Mitochondria and Reactive Oxygen Species: Physiology and Pathophysiology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
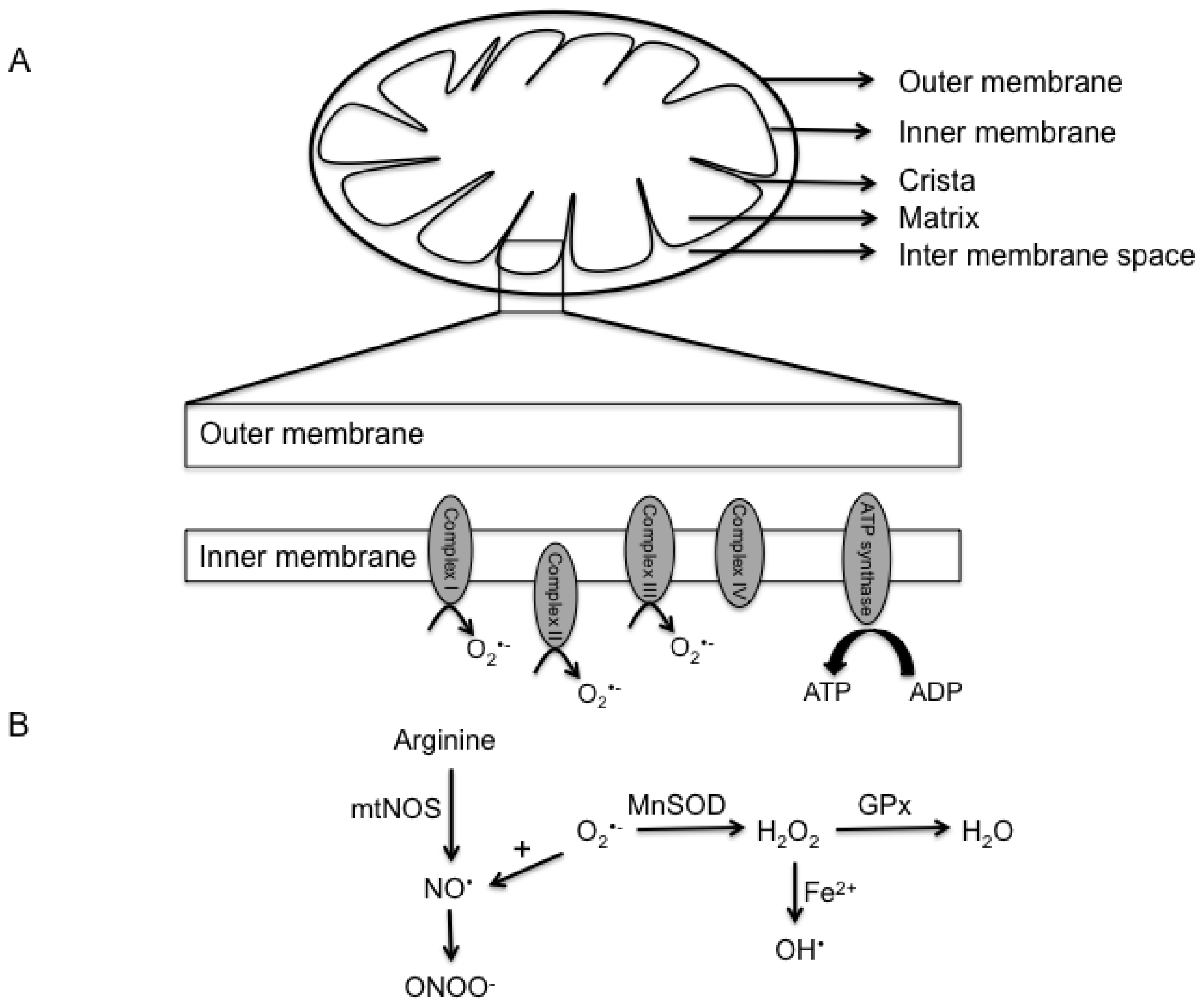
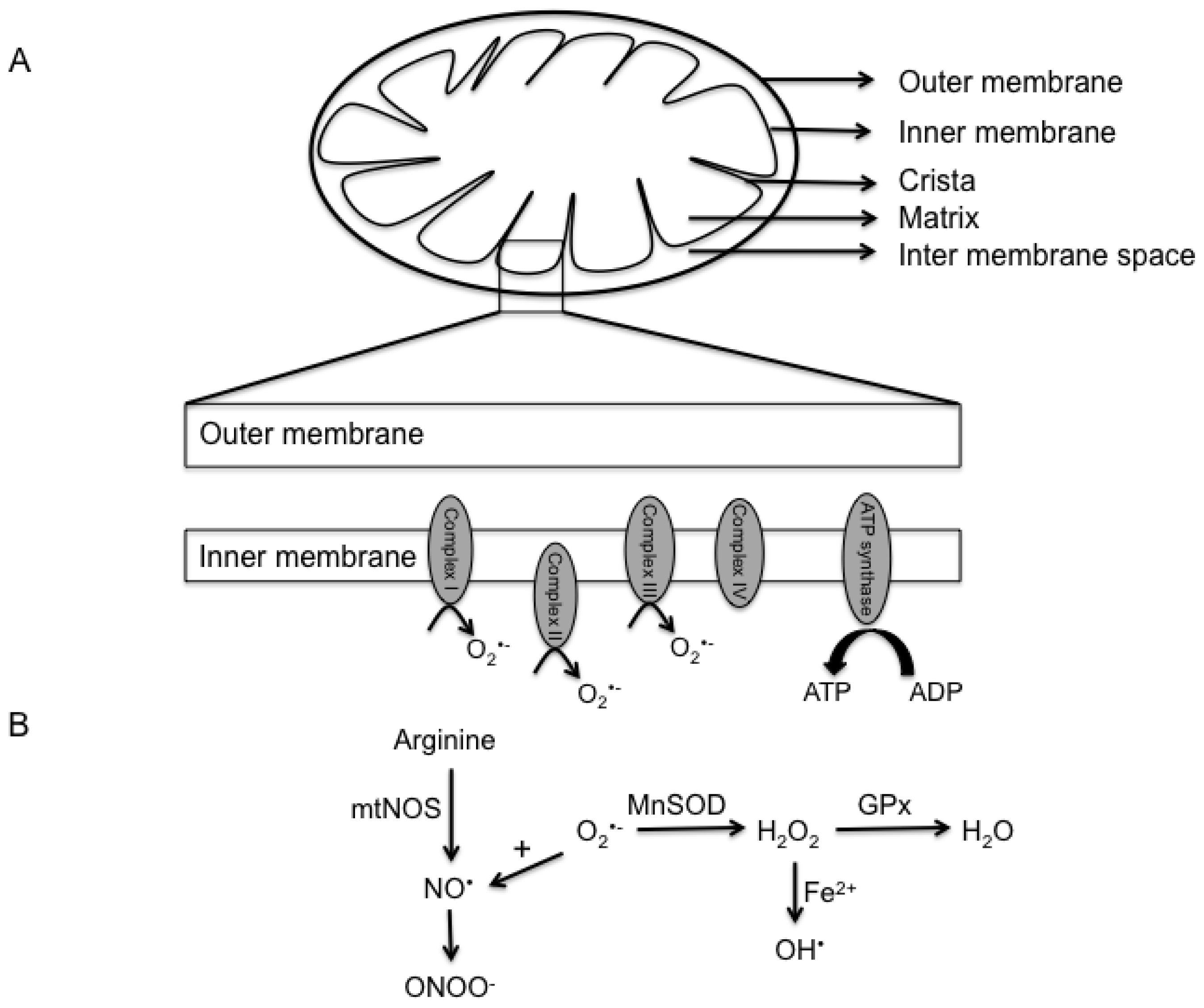
2. Mitochondria Structure and Function
3. Reactive Oxygen and Nitrogen Species
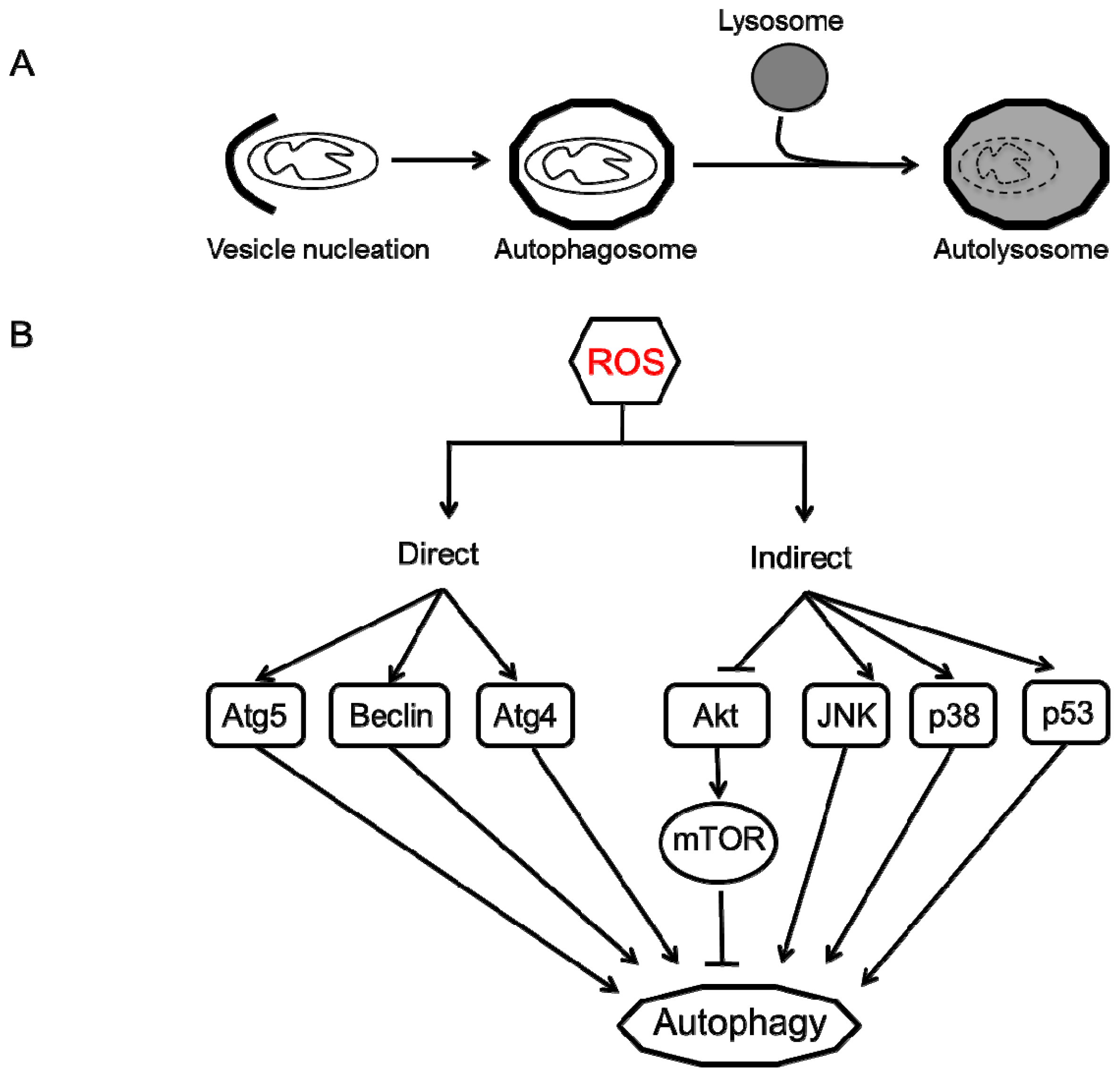
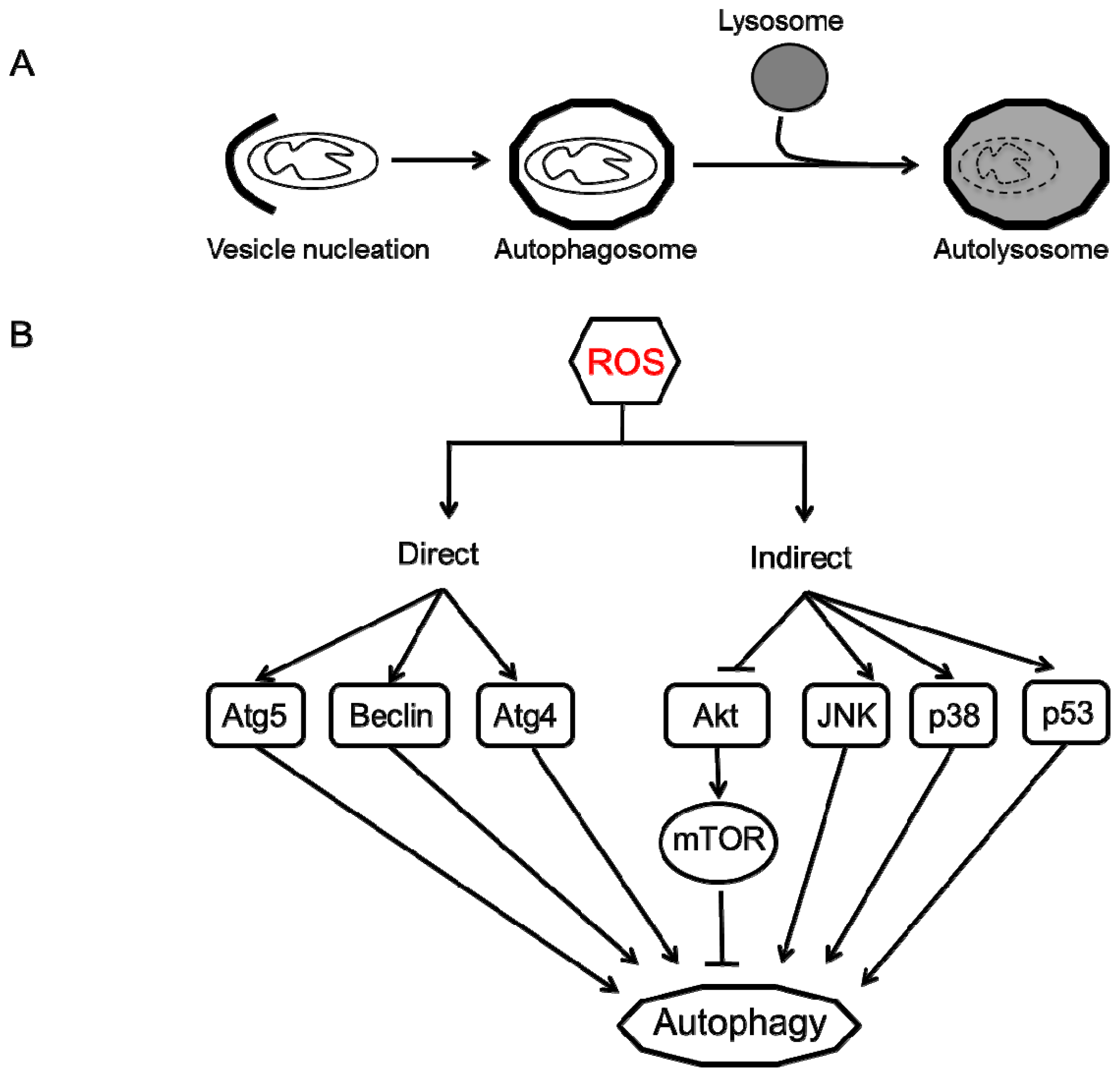
4. Autophagy
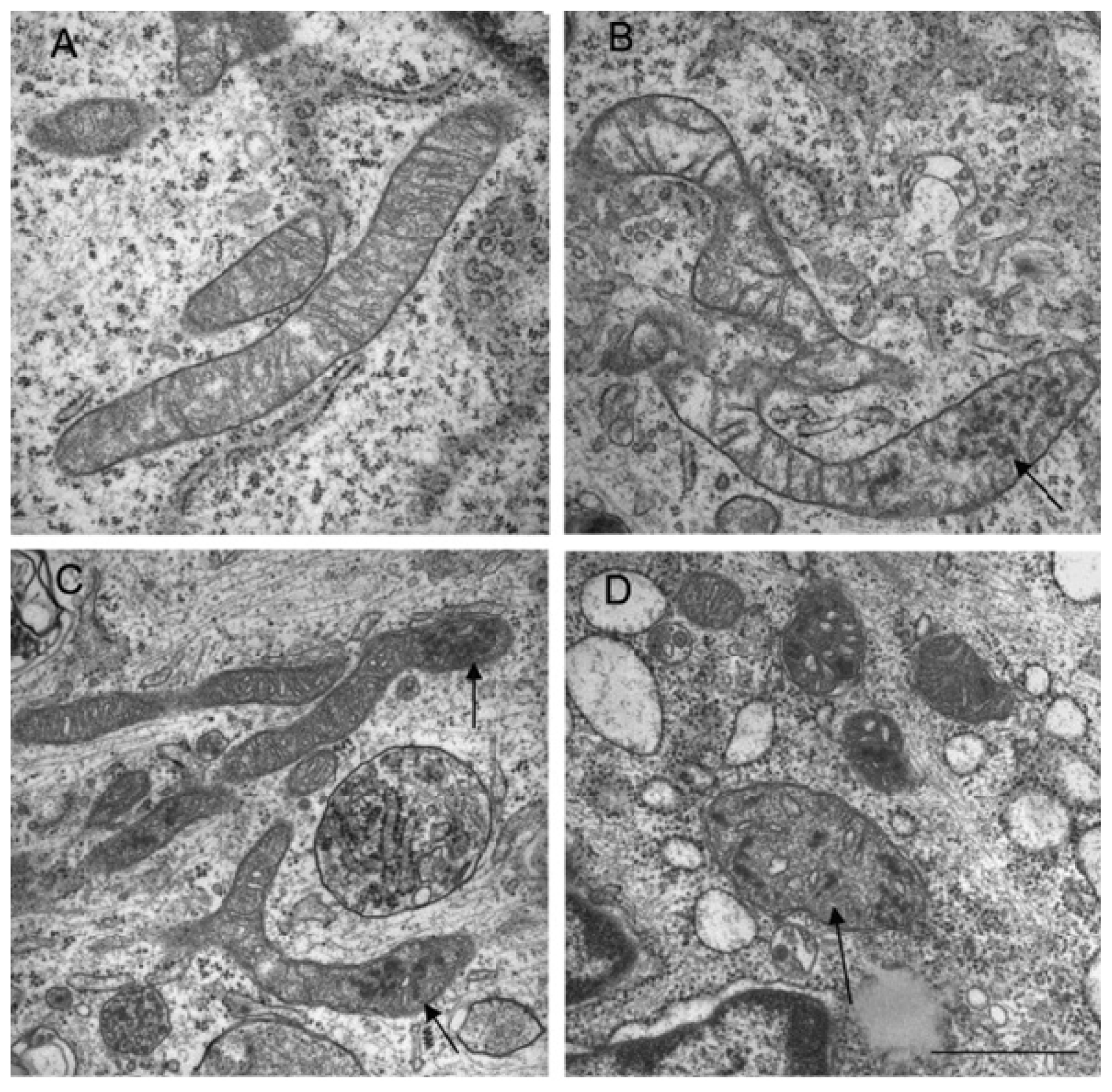
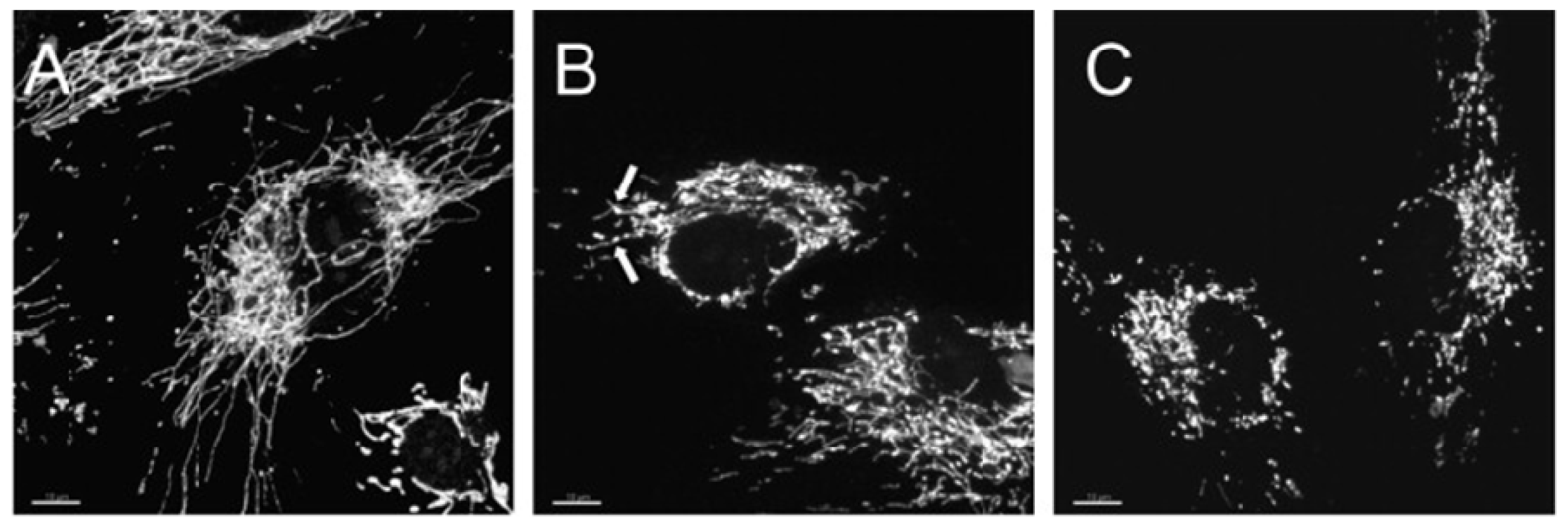
5. Mitochondria Network
6. Signaling
7. Modulation of Nrf2 by Reactive Oxygen and Nitrogen Species
8. NFκB Activation by Reactive Species
9. Other Signaling Pathways
10. Antioxidants
11. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol 1981, 91, 227s–255s. [Google Scholar]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci 2005, 118, 5411–5419. [Google Scholar]
- Lenaz, G. The mitochondrial production of reactive oxygen species: Mechanisms and implications in human pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar]
- Swerdlow, R.H. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 2011, 1812, 1630–1639. [Google Scholar]
- Griffiths, E.J. Mitochondria and heart disease. Adv. Exp. Med. Biol 2012, 942, 249–267. [Google Scholar]
- Birch-Machin, M.A. Mitochondria and skin disease. Clin. Exp. Dermatol 2000, 25, 141–146. [Google Scholar]
- Frohman, M.A. Mitochondria as integrators of signal transduction and energy production in cardiac physiology and disease. J. Mol. Med. (Berl. ) 2010, 88, 967–970. [Google Scholar]
- Grattagliano, I.; Russmann, S.; Diogo, C.; Bonfrate, L.; Oliveira, P.J.; Wang, D.Q.; Portincasa, P. Mitochondria in chronic liver disease. Curr. Drug Targets 2011, 12, 879–893. [Google Scholar]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Aspects Med 2004, 25, 365–451. [Google Scholar]
- Smith, R.A.; Adlam, V.J.; Blaikie, F.H.; Manas, A.R.; Porteous, C.M.; James, A.M.; Ross, M.F.; Logan, A.; Cocheme, H.M.; Trnka, J.; et al. Mitochondria-targeted antioxidants in the treatment of disease. Ann. N. Y. Acad. Sci 2008, 1147, 105–111. [Google Scholar]
- Gobe, G.; Crane, D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol. Lett 2010, 198, 49–55. [Google Scholar]
- Cloonan, S.M.; Choi, A.M. Mitochondria: Commanders of innate immunity and disease? Curr. Opin. Immunol 2012, 24, 32–40. [Google Scholar]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar]
- Schapira, A.H. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar]
- Armstrong, J.S. Mitochondrial medicine: Pharmacological targeting of mitochondria in disease. Br. J. Pharmacol 2007, 151, 1154–1165. [Google Scholar]
- Minocherhomji, S.; Tollefsbol, T.O.; Singh, K.K. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 2012, 7, 326–334. [Google Scholar]
- Rocha, M.; Apostolova, N.; Hernandez-Mijares, A.; Herance, R.; Victor, V.M. Oxidative stress and endothelial dysfunction in cardiovascular disease: Mitochondria-targeted therapeutics. Curr. Med. Chem 2010, 17, 3827–3841. [Google Scholar]
- Diogo, C.V.; Grattagliano, I.; Oliveira, P.J.; Bonfrate, L.; Portincasa, P. Re-wiring the circuit: Mitochondria as a pharmacological target in liver disease. Curr. Med. Chem 2011, 18, 5448–5465. [Google Scholar]
- Johannsen, D.L.; Ravussin, E. The role of mitochondria in health and disease. Curr. Opin. Pharmacol 2009, 9, 780–786. [Google Scholar]
- Duchen, M.R. Roles of mitochondria in health and disease. Diabetes 2004, 53, S96–S102. [Google Scholar]
- Duchen, M.R.; Szabadkai, G. Roles of mitochondria in human disease. Essays Biochem 2010, 47, 115–137. [Google Scholar]
- Hedskog, L.; Zhang, S.; Ankarcrona, M. Strategic role for mitochondria in Alzheimer’s disease and cancer. Antioxid. Redox Signal 2012, 16, 1476–1491. [Google Scholar]
- Watson, K.; Haslam, J.M.; Linnane, A.W. Biogenesis of mitochondria. 13. The isolation of mitochondrial structures from anaerobically grown Saccharomyces cerevisiae. J. Cell Biol 1970, 46, 88–96. [Google Scholar]
- Green, D.E.; Oda, T. On the unit of mitochondrial structure and function. J. Biochem 1961, 49, 742–757. [Google Scholar]
- Neupert, W. Protein import into mitochondria. Annu. Rev. Biochem 1997, 66, 863–917. [Google Scholar]
- Pfanner, N.; Craig, E.A.; Honlinger, A. Mitochondrial preprotein translocase. Annu. Rev. Cell Dev. Biol 1997, 13, 25–51. [Google Scholar]
- Dekker, P.J.; Ryan, M.T.; Brix, J.; Muller, H.; Honlinger, A.; Pfanner, N. Preprotein translocase of the outer mitochondrial membrane: Molecular dissection and assembly of the general import pore complex. Mol. Cell. Biol 1998, 18, 6515–6524. [Google Scholar]
- Yamamoto, H.; Itoh, N.; Kawano, S.; Yatsukawa, Y.; Momose, T.; Makio, T.; Matsunaga, M.; Yokota, M.; Esaki, M.; Shodai, T.; et al. Dual role of the receptor Tom20 in specificity and efficiency of protein import into mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 91–96. [Google Scholar]
- Herrmann, J.M.; Riemer, J. The intermembrane space of mitochondria. Antioxid. Redox Signal 2010, 13, 1341–1358. [Google Scholar]
- Davies, K.M.; Strauss, M.; Daum, B.; Kief, J.H.; Osiewacz, H.D.; Rycovska, A.; Zickermann, V.; Kuhlbrandt, W. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 14121–14126. [Google Scholar]
- O’Rourke, B. Mitochondrial ion channels. Annu. Rev. Physiol 2007, 69, 19–49. [Google Scholar]
- Asin-Cayuela, J.; Gustafsson, C.M. Mitochondrial transcription and its regulation in mammalian cells. Trends Biochem. Sci 2007, 32, 111–117. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J 1984, 219, 1–14. [Google Scholar]
