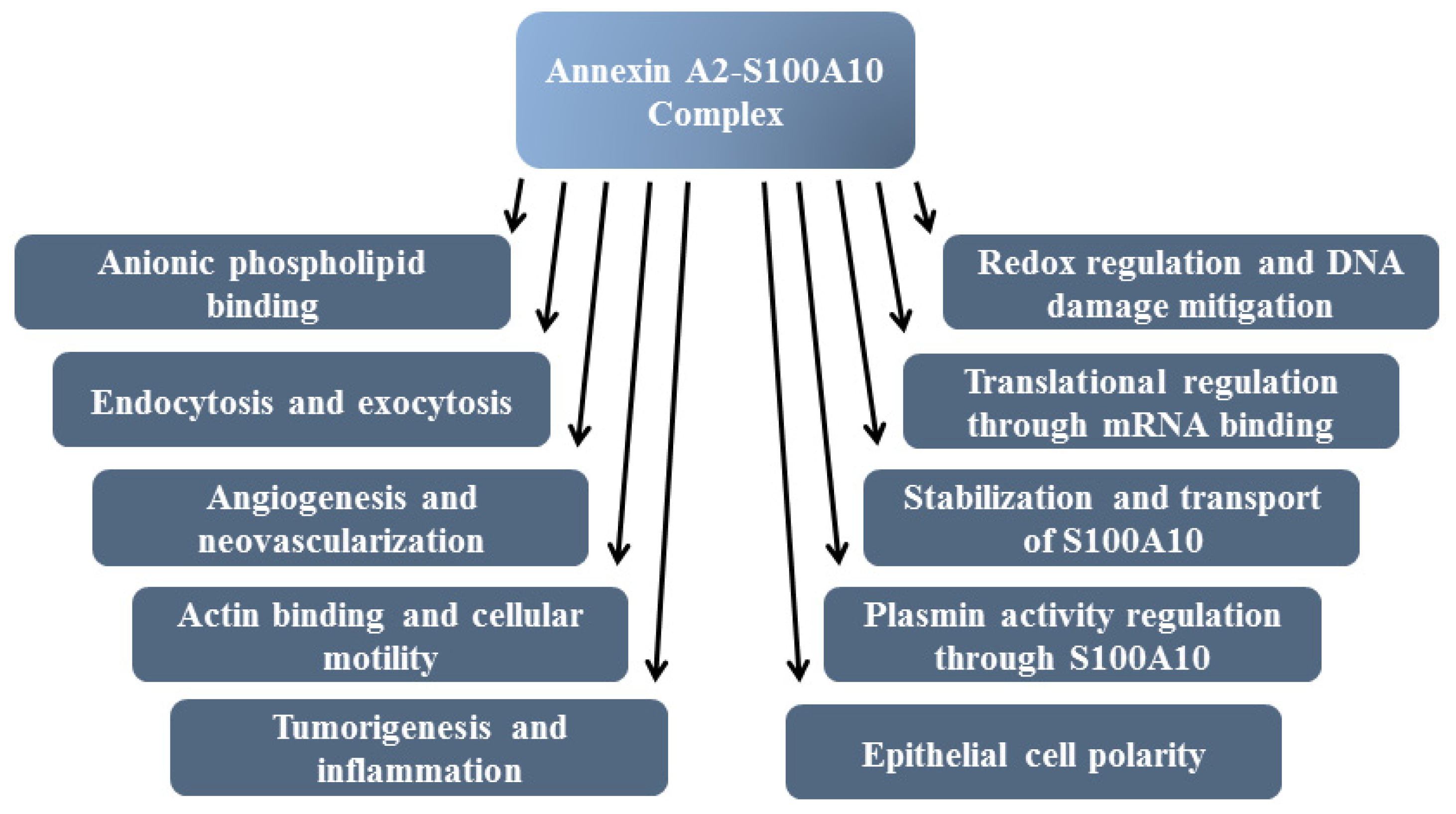
Annexin A2 Heterotetramer: Structure and Function


Abstract
:
1. Introduction
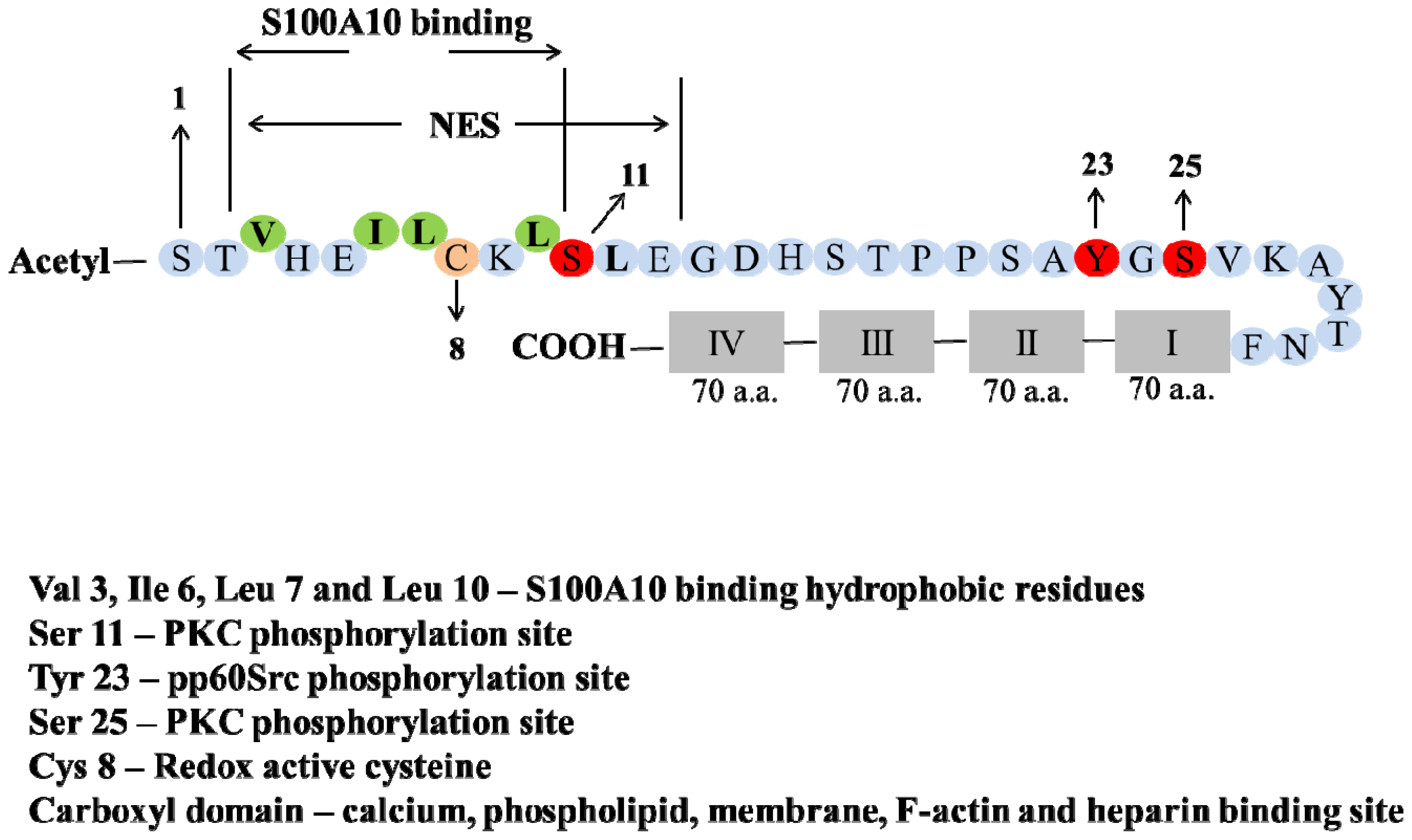
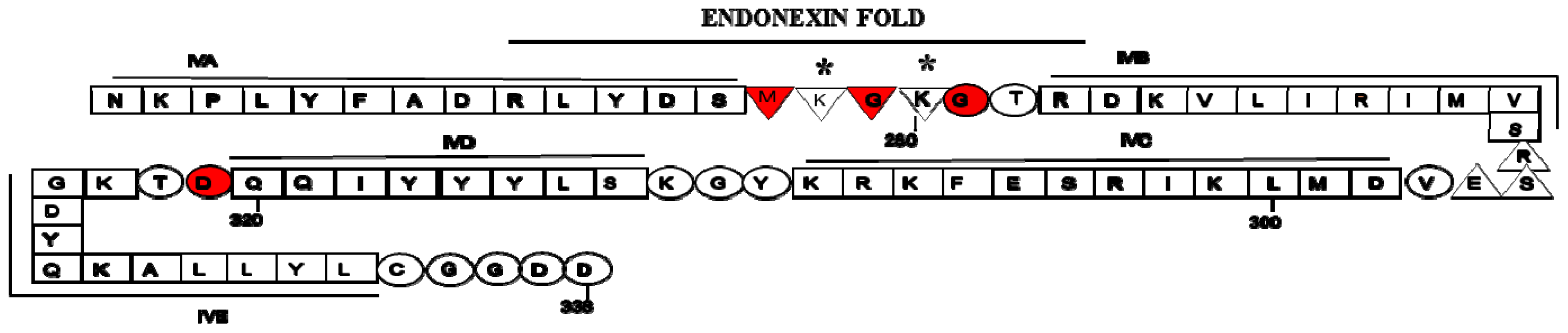
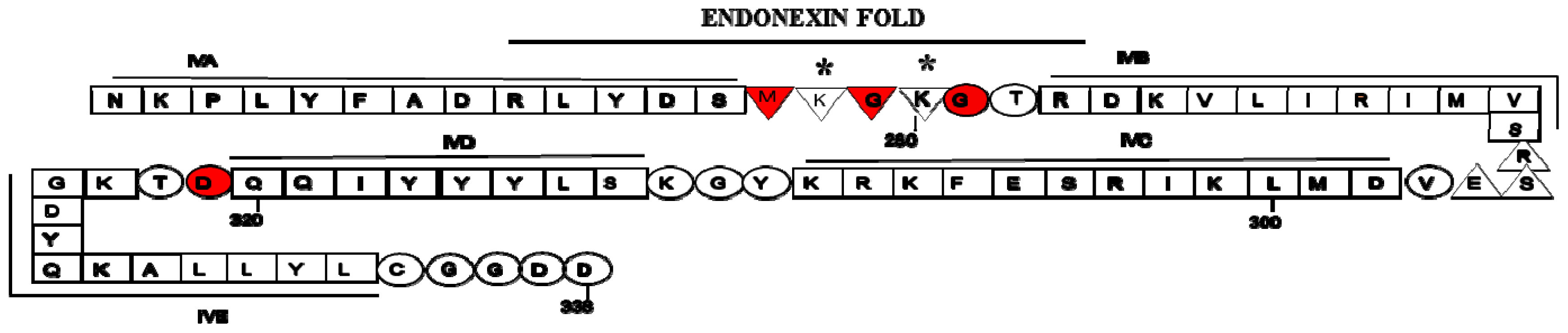
2. Structure of Annexin A2 and AIIt Heterotetramer
3. Annexin A2 Binds to Anionic Phospholipids
4. Transcriptional Regulation of Annexin A2
5. Post-Translation Modification and Regulation of Annexin A2
6. Phosphorylation
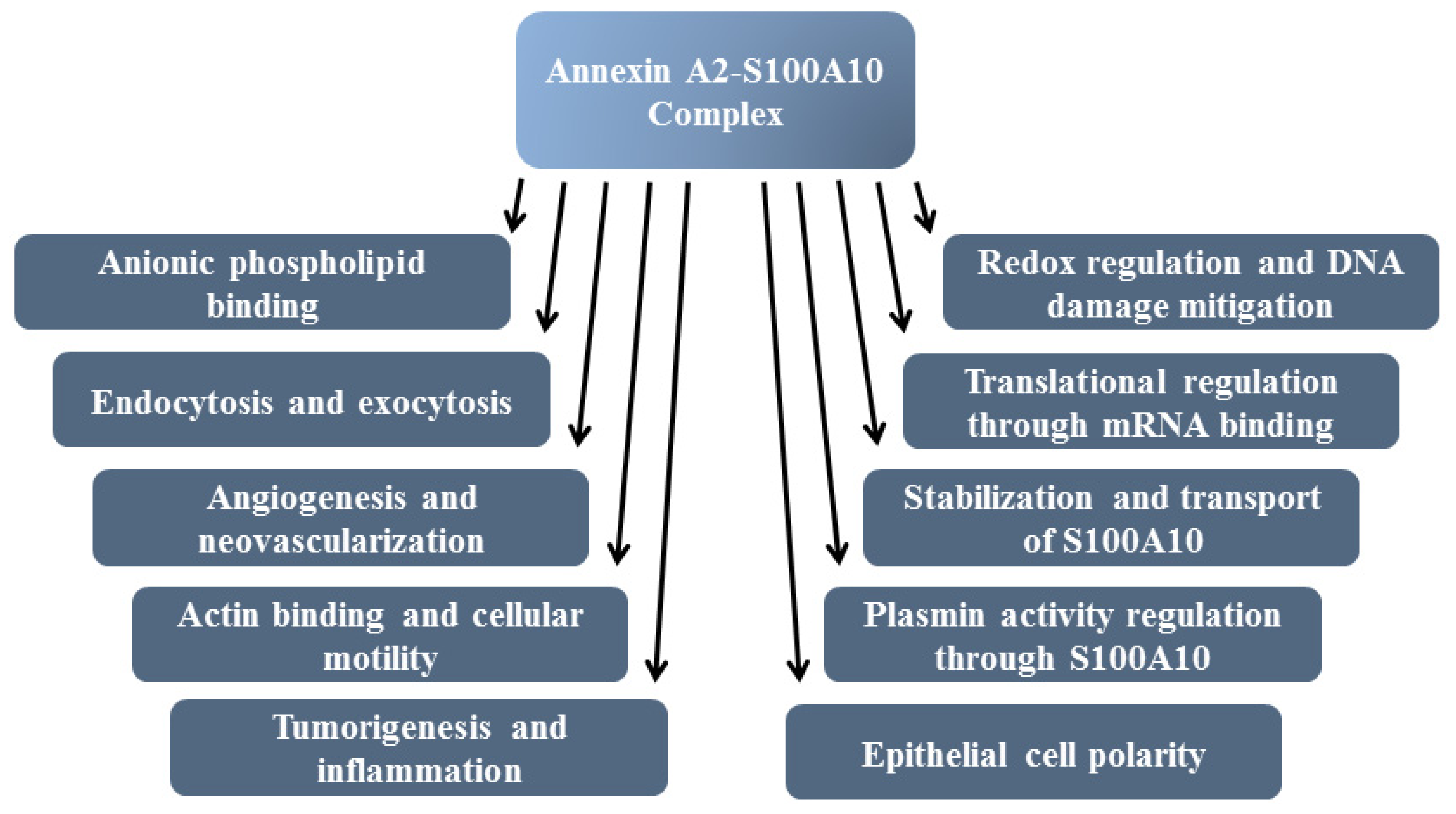
7. Redox Regulation
8. Functions of Annexin A2
8.1. F-Actin Binding
8.2. Exocytosis and Endocytosis
8.3. Epithelial and Endothelial Cell Polarity
8.4. mRNA Binding
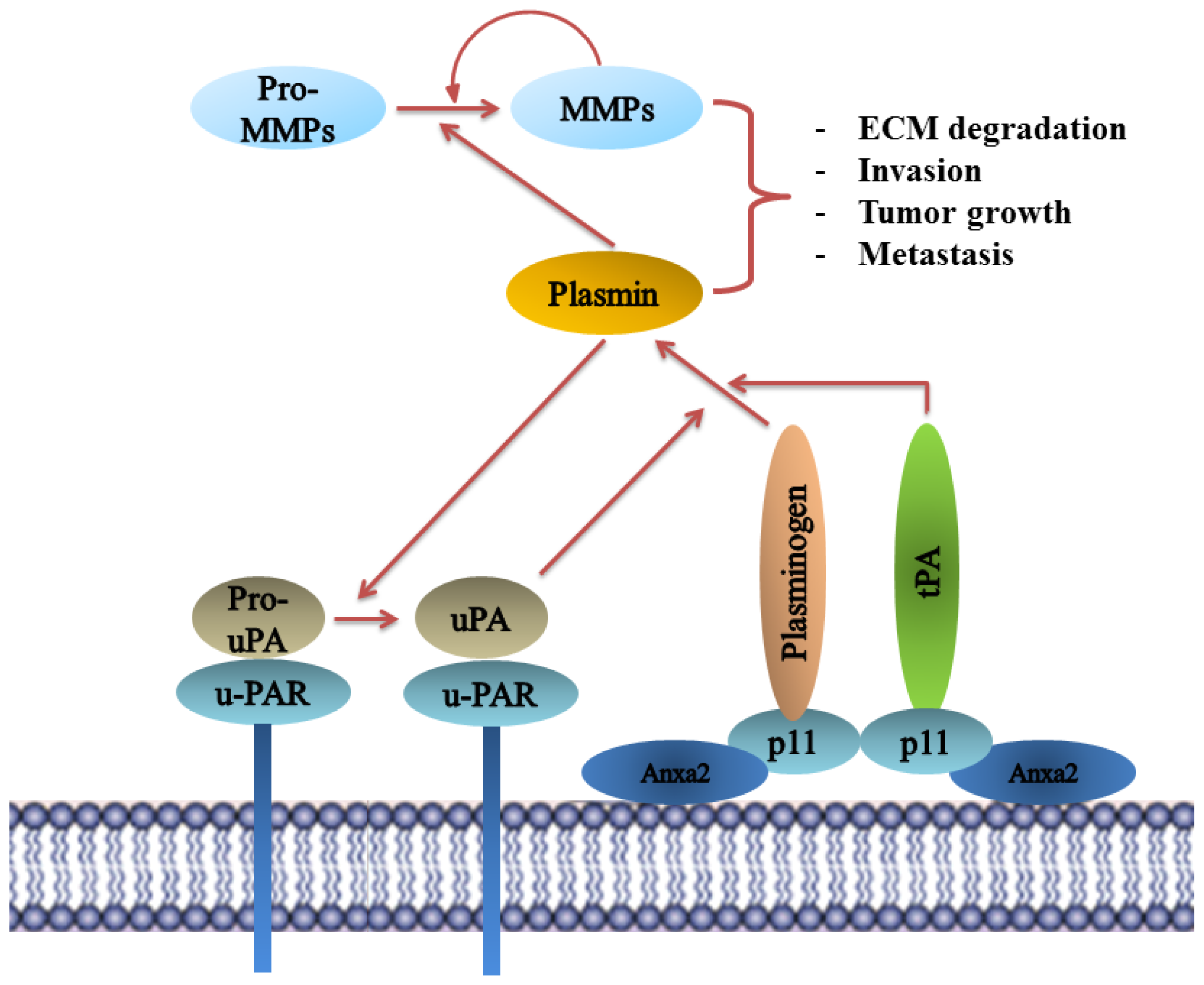
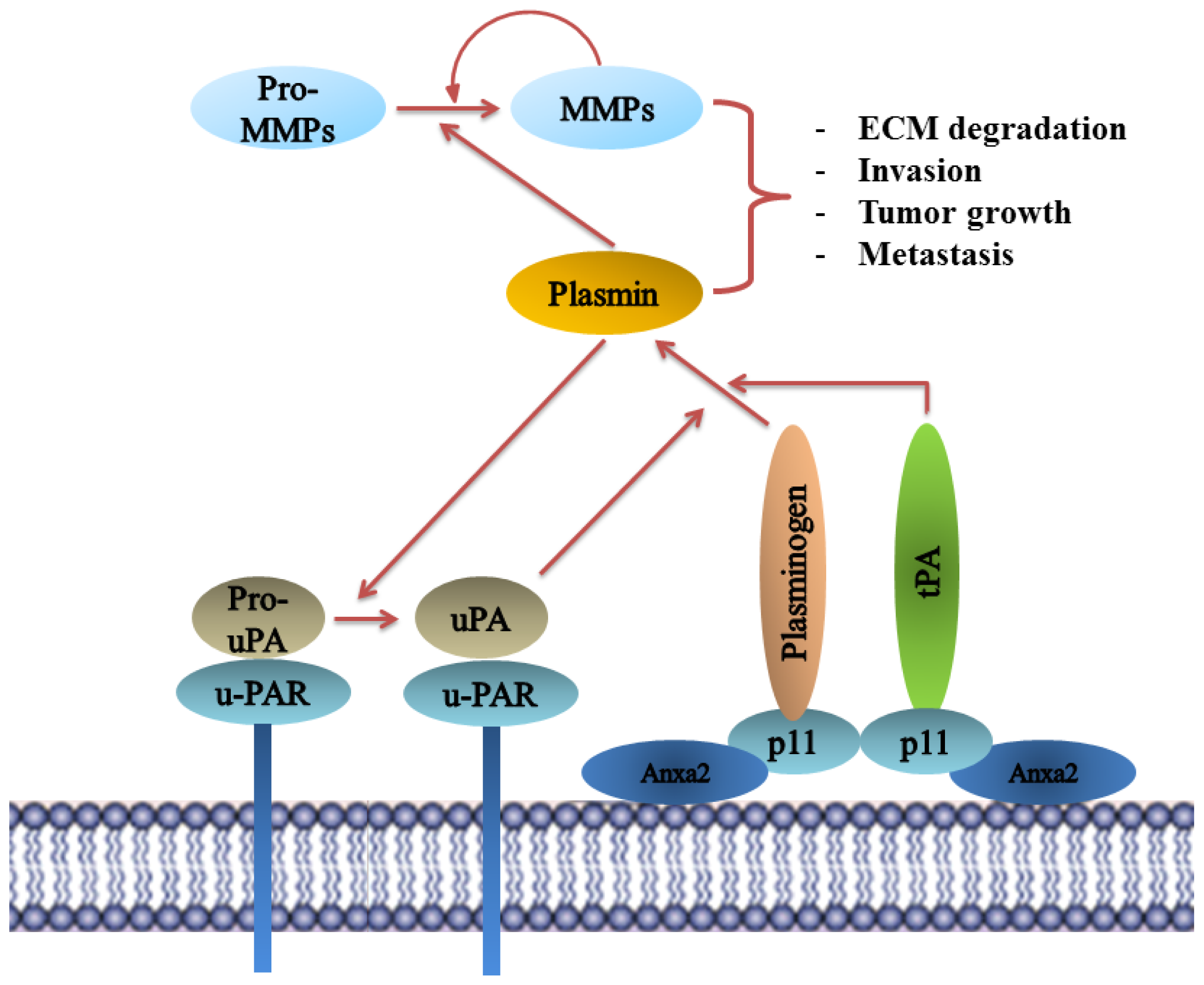
8.5. Plasminogen Receptor—S100A10, and Not Annexin A2, Binds Plasminogen and tPA and Regulates Plasmin Generation
9. Role of Annexin A2 in Diseases
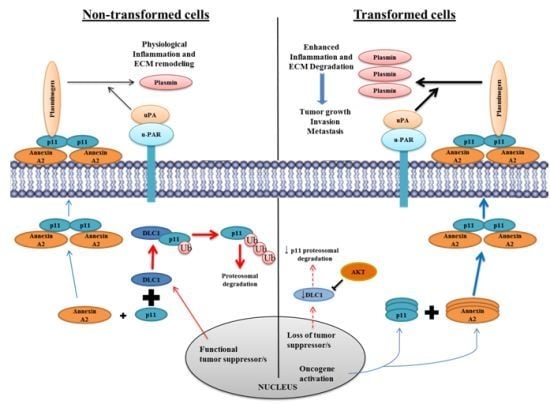
9.1. Annexin A2 in Cancer Progression
9.2. Annexin A2-S100A10 Complex and Not Monomeric Annexin A2 Mediates Invasion, Metastasis and Drug Resistance in Breast Cancer and May Act as Pathological Predictor
9.3. Annexin A2 Promotes Invasion and Metastasis in Pancreatic Ductal Adenocarcinoma (PDAC) and Is Predictive of Post-Operative Recurrence and Patient Survival
9.4. Annexin A2 Is a Metastatic Marker in Renal Cell Carcinoma (RCC)
9.5. Annexin A2 Expression Correlates with TNC Expression and Is a Potential Prognostic Marker of Advanced Colorectal Carcinoma (CRC)
9.6. Annexin A2 Is a Differential Diagnostic Tissue and Serum Marker in Hepatocellular Carcinoma (HCC)
9.7. Annexin A2 May Promote Hyperfibrinolysis and Acute Bleeding in Acute Promyelocytic Leukemia (APL)
9.8. Annexin A2 Expression Presents Conflicting Results in Prostate Cancer
9.9. Annexin A2 Expression Is Prognostic of Histological Grade and Metastasis in Most Head and Neck Cancers
9.10. Annexin A2 Is a Promising Therapeutic Target in Cancer
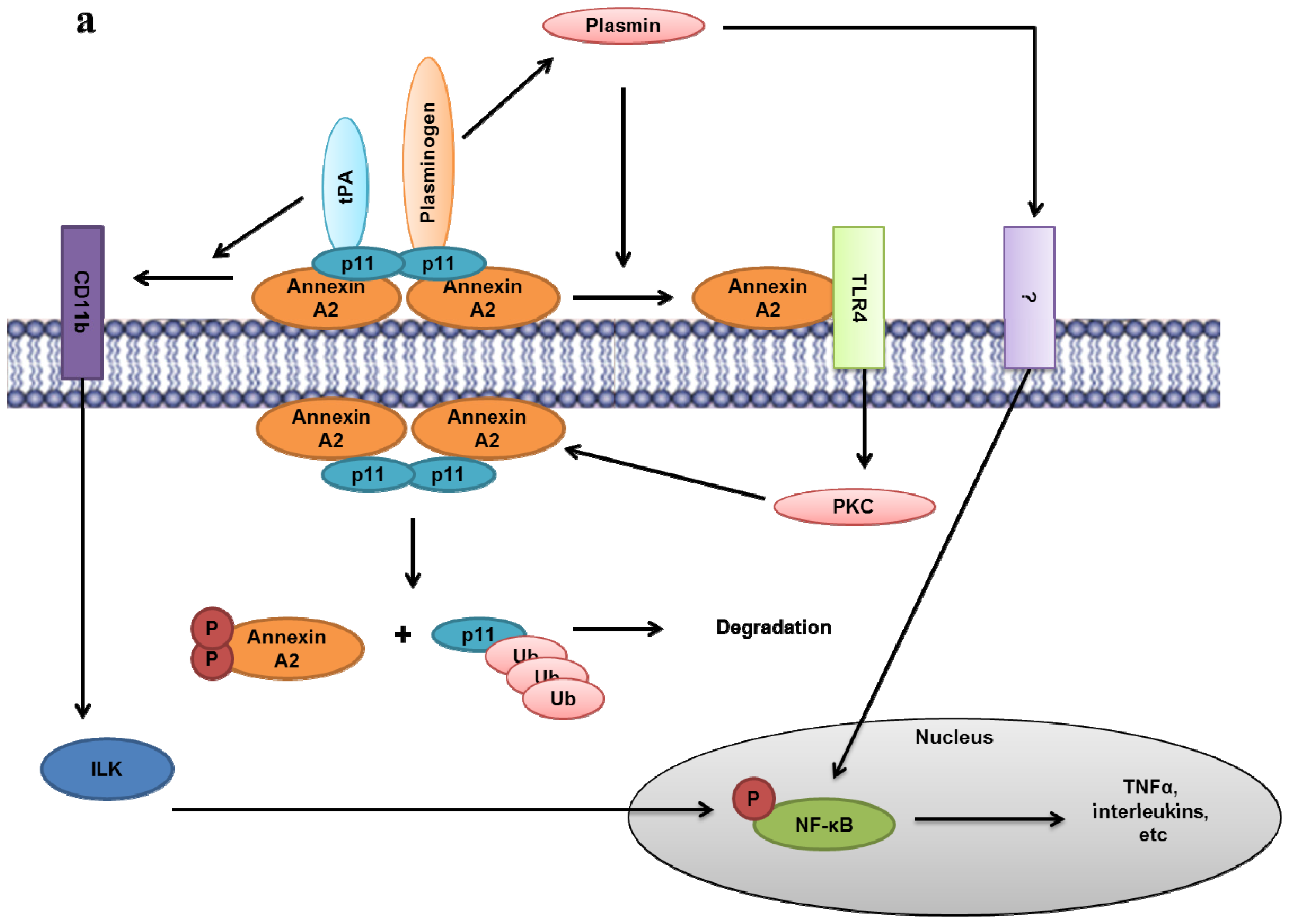
9.11. Role of the Extracellular Annexin A2-S100A10 Complex in Inflammation
9.12. Annexin A2 in Anti-Phospholipid Syndrome
10. Concluding Comments
Acknowledgements
References
- Clark, G.B.; Morgan, R.O.; Fernandez, M.P.; Roux, S.J. Evolutionary adaptation of plant annexins has diversified their molecular structures, interactions and functional roles. New Phytol 2012, 196, 695–712. [Google Scholar]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol 2005, 6, 449–461. [Google Scholar]
- Gerke, V.; Moss, S.E. Annexins: From structure to function. Physiol. Rev 2002, 82, 331–371. [Google Scholar]
- Moss, S.E.; Morgan, R.O. The annexins. Genome Biol 2004, 5, 219. [Google Scholar]
- Seaton, B.A.; Dedman, J.R. Annexins. Biometals 1998, 11, 399–404. [Google Scholar]
- Rescher, U.; Gerke, V. Annexins—Unique membrane binding proteins with diverse functions. J. Cell Sci 2004, 117, 2631–2639. [Google Scholar]
- Creutz, C.E.; Pazoles, C.J.; Pollard, H.B. Identification and purification of an adrenal medullary protein (synexin) that causes calcium-dependent aggregation of isolated chromaffin granules. J. Biol. Chem 1978, 253, 2858–2866. [Google Scholar]
- Luecke, H.; Chang, B.T.; Mailliard, W.S.; Schlaepfer, D.D.; Haigler, H.T. Crystal structure of the annexin XII hexamer and implications for bilayer insertion. Nature 1995, 378, 512–515. [Google Scholar]
- Domon, M.; Nasir, M.N.; Matar, G.; Pikula, S.; Besson, F.; Bandorowicz-Pikula, J. Annexins as organizers of cholesterol- and sphingomyelin-enriched membrane microdomains in Niemann-Pick type C disease. Cell Mol. Life Sci 2012, 69, 1773–1785. [Google Scholar]
- Erikson, E.; Erikson, R.L. Identification of a cellular protein substrate phosphorylated by the avian sarcoma virus-transforming gene product. Cell 1980, 21, 829–836. [Google Scholar]
- Waisman, D.M. Annexin II tetramer: Structure and function. Mol. Cell Biochem. 1995, 149–150, 301–322. [Google Scholar]
- Gladwin, M.T.; Yao, X.L.; Cowan, M.; Huang, X.L.; Schneider, R.; Grant, L.R.; Logun, C.; Shelhamer, J.H. Retinoic acid reduces p11 protein levels in bronchial epithelial cells by a posttranslational mechanism. Am. J. Physiol. Lung Cell Mol. Physiol 2000, 279, L1103–L1109. [Google Scholar]
- He, K.L.; Deora, A.B.; Xiong, H.; Ling, Q.; Weksler, B.B.; Niesvizky, R.; Hajjar, K.A. Endothelial cell annexin A2 regulates polyubiquitination and degradation of its binding partner S100A10/p11. J. Biol. Chem 2008, 283, 19192–19200. [Google Scholar]
- Hou, Y.; Yang, L.; Mou, M.; Zhang, A.; Pan, N.; Qiang, R.; Wei, L.; Zhang, N. Annexin A2 regulates the levels of plasmin, S100A10 and Fascin in L5178Y cells. Cancer Invest 2008, 26, 809–815. [Google Scholar]
- Puisieux, A.; Ji, J.; Ozturk, M. Annexin II up-regulates cellular levels of p11 protein by a post-translational mechanisms. Biochem. J. 1996, 313(Pt. 1), 51–55. [Google Scholar]
- Yang, X.; Popescu, N.C.; Zimonjic, D.B. DLC1 interaction with S100A10 mediates inhibition of in vitro cell invasion and tumorigenicity of lung cancer cells through a RhoGAP-independent mechanism. Cancer Res 2011, 71, 2916–2925. [Google Scholar]
- Zhang, J.; Guo, B.; Zhang, Y.; Cao, J.; Chen, T. Silencing of the annexin II gene down-regulates the levels of S100A10, c-Myc, and plasmin and inhibits breast cancer cell proliferation and invasion. Saudi Med. J 2010, 31, 374–381. [Google Scholar]
- Madureira, P.A.; Surette, A.P.; Phipps, K.D.; Taboski, M.A.; Miller, V.A.; Waisman, D.M. The role of the annexin A2 heterotetramer in vascular fibrinolysis. Blood 2011, 118, 4789–4797. [Google Scholar]
- Madureira, P.A.; O’Connell, P.A.; Surette, A.P.; Miller, V.A.; Waisman, D.M. The biochemistry and regulation of S100A10: A multifunctional plasminogen receptor involved in oncogenesis. J. Biomed. Biotechnol 2012, 2012, 353687. [Google Scholar]
- Phipps, K.D.; Surette, A.P.; O’Connell, P.A.; Waisman, D.M. Plasminogen receptor S100A10 is essential for the migration of tumor-promoting macrophages into tumor sites. Cancer Res 2011, 71, 6676–6683. [Google Scholar]
- Kwon, M.; MacLeod, T.J.; Zhang, Y.; Waisman, D.M. S100A10, annexin A2, and annexin a2 heterotetramer as candidate plasminogen receptors. Front. Biosci 2005, 10, 300–325. [Google Scholar]
