Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy



Abstract
:
1. Introduction
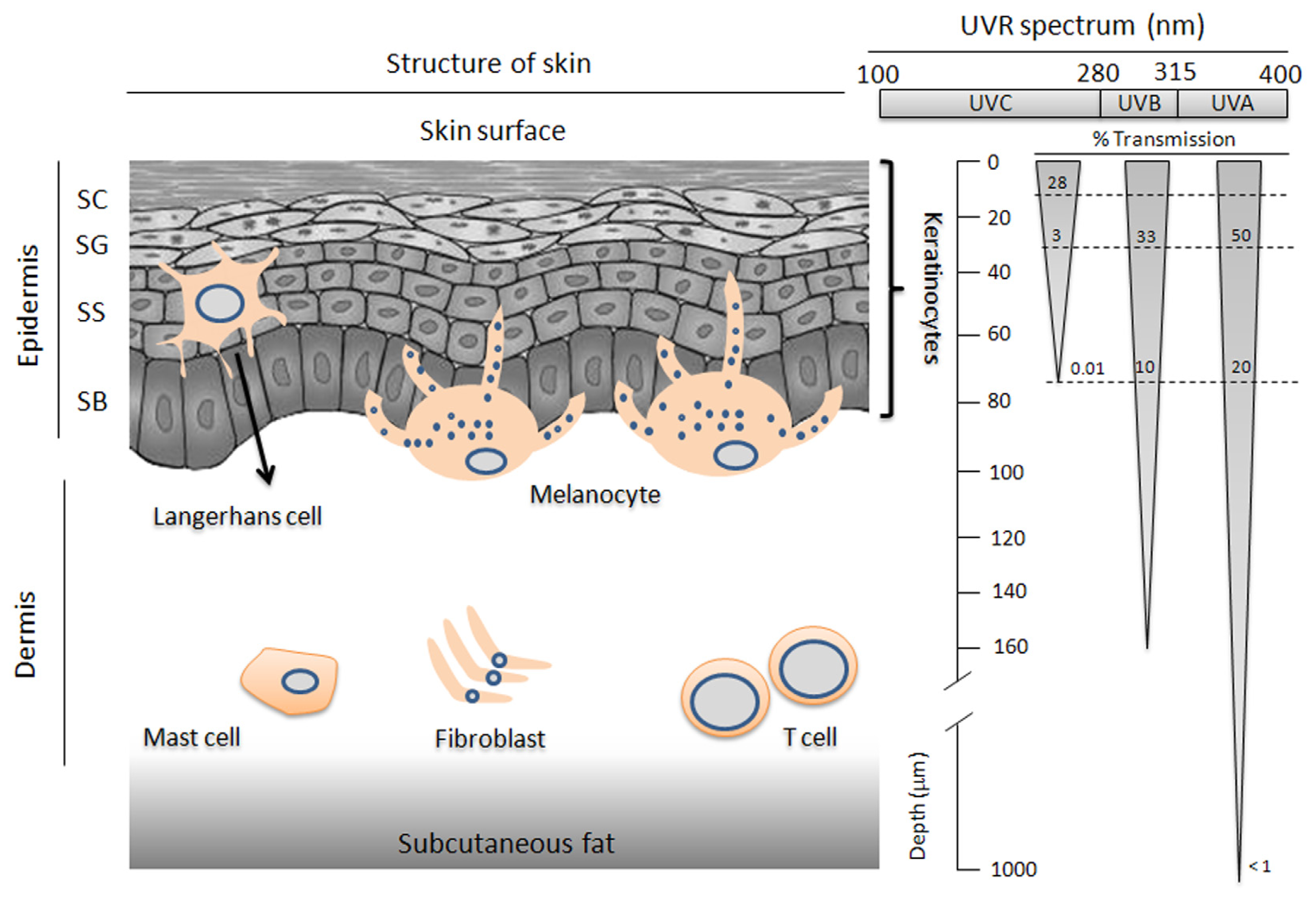
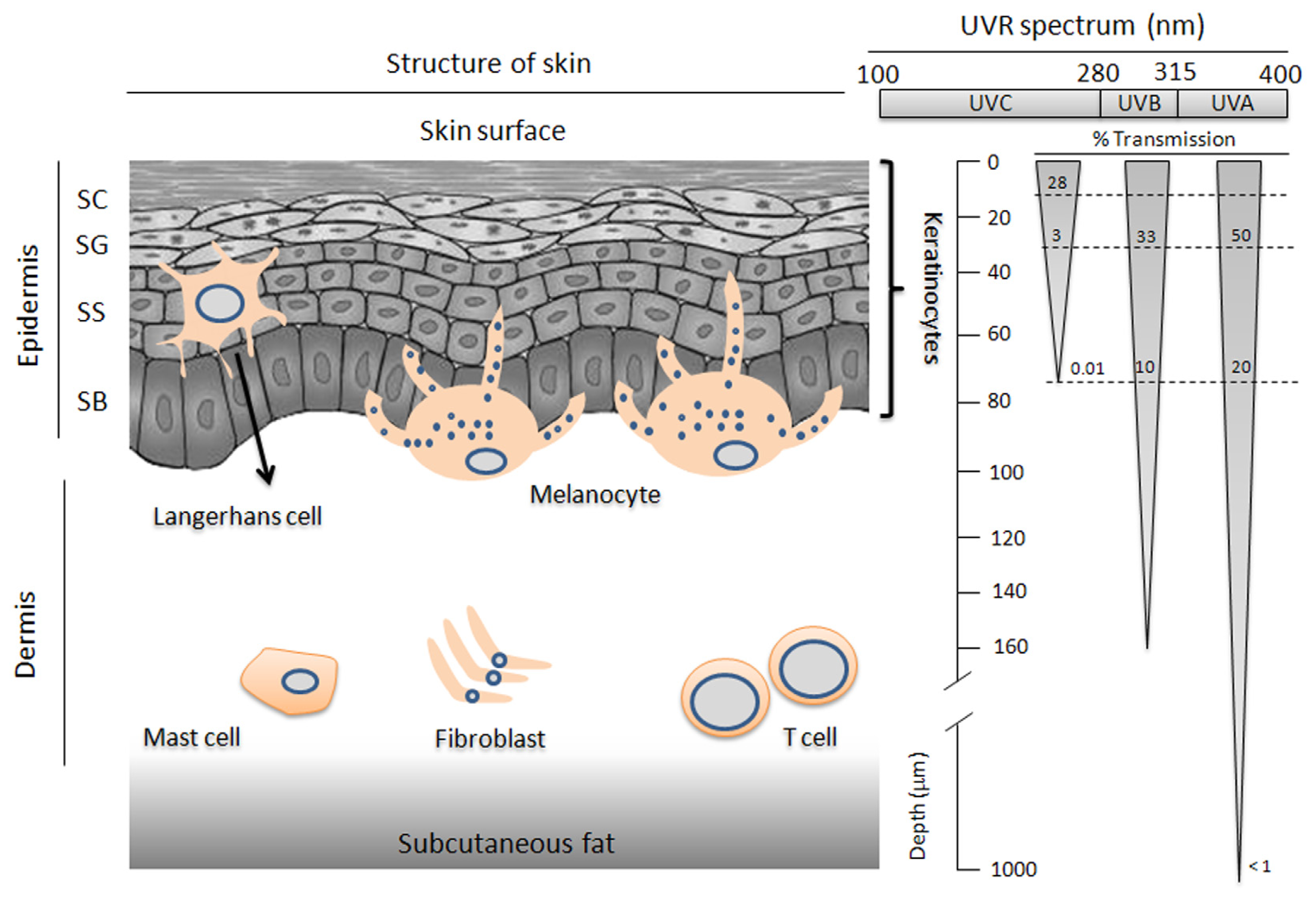
2. Skin Physiology
3. The Biological Relevance of UVR to Skin
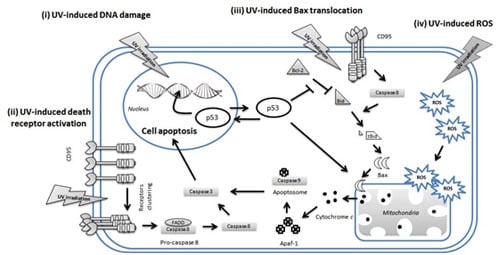
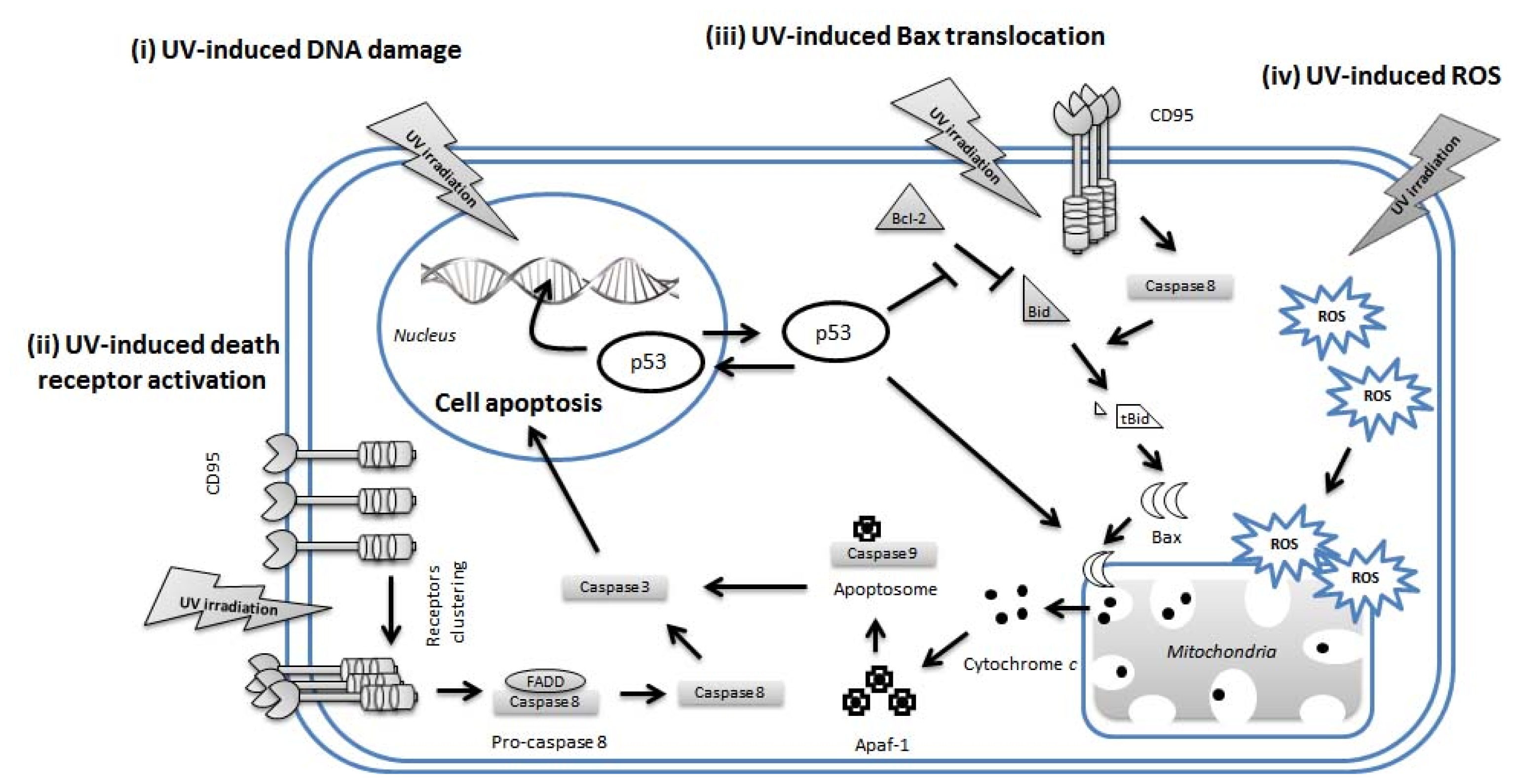
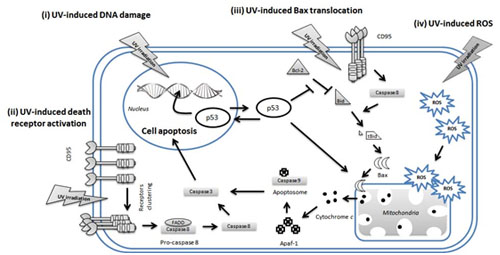
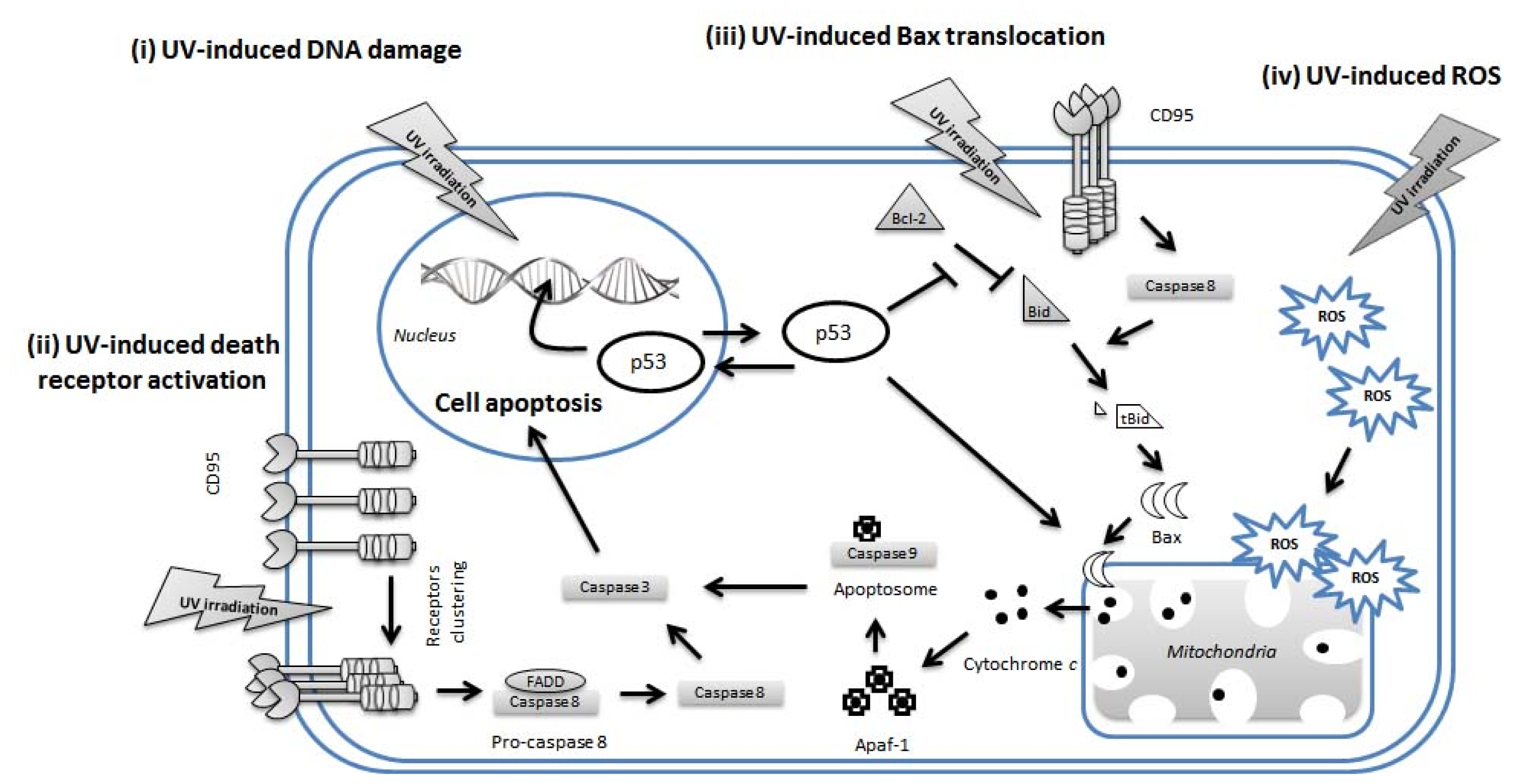
4. UV-Induced Apoptosis of Keratinocytes
4.1. p53 in UVR-Induced Apoptosis of Keratinocytes
4.2. Extrinsic Pathways in UV-Induced Apoptosis
4.3. Intrinsic Pathways in UV-Induced Apoptosis
5. The Modulation of Immune Responses by UVR through Apoptosis
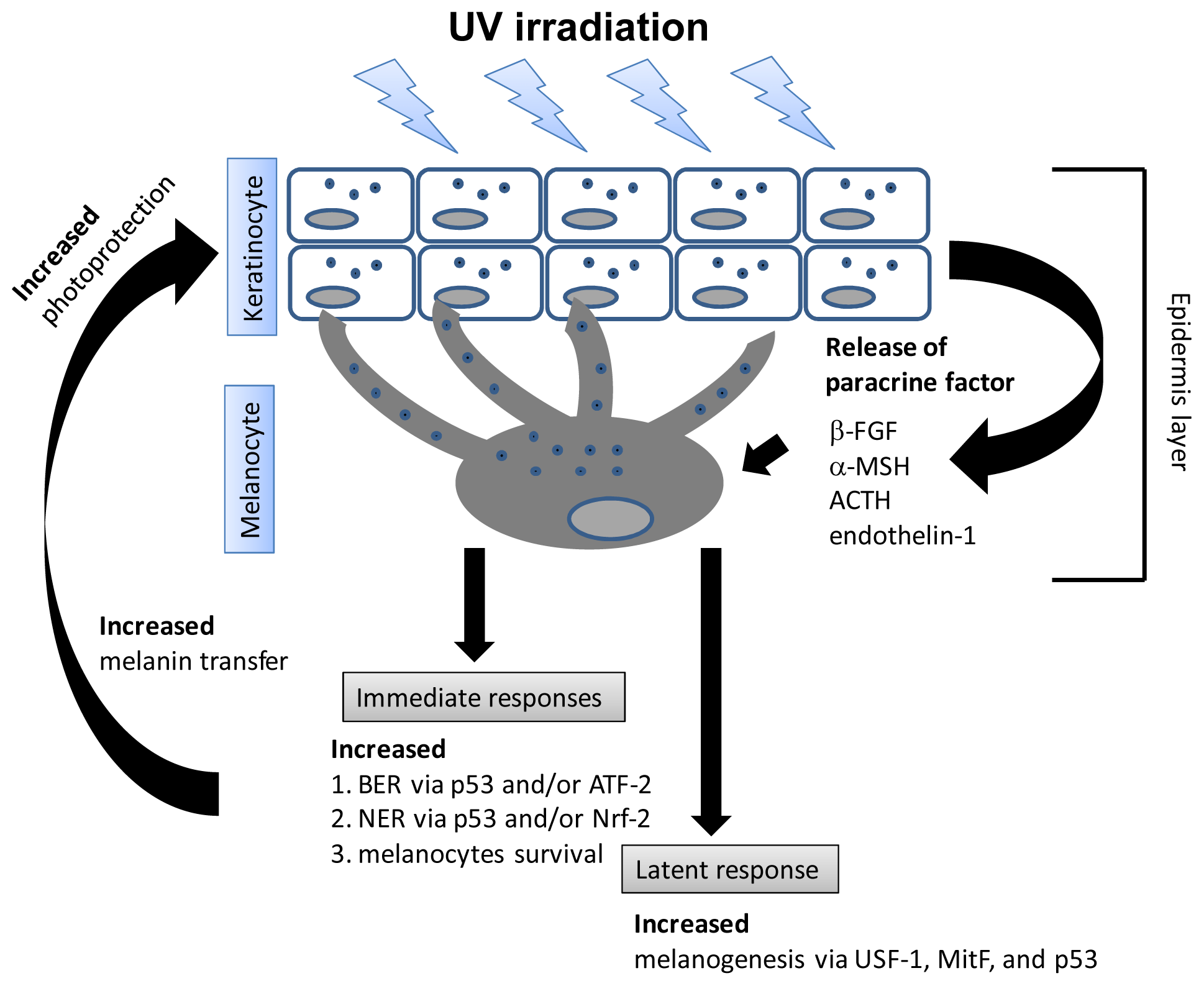
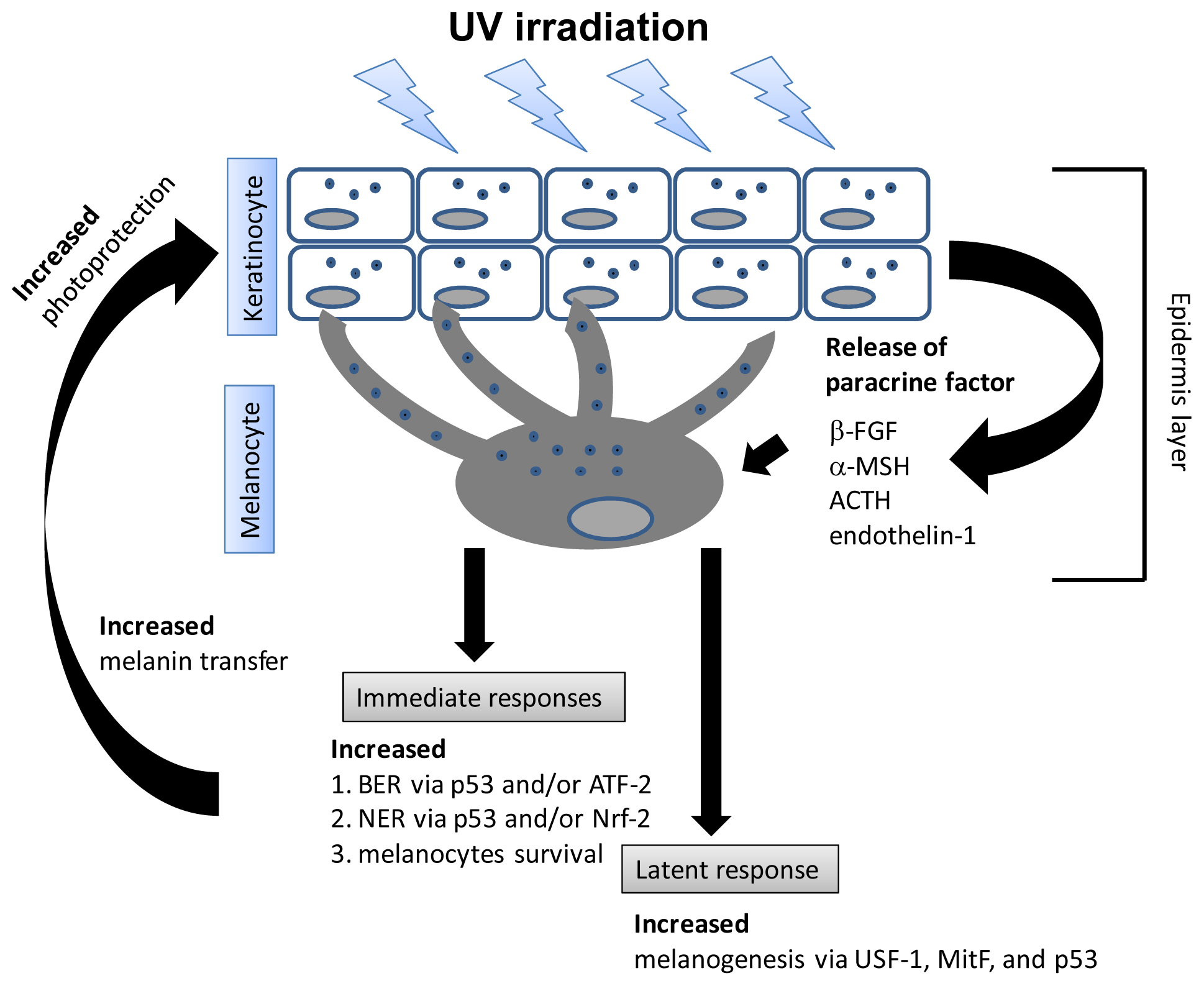
6. UV Induces Melanogenesis and Apoptosis in Melanocytes Differentially Based on Wavelength and Dose
7. UV and Vitamin D Synthesis in Skin: Immunological Modulations
8. Application of Phototherapy: UV-Induced Apoptosis and Biological Consequences
9. Clinical Implications of UV-Induced Apoptosis
9.1. Keratinocytes
9.2. Melanocytes
9.3. Skin-Associated Lymphoid Tissues
10. Conclusions
11. Perspectives
Acknowledgments
Conflict of Interest
References
- Swann, G. The skin is the body’s largest organ. J. Vis. Commun. Med 2010, 33, 148–149. [Google Scholar]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol 2011, 9, 244–253. [Google Scholar]
- Madison, K.C. Barrier function of the skin: “La raison d’etre” of the epidermis. J. Invest. Dermatol 2003, 121, 231–241. [Google Scholar]
- Pincelli, C.; Marconi, A. Keratinocyte stem cells: Friends and foes. J. Cell Physiol 2010, 225, 310–315. [Google Scholar] [Green Version]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar]
- Koch, S.; Kohl, K.; Klein, E.; von Bubnoff, D.; Bieber, T. Skin homing of Langerhans cell precursors: Adhesion, chemotaxis, and migration. J. Allergy Clin. Immunol 2006, 117, 163–168. [Google Scholar]
- Bennett, C.L.; Noordegraaf, M.; Martina, C.A.; Clausen, B.E. Langerhans cells are required for efficient presentation of topically applied hapten to T cells. J. Immunol 2007, 179, 6830–6835. [Google Scholar]
- Lugo, L.M.; Lei, P.; Andreadis, S.T. Vascularization of the dermal support enhances wound re-epithelialization by in situ delivery of epidermal keratinocytes. Tissue Eng. Part A 2011, 17, 665–675. [Google Scholar]
- Hayward, M.G.; Keatinge, W.R. Roles of subcutaneous fat and thermoregulatory reflexes in determining ability to stabilize body temperature in water. J. Physiol 1981, 320, 229–251. [Google Scholar]
- Diffey, B.L. Sources and measurement of ultraviolet radiation. Methods 2002, 28, 4–13. [Google Scholar]
- Krutmann, J. The role of UVA rays in skin aging. Eur. J. Dermatol 2001, 11, 170–171. [Google Scholar]
- Coelho, S.G.; Hearing, V.J. UVA tanning is involved in the increased incidence of skin cancers in fair-skinned young women. Pigment Cell Melanoma Res 2010, 23, 57–63. [Google Scholar]
- Krutmann, J. Ultraviolet A radiation-induced biological effects in human skin: Relevance for photoaging and photodermatosis. J. Dermatol. Sci 2000, 23, S22–S26. [Google Scholar]
- Phan, T.A.; Halliday, G.M.; Barnetson, R.S.; Damian, D.L. Spectral and dose dependence of ultraviolet radiation-induced immunosuppression. Front. Biosci 2006, 11, 394–411. [Google Scholar]
- Batista, L.F.; Kaina, B.; Meneghini, R.; Menck, C.F. How DNA lesions are turned into powerful killing structures: Insights from UV-induced apoptosis. Mutat. Res 2009, 681, 197–208. [Google Scholar]
- Setlow, R.B.; Carrier, W.L. Pyrimidine dimers in ultraviolet-irradiated DNA’s. J. Mol. Biol 1966, 17, 237–254. [Google Scholar]