- Cline, M.J.; Lehrer, R.I. d-amino acid oxidase in leukocytes: A possible d-amino-acid-linked antimicrobial system. Proc. Natl. Acad. Sci. USA 1969, 62, 756–763. [Google Scholar]
- Ji, L.L. Antioxidant signaling in skeletal muscle: A brief review. Exp. Gerontol 2007, 42, 582–593. [Google Scholar]
- Kim, C.; Kim, J.Y.; Kim, J.H. Cytosolic phospholipase A2, lipoxygenase metabolites, and reactive oxygen species. BMB Rep 2008, 41, 555–559. [Google Scholar]
- Nishino, T. The conversion of xanthine dehydrogenase to xanthine oxidase and the role of the enzyme in reperfusion injury. J. Biochem 1994, 116, 1–6. [Google Scholar]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J 2008, 275, 3278–3289. [Google Scholar]
- Sumimoto, H.; Miyano, K.; Takeya, R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem. Biophys. Res. Commun 2005, 338, 677–686. [Google Scholar]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol 2004, 199, 316–331. [Google Scholar]
- Brown, G.C. Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem. J 1992, 284, 1–13. [Google Scholar]
- Paul, T. Effect of a prolonged superoxide flux on transferrin and ferritin. Arch. Biochem. Biophys 2000, 382, 253–261. [Google Scholar]
- Biemond, P.; Swaak, A.J.; van Eijk, H.G.; Koster, J.F. Superoxide dependent iron release from ferritin in inflammatory diseases. Free Radic. Biol. Med 1988, 4, 185–198. [Google Scholar]
- Liochev, S.L. The role of iron-sulfur clusters in in vivo hydroxyl radical production. Free Radic. Res 1996, 25, 369–384. [Google Scholar]
- Flint, D.H.; Tuminello, J.F.; Emptage, M.H. The inactivation of Fe–S cluster containing hydro-lyases by superoxide. J. Biol. Chem 1993, 268, 22369–22376. [Google Scholar]
- Lenaz, G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv. Exp. Med. Biol 2012, 942, 93–136. [Google Scholar]
- McLennan, H.R.; Esposti, M.D. The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J. Bioenerg. Biomembr 2000, 32, 153–162. [Google Scholar]
- Fato, R.; Bergamini, C.; Leoni, S.; Strocchi, P.; Lenaz, G. Generation of reactive oxygen species by mitochondrial complex I: Implications in neurodegeneration. Neurochem. Res 2008, 33, 2487–2501. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J 2009, 417, 1–13. [Google Scholar]
- Yankovskaya, V.; Horsefield, R.; Tornroth, S.; Luna-Chavez, C.; Miyoshi, H.; Leger, C.; Byrne, B.; Cecchini, G.; Iwata, S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 2003, 299, 700–704. [Google Scholar]
- Casteilla, L.; Rigoulet, M.; Penicaud, L. Mitochondrial ROS metabolism: Modulation by uncoupling proteins. IUBMB Life 2001, 52, 181–188. [Google Scholar]
- Starkov, A.A.; Fiskum, G. Myxothiazol induces H2O2 production from mitochondrial respiratory chain. Biochem. Biophys. Res. Commun 2001, 281, 645–650. [Google Scholar]
- Barja, G. Mitochondrial oxygen radical generation and leak: Sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg. Biomembr 1999, 31, 347–366. [Google Scholar]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J 2002, 368, 545–553. [Google Scholar]
- Barja, G.; Herrero, A. Localization at complex I and mechanism of the higher free radical production of brain nonsynaptic mitochondria in the short-lived rat than in the longevous pigeon. J. Bioenerg. Biomembr 1998, 30, 235–243. [Google Scholar]
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J 1980, 191, 421–427. [Google Scholar]
- Miwa, S.; St-Pierre, J.; Partridge, L.; Brand, M.D. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic. Biol. Med 2003, 35, 938–948. [Google Scholar]
- Drahota, Z.; Chowdhury, S.K.; Floryk, D.; Mracek, T.; Wilhelm, J.; Rauchova, H.; Lenaz, G.; Houstek, J. Glycerophosphate-dependent hydrogen peroxide production by brown adipose tissue mitochondria and its activation by ferricyanide. J. Bioenerg. Biomembr 2002, 34, 105–113. [Google Scholar]
- Seifert, E.L.; Estey, C.; Xuan, J.Y.; Harper, M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem 2010, 285, 5748–5758. [Google Scholar]
- Maurel, A.; Hernandez, C.; Kunduzova, O.; Bompart, G.; Cambon, C.; Parini, A.; Frances, B. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am. J. Physiol. Heart Circ. Physiol 2003, 284, H1460–H1467. [Google Scholar]
- Starkov, A.A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci 2004, 24, 7779–7788. [Google Scholar]
- Tretter, L.; Adam-Vizi, V. Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J. Neurosci 2004, 24, 7771–7778. [Google Scholar]
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar]
- Lacza, Z.; Snipes, J.A.; Zhang, J.; Horvath, E.M.; Figueroa, J.P.; Szabo, C.; Busija, D.W. Mitochondrial nitric oxide synthase is not eNOS, nNOS or iNOS. Free Radic. Biol. Med 2003, 35, 1217–1228. [Google Scholar]
- Ghafourifar, P.; Richter, C. Nitric oxide synthase activity in mitochondria. FEBS Lett 1997, 418, 291–296. [Google Scholar]
- Giulivi, C.; Poderoso, J.J.; Boveris, A. Production of nitric oxide by mitochondria. J. Biol. Chem 1998, 273, 11038–11043. [Google Scholar]
- Alvarez, S.; Valdez, L.B.; Zaobornyj, T.; Boveris, A. Oxygen dependence of mitochondrial nitric oxide synthase activity. Biochem. Biophys. Res. Commun 2003, 305, 771–775. [Google Scholar]
- Lacza, Z.; Puskar, M.; Figueroa, J.P.; Zhang, J.; Rajapakse, N.; Busija, D.W. Mitochondrial nitric oxide synthase is constitutively active and is functionally upregulated in hypoxia. Free Radic. Biol. Med 2001, 31, 1609–1615. [Google Scholar]