- Roth, D.; Morgan, A.; Burgoyne, R.D. Identification of a key domain in annexin and 14-3-3 proteins that stimulate calcium-dependent exocytosis in permeabilized adrenal chromaffin cells. FEBS Lett 1993, 320, 207–210. [Google Scholar]
- Liemann, S.; Huber, R. Three-dimensional structure of annexins. Cell Mol. Life Sci 1997, 53, 516–521. [Google Scholar]
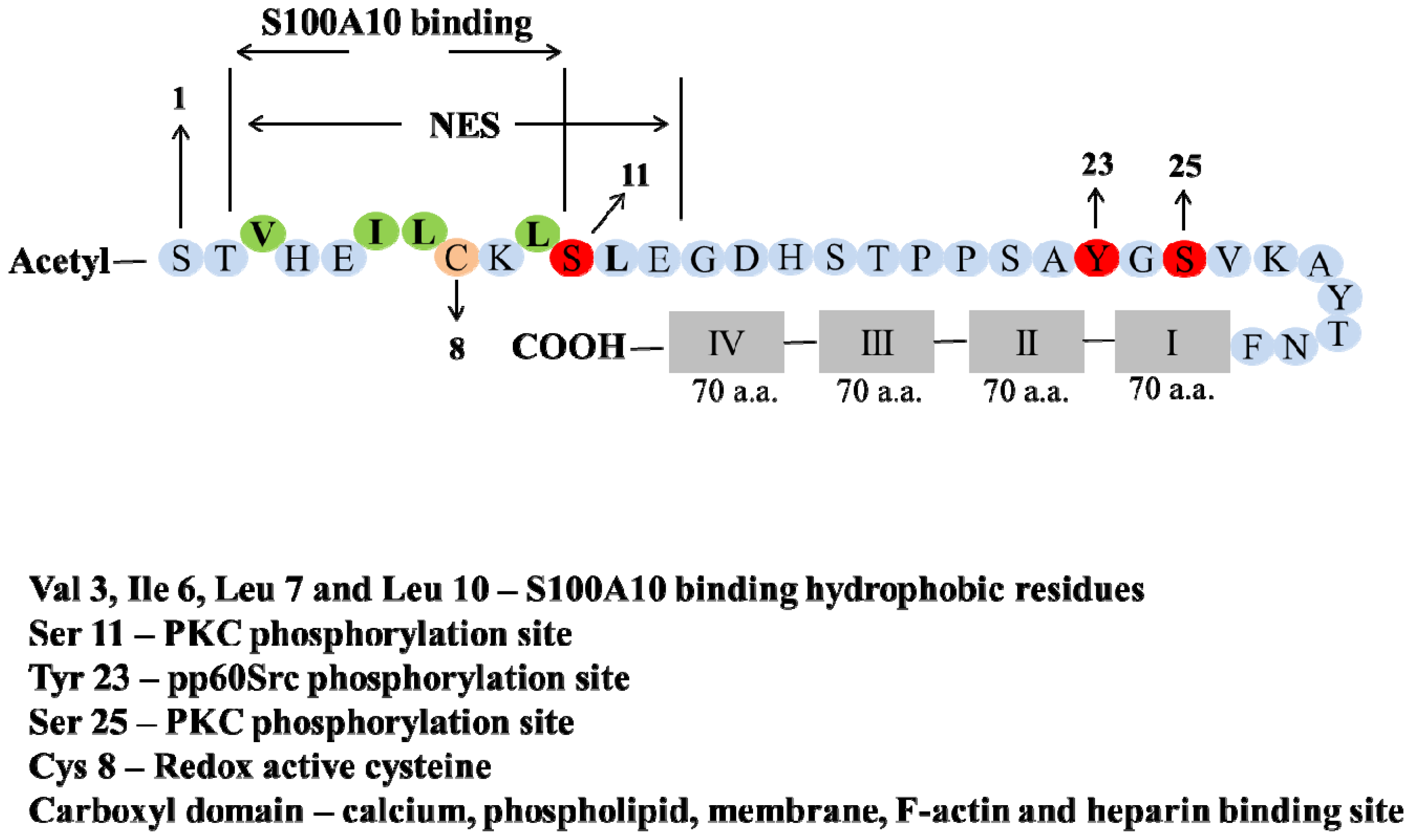
- Becker, T.; Weber, K.; Johnsson, N. Protein-protein recognition via short amphiphilic helices; a mutational analysis of the binding site of annexin II for p11. EMBO J 1990, 9, 4207–4213. [Google Scholar]
- Johnsson, N.; Marriott, G.; Weber, K. p36, the major cytoplasmic substrate of src tyrosine protein kinase, binds to its p11 regulatory subunit via a short amino-terminal amphiphatic helix. EMBO J 1988, 7, 2435–2442. [Google Scholar]
- Rety, S.; Sopkova, J.; Renouard, M.; Osterloh, D.; Gerke, V.; Tabaries, S.; Russo-Marie, F.; Lewit-Bentley, A. The crystal structure of a complex of p11 with the annexin II N-terminal peptide. Nat. Struct. Biol 1999, 6, 89–95. [Google Scholar]
- Rezvanpour, A.; Shaw, G.S. Unique S100 target protein interactions. Gen. Physiol. Biophys. 2009, 28(Focus Issue), F39–46. [Google Scholar]
- Lambert, O.; Gerke, V.; Bader, M.F.; Porte, F.; Brisson, A. Structural analysis of junctions formed between lipid membranes and several annexins by cryo-electron microscopy. J. Mol. Biol 1997, 272, 42–55. [Google Scholar]
- Menke, M.; Ross, M.; Gerke, V.; Steinem, C. The molecular arrangement of membrane-bound annexin A2-S100A10 tetramer as revealed by scanning force microscopy. Chembiochem 2004, 5, 1003–1006. [Google Scholar]
- Schulz, D.M.; Kalkhof, S.; Schmidt, A.; Ihling, C.; Stingl, C.; Mechtler, K.; Zschornig, O.; Sinz, A. Annexin A2/P11 interaction: New insights into annexin A2 tetramer structure by chemical crosslinking, high-resolution mass spectrometry, and computational modeling. Proteins 2007, 69, 254–269. [Google Scholar]
- Glenney, J.R., Jr; Tack, B.F. Amino-terminal sequence of p36 and associated p10: Identification of the site of tyrosine phosphorylation and homology with S-100. Proc. Natl. Acad. Sci. USA 1985, 82, 7884–7888. [Google Scholar]
- Kwon, M.; Yoon, C.S.; Jeong, W.; Rhee, S.G.; Waisman, D.M. Annexin A2-S100A10 heterotetramer, a novel substrate of thioredoxin. J. Biol. Chem 2005, 280, 23584–23592. [Google Scholar]
- Eberhard, D.A.; Karns, L.R.; VandenBerg, S.R.; Creutz, C.E. Control of the nuclear-cytoplasmic partitioning of annexin II by a nuclear export signal and by p11 binding. J. Cell Sci 2001, 114, 3155–3166. [Google Scholar]
- Concha, N.O.; Head, J.F.; Kaetzel, M.A.; Dedman, J.R.; Seaton, B.A. Rat annexin V crystal structure: Ca(2+)-induced conformational changes. Science 1993, 261, 1321–1324. [Google Scholar]
- Jost, M.; Thiel, C.; Weber, K.; Gerke, V. Mapping of three unique Ca(2+)-binding sites in human annexin II. Eur. J. Biochem 1992, 207, 923–930. [Google Scholar]
- Jost, M.; Weber, K.; Gerke, V. Annexin II contains two types of Ca(2+)-binding sites. Biochem. J. 1994, 298(Pt. 3), 553–559. [Google Scholar]
- Sopkova, J.; Renouard, M.; Lewit-Bentley, A. The crystal structure of a new high-calcium form of annexin V. J. Mol. Biol 1993, 234, 816–825. [Google Scholar]
- Weng, X.; Luecke, H.; Song, I.S.; Kang, D.S.; Kim, S.H.; Huber, R. Crystal structure of human annexin I at 2.5 A resolution. Protein Sci 1993, 2, 448–458. [Google Scholar]
- Shao, C.; Zhang, F.; Kemp, M.M.; Linhardt, R.J.; Waisman, D.M.; Head, J.F.; Seaton, B.A. Crystallographic analysis of calcium-dependent heparin binding to annexin A2. J. Biol. Chem 2006, 281, 31689–31695. [Google Scholar]
- Filipenko, N.R.; Kang, H.M.; Waisman, D.M. Characterization of the Ca2+-binding sites of annexin II tetramer. J. Biol. Chem 2000, 275, 38877–38884. [Google Scholar]
- Blackwood, R.A.; Ernst, J.D. Characterization of Ca2(+)-dependent phospholipid binding, vesicle aggregation and membrane fusion by annexins. Biochem. J 1990, 266, 195–200. [Google Scholar]
- Menke, M.; Gerke, V.; Steinem, C. Phosphatidylserine membrane domain clustering induced by annexin A2/S100A10 heterotetramer. Biochemistry 2005, 44, 15296–15303. [Google Scholar]
- Babiychuk, E.B.; Draeger, A. Annexins in cell membrane dynamics. Ca(2+)-regulated association of lipid microdomains. J. Cell Biol 2000, 150, 1113–1124. [Google Scholar]
- Oliferenko, S.; Paiha, K.; Harder, T.; Gerke, V.; Schwarzler, C.; Schwarz, H.; Beug, H.; Gunthert, U.; Huber, L.A. Analysis of CD44-containing lipid rafts: Recruitment of annexin II and stabilization by the actin cytoskeleton. J. Cell Biol 1999, 146, 843–854. [Google Scholar]
- Benaud, C.; Gentil, B.J.; Assard, N.; Court, M.; Garin, J.; Delphin, C.; Baudier, J. AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J. Cell Biol 2004, 164, 133–144. [Google Scholar]
- Harder, T.; Kellner, R.; Parton, R.G.; Gruenberg, J. Specific release of membrane-bound annexin II and cortical cytoskeletal elements by sequestration of membrane cholesterol. Mol. Biol. Cell 1997, 8, 533–545. [Google Scholar]
- Merrifield, C.J.; Rescher, U.; Almers, W.; Proust, J.; Gerke, V.; Sechi, A.S.; Moss, S.E. Annexin 2 has an essential role in actin-based macropinocytic rocketing. Curr. Biol 2001, 11, 1136–1141. [Google Scholar]
- Zobiack, N.; Rescher, U.; Laarmann, S.; Michgehl, S.; Schmidt, M.A.; Gerke, V. Cell-surface attachment of pedestal-forming enteropathogenic E. coli induces a clustering of raft components and a recruitment of annexin 2. J. Cell Sci 2002, 115, 91–98. [Google Scholar]
- Gokhale, N.A.; Abraham, A.; Digman, M.A.; Gratton, E.; Cho, W. Phosphoinositide specificity of and mechanism of lipid domain formation by annexin A2-p11 heterotetramer. J. Biol. Chem 2005, 280, 42831–42840. [Google Scholar]
- Hayes, M.J.; Merrifield, C.J.; Shao, D.; Ayala-Sanmartin, J.; Schorey, C.D.; Levine, T.P.; Proust, J.; Curran, J.; Bailly, M.; Moss, S.E. Annexin 2 binding to phosphatidylinositol 4,5-bisphosphate on endocytic vesicles is regulated by the stress response pathway. J. Biol. Chem 2004, 279, 14157–14164. [Google Scholar]
- Ayala-Sanmartin, J.; Henry, J.P.; Pradel, L.A. Cholesterol regulates membrane binding and aggregation by annexin 2 at submicromolar Ca(2+) concentration. Biochim. Biophys. Acta 2001, 1510, 18–28. [Google Scholar]
- Mayran, N.; Parton, R.G.; Gruenberg, J. Annexin II regulates multivesicular endosome biogenesis in the degradation pathway of animal cells. EMBO J 2003, 22, 3242–3253. [Google Scholar]