- D’Errico, M.; Lemma, T.; Calcagnile, A.; de Santis, L.P.; Dogliotti, E. Cell type and DNA damage specific response of human skin cells to environmental agents. Mutat. Res 2007, 614, 37–47. [Google Scholar]
- Pfeifer, G.P. Formation and processing of UV photoproducts: Effects of DNA sequence and chromatin environment. Photochem. Photobiol 1997, 65, 270–283. [Google Scholar]
- Ziegler, A.; Jonason, A.S.; Leffell, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar]
- Pierceall, W.E.; Kripke, M.L.; Ananthaswamy, H.N. N-ras mutation in ultraviolet radiation-induced murine skin cancers. Cancer Res 1992, 52, 3946–3951. [Google Scholar]
- Jiveskog, S.; Ragnarsson-Olding, B.; Platz, A.; Ringborg, U. N-ras mutations are common in melanomas from sun-exposed skin of humans but rare in mucosal membranes or unexposed skin. J. Invest. Dermatol 1998, 111, 757–761. [Google Scholar]
- Kulms, D.; Schwarz, T. Molecular mechanisms of UV-induced apoptosis. Photodermatol. Photoimmunol. Photomed 2000, 16, 195–201. [Google Scholar]
- Assefa, Z.; van Laethem, A.; Garmyn, M.; Agostinis, P. Ultraviolet radiation-induced apoptosis in keratinocytes: On the role of cytosolic factors. Biochim. Biophys. Acta 2005, 1755, 90–106. [Google Scholar]
- Green, E.A.; Choi, Y.; Flavell, R.A. Pancreatic lymph node-derived CD4(+)CD25(+) Treg cells: Highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity 2002, 16, 183–191. [Google Scholar]
- Takasawa, R.; Nakamura, H.; Mori, T.; Tanuma, S. Differential apoptotic pathways in human keratinocyte HaCaT cells exposed to UVB and UVC. Apoptosis 2005, 10, 1121–1130. [Google Scholar]
- Mouret, S.; Philippe, C.; Gracia-Chantegrel, J.; Banyasz, A.; Karpati, S.; Markovitsi, D.; Douki, T. UVA-induced cyclobutane pyrimidine dimers in DNA: A direct photochemical mechanism? Org. Biomol. Chem 2010, 8, 1706–1711. [Google Scholar]
- Courdavault, S.; Baudouin, C.; Charveron, M.; Canguilhem, B.; Favier, A.; Cadet, J.; Douki, T. Repair of the three main types of bipyrimidine DNA photoproducts in human keratinocytes exposed to UVB and UVA radiations. DNA Repair 2005, 4, 836–844. [Google Scholar]
- Greinert, R.; Volkmer, B.; Henning, S.; Breitbart, E.W.; Greulich, K.O.; Cardoso, M.C.; Rapp, A. UVA-induced DNA double-strand breaks result from the repair of clustered oxidative DNA damages. Nucleic Acids Res 2012, 40, 10263–10273. [Google Scholar]
- Zhan, Q.; Fan, S.; Smith, M.L.; Bae, I.; Yu, K.; Alamo, I., Jr; O’Connor, P.M.; Fornace, A.J., Jr. Abrogation of p53 function affects gadd gene responses to DNA base-damaging agents and starvation. DNA Cell Biol. 1996, 15, 805–815. [Google Scholar]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl J. Med 2009, 361, 1475–1485. [Google Scholar]
- Schoppy, D.W.; Ruzankina, Y.; Brown, E.J. Removing all obstacles: A critical role for p53 in promoting tissue renewal. Cell Cycle 2010, 9, 1313–1319. [Google Scholar]
- Tron, V.A.; Trotter, M.J.; Tang, L.; Krajewska, M.; Reed, J.C.; Ho, V.C.; Li, G. p53-regulated apoptosis is differentiation dependent in ultraviolet B-irradiated mouse keratinocytes. Am. J. Pathol 1998, 153, 579–585. [Google Scholar]
- Rodust, P.M.; Stockfleth, E.; Ulrich, C.; Leverkus, M.; Eberle, J. UV-induced squamous cell carcinoma—A role for antiapoptotic signalling pathways. Br. J. Dermatol 2009, 161, 107–115. [Google Scholar]
- Bang, B.; Gniadecki, R.; Larsen, J.K.; Baadsgaard, O.; Skov, L. In vivo UVB irradiation induces clustering of Fas (CD95) on human epidermal cells. Exp. Dermatol 2003, 12, 791–798. [Google Scholar]
- Aragane, Y.; Kulms, D.; Metze, D.; Wilkes, G.; Poppelmann, B.; Luger, T.A.; Schwarz, T. Ultraviolet light induces apoptosis via direct activation of CD95 (Fas/APO-1) independently of its ligand CD95L. J. Cell Biol 1998, 140, 171–182. [Google Scholar]