- Bates, T.E.; Loesch, A.; Burnstock, G.; Clark, J.B. Immunocytochemical evidence for a mitochondrially located nitric oxide synthase in brain and liver. Biochem. Biophys. Res. Commun 1995, 213, 896–900. [Google Scholar]
- Haynes, V.; Elfering, S.; Traaseth, N.; Giulivi, C. Mitochondrial nitric-oxide synthase: Enzyme expression, characterization, and regulation. J. Bioenerg. Biomembr 2004, 36, 341–346. [Google Scholar]
- Giulivi, C. Mitochondria as generators and targets of nitric oxide. Novartis Found. Symp. 2007, 287, 92–100, discussion 100–104. [Google Scholar]
- Lacza, Z.; Kozlov, A.V.; Pankotai, E.; Csordas, A.; Wolf, G.; Redl, H.; Kollai, M.; Szabo, C.; Busija, D.W.; Horn, T.F. Mitochondria produce reactive nitrogen species via an arginine-independent pathway. Free Radic. Res 2006, 40, 369–378. [Google Scholar]
- Kozlov, A.V.; Staniek, K.; Nohl, H. Nitrite reductase activity is a novel function of mammalian mitochondria. FEBS Lett 1999, 454, 127–130. [Google Scholar]
- Lacza, Z.; Pankotai, E.; Csordas, A.; Gero, D.; Kiss, L.; Horvath, E.M.; Kollai, M.; Busija, D.W.; Szabo, C. Mitochondrial NO and reactive nitrogen species production: Does mtNOS exist? Nitric. Oxide 2006, 14, 162–168. [Google Scholar]
- Mason, M.G.; Nicholls, P.; Wilson, M.T.; Cooper, C.E. Nitric oxide inhibition of respiration involves both competitive (heme) and noncompetitive (copper) binding to cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2006, 103, 708–713. [Google Scholar]
- Cooper, C.E.; Giulivi, C. Nitric oxide regulation of mitochondrial oxygen consumption II: Molecular mechanism and tissue physiology. Am. J. Physiol. Cell Physiol 2007, 292, C1993–C2003. [Google Scholar]
- Gladwin, M.T.; Shiva, S. The ligand binding battle at cytochrome c oxidase: How NO regulates oxygen gradients in tissue. Circ. Res 2009, 104, 1136–1138. [Google Scholar]
- Brunori, M.; Forte, E.; Arese, M.; Mastronicola, D.; Giuffre, A.; Sarti, P. Nitric oxide and the respiratory enzyme. Biochim. Biophys. Acta 2006, 1757, 1144–1154. [Google Scholar]
- Sarti, P.; Forte, E.; Giuffre, A.; Mastronicola, D.; Magnifico, M.C.; Arese, M. The chemical interplay between nitric oxide and mitochondrial cytochrome c oxidase: Reactions, effectors and pathophysiology. Int. J. Cell Biol 2012, 2012, 571067. [Google Scholar]
- Poderoso, J.J.; Carreras, M.C.; Lisdero, C.; Riobo, N.; Schopfer, F.; Boveris, A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch. Biochem. Biophys 1996, 328, 85–92. [Google Scholar]
- Brown, G.C.; Borutaite, V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. Biophys. Acta 2004, 1658, 44–49. [Google Scholar]
- Nisoli, E.; Falcone, S.; Tonello, C.; Cozzi, V.; Palomba, L.; Fiorani, M.; Pisconti, A.; Brunelli, S.; Cardile, A.; Francolini, M.; et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. USA 2004, 101, 16507–16512. [Google Scholar]
- Shen, W.; Hintze, T.H.; Wolin, M.S. Nitric oxide. An important signaling mechanism between vascular endothelium and parenchymal cells in the regulation of oxygen consumption. Circulation 1995, 92, 3505–3512. [Google Scholar]
- Brown, G.C. Nitric oxide and mitochondrial respiration. Biochim. Biophys. Acta 1999, 1411, 351–369. [Google Scholar]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar]
- Xie, Y.W.; Kaminski, P.M.; Wolin, M.S. Inhibition of rat cardiac muscle contraction and mitochondrial respiration by endogenous peroxynitrite formation during posthypoxic reoxygenation. Circ. Res 1998, 82, 891–897. [Google Scholar]
- Radi, R.; Rodriguez, M.; Castro, L.; Telleri, R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch. Biochem. Biophys 1994, 308, 89–95. [Google Scholar]
- Piantadosi, C.A.; Suliman, H.B. Redox regulation of mitochondrial biogenesis. Free Radic. Biol. Med 2012, 53, 2043–2053. [Google Scholar]
- Bourens, M.; Fontanesi, F.; Soto, I.C.; Liu, J.; Barrientos, A. Redox and reactive oxygen species regulation of mitochondrial cytochrome c oxidase biogenesis. Antioxid. Redox Signal. 2012. [Google Scholar] [CrossRef]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar]
- Nisoli, E.; Clementi, E.; Moncada, S.; Carruba, M.O. Mitochondrial biogenesis as a cellular signaling framework. Biochem. Pharmacol 2004, 67, 1–15. [Google Scholar]
- Lee, H.C.; Wei, Y.H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol 2005, 37, 822–834. [Google Scholar]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 2010, 5, e11707. [Google Scholar]
- Piantadosi, C.A.; Carraway, M.S.; Haden, D.W.; Suliman, H.B. Protecting the permeability pore and mitochondrial biogenesis. Novartis Found. Symp. 2007, 280, 266–276, discussion 276–280. [Google Scholar]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal 2012, 16, 1323–1367. [Google Scholar]
- Le Bras, M.; Clement, M.V.; Pervaiz, S.; Brenner, C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol 2005, 20, 205–219. [Google Scholar]
- Ma, Z.A. The role of peroxidation of mitochondrial membrane phospholipids in pancreatic beta-cell failure. Curr. Diabetes Rev 2012, 8, 69–75. [Google Scholar]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar]
- Mizushima, N. Autophagy: Process and function. Genes Dev 2007, 21, 2861–2873. [Google Scholar]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid. Redox Signal 2006, 8, 152–162. [Google Scholar]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr 2007, 27, 19–40. [Google Scholar]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol 2009, 10, 458–467. [Google Scholar]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. Cell 2003, 5, 539–545.