- Pike, L.J.; Casey, L. Localization and turnover of phosphatidylinositol 4,5-bisphosphate in caveolin-enriched membrane domains. J. Biol. Chem 1996, 271, 26453–26456. [Google Scholar]
- Campos, B.; Mo, Y.D.; Mealy, T.R.; Li, C.W.; Swairjo, M.A.; Balch, C.; Head, J.F.; Retzinger, G.; Dedman, J.R.; Seaton, B.A. Mutational and crystallographic analyses of interfacial residues in annexin V suggest direct interactions with phospholipid membrane components. Biochemistry 1998, 37, 8004–8010. [Google Scholar]
- Janmey, P.A.; Lamb, J.; Allen, P.G.; Matsudaira, P.T. Phosphoinositide-binding peptides derived from the sequences of gelsolin and villin. J. Biol. Chem 1992, 267, 11818–11823. [Google Scholar]
- Keutzer, J.C.; Hirschhorn, R.R. The growth-regulated gene 1B6 is identified as the heavy chain of calpactin I. Exp. Cell Res 1990, 188, 153–159. [Google Scholar]
- Ozaki, T.; Sakiyama, S. Molecular cloning of rat calpactin I heavy-chain cDNA whose expression is induced in v-src-transformed rat culture cell lines. Oncogene 1993, 8, 1707–1710. [Google Scholar]
- Braselmann, S.; Bergers, G.; Wrighton, C.; Graninger, P.; Superti-Furga, G.; Busslinger, M. Identification of Fos target genes by the use of selective induction systems. J. Cell Sci. Suppl 1992, 16, 97–109. [Google Scholar]
- Genetos, D.C.; Wong, A.; Watari, S.; Yellowley, C.E. Hypoxia increases Annexin A2 expression in osteoblastic cells via VEGF and ERK. Bone 2010, 47, 1013–1019. [Google Scholar]
- Zhao, S.; Huang, L.; Wu, J.; Zhang, Y.; Pan, D.; Liu, X. Vascular endothelial growth factor upregulates expression of annexin A2 in vitro and in a mouse model of ischemic retinopathy. Mol. Vis 2009, 15, 1231–1242. [Google Scholar]
- Konig, J.; Prenen, J.; Nilius, B.; Gerke, V. The annexin II-p11 complex is involved in regulated exocytosis in bovine pulmonary artery endothelial cells. J. Biol. Chem 1998, 273, 19679–19684. [Google Scholar]
- Fey, M.F.; Moffat, G.J.; Vik, D.P.; Meisenhelder, J.; Saris, C.J.; Hunter, T.; Tack, B.F. Complete structure of the murine p36 (annexin II) gene. Identification of mRNAs for both the murine and the human gene with alternatively spliced 5′ noncoding exons. Biochim. Biophys. Acta 1996, 1306, 160–170. [Google Scholar]
- Upton, A.L.; Moss, S.E. Molecular cloning of a novel N-terminal variant of annexin II from rat basophilic leukaemia cells. Biochem. J. 1994, 302(Pt. 2), 425–428. [Google Scholar]
- Hollas, H.; Aukrust, I.; Grimmer, S.; Strand, E.; Flatmark, T.; Vedeler, A. Annexin A2 recognises a specific region in the 3′-UTR of its cognate messenger RNA. Biochim. Biophys. Acta 2006, 1763, 1325–1334. [Google Scholar]
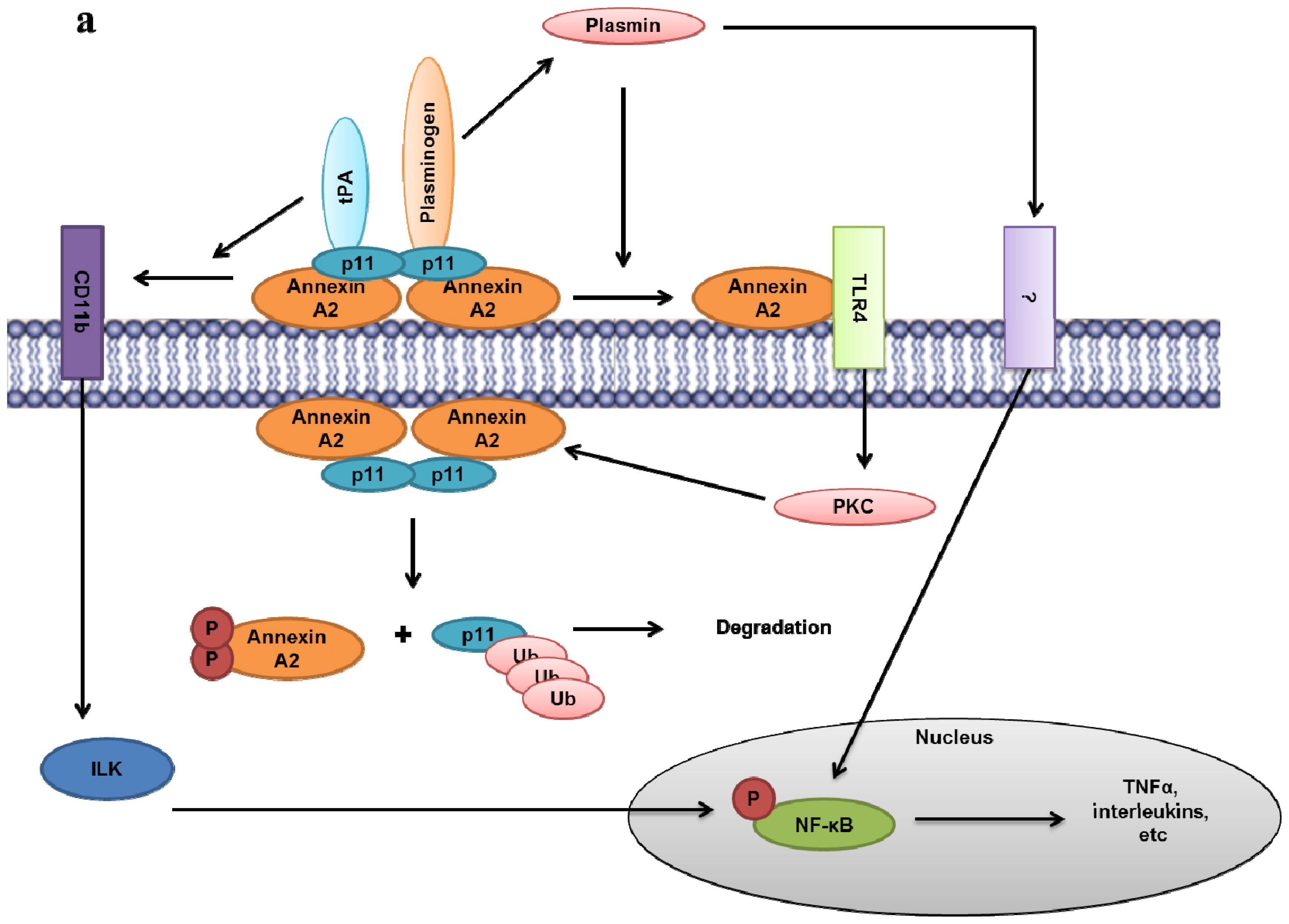
- Hubaishy, I.; Jones, P.G.; Bjorge, J.; Bellagamba, C.; Fitzpatrick, S.; Fujita, D.J.; Waisman, D.M. Modulation of annexin II tetramer by tyrosine phosphorylation. Biochemistry 1995, 34, 14527–14534. [Google Scholar]
- Lauvrak, S.U.; Hollas, H.; Doskeland, A.P.; Aukrust, I.; Flatmark, T.; Vedeler, A. Ubiquitinated annexin A2 is enriched in the cytoskeleton fraction. FEBS Lett 2005, 579, 203–206. [Google Scholar]
- Deng, S.; Jing, B.; Xing, T.; Hou, L.; Yang, Z. Overexpression of annexin A2 is associated with abnormal ubiquitination in breast cancer. Genomics Proteomics Bioinforma 2012, 10, 153–157. [Google Scholar]
- Giuliani, F.; Grieve, A.; Rabouille, C. Unconventional secretion: A stress on GRASP. Curr. Opin. Cell Biol 2011, 23, 498–504. [Google Scholar]
- Nickel, W. The unconventional secretory machinery of fibroblast growth factor 2. Traffic 2011, 12, 799–805. [Google Scholar]
- Temmerman, K.; Ebert, A.D.; Muller, H.M.; Sinning, I.; Tews, I.; Nickel, W. A direct role for phosphatidylinositol-4,5-bisphosphate in unconventional secretion of fibroblast growth factor 2. Traffic 2008, 9, 1204–1217. [Google Scholar]
- Deora, A.B.; Kreitzer, G.; Jacovina, A.T.; Hajjar, K.A. An annexin 2 phosphorylation switch mediates p11-dependent translocation of annexin 2 to the cell surface. J. Biol. Chem 2004, 279, 43411–43418. [Google Scholar]
- Valapala, M.; Vishwanatha, J.K. Lipid raft endocytosis and exosomal transport facilitate extracellular trafficking of annexin A2. J. Biol. Chem 2011, 286, 30911–30925. [Google Scholar]
- Danielsen, E.M.; van Deurs, B.; Hansen, G.H. “Nonclassical” secretion of annexin A2 to the lumenal side of the enterocyte brush border membrane”. Biochemistry 2003, 42, 14670–14676. [Google Scholar]
- Delouche, B.; Pradel, L.A.; Henry, J.P. Phosphorylation by protein kinase C of annexin 2 in chromaffin cells stimulated by nicotine. J. Neurochem 1997, 68, 1720–1727. [Google Scholar]
- Sarafian, T.; Pradel, L.A.; Henry, J.P.; Aunis, D.; Bader, M.F. The participation of annexin II (calpactin I) in calcium-evoked exocytosis requires protein kinase C. J. Cell Biol 1991, 114, 1135–1147. [Google Scholar]
- Cheng, Y.S.; Chen, L.B. Detection of phosphotyrosine-containing 34,000-dalton protein in the framework of cells transformed with Rous sarcoma virus. Proc. Natl. Acad. Sci. USA 1981, 78, 2388–2392. [Google Scholar]
- Radke, K.; Gilmore, T.; Martin, G.S. Transformation by Rous sarcoma virus: A cellular substrate for transformation-specific protein phosphorylation contains phosphotyrosine. Cell 1980, 21, 821–828. [Google Scholar]
- Johnstone, S.A.; Hubaishy, I.; Waisman, D.M. Phosphorylation of annexin II tetramer by protein kinase C inhibits aggregation of lipid vesicles by the protein. J. Biol. Chem 1992, 267, 25976–25981. [Google Scholar]
- Powell, M.A.; Glenney, J.R. Regulation of calpactin I phospholipid binding by calpactin I light-chain binding and phosphorylation by p60v-src. Biochem. J 1987, 247, 321–328. [Google Scholar]
- Jost, M.; Gerke, V. Mapping of a regulatory important site for protein kinase C phosphorylation in the N-terminal domain of annexin II. Biochim. Biophys. Acta 1996, 1313, 283–289. [Google Scholar]
- Regnouf, F.; Sagot, I.; Delouche, B.; Devilliers, G.; Cartaud, J.; Henry, J.P.; Pradel, L.A. “In vitro” phosphorylation of annexin 2 heterotetramer by protein kinase C. Comparative properties of the unphosphorylated and phosphorylated annexin 2 on the aggregation and fusion of chromaffin granule membranes. J. Biol. Chem 1995, 270, 27143–27150. [Google Scholar]