- Tobin, D.; van Hogerlinden, M.; Toftgard, R. UVB-induced association of tumor necrosis factor (TNF) receptor 1/TNF receptor-associated factor-2 mediates activation of Rel proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 565–569. [Google Scholar]
- Qin, J.Z.; Bacon, P.E.; Chaturvedi, V.; Bonish, B.; Nickoloff, B.J. Pathways involved in proliferating, senescent and immortalized keratinocyte cell death mediated by two different TRAIL preparations. Exp. Dermatol 2002, 11, 573–583. [Google Scholar]
- Qin, J.Z.; Bacon, P.; Panella, J.; Sitailo, L.A.; Denning, M.F.; Nickoloff, B.J. Low-dose UV-radiation sensitizes keratinocytes to TRAIL-induced apoptosis. J. Cell Physiol 2004, 200, 155–166. [Google Scholar]
- Kroemer, G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med 1997, 3, 614–620. [Google Scholar]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol 2007, 8, 405–413. [Google Scholar]
- Assefa, Z.; Garmyn, M.; Vantieghem, A.; Declercq, W.; Vandenabeele, P.; Vandenheede, J.R.; Agostinis, P. Ultraviolet B radiation-induced apoptosis in human keratinocytes: Cytosolic activation of procaspase-8 and the role of Bcl-2. FEBS Lett 2003, 540, 125–132. [Google Scholar]
- Van Laethem, A.; van Kelst, S.; Lippens, S.; Declercq, W.; Vandenabeele, P.; Janssens, S.; Vandenheede, J.R.; Garmyn, M.; Agostinis, P. Activation of p38 MAPK is required for Bax translocation to mitochondria, cytochrome c release and apoptosis induced by UVB irradiation in human keratinocytes. FASEB J 2004, 18, 1946–1948. [Google Scholar]
- Lee, C.H.; Yu, C.L.; Liao, W.T.; Kao, Y.H.; Chai, C.Y.; Chen, G.S.; Yu, H.S. Effects and interactions of low doses of arsenic and UVB on keratinocyte apoptosis. Chem. Res. Toxicol 2004, 17, 1199–1205. [Google Scholar]
- Lee, C.H.; Liao, W.T.; Yu, H.S. Aberrant immune responses in arsenical skin cancers. Kaohsiung J. Med. Sci 2011, 27, 396–401. [Google Scholar]
- Yu, H.S.; Liao, W.T.; Chai, C.Y. Arsenic carcinogenesis in the skin. J. Biomed. Sci 2006, 13, 657–666. [Google Scholar]
- Holley, A.K.; St. Clair, D.K. Watching the watcher: Regulation of p53 by mitochondria. Future Oncol 2009, 5, 117–130. [Google Scholar]
- Wu, Y.; Xing, D.; Liu, L.; Gao, B. Regulation of Bax activation and apoptotic response to UV irradiation by p53 transcription-dependent and -independent pathways. Cancer Lett 2008, 271, 231–239. [Google Scholar]
- Moysan, A.; Clement-Lacroix, P.; Michel, L.; Dubertret, L.; Morliere, P. Effects of ultraviolet A and antioxidant defense in cultured fibroblasts and keratinocytes. Photodermatol. Photoimmunol. Photomed 1996, 11, 192–197. [Google Scholar]
- Podda, M.; Traber, M.G.; Weber, C.; Yan, L.J.; Packer, L. UV-irradiation depletes antioxidants and causes oxidative damage in a model of human skin. Free Radic Biol. Med 1998, 24, 55–65. [Google Scholar]
- Halliday, G.M. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat. Res 2005, 571, 107–120. [Google Scholar]
- Birch-Machin, M.A.; Swalwell, H. How mitochondria record the effects of UV exposure and oxidative stress using human skin as a model tissue. Mutagenesis 2010, 25, 101–107. [Google Scholar]
- Lai, W.W.; Hsiao, Y.P.; Chung, J.G.; Wei, Y.H.; Cheng, Y.W.; Yang, J.H. Synergistic phototoxic effects of glycolic acid in a human keratinocyte cell line (HaCaT). J. Dermatol. Sci 2011, 64, 191–198. [Google Scholar]
- Liu, C.Y.; Lee, C.F.; Wei, Y.H. Quantitative effect of 4977 bp deletion of mitochondrial DNA on the susceptibility of human cells to UV-induced apoptosis. Mitochondrion 2007, 7, 89–95. [Google Scholar]
- Liu, C.Y.; Lee, C.F.; Wei, Y.H. Role of reactive oxygen species-elicited apoptosis in the pathophysiology of mitochondrial and neurodegenerative diseases associated with mitochondrial DNA mutations. J. Formos Med. Assoc 2009, 108, 599–611. [Google Scholar]