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal 2009, 11, 777–790. [Google Scholar]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007, 26, 1749–1760. [Google Scholar]
- Mathew, R.; White, E. Autophagy, stress, and cancer metabolism: What doesn’t kill you makes you stronger. Cold Spring Harb. Symp. Quant. Biol 2011, 76, 389–396. [Google Scholar]
- Li, Z.Y.; Yang, Y.; Ming, M.; Liu, B. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem. Biophys. Res. Commun 2011, 414, 5–8. [Google Scholar]
- Ishdorj, G.; Li, L.; Gibson, S.B. Regulation of autophagy in hematological malignancies: Role of reactive oxygen species. Leuk Lymphoma 2012, 53, 26–33. [Google Scholar]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar]
- Cuervo, A.M. Autophagy and aging—When “all you can eat” is yourself. Sci. Aging Knowl. Environ 2003, 2003, 25. [Google Scholar]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ 2005, 12, 1509–1518. [Google Scholar]
- Yuan, H.; Perry, C.N.; Huang, C.; Iwai-Kanai, E.; Carreira, R.S.; Glembotski, C.C.; Gottlieb, R.A. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am. J. Physiol. Heart Circ. Physiol 2009, 296, H470–H479. [Google Scholar]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 2008, 15, 171–182. [Google Scholar]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci 2007, 120, 4155–4166. [Google Scholar]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009, 16, 1040–1052. [Google Scholar]
- Yang, J.; Wu, L.J.; Tashino, S.; Onodera, S.; Ikejima, T. Reactive oxygen species and nitric oxide regulate mitochondria-dependent apoptosis and autophagy in evodiamine-treated human cervix carcinoma HeLa cells. Free Radic. Res 2008, 42, 492–504. [Google Scholar]
- Bolisetty, S.; Traylor, A.M.; Kim, J.; Joseph, R.; Ricart, K.; Landar, A.; Agarwal, A. Heme oxygenase-1 inhibits renal tubular macroautophagy in acute kidney injury. J. Am. Soc. Nephrol 2010, 21, 1702–1712. [Google Scholar]
- Morse, D.; Lin, L.; Choi, A.M.; Ryter, S.W. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury and disease. Free Radic. Biol. Med 2009, 47, 1–12. [Google Scholar]
- Byun, Y.J.; Kim, S.K.; Kim, Y.M.; Chae, G.T.; Jeong, S.W.; Lee, S.B. Hydrogen peroxide induces autophagic cell death in C6 glioma cells via BNIP3-mediated suppression of the mTOR pathway. Neurosci. Lett 2009, 461, 131–135. [Google Scholar]
- Zhang, H.; Kong, X.; Kang, J.; Su, J.; Li, Y.; Zhong, J.; Sun, L. Oxidative stress induces parallel autophagy and mitochondria dysfunction in human glioma U251 cells. Toxicol. Sci 2009, 110, 376–388. [Google Scholar]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell Physiol 2010, 298, C542–549. [Google Scholar]
- Wong, C.H.; Iskandar, K.B.; Yadav, S.K.; Hirpara, J.L.; Loh, T.; Pervaiz, S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS One 2010, 5, e9996. [Google Scholar]
- Wang, S.H.; Shih, Y.L.; Kuo, T.C.; Ko, W.C.; Shih, C.M. Cadmium toxicity toward autophagy through ROS-activated GSK-3beta in mesangial cells. Toxicol. Sci 2009, 108, 124–131. [Google Scholar]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar]
- Mitroulis, I.; Kourtzelis, I.; Kambas, K.; Rafail, S.; Chrysanthopoulou, A.; Speletas, M.; Ritis, K. Regulation of the autophagic machinery in human neutrophils. Eur. J. Immunol 2010, 40, 1461–1472. [Google Scholar]
- Huang, J.; Brumell, J.H. NADPH oxidases contribute to autophagy regulation. Autophagy 2009, 5, 887–889. [Google Scholar]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar]
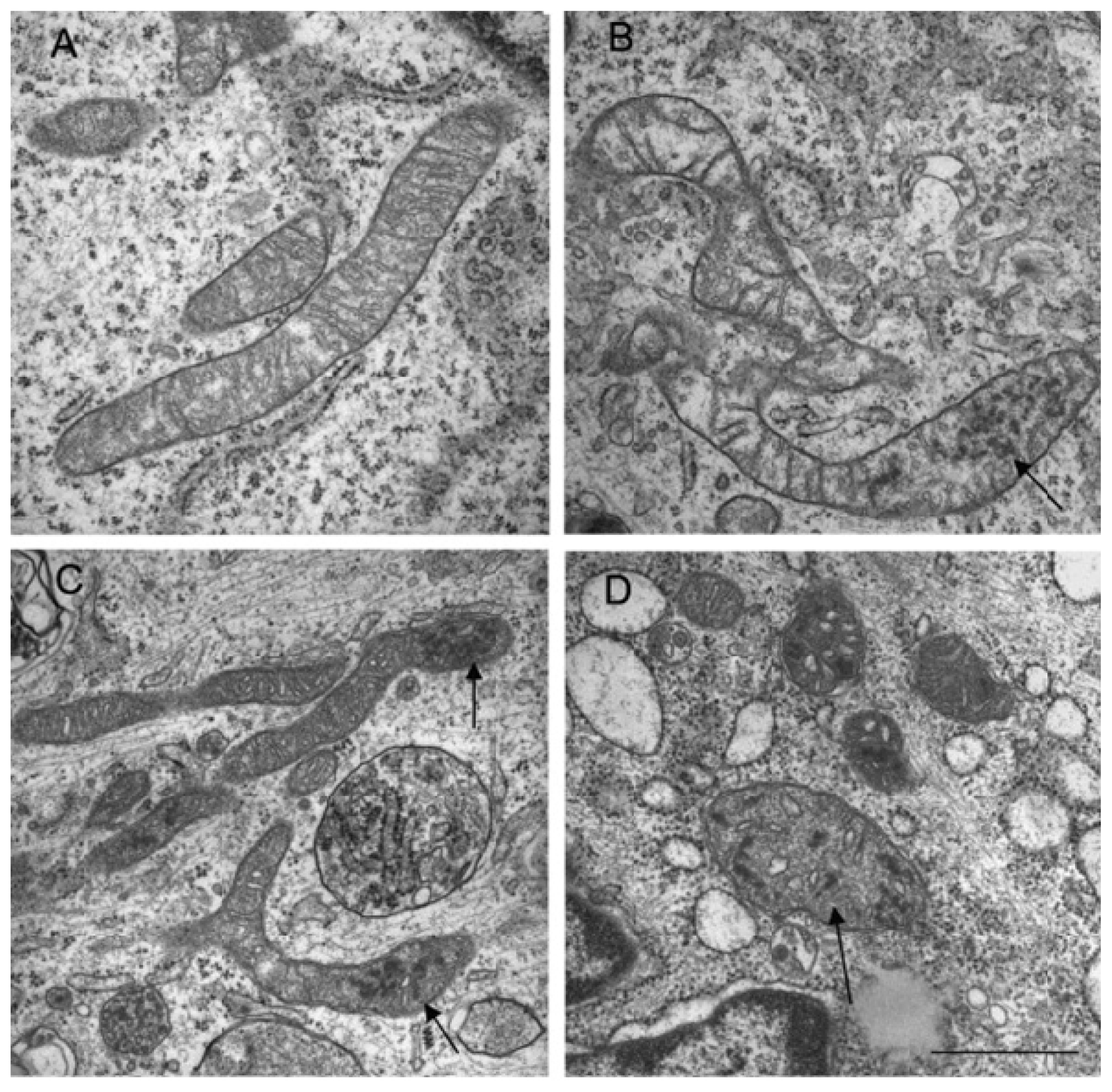
- Kissova, I.; Deffieu, M.; Manon, S.; Camougrand, N. Uth1p is involved in the autophagic degradation of mitochondria. J. Biol. Chem 2004, 279, 39068–39074. [Google Scholar]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005, 8, 3–5. [Google Scholar]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010, 8, e1000298. [Google Scholar]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol 2010, 191, 933–942. [Google Scholar]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol 2008, 183, 795–803. [Google Scholar]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol 2010, 189, 211–221. [Google Scholar]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol 2010, 12, 119–131. [Google Scholar]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar]
- Nishida, K.; Yamaguchi, O.; Otsu, K. Crosstalk between autophagy and apoptosis in heart disease. Circ. Res 2008, 103, 343–351. [Google Scholar]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar]
- Benard, G.; Karbowski, M. Mitochondrial fusion and division: Regulation and role in cell viability. Semin. Cell Dev. Biol 2009, 20, 365–374. [Google Scholar]
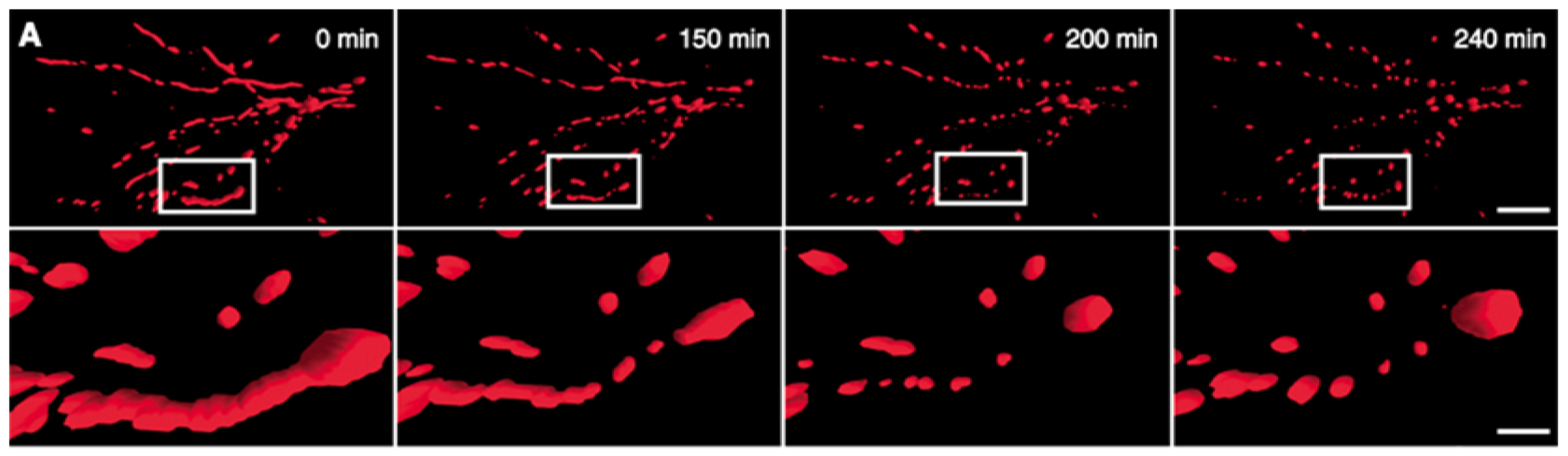
- Pletjushkina, O.Y.; Lyamzaev, K.G.; Popova, E.N.; Nepryakhina, O.K.; Ivanova, O.Y.; Domnina, L.V.; Chernyak, B.V.; Skulachev, V.P. Effect of oxidative stress on dynamics of mitochondrial reticulum. Biochim. Biophys. Acta 2006, 1757, 518–524. [Google Scholar]
- Fan, X.; Hussien, R.; Brooks, G.A. H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic. Biol. Med 2010, 49, 1646–1654. [Google Scholar]
- Jendrach, M.; Mai, S.; Pohl, S.; Voth, M.; Bereiter-Hahn, J. Short- and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion 2008, 8, 293–304. [Google Scholar]
- Makino, A.; Scott, B.T.; Dillmann, W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010, 53, 1783–1794. [Google Scholar]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Graber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 2006, 25, 3900–3911. [Google Scholar]
- Liot, G.; Bossy, B.; Lubitz, S.; Kushnareva, Y.; Sejbuk, N.; Bossy-Wetzel, E. Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ 2009, 16, 899–909. [Google Scholar]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Invest 2009, 119, 1275–1285. [Google Scholar]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Renal Physiol 2012, 302, F853–F864. [Google Scholar]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res 2008, 79, 341–351. [Google Scholar]
- Giedt, R.J.; Yang, C.; Zweier, J.L.; Matzavinos, A.; Alevriadou, B.R. Mitochondrial fission in endothelial cells after simulated ischemia/reperfusion: Role of nitric oxide and reactive oxygen species. Free Radic. Biol. Med 2012, 52, 348–356. [Google Scholar]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Del Mar Arroyo-Jimenez, M.; Jordan, J.; Galindo, M.F. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim. Biophys. Acta 2012, 1832, 174–182. [Google Scholar]
- Estaquier, J.; Arnoult, D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ 2007, 14, 1086–1094. [Google Scholar]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar]
- Lee, Y.J.; Jeong, S.Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar]
- Sugioka, R.; Shimizu, S.; Tsujimoto, Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J. Biol. Chem 2004, 279, 52726–52734. [Google Scholar]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem 2003, 278, 36373–36379. [Google Scholar]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem 2003, 278, 7743–7746. [Google Scholar]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.C. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One 2008, 3, e3257. [Google Scholar]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci 2007, 120, 838–848. [Google Scholar]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 2012, 1823, 2297–2310. [Google Scholar]
- Park, J.; Lee, J.; Choi, C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS One 2011, 6, e23211. [Google Scholar]
- Jou, M.J. Pathophysiological and pharmacological implications of mitochondria-targeted reactive oxygen species generation in astrocytes. Adv. Drug Deliv. Rev 2008, 60, 1512–1526. [Google Scholar]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial dynamics in diabetes. Antioxid. Redox Signal 2011, 14, 439–457. [Google Scholar]
- Duncan, J.G. Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim. Biophys. Acta 2011, 1813, 1351–1359. [Google Scholar]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res 2010, 88, 16–29. [Google Scholar]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J 2010, 29, 1774–1785. [Google Scholar]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res 2009, 84, 91–99. [Google Scholar]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem 2011, 286, 20710–20726. [Google Scholar]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci 2009, 29, 9090–9103. [Google Scholar]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Invest 1973, 52, 741–744. [Google Scholar]
- Babior, B.M.; Peters, W.A. The O2-producing enzyme of human neutrophils. Further properties. J. Biol. Chem 1981, 256, 2321–2323. [Google Scholar]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar]
- Gruetter, C.A.; Barry, B.K.; McNamara, D.B.; Gruetter, D.Y.; Kadowitz, P.J.; Ignarro, L. Relaxation of bovine coronary artery and activation of coronary arterial guanylate cyclase by nitric oxide, nitroprusside and a carcinogenic nitrosoamine. J. Cyclic Nucleotide Res 1979, 5, 211–224. [Google Scholar]
- Furchgott, R.F.; Carvalho, M.H.; Khan, M.T.; Matsunaga, K. Evidence for endothelium-dependent vasodilation of resistance vessels by acetylcholine. Blood Vessels 1987, 24, 145–149. [Google Scholar]
- Leclercq, B.; Jaimes, E.A.; Raij, L. Nitric oxide synthase and hypertension. Curr. Opin. Nephrol. Hypertens 2002, 11, 185–189. [Google Scholar]
