheble, V.; Menon, R.; Reischl, M.; Mikut, R.; Rubin, M.A.; Fend, F.; et al. Frequency and clinicopathologic correlates of KRAS amplification in non-small cell lung carcinoma. Lung Cancer 2011, 74, 118–123. [Google Scholar]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar]
- Singer, G.; Oldt, R., 3rd; Cohen, Y.; Wang, B.G.; Sidransky, D.; Kurman, R.J.; Shih, le-M. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar]
- Nakayama, K.; Miyazaki, K.; Kanzaki, A.; Fukumoto, M.; Takebayashi, Y. Expression and cisplatin sensitivity of copper-transporting P-type adenosine triphosphatase (ATP7B) in human solid carcinoma cell lines. Oncol. Rep 2001, 8, 1285–1287. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p-ERK negative | p-ERK positive | |
|---|---|---|
| KRAS/MAPK1 normal | 29 | 26 |
| KRAS/MAPK1 amplification | 5 | 8 |
| p = 0.5387 |
| Factors | Patients | KRAS FISH | p-value | MAPK1 FISH | p-value | p-ERK imunostaining | p-value | |||
|---|---|---|---|---|---|---|---|---|---|---|
| (n) | Normal | Amplification | Normal | Amplification | Negative | Positive | ||||
| FIGO stage | ||||||||||
| I, II | 31 | 26 | 5 | 0.5192 | 30 | 1 | 0.2327 | 14 | 17 | 0.4651 |
| III, IV | 37 | 33 | 4 | 33 | 4 | 20 | 17 | |||
| Age (years) | ||||||||||
| <60 | 32 | 26 | 6 | 0.2058 | 28 | 4 | 0.1252 | 15 | 17 | 0.627 |
| ≥60 | 36 | 33 | 3 | 35 | 1 | 19 | 17 | |||
| Residual tumor | ||||||||||
| <1 cm | 39 | 32 | 7 | 0.1835 | 38 | 1 | 0.0793 | 19 | 20 | 0.8063 |
| ≥1 cm | 29 | 27 | 2 | 25 | 4 | 15 | 14 | |||
| Factors | Patients | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|---|
| hazard ratio | 95% CI | p-value | hazard ratio | 95% CI | p-value | ||
| FIGO stage | |||||||
| I, II | 31 | 7.1 | 2.1–24.1 | 0.0015 | 1.3 | 0.3–5.8 | 0.7511 |
| III, IV | 37 | ||||||
| Age (years) | |||||||
| <60 | 32 | 2.5 | 1.0–6.0 | 0.0499 | 2.8 | 1.0–7.7 | 0.0438 |
| ≥60 | 36 | ||||||
| Residual tumor | |||||||
| <1 cm | 39 | 14.2 | 4.1–48.2 | <0.0001 | 10.2 | 2.2–46.7 | 0.0028 |
| ≥1 cm | 29 | ||||||
| MAPK1 FISH | |||||||
| Amplification | 5 | 4.4 | 1.4–1.3 | 0.0098 | 4.2 | 1.2–15.0 | 0.0285 |
| Normal | 63 | ||||||
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rahman, M.T.; Nakayama, K.; Rahman, M.; Katagiri, H.; Katagiri, A.; Ishibashi, T.; Ishikawa, M.; Sato, E.; Iida, K.; Nakayama, N.; et al. KRAS and MAPK1 Gene Amplification in Type II Ovarian Carcinomas. Int. J. Mol. Sci. 2013, 14, 13748-13762. https://doi.org/10.3390/ijms140713748
Rahman MT, Nakayama K, Rahman M, Katagiri H, Katagiri A, Ishibashi T, Ishikawa M, Sato E, Iida K, Nakayama N, et al. KRAS and MAPK1 Gene Amplification in Type II Ovarian Carcinomas. International Journal of Molecular Sciences. 2013; 14(7):13748-13762. https://doi.org/10.3390/ijms140713748
Chicago/Turabian StyleRahman, Mohammed Tanjimur, Kentaro Nakayama, Munmun Rahman, Hiroshi Katagiri, Atsuko Katagiri, Tomoka Ishibashi, Masako Ishikawa, Emi Sato, Kouji Iida, Naomi Nakayama, and et al. 2013. "KRAS and MAPK1 Gene Amplification in Type II Ovarian Carcinomas" International Journal of Molecular Sciences 14, no. 7: 13748-13762. https://doi.org/10.3390/ijms140713748
APA StyleRahman, M. T., Nakayama, K., Rahman, M., Katagiri, H., Katagiri, A., Ishibashi, T., Ishikawa, M., Sato, E., Iida, K., Nakayama, N., Ishikawa, N., & Miyazaki, K. (2013). KRAS and MAPK1 Gene Amplification in Type II Ovarian Carcinomas. International Journal of Molecular Sciences, 14(7), 13748-13762. https://doi.org/10.3390/ijms140713748