- Paz, M.L.; Gonzalez Maglio, D.H.; Weill, F.S.; Bustamante, J.; Leoni, J. Mitochondrial dysfunction and cellular stress progression after ultraviolet B irradiation in human keratinocytes. Photodermatol. Photoimmunol. Photomed 2008, 24, 115–122. [Google Scholar]
- Schroeder, P.; Gremmel, T.; Berneburg, M.; Krutmann, J. Partial depletion of mitochondrial DNA from human skin fibroblasts induces a gene expression profile reminiscent of photoaged skin. J. Invest. Dermatol 2008, 128, 2297–2303. [Google Scholar]
- Timares, L.; Katiyar, S.K.; Elmets, C.A. DNA damage, apoptosis and langerhans cells—Activators of UV-induced immune tolerance. Photochem. Photobiol 2008, 84, 422–436. [Google Scholar]
- Beissert, S.; Ruhlemann, D.; Mohammad, T.; Grabbe, S.; El-Ghorr, A.; Norval, M.; Morrison, H.; Granstein, R.D.; Schwarz, T. IL-12 prevents the inhibitory effects of cis-urocanic acid on tumor antigen presentation by Langerhans cells: Implications for photocarcinogenesis. J. Immunol 2001, 167, 6232–6238. [Google Scholar]
- Sreevidya, C.S.; Fukunaga, A.; Khaskhely, N.M.; Masaki, T.; Ono, R.; Nishigori, C.; Ullrich, S.E. Agents that reverse UV-Induced immune suppression and photocarcinogenesis affect DNA repair. J. Invest. Dermatol 2010, 130, 1428–1437. [Google Scholar]
- Hart, P.H.; Gorman, S.; Finlay-Jones, J.J. Modulation of the immune system by UV radiation: More than just the effects of vitamin D? Nat. Rev. Immunol 2011, 11, 584–596. [Google Scholar]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar]
- Kolgen, W.; Both, H.; van Weelden, H.; Guikers, K.L.; Bruijnzeel-Koomen, C.A.; Knol, E.F.; van Vloten, W.A.; de Gruijl, F.R. Epidermal langerhans cell depletion after artificial ultraviolet B irradiation of human skin in vivo: Apoptosis versus migration. J. Invest. Dermatol 2002, 118, 812–817. [Google Scholar]
- Nishigori, C.; Yarosh, D.; O’Connor, A.; Shreedhar, V.K.; Ullrich, S.E.; Cox, P.; Kripke, M.L. HindIII liposomes suppress delayed-type hypersensitivity responses in vivo and induce epidermal IL-10 in vitro. J. Immunol 1998, 161, 2684–2691. [Google Scholar]
- Kripke, M.L.; Cox, P.A.; Bucana, C.; Vink, A.A.; Alas, L.; Yarosh, D.B. Role of DNA damage in local suppression of contact hypersensitivity in mice by UV radiation. Exp. Dermatol 1996, 5, 173–180. [Google Scholar]
- Loser, K.; Mehling, A.; Loeser, S.; Apelt, J.; Kuhn, A.; Grabbe, S.; Schwarz, T.; Penninger, J.M.; Beissert, S. Epidermal RANKL controls regulatory T-cell numbers via activation of dendritic cells. Nat. Med 2006, 12, 1372–1379. [Google Scholar]
- Elmets, C.A.; Bergstresser, P.R.; Tigelaar, R.E.; Wood, P.J.; Streilein, J.W. Analysis of the mechanism of unresponsiveness produced by haptens painted on skin exposed to low dose ultraviolet radiation. J. Exp. Med 1983, 158, 781–794. [Google Scholar]
- Chen, A.; Xu, H.; Choi, Y.; Wang, B.; Zheng, G. TRANCE counteracts FasL-mediated apoptosis of murine bone marrow-derived dendritic cells. Cell Immunol 2004, 231, 40–48. [Google Scholar]
- Cremer, I.; Dieu-Nosjean, M.C.; Marechal, S.; Dezutter-Dambuyant, C.; Goddard, S.; Adams, D.; Winter, N.; Menetrier-Caux, C.; Sautes-Fridman, C.; Fridman, W.H.; et al. Long-lived immature dendritic cells mediated by TRANCE-RANK interaction. Blood 2002, 100, 3646–3655. [Google Scholar]
- Josien, R.; Li, H.L.; Ingulli, E.; Sarma, S.; Wong, B.R.; Vologodskaia, M.; Steinman, R.M.; Choi, Y. TRANCE, a tumor necrosis factor family member, enhances the longevity and adjuvant properties of dendritic cells in vivo. J. Exp. Med 2000, 191, 495–502. [Google Scholar]