- Tabima, D.M.; Frizzell, S.; Gladwin, M.T. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic. Biol. Med 2012, 52, 1970–1986. [Google Scholar]
- Tousoulis, D.; Kampoli, A.M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol 2012, 10, 4–18. [Google Scholar]
- Jaimes, E.A.; Hua, P.; Tian, R.X.; Raij, L. Human glomerular endothelium: Interplay among glucose, free fatty acids, angiotensin II, and oxidative stress. Am. J. Physiol. Renal Physiol 2010, 298, F125–F132. [Google Scholar]
- Radomski, M.W.; Moncada, S. Regulation of vascular homeostasis by nitric oxide. Thromb Haemost 1993, 70, 36–41. [Google Scholar]
- Frazziano, G.; Champion, H.C.; Pagano, P.J. NADPH oxidase-derived ROS and the regulation of pulmonary vessel tone. Am. J. Physiol. Heart Circ. Physiol 2012, 302, H2166–H2177. [Google Scholar]
- Satoh, K.; Berk, B.C.; Shimokawa, H. Vascular-derived reactive oxygen species for homeostasis and diseases. Nitric Oxide 2011, 25, 211–215. [Google Scholar]
- Erusalimsky, J.D.; Moncada, S. Nitric oxide and mitochondrial signaling: From physiology to pathophysiology. Arterioscler. Thromb. Vasc. Biol 2007, 27, 2524–2531. [Google Scholar]
- Napoli, C.; Ignarro, L.J. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch. Pharm. Res 2009, 32, 1103–1108. [Google Scholar]
- Zhou, M.S.; Jaimes, E.A.; Raij, L. Vascular but not cardiac remodeling is associated with superoxide production in angiotensin II hypertension. J. Hypertens 2005, 23, 1737–1743. [Google Scholar]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H2O2 signaling. Antioxid. Redox Signal 2011, 14, 459–468. [Google Scholar]
- Groeger, G.; Quiney, C.; Cotter, T.G. Hydrogen peroxide as a cell-survival signaling molecule. Antioxid. Redox Signal 2009, 11, 2655–2671. [Google Scholar]
- Miller, A.A.; Drummond, G.R.; Sobey, C.G. Reactive oxygen species in the cerebral circulation: Are they all bad? Antioxid. Redox Signal 2006, 8, 1113–1120. [Google Scholar]
- Kamsler, A.; Segal, M. Hydrogen peroxide as a diffusible signal molecule in synaptic plasticity. Mol. NeuroBiol 2004, 29, 167–178. [Google Scholar]
- Kochevar, I.E. Singlet oxygen signaling: From intimate to global. Sci. STKE 2004, 2004, pe7. [Google Scholar]
- Turpaev, K.T. Reactive oxygen species and regulation of gene expression. Biochemistry (Mosc. ) 2002, 67, 281–292. [Google Scholar]
- Kim, P.K.; Zamora, R.; Petrosko, P.; Billiar, T.R. The regulatory role of nitric oxide in apoptosis. Int. Immunopharmacol 2001, 1, 1421–1441. [Google Scholar]
- Martin, L.D.; Krunkosky, T.M.; Voynow, J.A.; Adler, K.B. The role of reactive oxygen and nitrogen species in airway epithelial gene expression. Environ. Health Perspect 1998, 106, 1197–1203. [Google Scholar]
- Ho, J.J.; Man, H.S.; Marsden, P.A. Nitric oxide signaling in hypoxia. J. Mol. Med. (Berl. ) 2012, 90, 217–231. [Google Scholar]
- Martinez-Ruiz, A.; Cadenas, S.; Lamas, S. Nitric oxide signaling: Classical, less classical, and nonclassical mechanisms. Free Radic. Biol. Med 2011, 51, 17–29. [Google Scholar]
- Bryan, N.S.; Bian, K.; Murad, F. Discovery of the nitric oxide signaling pathway and targets for drug development. Front. Biosci 2009, 14, 1–18. [Google Scholar]
- Afanas’ev, I.B. Signaling functions of free radicals superoxide & nitric oxide under physiological & pathological conditions. Mol. Biotechnol 2007, 37, 2–4. [Google Scholar]
- Buetler, T.M.; Krauskopf, A.; Ruegg, U.T. Role of superoxide as a signaling molecule. News Physiol. Sci 2004, 19, 120–123. [Google Scholar]
- Veal, E.; Day, A. Hydrogen peroxide as a signaling molecule. Antioxid. Redox Signal 2011, 15, 147–151. [Google Scholar]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal 2006, 8, 243–270. [Google Scholar]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med 1999, 31, 53–59. [Google Scholar]
- Lander, H.M. An essential role for free radicals and derived species in signal transduction. FASEB J 1997, 11, 118–124. [Google Scholar]
- Hensley, K.; Robinson, K.A.; Gabbita, S.P.; Salsman, S.; Floyd, R.A. Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med 2000, 28, 1456–1462. [Google Scholar]
- Chen, X.L.; Kunsch, C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: A new therapeutic approach for the treatment of inflammatory diseases. Curr. Pharm. Des 2004, 10, 879–891. [Google Scholar]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun 1997, 236, 313–322. [Google Scholar]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 2002, 62, 5196–5203. [Google Scholar]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res 2012, 40, 7416–7429. [Google Scholar]
- Taylor, R.C.; Acquaah-Mensah, G.; Singhal, M.; Malhotra, D.; Biswal, S. Network inference algorithms elucidate Nrf2 regulation of mouse lung oxidative stress. PLoS Comput. Biol 2008, 4, e1000166. [Google Scholar]
- Numazawa, S.; Yoshida, T. Nrf2-dependent gene expressions: A molecular toxicological aspect. J. Toxicol. Sci 2004, 29, 81–89. [Google Scholar]
- Reddy, N.M.; Kleeberger, S.R.; Yamamoto, M.; Kensler, T.W.; Scollick, C.; Biswal, S.; Reddy, S.P. Genetic dissection of the Nrf2-dependent redox signaling-regulated transcriptional programs of cell proliferation and cytoprotection. Physiol. Genomics 2007, 32, 74–81. [Google Scholar]
- Cheng, X.; Siow, R.C.; Mann, G.E. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: A role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid. Redox Signal 2011, 14, 469–487. [Google Scholar]
- Van Muiswinkel, F.L.; Kuiperij, H.B. The Nrf2-ARE Signalling pathway: Promising drug target to combat oxidative stress in neurodegenerative disorders. Curr. Drug Targets CNS Neurol. Disord 2005, 4, 267–281. [Google Scholar]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med 2009, 47, 1304–1309. [Google Scholar]
- Apopa, P.L.; He, X.; Ma, Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J. Biochem. Mol. Toxicol 2008, 22, 63–76. [Google Scholar]
- Numazawa, S.; Ishikawa, M.; Yoshida, A.; Tanaka, S.; Yoshida, T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am. J. Physiol. Cell Physiol 2003, 285, C334–C342. [Google Scholar]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999, 13, 76–86. [Google Scholar]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The role of Keap1 in cellular protective responses. Chem. Res. Toxicol 2005, 18, 1779–1791. [Google Scholar]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol 2004, 24, 7130–7139. [Google Scholar]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol 2004, 24, 10941–10953. [Google Scholar]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev 2006, 38, 769–789. [Google Scholar]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem 2006, 387, 1311–1320. [Google Scholar]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol 2007, 47, 89–116. [Google Scholar]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal 2010, 13, 1699–1712. [Google Scholar]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox Signal 2010, 13, 1665–1678. [Google Scholar]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol 2011, 85, 241–272. [Google Scholar]
- Tkachev, V.O.; Menshchikova, E.B.; Zenkov, N.K. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry (Mosc. ) 2011, 76, 407–422. [Google Scholar]
- Dhakshinamoorthy, S.; Porter, A.G. Nitric oxide-induced transcriptional up-regulation of protective genes by Nrf2 via the antioxidant response element counteracts apoptosis of neuroblastoma cells. J. Biol. Chem 2004, 279, 20096–20107. [Google Scholar]
- Wilson, L.A.; Gemin, A.; Espiritu, R.; Singh, G. ets-1 is transcriptionally up-regulated by H2O2 via an antioxidant response element. FASEB J 2005, 19, 2085–2087. [Google Scholar]
- Ashino, T.; Yamanaka, R.; Yamamoto, M.; Shimokawa, H.; Sekikawa, K.; Iwakura, Y.; Shioda, S.; Numazawa, S.; Yoshida, T. Negative feedback regulation of lipopolysaccharide-induced inducible nitric oxide synthase gene expression by heme oxygenase-1 induction in macrophages. Mol. Immunol 2008, 45, 2106–2115. [Google Scholar]
- Buckley, B.J.; Marshall, Z.M.; Whorton, A.R. Nitric oxide stimulates Nrf2 nuclear translocation in vascular endothelium. Biochem. Biophys. Res. Commun 2003, 307, 973–979. [Google Scholar]
- Fourquet, S.; Guerois, R.; Biard, D.; Toledano, M.B. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem 2010, 285, 8463–8471. [Google Scholar]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med 2004, 10, 549–557. [Google Scholar]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol 1998, 16, 225–260. [Google Scholar]
- Farrow, S.N. Death receptors, NF-kappa B activation and apoptosis: The potential for therapeutic intervention. Biochem. Soc. Trans 1999, 27, 812–814. [Google Scholar]
- Van Antwerp, D.J.; Martin, S.J.; Verma, I.M.; Green, D.R. Inhibition of TNF-induced apoptosis by NF-kappa B. Trends Cell Biol 1998, 8, 107–111. [Google Scholar]
- Sonenshein, G.E. Rel/NF-kappa B transcription factors and the control of apoptosis. Semin. Cancer Biol 1997, 8, 113–119. [Google Scholar]
- Francis, D.A.; Sen, R.; Rothstein, T.L. Receptor-specific regulation of NF-kappa B, c-Myc and Fas-mediated apoptosis in primary B cells. Curr. Top. MicroBiol. Immunol 1997, 224, 83–90. [Google Scholar]
- Wang, H.; Cho, C.H. Effect of NF-kappaB signaling on apoptosis in chronic inflammation-associated carcinogenesis. Curr. Cancer Drug Targets 2010, 10, 593–599. [Google Scholar]
- Piotrowska, A.; Izykowska, I.; Podhorska-Okolow, M.; Zabel, M.; Dziegiel, P. The structure of NF-kappaB family proteins and their role in apoptosis. Postepy Hig. Med. Dosw. (Online) 2008, 62, 64–74. [Google Scholar]