- De Graauw, M.; Tijdens, I.; Smeets, M.B.; Hensbergen, P.J.; Deelder, A.M.; van de Water, B. Annexin A2 phosphorylation mediates cell scattering and branching morphogenesis via cofilin Activation. Mol. Cell Biol 2008, 28, 1029–1040. [Google Scholar]
- Zheng, L.; Foley, K.; Huang, L.; Leubner, A.; Mo, G.; Olino, K.; Edil, B.H.; Mizuma, M.; Sharma, R.; Le, D.T.; et al. Tyrosine 23 phosphorylation-dependent cell-surface localization of annexin A2 is required for invasion and metastases of pancreatic cancer. PLoS One 2011, 6, e19390. [Google Scholar]
- Liu, J.; Rothermund, C.A.; Ayala-Sanmartin, J.; Vishwanatha, J.K. Nuclear annexin II negatively regulates growth of LNCaP cells and substitution of ser 11 and 25 to glu prevents nucleo-cytoplasmic shuttling of annexin II. BMC Biochem 2003, 4, 10. [Google Scholar]
- Karasik, A.; Pepinsky, R.B.; Shoelson, S.E.; Kahn, C.R. Lipocortins 1 and 2 as substrates for the insulin receptor kinase in rat liver. J. Biol. Chem 1988, 263, 11862–11867. [Google Scholar]
- Biener, Y.; Feinstein, R.; Mayak, M.; Kaburagi, Y.; Kadowaki, T.; Zick, Y. Annexin II is a novel player in insulin signal transduction. Possible association between annexin II phosphorylation and insulin receptor internalization. J. Biol. Chem 1996, 271, 29489–29496. [Google Scholar]
- Rescher, U.; Ludwig, C.; Konietzko, V.; Kharitonenkov, A.; Gerke, V. Tyrosine phosphorylation of annexin A2 regulates Rho-mediated actin rearrangement and cell adhesion. J. Cell Sci 2008, 121, 2177–2185. [Google Scholar]
- Jones, D.P. Redox sensing: Orthogonal control in cell cycle and apoptosis signalling. J. Intern. Med 2010, 268, 432–448. [Google Scholar]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar]
- Wang, Y.; Yang, J.; Yi, J. Redox sensing by proteins: Oxidative modifications on cysteines and the consequent events. Antioxid. Redox Signal 2012, 16, 649–657. [Google Scholar]
- Brandes, N.; Schmitt, S.; Jakob, U. Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal 2009, 11, 997–1014. [Google Scholar]
- Burger, A.; Berendes, R.; Liemann, S.; Benz, J.; Hofmann, A.; Gottig, P.; Huber, R.; Gerke, V.; Thiel, C.; Romisch, J.; et al. The crystal structure and ion channel activity of human annexin II, a peripheral membrane protein. J. Mol. Biol 1996, 257, 839–847. [Google Scholar]
- Kwon, M.; Caplan, J.F.; Filipenko, N.R.; Choi, K.S.; Fitzpatrick, S.L.; Zhang, L.; Waisman, D.M. Identification of annexin II heterotetramer as a plasmin reductase. J. Biol. Chem 2002, 277, 10903–10911. [Google Scholar]
- Sullivan, D.M.; Wehr, N.B.; Fergusson, M.M.; Levine, R.L.; Finkel, T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry 2000, 39, 11121–11128. [Google Scholar]
- Rowan, W.H., 3rd; Sun, P.; Liu, L. Nitration of annexin II tetramer. Biochemistry 2002, 41, 1409–1420. [Google Scholar]
- Singh, T.K.; Liu, L. Modification of cysteine residues by N-ethylmaleimide inhibits annexin II tetramer mediated liposome aggregation. Arch. Biochem. Biophys 2000, 381, 235–240. [Google Scholar]
- Caplan, J.F.; Filipenko, N.R.; Fitzpatrick, S.L.; Waisman, D.M. Regulation of annexin A2 by reversible glutathionylation. J. Biol. Chem 2004, 279, 7740–7750. [Google Scholar]
- Madureira, P.A.; Hill, R.; Miller, V.A.; Giacomantonio, C.; Lee, P.W.; Waisman, D.M. Annexin A2 is a novel cellular redox regulatory protein involved in tumorigenesis. Oncotarget 2011, 2, 1075–1093. [Google Scholar]
- Chuthapisith, S.; Bean, B.E.; Cowley, G.; Eremin, J.M.; Samphao, S.; Layfield, R.; Kerr, I.D.; Wiseman, J.; El-Sheemy, M.; Sreenivasan, T.; et al. Annexins in human breast cancer: Possible predictors of pathological response to neoadjuvant chemotherapy. Eur. J. Cancer 2009, 45, 1274–1281. [Google Scholar]
- Arrigo, A.P.; Darlix, J.L.; Spahr, P.F. A cellular protein phosphorylated by the avian sarcoma virus transforming gene product is associated with ribonucleoprotein particles. EMBO J 1983, 2, 309–315. [Google Scholar]
- Vishwanatha, J.K.; Jindal, H.K.; Davis, R.G. The role of primer recognition proteins in DNA replication: Association with nuclear matrix in HeLa cells. J. Cell Sci. 1992, 101(Pt. 1), 25–34. [Google Scholar]
- Filipenko, N.R.; MacLeod, T.J.; Yoon, C.S.; Waisman, D.M. Annexin A2 is a novel RNA-binding protein. J. Biol. Chem 2004, 279, 8723–8731. [Google Scholar]
- Jindal, H.K.; Chaney, W.G.; Anderson, C.W.; Davis, R.G.; Vishwanatha, J.K. The protein-tyrosine kinase substrate, calpactin I heavy chain (p36), is part of the primer recognition protein complex that interacts with DNA polymerase alpha. J. Biol. Chem 1991, 266, 5169–5176. [Google Scholar]
- Madureira, P.A.; Hill, R.; Lee, P.W.; Waisman, D.M. Genotoxic agents promote the nuclear accumulation of annexin A2: Role of annexin A2 in mitigating DNA damage. PLoS One 2012, 7, e50591. [Google Scholar]
- Waters, K.M.; Stenoien, D.L.; Sowa, M.B.; von Neubeck, C.H.; Chrisler, W.B.; Tan, R.; Sontag, R.L.; Weber, T.J. Annexin A2 modulates radiation-sensitive transcriptional programming and cell fate. Radiat. Res 2012, 179, 53–61. [Google Scholar]
- Gerke, V.; Weber, K. Identity of p36K phosphorylated upon Rous sarcoma virus transformation with a protein purified from brush borders, calcium-dependent binding to non-erythroid spectrin and F-actin. EMBO J 1984, 3, 227–233. [Google Scholar]
- Glenney, J. Two related but distinct forms of the Mr 36,000 tyrosine kinase substrate (calpactin) that interact with phospholipid and actin in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 1986, 83, 4258–4262. [Google Scholar]
- Ikebuchi, N.W.; Waisman, D.M. Calcium-dependent regulation of actin filament bundling by lipocortin-85. J. Biol. Chem 1990, 265, 3392–3400. [Google Scholar]
- Jones, P.G.; Moore, G.J.; Waisman, D.M. A nonapeptide to the putative F-actin binding site of annexin-II tetramer inhibits its calcium-dependent activation of actin filament bundling. J. Biol. Chem 1992, 267, 13993–13997. [Google Scholar]
- Filipenko, N.R.; Waisman, D.M. The C terminus of annexin II mediates binding to F-actin. J. Biol. Chem 2001, 276, 5310–5315. [Google Scholar]
- Hayes, M.J.; Shao, D.; Bailly, M.; Moss, S.E. Regulation of actin dynamics by annexin 2. EMBO J 2006, 25, 1816–1826. [Google Scholar]
- Sakai, E.; Miyamoto, H.; Okamoto, K.; Kato, Y.; Yamamoto, K.; Sakai, H. Characterization of phagosomal subpopulations along endocytic routes in osteoclasts and macrophages. J. Biochem 2001, 130, 823–831. [Google Scholar]