- Dudda, J.C.; Denfeld, R.W.; Simon, J.C.; Martin, S.F. UVB-irradiated dendritic cells fail to tolerize murine CD8 naive or effector T cells. J. Invest. Dermatol 2004, 122, 945–952. [Google Scholar]
- Garssen, J.; Vandebriel, R.J.; de Gruijl, F.R.; Wolvers, D.A.; van Dijk, M.; Fluitman, A.; van Loveren, H. UVB exposure-induced systemic modulation of Th1- and Th2-mediated immune responses. Immunology 1999, 97, 506–514. [Google Scholar]
- Johnson-Huang, L.M.; Suarez-Farinas, M.; Sullivan-Whalen, M.; Gilleaudeau, P.; Krueger, J.G.; Lowes, M.A. Effective narrow-band UVB radiation therapy suppresses the IL-23/IL-17 axis in normalized psoriasis plaques. J. Invest. Dermatol 2010, 130, 2654–2663. [Google Scholar]
- Walters, I.B.; Ozawa, M.; Cardinale, I.; Gilleaudeau, P.; Trepicchio, W.L.; Bliss, J.; Krueger, J.G. Narrowband (312-nm) UV-B suppresses interferon gamma and interleukin (IL) 12 and increases IL-4 transcripts: Differential regulation of cytokines at the single-cell level. Arch. Dermatol 2003, 139, 155–161. [Google Scholar]
- Pradhan, S.; Kim, H.K.; Thrash, C.J.; Cox, M.A.; Mantena, S.K.; Wu, J.H.; Athar, M.; Katiyar, S.K.; Elmets, C.A.; Timares, L. A critical role for the proapoptotic protein bid in ultraviolet-induced immune suppression and cutaneous apoptosis. J. Immunol 2008, 181, 3077–3088. [Google Scholar]
- Kobayashi, N.; Nakagawa, A.; Muramatsu, T.; Yamashina, Y.; Shirai, T.; Hashimoto, M.W.; Ishigaki, Y.; Ohnishi, T.; Mori, T. Supranuclear melanin caps reduce ultraviolet induced DNA photoproducts in human epidermis. J. Invest. Dermatol 1998, 110, 806–810. [Google Scholar]
- Abdel-Malek, Z.A.; Kadekaro, A.L.; Swope, V.B. Stepping up melanocytes to the challenge of UV exposure. Pigment. Cell Melanoma Res 2010, 23, 171–186. [Google Scholar]
- Smith, A.G.; Luk, N.; Newton, R.A.; Roberts, D.W.; Sturm, R.A.; Muscat, G.E. Melanocortin-1 receptor signaling markedly induces the expression of the NR4A nuclear receptor subgroup in melanocytic cells. J. Biol. Chem 2008, 283, 12564–12570. [Google Scholar]
- Song, X.; Mosby, N.; Yang, J.; Xu, A.; Abdel-Malek, Z.; Kadekaro, A.L. alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment. Cell Melanoma Res 2009, 22, 809–818. [Google Scholar]
- Ravnbak, M.H.; Wulf, H.C. Pigmentation after single and multiple UV-exposures depending on UV-spectrum. Arch. Dermatol. Res 2007, 299, 25–32. [Google Scholar]
- Miller, S.A.; Coelho, S.G.; Zmudzka, B.Z.; Bushar, H.F.; Yamaguchi, Y.; Hearing, V.J.; Beer, J.Z. Dynamics of pigmentation induction by repeated ultraviolet exposures: Dose, dose interval and ultraviolet spectrum dependence. Br. J. Dermatol 2008, 159, 921–930. [Google Scholar]
- MacLaughlin, J.A.; Anderson, R.R.; Holick, M.F. Spectral character of sunlight modulates photosynthesis of previtamin D3 and its photoisomers in human skin. Science 1982, 216, 1001–1003. [Google Scholar]
- Nesby-O’Dell, S.; Scanlon, K.S.; Cogswell, M.E.; Gillespie, C.; Hollis, B.W.; Looker, A.C.; Allen, C.; Doughertly, C.; Gunter, E.W.; Bowman, B.A. Hypovitaminosis D prevalence and determinants among African American and white women of reproductive age: Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Clin. Nutr 2002, 76, 187–192. [Google Scholar]
- Garland, C.F.; Garland, F.C.; Gorham, E.D.; Lipkin, M.; Newmark, H.; Mohr, S.B.; Holick, M.F. The role of vitamin D in cancer prevention. Am. J. Public Health 2006, 96, 252–261. [Google Scholar]
- Hewison, M. Vitamin D and the intracrinology of innate immunity. Mol. Cell Endocrinol 2010, 321, 103–111. [Google Scholar]
- Ellison, T.I.; Smith, M.K.; Gilliam, A.C.; MacDonald, P.N. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. J. Invest. Dermatol 2008, 128, 2508–2517. [Google Scholar]