- Sen, R. Control of B lymphocyte apoptosis by the transcription factor NF-kappaB. Immunity 2006, 25, 871–883. [Google Scholar]
- Kucharczak, J.; Simmons, M.J.; Fan, Y.; Gelinas, C. To be, or not to be: NF-kappaB is the answer—Role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene 2003, 22, 8961–8982. [Google Scholar]
- Barkett, M.; Gilmore, T.D. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6910–6924. [Google Scholar]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the link between inflammation and cancer. Immunol. Rev 2012, 246, 379–400. [Google Scholar]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol 2011, 12, 715–723. [Google Scholar]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab 2011, 13, 11–22. [Google Scholar]
- Sanz, A.B.; Sanchez-Nino, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-kappaB in renal inflammation. J. Am. Soc. Nephrol 2010, 21, 1254–1262. [Google Scholar]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol 2009, 1, a001651. [Google Scholar]
- Lawrence, T.; Fong, C. The resolution of inflammation: Anti-inflammatory roles for NF-kappaB. Int. J. Biochem. Cell Biol 2010, 42, 519–523. [Google Scholar]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Ho, W.; Banerjee, A. Unravelling the complexities of the NF-kappaB signalling pathway using mouse knockout and transgenic models. Oncogene 2006, 25, 6781–6799. [Google Scholar]
- Monaco, C.; Andreakos, E.; Kiriakidis, S.; Mauri, C.; Bicknell, C.; Foxwell, B.; Cheshire, N.; Paleolog, E.; Feldmann, M. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 5634–5639. [Google Scholar]
- Song, X.Q.; Lv, L.X.; Li, W.Q.; Hao, Y.H.; Zhao, J.P. The interaction of nuclear factor-kappa B and cytokines is associated with schizophrenia. Biol. Psychiatry 2009, 65, 481–488. [Google Scholar]
- Yamamoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J. Clin. Invest 2001, 107, 135–142. [Google Scholar]
- Yamamoto, Y.; Gaynor, R.B. Role of the NF-kappaB pathway in the pathogenesis of human disease states. Curr. Mol. Med 2001, 1, 287–296. [Google Scholar]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar]
- Yamamoto, Y.; Gaynor, R.B. IkappaB kinases: Key regulators of the NF-kappaB pathway. Trends Biochem. Sci 2004, 29, 72–79. [Google Scholar]
- Hansen, S.K.; Guerrini, L.; Blasi, F. Differential DNA sequence specificity and regulation of HIV-1 enhancer activity by cRel-RelA transcription factor. J. Biol. Chem 1994, 269, 22230–22237. [Google Scholar]
- Brown, A.M.; Linhoff, M.W.; Stein, B.; Wright, K.L.; Baldwin, A.S., Jr; Basta, P.V.; Ting, J.P. Function of NF-kappa B/Rel binding sites in the major histocompatibility complex class II invariant chain promoter is dependent on cell-specific binding of different NF-kappa B/Rel subunits. Mol. Cell Biol. 1994, 14, 2926–2935. [Google Scholar]
- Plaksin, D.; Baeuerle, P.A.; Eisenbach, L. KBF1 (p50 NF-kappa B homodimer) acts as a repressor of H-2Kb gene expression in metastatic tumor cells. J. Exp. Med 1993, 177, 1651–1662. [Google Scholar]
- Kone, B.C.; Schwobel, J.; Turner, P.; Mohaupt, M.G.; Cangro, C.B. Role of NF-kappa B in the regulation of inducible nitric oxide synthase in an MTAL cell line. Am. J. Physiol 1995, 269, F718–F729. [Google Scholar]
- Pahan, K.; Sheikh, F.G.; Liu, X.; Hilger, S.; McKinney, M.; Petro, T.M. Induction of nitric-oxide synthase and activation of NF-kappaB by interleukin-12 p40 in microglial cells. J. Biol. Chem 2001, 276, 7899–7905. [Google Scholar]
- Fouad, D.; Siendones, E.; Costan, G.; Muntane, J. Role of NF-kappaB activation and nitric oxide expression during PGE protection against d-galactosamine-induced cell death in cultured rat hepatocytes. Liver Int 2004, 24, 227–236. [Google Scholar]
- Griscavage, J.M.; Wilk, S.; Ignarro, L.J. Inhibitors of the proteasome pathway interfere with induction of nitric oxide synthase in macrophages by blocking activation of transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 1996, 93, 3308–3312. [Google Scholar]
- Lee, K.M.; Kang, B.S.; Lee, H.L.; Son, S.J.; Hwang, S.H.; Kim, D.S.; Park, J.S.; Cho, H.J. Spinal NF-kB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. Eur. J. NeuroSci 2004, 19, 3375–3381. [Google Scholar]
- Schmedtje, J.F., Jr; Ji, Y.S.; Liu, W.L.; DuBois, R.N.; Runge, M.S. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J. Biol. Chem. 1997, 272, 601–608. [Google Scholar]
- Jobin, C.; Morteau, O.; Han, D.S.; Balfour Sartor, R. Specific NF-kappaB blockade selectively inhibits tumour necrosis factor-alpha-induced COX-2 but not constitutive COX-1 gene expression in HT-29 cells. Immunology 1998, 95, 537–543. [Google Scholar]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J 1991, 10, 2247–2258. [Google Scholar]
- Schreck, R.; Meier, B.; Mannel, D.N.; Droge, W.; Baeuerle, P.A. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J. Exp. Med 1992, 175, 1181–1194. [Google Scholar]
- Schreck, R.; Grassmann, R.; Fleckenstein, B.; Baeuerle, P.A. Antioxidants selectively suppress activation of NF-kappa B by human T-cell leukemia virus type I Tax protein. J. Virol 1992, 66, 6288–6293. [Google Scholar]
- Lander, H.M.; Sehajpal, P.; Levine, D.M.; Novogrodsky, A. Activation of human peripheral blood mononuclear cells by nitric oxide-generating compounds. J. Immunol 1993, 150, 1509–1516. [Google Scholar]
- Shibanuma, M.; Kuroki, T.; Nose, K. Inhibition by N-acetyl-l-cysteine of interleukin-6 mRNA induction and activation of NF kappa B by tumor necrosis factor alpha in a mouse fibroblastic cell line, Balb/3T3. FEBS Lett 1994, 353, 62–66. [Google Scholar]
- Manna, S.K.; Zhang, H.J.; Yan, T.; Oberley, L.W.; Aggarwal, B.B. Overexpression of manganese superoxide dismutase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappaB and activated protein-1. J. Biol. Chem 1998, 273, 13245–13254. [Google Scholar]
- Meyer, M.; Schreck, R.; Baeuerle, P.A. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J 1993, 12, 2005–2015. [Google Scholar]
- Kim, S.J.; Hwang, S.G.; Shin, D.Y.; Kang, S.S.; Chun, J.S. p38 kinase regulates nitric oxide-induced apoptosis of articular chondrocytes by accumulating p53 via NFkappa B-dependent transcription and stabilization by serine 15 phosphorylation. J. Biol. Chem 2002, 277, 33501–33508. [Google Scholar]
- Lee, S.; Chung, J.; Ha, I.S.; Yi, K.; Lee, J.E.; Kang, H.G.; Choi, I.; Oh, K.H.; Kim, J.Y.; Surh, C.D.; et al. Hydrogen peroxide increases human leukocyte adhesion to porcine aortic endothelial cells via NFkappaB-dependent up-regulation of VCAM-1. Int. Immunol 2007, 19, 1349–1359. [Google Scholar]
- Zhang, J.; Johnston, G.; Stebler, B.; Keller, E.T. Hydrogen peroxide activates NFkappaB and the interleukin-6 promoter through NFkappaB-inducing kinase. Antioxid. Redox Signal 2001, 3, 493–504. [Google Scholar]
- Waldow, T.; Witt, W.; Weber, E.; Matschke, K. Nitric oxide donor-induced persistent inhibition of cell adhesion protein expression and NFkappaB activation in endothelial cells. Nitric Oxide 2006, 15, 103–113. [Google Scholar]
- Nam, H.Y.; Choi, B.H.; Lee, J.Y.; Lee, S.G.; Kim, Y.H.; Lee, K.H.; Yoon, H.K.; Song, J.S.; Kim, H.J.; Lim, Y. The role of nitric oxide in the particulate matter (PM2.5)-induced NFkappaB activation in lung epithelial cells. Toxicol. Lett 2004, 148, 95–102. [Google Scholar]
- Tsai, S.H.; Lin-Shiau, S.Y.; Lin, J.K. Suppression of nitric oxide synthase and the down-regulation of the activation of NFkappaB in macrophages by resveratrol. Br. J. Pharmacol 1999, 126, 673–680. [Google Scholar]
- Schreck, R.; Albermann, K.; Baeuerle, P.A. Nuclear factor kappa B: An oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic. Res. Commun 1992, 17, 221–237. [Google Scholar]
- Packer, L.; Suzuki, Y.J. Vitamin E and alpha-lipoate: Role in antioxidant recycling and activation of the NF-kappa B transcription factor. Mol. Aspects Med 1993, 14, 229–239. [Google Scholar]
- Suzuki, Y.J.; Packer, L. Inhibition of NF-kappa B DNA binding activity by alpha-tocopheryl succinate. Biochem. Mol. Biol. Int 1993, 31, 693–700. [Google Scholar]
- Kretz-Remy, C.; Mehlen, P.; Mirault, M.E.; Arrigo, A.P. Inhibition of I kappa B-alpha phosphorylation and degradation and subsequent NF-kappa B activation by glutathione peroxidase overexpression. J. Cell Biol 1996, 133, 1083–1093. [Google Scholar]
- Aravindan, N.; Mohan, S.; Herman, T.S.; Natarajan, M. Nitric oxide-mediated inhibition of NFkappaB regulates hyperthermia-induced apoptosis. J. Cell Biochem 2009, 106, 999–1009. [Google Scholar]
- Gilad, E.; Wong, H.R.; Zingarelli, B.; Virag, L.; O’Connor, M.; Salzman, A.L.; Szabo, C. Melatonin inhibits expression of the inducible isoform of nitric oxide synthase in murine macrophages: Role of inhibition of NFkappaB activation. FASEB J 1998, 12, 685–693. [Google Scholar]
- Matthews, J.R.; Botting, C.H.; Panico, M.; Morris, H.R.; Hay, R.T. Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Res 1996, 24, 2236–2242. [Google Scholar]
- Peng, H.B.; Libby, P.; Liao, J.K. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J. Biol. Chem 1995, 270, 14214–14219. [Google Scholar]
- Taylor, B.S.; Kim, Y.M.; Wang, Q.; Shapiro, R.A.; Billiar, T.R.; Geller, D.A. Nitric oxide down-regulates hepatocyte-inducible nitric oxide synthase gene expression. Arch. Surg 1997, 132, 1177–1183. [Google Scholar]
- Sekkai, D.; Aillet, F.; Israel, N.; Lepoivre, M. Inhibition of NF-kappaB and HIV-1 long terminal repeat transcriptional activation by inducible nitric oxide synthase 2 activity. J. Biol. Chem 1998, 273, 3895–3900. [Google Scholar]