- Higgs, H.N.; Pollard, T.D. Regulation of actin filament network formation through ARP2/3 complex: Activation by a diverse array of proteins. Annu. Rev. Biochem 2001, 70, 649–676. [Google Scholar]
- Hansen, M.D.; Ehrlich, J.S.; Nelson, W.J. Molecular mechanism for orienting membrane and actin dynamics to nascent cell-cell contacts in epithelial cells. J. Biol. Chem 2002, 277, 45371–45376. [Google Scholar]
- Jacob, R.; Heine, M.; Eikemeyer, J.; Frerker, N.; Zimmer, K.P.; Rescher, U.; Gerke, V.; Naim, H.Y. Annexin II is required for apical transport in polarized epithelial cells. J. Biol. Chem 2004, 279, 3680–3684. [Google Scholar]
- Ali, S.M.; Burgoyne, R.D. The stimulatory effect of calpactin (annexin II) on calcium-dependent exocytosis in chromaffin cells: Requirement for both the N-terminal and core domains of p36 and ATP. Cell. Signal 1990, 2, 265–276. [Google Scholar]
- Ali, S.M.; Geisow, M.J.; Burgoyne, R.D. A role for calpactin in calcium-dependent exocytosis in adrenal chromaffin cells. Nature 1989, 340, 313–315. [Google Scholar]
- Graham, M.E.; Gerke, V.; Burgoyne, R.D. Modification of annexin II expression in PC12 cell lines does not affect Ca(2+)-dependent exocytosis. Mol. Biol. Cell 1997, 8, 431–442. [Google Scholar]
- Chasserot-Golaz, S.; Vitale, N.; Sagot, I.; Delouche, B.; Dirrig, S.; Pradel, L.A.; Henry, J.P.; Aunis, D.; Bader, M.F. Annexin II in exocytosis: Catecholamine secretion requires the translocation of p36 to the subplasmalemmal region in chromaffin cells. J. Cell Biol 1996, 133, 1217–1236. [Google Scholar]
- Chasserot-Golaz, S.; Vitale, N.; Umbrecht-Jenck, E.; Knight, D.; Gerke, V.; Bader, M.F. Annexin 2 promotes the formation of lipid microdomains required for calcium-regulated exocytosis of dense-core vesicles. Mol. Biol. Cell 2005, 16, 1108–1119. [Google Scholar]
- Martin, T.F.; Loyet, K.M.; Barry, V.A.; Kowalchyk, J.A. The role of PtdIns(4,5)P2 in exocytotic membrane fusion. Biochem. Soc. Trans 1997, 25, 1137–1141. [Google Scholar]
- Emans, N.; Gorvel, J.P.; Walter, C.; Gerke, V.; Kellner, R.; Griffiths, G.; Gruenberg, J. Annexin II is a major component of fusogenic endosomal vesicles. J. Cell Biol 1993, 120, 1357–1369. [Google Scholar]
- Creutz, C.E.; Snyder, S.L. Interactions of annexins with the mu subunits of the clathrin assembly proteins. Biochemistry 2005, 44, 13795–13806. [Google Scholar]
- Urbanska, A.; Sadowski, L.; Kalaidzidis, Y.; Miaczynska, M. Biochemical characterization of APPL endosomes: The role of annexin A2 in APPL membrane recruitment. Traffic 2011, 12, 1227–1241. [Google Scholar]
- Bucci, C.; Parton, R.G.; Mather, I.H.; Stunnenberg, H.; Simons, K.; Hoflack, B.; Zerial, M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 1992, 70, 715–728. [Google Scholar]
- Denzer, K.; Kleijmeer, M.J.; Heijnen, H.F.; Stoorvogel, W.; Geuze, H.J. Exosome: From internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 2000, 113(Pt. 19), 3365–3374. [Google Scholar]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol 2002, 2, 569–579. [Google Scholar]
- Fang, Y.T.; Lin, C.F.; Wang, C.Y.; Anderson, R.; Lin, Y.S. Interferon-gamma stimulates p11-dependent surface expression of annexin A2 in lung epithelial cells to enhance phagocytosis. J. Cell Physiol 2012, 227, 2775–2787. [Google Scholar]
- Yang, C.; Robbins, P.D. The roles of tumor-derived exosomes in cancer pathogenesis. Clin. Dev. Immunol 2011, 2011, 842849. [Google Scholar]
- Mears, R.; Craven, R.A.; Hanrahan, S.; Totty, N.; Upton, C.; Young, S.L.; Patel, P.; Selby, P.J.; Banks, R.E. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics 2004, 4, 4019–4031. [Google Scholar]
- Xiao, D.; Ohlendorf, J.; Chen, Y.; Taylor, D.D.; Rai, S.N.; Waigel, S.; Zacharias, W.; Hao, H.; McMasters, K.M. Identifying mRNA, microRNA and protein profiles of melanoma exosomes. PLoS One 2012, 7, e46874. [Google Scholar]
- Drust, D.S.; Creutz, C.E. Aggregation of chromaffin granules by calpactin at micromolar levels of calcium. Nature 1988, 331, 88–91. [Google Scholar]
- Thiel, C.; Osborn, M.; Gerke, V. The tight association of the tyrosine kinase substrate annexin II with the submembranous cytoskeleton depends on intact p11- and Ca(2+)-binding sites. J. Cell Sci. 1992, 103(Pt. 3), 733–742. [Google Scholar]
- Zokas, L.; Glenney, J.R., Jr. The calpactin light chain is tightly linked to the cytoskeletal form of calpactin I: Studies using monoclonal antibodies to calpactin subunits. J. Cell Biol. 1987, 105, 2111–2121. [Google Scholar]
- Umbrecht-Jenck, E.; Demais, V.; Calco, V.; Bailly, Y.; Bader, M.F.; Chasserot-Golaz, S. S100A10-mediated translocation of annexin-A2 to SNARE proteins in adrenergic chromaffin cells undergoing exocytosis. Traffic 2010, 11, 958–971. [Google Scholar]
- Babbin, B.A.; Parkos, C.A.; Mandell, K.J.; Winfree, L.M.; Laur, O.; Ivanov, A.I.; Nusrat, A. Annexin 2 regulates intestinal epithelial cell spreading and wound closure through Rho-related signaling. Am. J. Pathol 2007, 170, 951–966. [Google Scholar]
- Barwe, S.P.; Anilkumar, G.; Moon, S.Y.; Zheng, Y.; Whitelegge, J.P.; Rajasekaran, S.A.; Rajasekaran, A.K. Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol. Biol. Cell 2005, 16, 1082–1094. [Google Scholar]
- Hall, A. G proteins and small GTPases: Distant relatives keep in touch. Science 1998, 280, 2074–2075. [Google Scholar]
- Tapon, N.; Hall, A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr. Opin. Cell Biol 1997, 9, 86–92. [Google Scholar]
- Yamada, A.; Irie, K.; Hirota, T.; Ooshio, T.; Fukuhara, A.; Takai, Y. Involvement of the annexin II-S100A10 complex in the formation of E-cadherin-based adherens junctions in Madin-Darby canine kidney cells. J. Biol. Chem 2005, 280, 6016–6027. [Google Scholar]
- Su, S.C.; Maxwell, S.A.; Bayless, K.J. Annexin 2 regulates endothelial morphogenesis by controlling AKT activation and junctional integrity. J. Biol. Chem 2010, 285, 40624–40634. [Google Scholar]
- Lee, D.B.; Jamgotchian, N.; Allen, S.G.; Kan, F.W.; Hale, I.L. Annexin A2 heterotetramer: Role in tight junction assembly. Am. J. Physiol. Renal Physiol 2004, 287, F481–F491. [Google Scholar]
- Krishna, P.; Kennedy, B.P.; Waisman, D.M.; van de Sande, J.H.; McGhee, J.D. Are many Z-DNA binding proteins actually phospholipid-binding proteins? Proc. Natl. Acad. Sci. USA 1990, 87, 1292–1295. [Google Scholar]