- Di Rosa, M.; Malaguarnera, M.; Nicoletti, F.; Malaguarnera, L. Vitamin D3: A helpful immuno-modulator. Immunology 2011, 134, 123–139. [Google Scholar]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar]
- Biggs, L.; Yu, C.; Fedoric, B.; Lopez, A.F.; Galli, S.J.; Grimbaldeston, M.A. Evidence that vitamin D(3) promotes mast cell-dependent reduction of chronic UVB-induced skin pathology in mice. J. Exp. Med 2010, 207, 455–463. [Google Scholar]
- Hart, P.H.; Grimbaldeston, M.A.; Swift, G.J.; Jaksic, A.; Noonan, F.P.; Finlay-Jones, J.J. Dermal mast cells determine susceptibility to ultraviolet B-induced systemic suppression of contact hypersensitivity responses in mice. J. Exp. Med 1998, 187, 2045–2053. [Google Scholar]
- Hart, P.H.; Townley, S.L.; Grimbaldeston, M.A.; Khalil, Z.; Finlay-Jones, J.J. Mast cells, neuropeptides, histamine, and prostaglandins in UV-induced systemic immunosuppression. Methods 2002, 28, 79–89. [Google Scholar]
- Obituary: Niels Ryberg Finsen, M.D. Brit. Med. J. 1904, 865–866.
- Moller, K.I.; Kongshoj, B.; Philipsen, P.A.; Thomsen, V.O.; Wulf, H.C. How Finsen’s light cured lupus vulgaris. Photodermatol. Photoimmunol. Photomed 2005, 21, 118–124. [Google Scholar]
- Martineau, A.R.; Honecker, F.U.; Wilkinson, R.J.; Griffiths, C.J. Vitamin D in the treatment of pulmonary tuberculosis. J. Steroid Biochem. Mol. Biol 2007, 103, 793–798. [Google Scholar]
- Maisels, M.J.; McDonagh, A.F. Phototherapy for neonatal jaundice. N. Engl. J. Med 2008, 358, 920–928. [Google Scholar]
- Roll, E.B.; Christensen, T. Formation of photoproducts and cytotoxicity of bilirubin irradiated with turquoise and blue phototherapy light. Acta Paediatr 2005, 94, 1448–1454. [Google Scholar]
- Tierney, E.P.; Hanke, C.W.; Petersen, J. Ablative fractionated CO2 laser treatment of photoaging: A clinical and histologic study. Dermatol. Surg 2012, 38, 1777–1789. [Google Scholar]
- Hunzeker, C.M.; Geronemus, R.G. Treatment of superficial infantile hemangiomas of the eyelid using the 595-nm pulsed dye laser. Dermatol. Surg 2010, 36, 590–597. [Google Scholar]
- Raulin, C.; Schonermark, M.P.; Greve, B.; Werner, S. Q-switched ruby laser treatment of tattoos and benign pigmented skin lesions: A critical review. Ann. Plast. Surg 1998, 41, 555–565. [Google Scholar]
- Breuckmann, F.; Appelhans, C.; Bastian, A.; Stuecker, M.; Altmeyer, P.; Kreuter, A. UVA1-induced decrease in dermal neuron-specific enolase (NSE) in acrosclerosis. Arch. Dermatol. Res 2004, 296, 182–184. [Google Scholar]
- Stern, R.S. Psoralen and ultraviolet a light therapy for psoriasis. N. Engl. J. Med 2007, 357, 682–690. [Google Scholar]
- Stadler, R. Optimal combination with PUVA: Rationale and clinical trial update. Oncology 2007, 21, 29–32. [Google Scholar]
- Wolff, K. Treatment of cutaneous mastocytosis. Int. Arch. Allergy Immunol 2002, 127, 156–159. [Google Scholar]
- Hu, W.P.; Wang, J.J.; Yu, C.L.; Lan, C.C.; Chen, G.S.; Yu, H.S. Helium-neon laser irradiation stimulates cell proliferation through photostimulatory effects in mitochondria. J. Invest. Dermatol 2007, 127, 2048–2057. [Google Scholar]
- Lan, C.C.; Wu, C.S.; Chiou, M.H.; Hsieh, P.C.; Yu, H.S. Low-energy helium-neon laser induces locomotion of the immature melanoblasts and promotes melanogenesis of the more differentiated melanoblasts: Recapitulation of vitiligo repigmentation in vitro. J. Invest. Dermatol 2006, 126, 2119–2126. [Google Scholar]
- Atta, M.; Papanicolaou, N.; Tsirigotis, P. The role of extracorporeal photopheresis in the treatment of cutaneous T-cell lymphomas. Transfus. Apher. Sci 2012, 46, 195–202. [Google Scholar]
- Lee, Y.; Baron, E.D. Photodynamic therapy: Current evidence and applications in dermatology. Semin. Cutan. Med. Surg 2011, 30, 199–209. [Google Scholar]