- Ohkita, M.; Takaoka, M.; Shiota, Y.; Nojiri, R.; Matsumura, Y. Nitric oxide inhibits endothelin-1 production through the suppression of nuclear factor kappa B. Clin. Sci. (Lond. ) 2002, 103, 68S–71S. [Google Scholar]
- Perkins, N.D.; Gilmore, T.D. Good cop, bad cop: The different faces of NF-kappaB. Cell Death Differ 2006, 13, 759–772. [Google Scholar]
- Xie, Q.W.; Kashiwabara, Y.; Nathan, C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J. Biol. Chem 1994, 269, 4705–4708. [Google Scholar]
- Muhl, H.; Pfeilschifter, J. Amplification of nitric oxide synthase expression by nitric oxide in interleukin 1 beta-stimulated rat mesangial cells. J. Clin. Invest 1995, 95, 1941–1946. [Google Scholar]
- Zouki, C.; Jozsef, L.; Ouellet, S.; Paquette, Y.; Filep, J.G. Peroxynitrite mediates cytokine-induced IL-8 gene expression and production by human leukocytes. J. Leukoc Biol 2001, 69, 815–824. [Google Scholar]
- Colasanti, M.; Persichini, T.; Menegazzi, M.; Mariotto, S.; Giordano, E.; Caldarera, C.M.; Sogos, V.; Lauro, G.M.; Suzuki, H. Induction of nitric oxide synthase mRNA expression. Suppression by exogenous nitric oxide. J. Biol. Chem 1995, 270, 26731–26733. [Google Scholar]
- Park, S.K.; Lin, H.L.; Murphy, S. Nitric oxide regulates nitric oxide synthase-2 gene expression by inhibiting NF-kappaB binding to DNA. Biochem. J 1997, 322, 609–613. [Google Scholar]
- Chen, F.; Lu, Y.; Castranova, V.; Rojanasakul, Y.; Miyahara, K.; Shizuta, Y.; Vallyathan, V.; Shi, X.; Demers, L.M. Nitric oxide inhibits HIV tat-induced NF-kappaB activation. Am. J. Pathol 1999, 155, 275–284. [Google Scholar]
- Matthews, J.R.; Wakasugi, N.; Virelizier, J.L.; Yodoi, J.; Hay, R.T. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res 1992, 20, 3821–3830. [Google Scholar]
- Mitomo, K.; Nakayama, K.; Fujimoto, K.; Sun, X.; Seki, S.; Yamamoto, K. Two different cellular redox systems regulate the DNA-binding activity of the p50 subunit of NF-kappa B in vitro. Gene 1994, 145, 197–203. [Google Scholar]
- Satriano, J.; Schlondorff, D. Activation and attenuation of transcription factor NF-κB in mouse glomerular mesangial cells in response to tumor necrosis factor-alpha, immunoglobulin G, and adenosine 3′:5′-cyclic monophosphate. Evidence for involvement of reactive oxygen species. J. Clin. Invest 1994, 94, 1629–1636. [Google Scholar]
- Schreck, R.; Baeuerle, P.A. Assessing oxygen radicals as mediators in activation of inducible eukaryotic transcription factor NF-kappa B. Methods Enzymol 1994, 234, 151–163. [Google Scholar]
- Kabe, Y.; Ando, K.; Hirao, S.; Yoshida, M.; Handa, H. Redox regulation of NF-kappaB activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox Signal 2005, 7, 395–403. [Google Scholar]
- Nishi, T.; Shimizu, N.; Hiramoto, M.; Sato, I.; Yamaguchi, Y.; Hasegawa, M.; Aizawa, S.; Tanaka, H.; Kataoka, K.; Watanabe, H.; et al. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J. Biol. Chem 2002, 277, 44548–44556. [Google Scholar]
- Toledano, M.B.; Leonard, W.J. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 4328–4332. [Google Scholar]
- Pineda-Molina, E.; Klatt, P.; Vazquez, J.; Marina, A.; Garcia de Lacoba, M.; Perez-Sala, D.; Lamas, S. Glutathionylation of the p50 subunit of NF-kappaB: A mechanism for redox-induced inhibition of DNA binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar]
- Kamata, H.; Manabe, T.; Oka, S.; Kamata, K.; Hirata, H. Hydrogen peroxide activates IkappaB kinases through phosphorylation of serine residues in the activation loops. FEBS Lett 2002, 519, 231–237. [Google Scholar]
- Vandermoere, F.; El Yazidi-Belkoura, I.; Adriaenssens, E.; Lemoine, J.; Hondermarck, H. The antiapoptotic effect of fibroblast growth factor-2 is mediated through nuclear factor-kappaB activation induced via interaction between Akt and IkappaB kinase-beta in breast cancer cells. Oncogene 2005, 24, 5482–5491. [Google Scholar]
- Jamaluddin, M.; Wang, S.; Boldogh, I.; Tian, B.; Brasier, A.R. TNF-alpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal 2007, 19, 1419–1433. [Google Scholar]
- Go, Y.M.; Gipp, J.J.; Mulcahy, R.T.; Jones, D.P. H2O2-dependent activation of GCLC-ARE4 reporter occurs by mitogen-activated protein kinase pathways without oxidation of cellular glutathione or thioredoxin-1. J. Biol. Chem 2004, 279, 5837–5845. [Google Scholar]
- Kamata, H.; Manabe, T.; Kakuta, J.; Oka, S.; Hirata, H. Multiple redox regulation of the cellular signaling system linked to AP-1 and NFkappaB: Effects of N-acetylcysteine and H2O2 on the receptor tyrosine kinases, the MAP kinase cascade, and IkappaB kinases. Ann. N. Y. Acad. Sci 2002, 973, 419–422. [Google Scholar]
- Vayalil, P.K.; Iles, K.E.; Choi, J.; Yi, A.K.; Postlethwait, E.M.; Liu, R.M. Glutathione suppresses TGF-beta-induced PAI-1 expression by inhibiting p38 and JNK MAPK and the binding of AP-1, SP-1, and Smad to the PAI-1 promoter. Am. J. Physiol. Lung Cell Mol. Physiol 2007, 293, L1281–L1292. [Google Scholar]
- Finocchietto, P.V.; Franco, M.C.; Holod, S.; Gonzalez, A.S.; Converso, D.P.; Antico Arciuch, V.G.; Serra, M.P.; Poderoso, J.J.; Carreras, M.C. Mitochondrial nitric oxide synthase: A masterpiece of metabolic adaptation, cell growth, transformation, and death. Exp. Biol. Med. (Maywood) 2009, 234, 1020–1028. [Google Scholar]
- Carreras, M.C.; Poderoso, J.J. Mitochondrial nitric oxide in the signaling of cell integrated responses. Am. J. Physiol. Cell Physiol 2007, 292, C1569–C1580. [Google Scholar]
- Gorlach, A.; Diebold, I.; Schini-Kerth, V.B.; Berchner-Pfannschmidt, U.; Roth, U.; Brandes, R.P.; Kietzmann, T.; Busse, R. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: Role of the p22(phox)-containing NADPH oxidase. Circ. Res 2001, 89, 47–54. [Google Scholar]
- Shatrov, V.A.; Sumbayev, V.V.; Zhou, J.; Brune, B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1alpha (HIF-1alpha) accumulation via redox-dependent mechanisms. Blood 2003, 101, 4847–4849. [Google Scholar]
- Li, C.Q.; Robles, A.I.; Hanigan, C.L.; Hofseth, L.J.; Trudel, L.J.; Harris, C.C.; Wogan, G.N. Apoptotic signaling pathways induced by nitric oxide in human lymphoblastoid cells expressing wild-type or mutant p53. Cancer Res 2004, 64, 3022–3029. [Google Scholar]
- Rainwater, R.; Parks, D.; Anderson, M.E.; Tegtmeyer, P.; Mann, K. Role of cysteine residues in regulation of p53 function. Mol. Cell Biol 1995, 15, 3892–3903. [Google Scholar]
- Yang, F.; von Knethen, A.; Brune, B. Modulation of nitric oxide-evoked apoptosis by the p53-downstream target p21(WAF1/CIP1). J. Leukoc. Biol 2000, 68, 916–922. [Google Scholar]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev 1998, 12, 2973–2983. [Google Scholar]
- Li, J.M.; Fan, L.M.; George, V.T.; Brooks, G. Nox2 regulates endothelial cell cycle arrest and apoptosis via p21cip1 and p53. Free Radic. Biol. Med 2007, 43, 976–986. [Google Scholar]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med 2008, 44, 1529–1535. [Google Scholar]
- Sun, Y.; Oberley, L.W. Redox regulation of transcriptional activators. Free Radic. Biol. Med 1996, 21, 335–348. [Google Scholar]
- Roebuck, K.A. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: Differential activation and binding of the transcription factors AP-1 and NF-kappaB (Review). Int. J. Mol. Med 1999, 4, 223–230. [Google Scholar]
- Wu, X.; Bishopric, N.H.; Discher, D.J.; Murphy, B.J.; Webster, K.A. Physical and functional sensitivity of zinc finger transcription factors to redox change. Mol. Cell. Biol 1996, 16, 1035–1046. [Google Scholar]
- Akiba, S.; Chiba, M.; Mukaida, Y.; Sato, T. Involvement of reactive oxygen species and SP-1 in fibronectin production by oxidized LDL. Biochem. Biophys. Res. Commun 2003, 310, 491–497. [Google Scholar]
- Hampton, M.B.; Orrenius, S. Dual regulation of caspase activity by hydrogen peroxide: Implications for apoptosis. FEBS Lett 1997, 414, 552–556. [Google Scholar]
- Higuchi, M.; Honda, T.; Proske, R.J.; Yeh, E.T. Regulation of reactive oxygen species-induced apoptosis and necrosis by caspase 3-like proteases. Oncogene 1998, 17, 2753–2760. [Google Scholar]
- Moungjaroen, J.; Nimmannit, U.; Callery, P.S.; Wang, L.; Azad, N.; Lipipun, V.; Chanvorachote, P.; Rojanasakul, Y. Reactive oxygen species mediate caspase activation and apoptosis induced by lipoic acid in human lung epithelial cancer cells through Bcl-2 down-regulation. J. Pharmacol. Exp. Ther 2006, 319, 1062–1069. [Google Scholar]
- Fadeel, B.; Ahlin, A.; Henter, J.I.; Orrenius, S.; Hampton, M.B. Involvement of caspases in neutrophil apoptosis: Regulation by reactive oxygen species. Blood 1998, 92, 4808–4818. [Google Scholar]
- Izeradjene, K.; Douglas, L.; Tillman, D.M.; Delaney, A.B.; Houghton, J.A. Reactive oxygen species regulate caspase activation in tumor necrosis factor-related apoptosis-inducing ligand-resistant human colon carcinoma cell lines. Cancer Res 2005, 65, 7436–7445. [Google Scholar]
- Vlahopoulos, S.; Boldogh, I.; Casola, A.; Brasier, A.R. Nuclear factor-kappaB-dependent induction of interleukin-8 gene expression by tumor necrosis factor alpha: Evidence for an antioxidant sensitive activating pathway distinct from nuclear translocation. Blood 1999, 94, 1878–1889. [Google Scholar]
- Kurdi, M.; Booz, G.W. Evidence that IL-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress: Parthenolide targets JAK1 activation by generating ROS. J. Cell Physiol 2007, 212, 424–431. [Google Scholar]