- Vedeler, A.; Hollas, H. Annexin II is associated with mRNAs which may constitute a distinct subpopulation. Biochem. J. 2000, 348(Pt. 3), 565–572. [Google Scholar]
- Aukrust, I.; Hollas, H.; Strand, E.; Evensen, L.; Trave, G.; Flatmark, T.; Vedeler, A. The mRNA-binding site of annexin A2 resides in helices C-D of its domain IV. J. Mol. Biol 2007, 368, 1367–1378. [Google Scholar]
- Mickleburgh, I.; Burtle, B.; Hollas, H.; Campbell, G.; Chrzanowska-Lightowlers, Z.; Vedeler, A.; Hesketh, J. Annexin A2 binds to the localization signal in the 3′ untranslated region of c-myc mRNA. FEBS J 2005, 272, 413–421. [Google Scholar]
- Bassell, G.; Singer, R.H. mRNA and cytoskeletal filaments. Curr. Opin. Cell Biol 1997, 9, 109–115. [Google Scholar]
- Miles, L.A.; Plow, E.F.; Waisman, D.M.; Parmer, R.J. Plasminogen receptors. J. Biomed. Biotechnol 2012, 2012, 130735. [Google Scholar]
- Ellis, V.; Behrendt, N.; Dano, K. Plasminogen activation by receptor-bound urokinase. A kinetic study with both cell-associated and isolated receptor. J. Biol. Chem 1991, 266, 12752–12758. [Google Scholar]
- Plow, E.F.; Freaney, D.E.; Plescia, J.; Miles, L.A. The plasminogen system and cell surfaces: Evidence for plasminogen and urokinase receptors on the same cell type. J. Cell Biol 1986, 103, 2411–2420. [Google Scholar]
- Felez, J.; Miles, L.A.; Fabregas, P.; Jardi, M.; Plow, E.F.; Lijnen, R.H. Characterization of cellular binding sites and interactive regions within reactants required for enhancement of plasminogen activation by tPA on the surface of leukocytic cells. Thromb. Haemost 1996, 76, 577–584. [Google Scholar]
- Hajjar, K.A.; Jacovina, A.T.; Chacko, J. An endothelial cell receptor for plasminogen/tissue plasminogen activator. I. Identity with annexin II. J. Biol. Chem 1994, 269, 21191–21197. [Google Scholar]
- O’Connell, P.A.; Surette, A.P.; Liwski, R.S.; Svenningsson, P.; Waisman, D.M. S100A10 regulates plasminogen-dependent macrophage invasion. Blood 2010, 116, 1136–1146. [Google Scholar]
- MacLeod, T.J.; Kwon, M.; Filipenko, N.R.; Waisman, D.M. Phospholipid-associated annexin A2-S100A10 heterotetramer and its subunits: Characterization of the interaction with tissue plasminogen activator, plasminogen, and plasmin. J. Biol. Chem 2003, 278, 25577–25584. [Google Scholar]
- Das, R.; Burke, T.; Plow, E.F. Histone H2B as a functionally important plasminogen receptor on macrophages. Blood 2007, 110, 3763–3772. [Google Scholar]
- Roda, O.; Valero, M.L.; Peiro, S.; Andreu, D.; Real, F.X.; Navarro, P. New insights into the tPA-annexin A2 interaction. Is annexin A2 CYS8 the sole requirement for this association? J. Biol. Chem 2003, 278, 5702–5709. [Google Scholar]
- Felez, J.; Chanquia, C.J.; Fabregas, P.; Plow, E.F.; Miles, L.A. Competition between plasminogen and tissue plasminogen activator for cellular binding sites. Blood 1993, 82, 2433–2441. [Google Scholar]
- Felez, J.; Chanquia, C.J.; Levin, E.G.; Miles, L.A.; Plow, E.F. Binding of tissue plasminogen activator to human monocytes and monocytoid cells. Blood 1991, 78, 2318–2327. [Google Scholar]
- Beebe, D.P.; Miles, L.A.; Plow, E.F. A linear amino acid sequence involved in the interaction of t-PA with its endothelial cell receptor. Blood 1989, 74, 2034–2037. [Google Scholar]
- Sinniger, V.; Merton, R.E.; Fabregas, P.; Felez, J.; Longstaff, C. Regulation of tissue plasminogen activator activity by cells. Domains responsible for binding and mechanism of stimulation. J. Biol. Chem 1999, 274, 12414–12422. [Google Scholar]
- Suzuki, Y.; Mogami, H.; Ihara, H.; Urano, T. Unique secretory dynamics of tissue plasminogen activator and its modulation by plasminogen activator inhibitor-1 in vascular endothelial cells. Blood 2009, 113, 470–478. [Google Scholar]
- Byeon, I.J.; Llinas, M. Solution structure of the tissue-type plasminogen activator kringle 2 domain complexed to 6-aminohexanoic acid an antifibrinolytic drug. J. Mol. Biol 1991, 222, 1035–1051. [Google Scholar]
- Galantai, R.; Modos, K.; Fidy, J.; Kolev, K.; Machovich, R. Structural basis of the cofactor function of denatured albumin in plasminogen activation by tissue-type plasminogen activator. Biochem. Biophys. Res. Commun 2006, 341, 736–741. [Google Scholar]
- Novokhatny, V.V.; Ingham, K.C.; Medved, L.V. Domain structure and domain-domain interactions of recombinant tissue plasminogen activator. J. Biol. Chem 1991, 266, 12994–13002. [Google Scholar]
- Kassam, G.; Choi, K.S.; Ghuman, J.; Kang, H.M.; Fitzpatrick, S.L.; Zackson, T.; Zackson, S.; Toba, M.; Shinomiya, A.; Waisman, D.M. The role of annexin II tetramer in the activation of plasminogen. J. Biol. Chem 1998, 273, 4790–4799. [Google Scholar]
- Nilius, B.; Gerke, V.; Prenen, J.; Szucs, G.; Heinke, S.; Weber, K.; Droogmans, G. Annexin II modulates volume-activated chloride currents in vascular endothelial cells. J. Biol. Chem 1996, 271, 30631–30636. [Google Scholar]
- Chetcuti, A.; Margan, S.H.; Russell, P.; Mann, S.; Millar, D.S.; Clark, S.J.; Rogers, J.; Handelsman, D.J.; Dong, Q. Loss of annexin II heavy and light chains in prostate cancer and its precursors. Cancer Res 2001, 61, 6331–6334. [Google Scholar]
- Zobiack, N.; Rescher, U.; Ludwig, C.; Zeuschner, D.; Gerke, V. The annexin 2/S100A10 complex controls the distribution of transferrin receptor-containing recycling endosomes. Mol. Biol. Cell 2003, 14, 4896–4908. [Google Scholar]
- Choi, K.S.; Fogg, D.K.; Yoon, C.S.; Waisman, D.M. p11 regulates extracellular plasmin production and invasiveness of HT1080 fibrosarcoma cells. FASEB J 2003, 17, 235–246. [Google Scholar]
- Surette, A.P.; Madureira, P.A.; Phipps, K.D.; Miller, V.A.; Svenningsson, P.; Waisman, D.M. Regulation of fibrinolysis by S100A10 in vivo. Blood 2011, 118, 3172–3181. [Google Scholar]
- Ling, Q.; Jacovina, A.T.; Deora, A.; Febbraio, M.; Simantov, R.; Silverstein, R.L.; Hempstead, B.; Mark, W.H.; Hajjar, K.A. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J. Clin. Invest 2004, 113, 38–48. [Google Scholar]
- Brownstein, C.; Deora, A.B.; Jacovina, A.T.; Weintraub, R.; Gertler, M.; Khan, K.M.; Falcone, D.J.; Hajjar, K.A. Annexin II mediates plasminogen-dependent matrix invasion by human monocytes: Enhanced expression by macrophages. Blood 2004, 103, 317–324. [Google Scholar]