- Bulat, V.; Situm, M.; Dediol, I.; Ljubicic, I.; Bradic, L. The mechanisms of action of phototherapy in the treatment of the most common dermatoses. Coll. Antropol 2011, 35, 147–151. [Google Scholar]
- Housman, T.S.; Rohrback, J.M.; Fleischer, A.B., Jr; Feldman, S.R. Phototherapy utilization for psoriasis is declining in the United States. J. Am. Acad. Dermatol. 2002, 46, 557–559. [Google Scholar]
- Wan, J.; Abuabara, K.; Troxel, A.B.; Shin, D.B.; van Voorhees, A.S.; Bebo, B.F., Jr; Krueger, G.G.; Callis Duffin, K.; Gelfand, J.M. Dermatologist preferences for first-line therapy of moderate to severe psoriasis in healthy adult patients. J. Am. Acad. Dermatol. 2012, 66, 376–386. [Google Scholar]
- Lapolla, W.; Yentzer, B.A.; Bagel, J.; Halvorson, C.R.; Feldman, S.R. A review of phototherapy protocols for psoriasis treatment. J. Am. Acad. Dermatol 2011, 64, 936–949. [Google Scholar]
- Tewari, A.; Sarkany, R.P.; Young, A.R. UVA1 induces cyclobutane pyrimidine dimers but not 6-4 photoproducts in human skin in vivo. J. Invest. Dermatol 2012, 132, 394–400. [Google Scholar]
- Epstein, J.H. Phototherapy and photochemotherapy. N. Engl. J. Med 1990, 322, 1149–1151. [Google Scholar]
- Mermelstein, F.H.; Abidi, T.F.; Laskin, J.D. Inhibition of epidermal growth factor receptor tyrosine kinase activity in A431 human epidermoid cells following psoralen/ultraviolet light treatment. Mol. Pharmacol 1989, 36, 848–855. [Google Scholar]
- Averbeck, D. Recent advances in psoralen phototoxicity mechanism. Photochem. Photobiol 1989, 50, 859–882. [Google Scholar]
- Volkmar, C.M.; Vukadinovic-Walter, B.; Oplander, C.; Bozkurt, A.; Korth, H.G.; Kirsch, M.; Mahotka, C.; Pallua, N.; Suschek, C.V. UVA-induced phenoxyl radical formation: A new cytotoxic principle in photodynamic therapy. Free Radic Biol. Med 2010, 49, 1129–1137. [Google Scholar]
- Lehmann, P.; Homey, B. Clinic and pathophysiology of photosensitivity in lupus erythematosus. Autoimmun. Rev 2009, 8, 456–461. [Google Scholar]
- Weatherhead, S.C.; Farr, P.M.; Reynolds, N.J. Spectral effects of UV on psoriasis. Photochem. Photobiol. Sci 2013, 12, 47–53. [Google Scholar]
- Yamaguchi, Y.; Takahashi, K.; Zmudzka, B.Z.; Kornhauser, A.; Miller, S.A.; Tadokoro, T.; Berens, W.; Beer, J.Z.; Hearing, V.J. Human skin responses to UV radiation: Pigment in the upper epidermis protects against DNA damage in the lower epidermis and facilitates apoptosis. FASEB J 2006, 20, 1486–1488. [Google Scholar]
- Bivik, C.A.; Larsson, P.K.; Kagedal, K.M.; Rosdahl, I.K.; Ollinger, K.M. UVA/B-induced apoptosis in human melanocytes involves translocation of cathepsins and Bcl-2 family members. J. Invest. Dermatol 2006, 126, 1119–1127. [Google Scholar]
- Yoshiki, R.; Nakamura, M.; Tokura, Y. The biological role of UVB-induced cutaneous immunosuppression. J. UOEH 2012, 34, 77–83. [Google Scholar]
- Dong, D.; Jiang, M.; Xu, X.; Guan, M.; Wu, J.; Chen, Q.; Xiang, L. The effects of NB-UVB on the hair follicle-derived neural crest stem cells differentiating into melanocyte lineage in vitro. J. Dermatol. Sci 2012, 66, 20–28. [Google Scholar]
- Wu, C.S.; Yu, C.L.; Lan, C.C.; Yu, H.S. Narrow-band ultraviolet-B stimulates proliferation and migration of cultured melanocytes. Exp. Dermatol 2004, 13, 755–763. [Google Scholar]
- Euvrard, S.; Kanitakis, J.; Claudy, A. Skin cancers after organ transplantation. N. Engl. J. Med 2003, 348, 1681–1691. [Google Scholar]
- Kripke, M.L. Reflections on the field of photoimmunology. J. Invest. Dermatol 2013, 133, 27–30. [Google Scholar]
- Wang, L.; Jameson, S.C.; Hogquist, K.A. Epidermal Langerhans cells are not required for UV-induced immunosuppression. J. Immunol 2009, 183, 5548–5553. [Google Scholar]