- Schulze-Osthoff, K.; Beyaert, R.; Vandevoorde, V.; Haegeman, G.; Fiers, W. Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. EMBO J 1993, 12, 3095–3104. [Google Scholar]
- De Keulenaer, G.W.; Ushio-Fukai, M.; Yin, Q.; Chung, A.B.; Lyons, P.R.; Ishizaka, N.; Rengarajan, K.; Taylor, W.R.; Alexander, R.W.; Griendling, K.K. Convergence of redox-sensitive and mitogen-activated protein kinase signaling pathways in tumor necrosis factor-alpha-mediated monocyte chemoattractant protein-1 induction in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol 2000, 20, 385–391. [Google Scholar]
- Shimada, T.; Watanabe, N.; Hiraishi, H.; Terano, A. Redox regulation of interleukin-8 expression in MKN28 cells. Dig. Dis. Sci 1999, 44, 266–273. [Google Scholar]
- Lo, Y.Y.; Cruz, T.F. Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. J. Biol. Chem 1995, 270, 11727–11730. [Google Scholar]
- Maulik, N.; Das, D.K. Redox signaling in vascular angiogenesis. Free Radic. Biol. Med 2002, 33, 1047–1060. [Google Scholar]
- Eyries, M.; Collins, T.; Khachigian, L.M. Modulation of growth factor gene expression in vascular cells by oxidative stress. Endothelium 2004, 11, 133–139. [Google Scholar]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar]
- Morgan, M.J.; Liu, Z.G. Reactive oxygen species in TNFalpha-induced signaling and cell death. Mol. Cells 2010, 30, 1–12. [Google Scholar]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med 2010, 48, 749–762. [Google Scholar]
- Fruehauf, J.P.; Meyskens, F.L., Jr. Reactive oxygen species: A breath of life or death? Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar]
- Kizaki, M.; Xian, M.; Sagawa, M.; Ikeda, Y. Induction of apoptosis via the modulation of reactive oxygen species (ROS) production in the treatment of myeloid leukemia. Curr. Pharm. Biotechnol 2006, 7, 323–329. [Google Scholar]
- Leon, L.; Jeannin, J.F.; Bettaieb, A. Post-translational modifications induced by nitric oxide (NO): Implication in cancer cells apoptosis. Nitric. Oxide 2008, 19, 77–83. [Google Scholar]
- Tarr, J.M.; Eggleton, P.; Winyard, P.G. Nitric oxide and the regulation of apoptosis in tumour cells. Curr. Pharm. Des 2006, 12, 4445–4468. [Google Scholar]
- Li, C.Q.; Wogan, G.N. Nitric oxide as a modulator of apoptosis. Cancer Lett 2005, 226, 1–15. [Google Scholar]
- Brune, B. The intimate relation between nitric oxide and superoxide in apoptosis and cell survival. Antioxid. Redox Signal 2005, 7, 497–507. [Google Scholar]
- Vieira, H.; Kroemer, G. Mitochondria as targets of apoptosis regulation by nitric oxide. IUBMB Life 2003, 55, 613–616. [Google Scholar]
- Brune, B.; Zhou, J.; von Knethen, A. Nitric oxide, oxidative stress, and apoptosis. Kidney Int. Suppl. 2003, S22–24. [Google Scholar]
- Park, J.B. Phagocytosis induces superoxide formation and apoptosis in macrophages. Exp. Mol. Med 2003, 35, 325–335. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol 2007, 39, 44–84. [Google Scholar]
- Ghosh, N.; Ghosh, R.; Mandal, S.C. Antioxidant protection: A promising therapeutic intervention in neurodegenerative disease. Free Radic. Res 2011, 45, 888–905. [Google Scholar]
- Iborra, M.; Moret, I.; Rausell, F.; Bastida, G.; Aguas, M.; Cerrillo, E.; Nos, P.; Beltran, B. Role of oxidative stress and antioxidant enzymes in Crohn’s disease. Biochem. Soc. Trans 2011, 39, 1102–1106. [Google Scholar]
- Dumont, M.; Lin, M.T.; Beal, M.F. Mitochondria and antioxidant targeted therapeutic strategies for Alzheimer’s disease. J. Alzheimers Dis 2010, 20, S633–S643. [Google Scholar]
- De Vries, H.E.; Witte, M.; Hondius, D.; Rozemuller, A.J.; Drukarch, B.; Hoozemans, J.; van Horssen, J. Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic. Biol. Med 2008, 45, 1375–1383. [Google Scholar]
- De Araujo, D.P.; de Lobato, R.F.; Cavalcanti, J.R.; Sampaio, L.R.; Araujo, P.V.; Silva, M.C.; Neves, K.R.; Fonteles, M.M.; Sousa, F.C.; Vasconcelos, S.M. The contributions of antioxidant activity of lipoic acid in reducing neurogenerative progression of Parkinson’s disease: A review. Int. J. NeuroSci 2011, 121, 51–57. [Google Scholar]
- Aliev, G.; Obrenovich, M.E.; Reddy, V.P.; Shenk, J.C.; Moreira, P.I.; Nunomura, A.; Zhu, X.; Smith, M.A.; Perry, G. Antioxidant therapy in Alzheimer’s disease: Theory and practice. Mini Rev. Med. Chem 2008, 8, 1395–1406. [Google Scholar]
- Zarjou, A.; Sanders, P.W.; Mehta, R.L.; Agarwal, A. Enabling innovative translational research in acute kidney injury. Clin. Transl. Sci 2012, 5, 93–101. [Google Scholar]
- Aziz, M.H.; Kumar, R.; Ahmad, N. Cancer chemoprevention by resveratrol: In vitro and in vivo studies and the underlying mechanisms (review). Int. J. Oncol 2003, 23, 17–28. [Google Scholar]
- Turan, B.; Vassort, G. Vitamin E in oxidant stress-related cardiovascular pathologies: Focus on experimental studies. Curr. Pharm. Des 2011, 17, 2155–2169. [Google Scholar]
- Turan, B. Role of antioxidants in redox regulation of diabetic cardiovascular complications. Curr. Pharm. Biotechnol 2010, 11, 819–836. [Google Scholar]
- Violi, F.; Cangemi, R. Antioxidants and cardiovascular disease. Curr. Opin. Investig Drugs 2005, 6, 895–900. [Google Scholar]
- Maxwell, S.R.; Lip, G.Y. Free radicals and antioxidants in cardiovascular disease. Br. J. Clin. Pharmacol 1997, 44, 307–317. [Google Scholar]
- Tylicki, L.; Rutkowski, B.; Horl, W.H. Antioxidants: A possible role in kidney protection. Kidney Blood Press Res 2003, 26, 303–314. [Google Scholar]
- Day, B.J. Antioxidants as potential therapeutics for lung fibrosis. Antioxid. Redox Signal 2008, 10, 355–370. [Google Scholar]
- Kinnula, V.L.; Crapo, J.D. Superoxide dismutases in the lung and human lung diseases. Am. J. Respir Crit. Care Med 2003, 167, 1600–1619. [Google Scholar]
- Russell, G.A. Antioxidants and neonatal lung disease. Eur. J. Pediatr 1994, 153, S36–S41. [Google Scholar]
- Kamat, C.D.; Gadal, S.; Mhatre, M.; Williamson, K.S.; Pye, Q.N.; Hensley, K. Antioxidants in central nervous system diseases: Preclinical promise and translational challenges. J. Alzheimers Dis 2008, 15, 473–493. [Google Scholar]
- Durante, W. Targeting heme oxygenase-1 in vascular disease. Curr. Drug Targets 2010, 11, 1504–1516. [Google Scholar]
- Subramanian, S.; Kalyanaraman, B.; Migrino, R.Q. Mitochondrially targeted antioxidants for the treatment of cardiovascular diseases. Recent Pat. Cardiovasc. Drug Discov 2010, 5, 54–65. [Google Scholar]
- Sawyer, D.B. Oxidative stress in heart failure: What are we missing? Am. J. Med. Sci 2011, 342, 120–124. [Google Scholar]
- Wu, J.; Hecker, J.G.; Chiamvimonvat, N. Antioxidant enzyme gene transfer for ischemic diseases. Adv. Drug Deliv. Rev 2009, 61, 351–363. [Google Scholar]
- Golden, T.R.; Patel, M. Catalytic antioxidants and neurodegeneration. Antioxid. Redox Signal 2009, 11, 555–570. [Google Scholar]
- Poljsak, B.; Milisav, I. The neglected significance of “antioxidative stress”. Oxid. Med. Cell Longev 2012, 2012, 480895. [Google Scholar]
- Firuzi, O.; Miri, R.; Tavakkoli, M.; Saso, L. Antioxidant therapy: Current status and future prospects. Curr. Med. Chem 2011, 18, 3871–3888. [Google Scholar]
- Mecocci, P.; Polidori, M.C. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 631–638. [Google Scholar]
- Shamseer, L.; Adams, D.; Brown, N.; Johnson, J.A.; Vohra, S. Antioxidant micronutrients for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef]
- Nunez-Cordoba, J.M.; Martinez-Gonzalez, M.A. Antioxidant vitamins and cardiovascular disease. Curr. Top. Med. Chem 2011, 11, 1861–1869. [Google Scholar]
- Bonnefont-Rousselot, D.; Collin, F. Melatonin: Action as antioxidant and potential applications in human disease and aging. Toxicology 2010, 278, 55–67. [Google Scholar]
- Lee, H.P.; Zhu, X.; Casadesus, G.; Castellani, R.J.; Nunomura, A.; Smith, M.A.; Lee, H.G.; Perry, G. Antioxidant approaches for the treatment of Alzheimer’s disease. Expert Rev. Neurother 2010, 10, 1201–1208. [Google Scholar]
- Farbstein, D.; Kozak-Blickstein, A.; Levy, A.P. Antioxidant vitamins and their use in preventing cardiovascular disease. Molecules 2010, 15, 8098–8110. [Google Scholar]
- Levonen, A.L.; Vahakangas, E.; Koponen, J.K.; Yla-Herttuala, S. Antioxidant gene therapy for cardiovascular disease: Current status and future perspectives. Circulation 2008, 117, 2142–2150. [Google Scholar]
- Davis, C.A.; Nick, H.S.; Agarwal, A. Manganese superoxide dismutase attenuates Cisplatin-induced renal injury: Importance of superoxide. J. Am. Soc. Nephrol 2001, 12, 2683–2690. [Google Scholar]
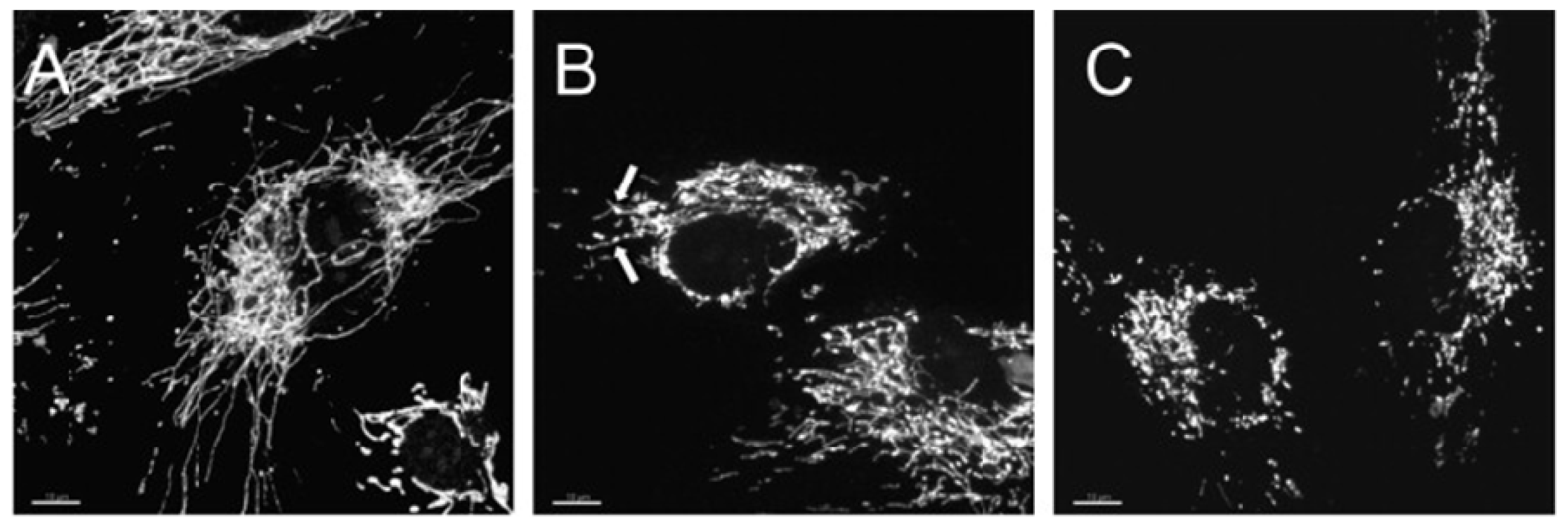

© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bolisetty, S.; Jaimes, E.A. Mitochondria and Reactive Oxygen Species: Physiology and Pathophysiology. Int. J. Mol. Sci. 2013, 14, 6306-6344. https://doi.org/10.3390/ijms14036306
Bolisetty S, Jaimes EA. Mitochondria and Reactive Oxygen Species: Physiology and Pathophysiology. International Journal of Molecular Sciences. 2013; 14(3):6306-6344. https://doi.org/10.3390/ijms14036306
Chicago/Turabian StyleBolisetty, Subhashini, and Edgar A. Jaimes. 2013. "Mitochondria and Reactive Oxygen Species: Physiology and Pathophysiology" International Journal of Molecular Sciences 14, no. 3: 6306-6344. https://doi.org/10.3390/ijms14036306
APA StyleBolisetty, S., & Jaimes, E. A. (2013). Mitochondria and Reactive Oxygen Species: Physiology and Pathophysiology. International Journal of Molecular Sciences, 14(3), 6306-6344. https://doi.org/10.3390/ijms14036306