- Falcone, D.J.; Borth, W.; Khan, K.M.; Hajjar, K.A. Plasminogen-mediated matrix invasion and degradation by macrophages is dependent on surface expression of annexin II. Blood 2001, 97, 777–784. [Google Scholar]
- Gou, D.; Mishra, A.; Weng, T.; Su, L.; Chintagari, N.R.; Wang, Z.; Zhang, H.; Gao, L.; Wang, P.; Stricker, H.M.; Liu, L. Annexin A2 interactions with Rab14 in alveolar type II cells. J. Biol. Chem 2008, 283, 13156–13164. [Google Scholar]
- Gong, Y.; Hart, E.; Shchurin, A.; Hoover-Plow, J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J. Clin. Invest 2008, 118, 3012–3024. [Google Scholar]
- Mai, J.; Finley, R.L., Jr; Waisman, D.M.; Sloane, B.F. Human procathepsin B interacts with the annexin II tetramer on the surface of tumor cells. J. Biol. Chem. 2000, 275, 12806–12812. [Google Scholar]
- McColl, B.K.; Baldwin, M.E.; Roufail, S.; Freeman, C.; Moritz, R.L.; Simpson, R.J.; Alitalo, K.; Stacker, S.A.; Achen, M.G. Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J. Exp. Med 2003, 198, 863–868. [Google Scholar]
- Chiang, Y.; Schneiderman, M.H.; Vishwanatha, J.K. Annexin II expression is regulated during mammalian cell cycle. Cancer Res 1993, 53, 6017–6021. [Google Scholar]
- Sharma, M.R.; Rothman, V.; Tuszynski, G.P.; Sharma, M.C. Antibody-directed targeting of angiostatin’s receptor annexin II inhibits Lewis Lung Carcinoma tumor growth via blocking of plasminogen activation: Possible biochemical mechanism of angiostatin’s action. Exp. Mol. Pathol 2006, 81, 136–145. [Google Scholar]
- Tuszynski, G.P.; Sharma, M.R.; Rothman, V.L.; Sharma, M.C. Angiostatin binds to tyrosine kinase substrate annexin II through the lysine-binding domain in endothelial cells. Microvasc. Res 2002, 64, 448–462. [Google Scholar]
- Sharma, M.; Ownbey, R.T.; Sharma, M.C. Breast cancer cell surface annexin II induces cell migration and neoangiogenesis via tPA dependent plasmin generation. Exp. Mol. Pathol 2010, 88, 278–286. [Google Scholar]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med 2011, 17, 867–874. [Google Scholar]
- Chuthapisith, S.; Layfield, R.; Kerr, I.D.; Hughes, C.; Eremin, O. Proteomic profiling of MCF-7 breast cancer cells with chemoresistance to different types of anti-cancer drugs. Int. J. Oncol 2007, 30, 1545–1551. [Google Scholar]
- Esposito, I.; Penzel, R.; Chaib-Harrireche, M.; Barcena, U.; Bergmann, F.; Riedl, S.; Kayed, H.; Giese, N.; Kleeff, J.; Friess, H.; Schirmacher, P. Tenascin C and annexin II expression in the process of pancreatic carcinogenesis. J. Pathol 2006, 208, 673–685. [Google Scholar]
- Sitek, B.; Sipos, B.; Alkatout, I.; Poschmann, G.; Stephan, C.; Schulenborg, T.; Marcus, K.; Luttges, J.; Dittert, D.D.; Baretton, G.; et al. Analysis of the pancreatic tumor progression by a quantitative proteomic approach and immunhistochemical validation. J. Proteome Res 2009, 8, 1647–1656. [Google Scholar]
- Diaz, V.M.; Hurtado, M.; Thomson, T.M.; Reventos, J.; Paciucci, R. Specific interaction of tissue-type plasminogen activator (t-PA) with annexin II on the membrane of pancreatic cancer cells activates plasminogen and promotes invasion in vitro. Gut 2004, 53, 993–1000. [Google Scholar]
- Tian, R.; Wei, L.M.; Qin, R.Y.; Li, Y.; Du, Z.Y.; Xia, W.; Shi, C.J.; Jin, H. Proteome analysis of human pancreatic ductal adenocarcinoma tissue using two-dimensional gel electrophoresis and tandem mass spectrometry for identification of disease-related proteins. Dig. Dis. Sci 2008, 53, 65–72. [Google Scholar]
- Chung, C.Y.; Murphy-Ullrich, J.E.; Erickson, H.P. Mitogenesis, cell migration, and loss of focal adhesions induced by tenascin-C interacting with its cell surface receptor, annexin II. Mol. Biol. Cell 1996, 7, 883–892. [Google Scholar]
- Takano, S.; Togawa, A.; Yoshitomi, H.; Shida, T.; Kimura, F.; Shimizu, H.; Yoshidome, H.; Ohtsuka, M.; Kato, A.; Tomonaga, T.; et al. Annexin II overexpression predicts rapid recurrence after surgery in pancreatic cancer patients undergoing gemcitabine-adjuvant chemotherapy. Ann. Surg. Oncol 2008, 15, 3157–3168. [Google Scholar]
- Domoto, T.; Miyama, Y.; Suzuki, H.; Teratani, T.; Arai, K.; Sugiyama, T.; Takayama, T.; Mugiya, S.; Ozono, S.; Nozawa, R. Evaluation of S100A10, annexin II and B-FABP expression as markers for renal cell carcinoma. Cancer Sci 2007, 98, 77–82. [Google Scholar]
- Ohno, Y.; Izumi, M.; Kawamura, T.; Nishimura, T.; Mukai, K.; Tachibana, M. Annexin II represents metastatic potential in clear-cell renal cell carcinoma. Br. J. Cancer 2009, 101, 287–294. [Google Scholar]
- Emoto, K.; Yamada, Y.; Sawada, H.; Fujimoto, H.; Ueno, M.; Takayama, T.; Kamada, K.; Naito, A.; Hirao, S.; Nakajima, Y. Annexin II overexpression correlates with stromal tenascin-C overexpression: A prognostic marker in colorectal carcinoma. Cancer 2001, 92, 1419–1426. [Google Scholar]
- Pei, H.; Zhu, H.; Zeng, S.; Li, Y.; Yang, H.; Shen, L.; Chen, J.; Zeng, L.; Fan, J.; Li, X.; et al. Proteome analysis and tissue microarray for profiling protein markers associated with lymph node metastasis in colorectal cancer. J. Proteome Res 2007, 6, 2495–2501. [Google Scholar]
- Zhang, L.; Fogg, D.K.; Waisman, D.M. RNA interference-mediated silencing of the S100A10 gene attenuates plasmin generation and invasiveness of Colo 222 colorectal cancer cells. J. Biol. Chem 2004, 279, 2053–2062. [Google Scholar]
- Giraldez, M.D.; Lozano, J.J.; Cuatrecasas, M.; Alonso-Espinaco, V.; Maurel, J.; Marmol, M.; Horndler, C.; Ortego, J.; Alonso, V.; Escudero, P.; et al. Gene-expression signature of tumor recurrence in patients with stage II and III colon cancer treated with 5′ fluoruracil-based adjuvant chemotherapy. Int. J. Cancer 2013, 132, 1090–1097. [Google Scholar]
- Frohlich, M.; Motte, P.; Galvin, K.; Takahashi, H.; Wands, J.; Ozturk, M. Enhanced expression of the protein kinase substrate p36 in human hepatocellular carcinoma. Mol. Cell. Biol 1990, 10, 3216–3223. [Google Scholar]
- Swisher, J.F.; Khatri, U.; Feldman, G.M. Annexin A2 is a soluble mediator of macrophage activation. J. Leukoc. Biol 2007, 82, 1174–1184. [Google Scholar]
- Mohammad, H.S.; Kurokohchi, K.; Yoneyama, H.; Tokuda, M.; Morishita, A.; Jian, G.; Shi, L.; Murota, M.; Tani, J.; Kato, K.; et al. Annexin A2 expression and phosphorylation are up-regulated in hepatocellular carcinoma. Int. J. Oncol 2008, 33, 1157–1163. [Google Scholar]