- Erkin, G.; Ugur, Y.; Gurer, C.K.; Asan, E.; Korkusuz, P.; Sahin, S.; Kolemen, F. Effect of PUVA, narrow-band UVB and cyclosporin on inflammatory cells of the psoriatic plaque. J. Cutan. Pathol 2007, 34, 213–219. [Google Scholar]
- Hamakawa, M.; Sugihara, A.; Okamoto, H.; Horio, T. Ultraviolet B radiation suppresses Langerhans cell migration in the dermis by down-regulation of alpha4 integrin. Photodermatol. Photoimmunol. Photomed 2006, 22, 116–123. [Google Scholar]
- Tang, A.; Udey, M.C. Effects of ultraviolet radiation on murine epidermal Langerhans cells: Doses of ultraviolet radiation that modulate ICAM-1 (CD54) expression and inhibit Langerhans cell function cause delayed cytotoxicity in vitro. J. Invest. Dermatol 1992, 99, 83–89. [Google Scholar]
- Zandi, S.; Kalia, S.; Lui, H. UVA1 phototherapy: A concise and practical review. Skin Therapy Lett 2012, 17, 1–4. [Google Scholar]
- Howes, R.A.; Halliday, G.M.; Barnetson, R.S.; Friedmann, A.C.; Damian, D.L. Topical capsaicin reduces ultraviolet radiation-induced suppression of Mantoux reactions in humans. J. Dermatol. Sci 2006, 44, 113–115. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Light source | Targets | Mechanisms | Diseases |
|---|---|---|---|
| 1. Ablation | |||
| CO2 laser (10,800 nm) Er-YAG laser (3850 nm) | Water in and outside the cells | Evaporation | Superficial skin tumors [94] |
| 2. Non-ablation | |||
| Dye laser | Hemoglobin | Photoselective thermolysis | Vascular lesions: Hemangioma and telangiectasia [95] |
| Ruby laser (694 nm) Alexander laser (700–820 nm) | Melanin | Photoselective thermolysis | Pigmentary lesions: Lentigenes Freckles melanocytic nevi [96] |
| 3. UV | |||
| UVA1 (340–400 nm) | Chormophores | T cell apoptosis Collagenase induction Angiogenesis Tissue remodeling | Atopic dermatitis Localized scleroderma [97] |
| UVB | |||
| (Broadband and narrowband) (Excimer light/lasers) | Chromophores | Anti-inflammation, Melanogenesis Apoptosis | Psoriasis vitiligo Atopic dermatitis Intractable pruritus [98] |
| 4. Chemophototherapy | |||
| PUVA (Psoralen + UVA) | Psoralen, DNA | ROS production DNA replication inhibition Cell cycle arrest Apoptosis Anti-inflammation Melanogenesis | Cutaneous T cell lymphoma Skin mastocytosis [99,100] |
| 5. Low power lasers/light/LEDs | |||
| IR or visible light | Chromophores | Immunomodulation Tissue remodeling Melanogenesis Analgesic | Vitiligo Chronic wound Neuralgia [101,102] |
| Blue-Green visible light | Bilirubin | Photoisomerization Photodegradation | Neonatal jaundice [92] |
| 6. Other Phototherapies | |||
| Extra-corporeal photopheresis | Chromophore | T cells depletion | Erythrodermic cutaneous T cell lymphoma [103] |
| Photodynamic therapy | Photosensitizers | ROS production Apoptosis | Superficial skin cancer [104] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, C.-H.; Wu, S.-B.; Hong, C.-H.; Yu, H.-S.; Wei, Y.-H. Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy. Int. J. Mol. Sci. 2013, 14, 6414-6435. https://doi.org/10.3390/ijms14036414
Lee C-H, Wu S-B, Hong C-H, Yu H-S, Wei Y-H. Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy. International Journal of Molecular Sciences. 2013; 14(3):6414-6435. https://doi.org/10.3390/ijms14036414
Chicago/Turabian StyleLee, Chih-Hung, Shi-Bei Wu, Chien-Hui Hong, Hsin-Su Yu, and Yau-Huei Wei. 2013. "Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy" International Journal of Molecular Sciences 14, no. 3: 6414-6435. https://doi.org/10.3390/ijms14036414
APA StyleLee, C.-H., Wu, S.-B., Hong, C.-H., Yu, H.-S., & Wei, Y.-H. (2013). Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy. International Journal of Molecular Sciences, 14(3), 6414-6435. https://doi.org/10.3390/ijms14036414