- Longerich, T.; Haller, M.T.; Mogler, C.; Aulmann, S.; Lohmann, V.; Schirmacher, P.; Brand, K. Annexin A2 as a differential diagnostic marker of hepatocellular tumors. Pathol. Res. Pract 2011, 207, 8–14. [Google Scholar]
- Kittaka, N.; Takemasa, I.; Takeda, Y.; Marubashi, S.; Nagano, H.; Umeshita, K.; Dono, K.; Matsubara, K.; Matsuura, N.; Monden, M. Molecular mapping of human hepatocellular carcinoma provides deeper biological insight from genomic data. Eur. J. Cancer 2008, 44, 885–897. [Google Scholar]
- Sun, Y.; Gao, G.; Cai, J.; Wang, Y.; Qu, X.; He, L.; Liu, F.; Zhang, Y.; Lin, K.; Ma, S.; et al. Annexin A2 is a discriminative serological candidate in early hepatocellular carcinoma. Carcinogenesis 2013, 34, 595–604. [Google Scholar]
- Menell, J.S.; Cesarman, G.M.; Jacovina, A.T.; McLaughlin, M.A.; Lev, E.A.; Hajjar, K.A. Annexin II and bleeding in acute promyelocytic leukemia. N. Engl. J. Med 1999, 340, 994–1004. [Google Scholar]
- Liu, Y.; Wang, Z.; Jiang, M.; Dai, L.; Zhang, W.; Wu, D.; Ruan, C. The expression of annexin II and its role in the fibrinolytic activity in acute promyelocytic leukemia. Leuk. Res 2011, 35, 879–884. [Google Scholar]
- O’Connell, P.A.; Madureira, P.A.; Berman, J.N.; Liwski, R.S.; Waisman, D.M. Regulation of S100A10 by the PML-RAR-alpha oncoprotein. Blood 2011, 117, 4095–4105. [Google Scholar]
- Zhao, W.; Wang, X.; Guo, W.; Qu, B.; Wang, H.; Shen, Z.; Chen, Z.; Wang, Z. Hemostatic abnormalities associated with acute promyelocytic leukemia and corrective effects of all-trans-retinoic acid or arsenic trioxide treatment. Chin. Med. J. (Engl. ) 2000, 113, 236–240. [Google Scholar]
- Xie, Y.; Wang, Z.Y.; Zhang, W.; Dai, L.; Bai, X. The fibrinolytic activity in leukemic cell lines and its alteration on all-trans retinoic acid treatment (in Chinese). Zhonghua Xue Ye Xue Za Zhi 2006, 27, 588–592. [Google Scholar]
- Zhang, X.H.; Hu, Y.; Bao, L.; Jiang, Q.; Yang, L.H.; Lu, X.J.; Hong, M.; Xia, L.H.; Guo, T.; Shen, G.X.; et al. Arsenic trioxide downregulates the expression of annexin II in bone marrow cells from patients with acute myelogenous leukemia. Chin. Med. J. (Engl. ) 2009, 122, 1969–1973. [Google Scholar]
- Yee, D.S.; Narula, N.; Ramzy, I.; Boker, J.; Ahlering, T.E.; Skarecky, D.W.; Ornstein, D.K. Reduced annexin II protein expression in high-grade prostatic intraepithelial neoplasia and prostate cancer. Arch. Pathol. Lab. Med 2007, 131, 902–908. [Google Scholar]
- Xin, W.; Rhodes, D.R.; Ingold, C.; Chinnaiyan, A.M.; Rubin, M.A. Dysregulation of the annexin family protein family is associated with prostate cancer progression. Am. J. Pathol 2003, 162, 255–261. [Google Scholar]
- Banerjee, A.G.; Liu, J.; Yuan, Y.; Gopalakrishnan, V.K.; Johansson, S.L.; Dinda, A.K.; Gupta, N.P.; Trevino, L.; Vishwanatha, J.K. Expression of biomarkers modulating prostate cancer angiogenesis: Differential expression of annexin II in prostate carcinomas from India and USA. Mol. Cancer 2003, 2, 34. [Google Scholar]
- Inokuchi, J.; Narula, N.; Yee, D.S.; Skarecky, D.W.; Lau, A.; Ornstein, D.K.; Tyson, D.R. Annexin A2 positively contributes to the malignant phenotype and secretion of IL-6 in DU145 prostate cancer cells. Int. J. Cancer 2009, 124, 68–74. [Google Scholar]
- Shiozawa, Y.; Havens, A.M.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Lu, G.; Roodman, G.D.; Loberg, R.D.; Pienta, K.J.; et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell. Biochem 2008, 105, 370–380. [Google Scholar]
- Qi, Y.J.; Wang, L.D.; Jiao, X.Y.; Feng, X.S.; Fan, Z.M.; Gao, S.S.; He, X.; Li, J.L.; Chang, F.B. Dysregulation of Annexin II expression in esophageal squamous cell cancer and adjacent tissues from a high-incidence area for esophageal cancer in Henan province (in Chinese). Ai Zheng 2007, 26, 730–736. [Google Scholar]
- Chan, C.M.; Wong, S.C.; Lam, M.Y.; Hui, E.P.; Chan, J.K.; Lo, E.S.; Cheuk, W.; Wong, M.C.; Tsao, S.W.; Chan, A.T. Proteomic comparison of nasopharyngeal cancer cell lines C666–1 and NP69 identifies down-regulation of annexin II and beta2-tubulin for nasopharyngeal carcinoma. Arch. Pathol. Lab. Med 2008, 132, 675–683. [Google Scholar]
- Pena-Alonso, E.; Rodrigo, J.P.; Parra, I.C.; Pedrero, J.M.; Meana, M.V.; Nieto, C.S.; Fresno, M.F.; Morgan, R.O.; Fernandez, M.P. Annexin A2 localizes to the basal epithelial layer and is down-regulated in dysplasia and head and neck squamous cell carcinoma. Cancer Lett 2008, 263, 89–98. [Google Scholar]
- Wu, W.; Tang, X.; Hu, W.; Lotan, R.; Hong, W.K.; Mao, L. Identification and validation of metastasis-associated proteins in head and neck cancer cell lines by two-dimensional electrophoresis and mass spectrometry. Clin. Exp. Metastasis 2002, 19, 319–326. [Google Scholar]
- Kesavan, K.; Ratliff, J.; Johnson, E.W.; Dahlberg, W.; Asara, J.M.; Misra, P.; Frangioni, J.V.; Jacoby, D.B. Annexin A2 is a molecular target for TM601, a peptide with tumor-targeting and anti-angiogenic effects. J. Biol. Chem 2010, 285, 4366–4374. [Google Scholar]
- Braden, A.R.; Kafka, M.T.; Cunningham, L.; Jones, H.; Vishwanatha, J.K. Polymeric nanoparticles for sustained down-regulation of annexin A2 inhibit prostate tumor growth. J. Nanosci. Nanotechnol 2009, 9, 2856–2865. [Google Scholar]
- Song, C.; Zhou, X.; Dong, Q.; Fan, R.; Wu, G.; Ji, B.; Meng, Q.; Zheng, M. Regulation of inflammatory response in human chondrocytes by lentiviral mediated RNA interference against S100A10. Inflamm. Res 2012, 61, 1219–1227. [Google Scholar]
- Lin, L.; Wu, C.; Hu, K. Tissue plasminogen activator activates NF-kappaB through a pathway involving annexin A2/CD11b and integrin-linked kinase. J. Am. Soc. Nephrol 2012, 23, 1329–1338. [Google Scholar]
- Laumonnier, Y.; Syrovets, T.; Burysek, L.; Simmet, T. Identification of the annexin A2 heterotetramer as a receptor for the plasmin-induced signaling in human peripheral monocytes. Blood 2006, 107, 3342–3349. [Google Scholar]
- Li, Q.; Laumonnier, Y.; Syrovets, T.; Simmet, T. Plasmin triggers cytokine induction in human monocyte-derived macrophages. Arterioscler. Thromb. Vasc. Biol 2007, 27, 1383–1389. [Google Scholar]
- He, K.L.; Sui, G.; Xiong, H.; Broekman, M.J.; Huang, B.; Marcus, A.J.; Hajjar, K.A. Feedback regulation of endothelial cell surface plasmin generation by PKC-dependent phosphorylation of annexin A2. J. Biol. Chem 2011, 286, 15428–15439. [Google Scholar]
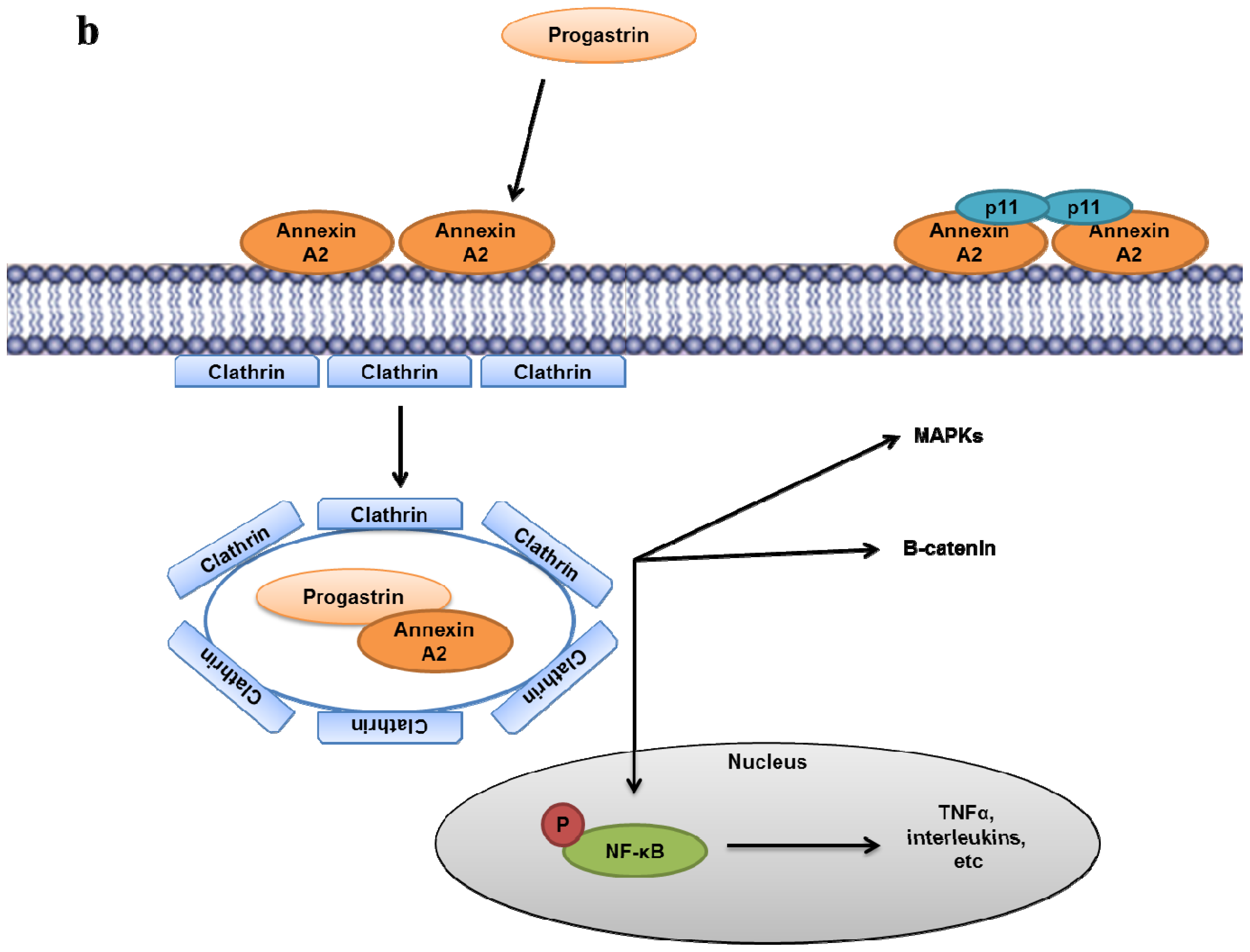
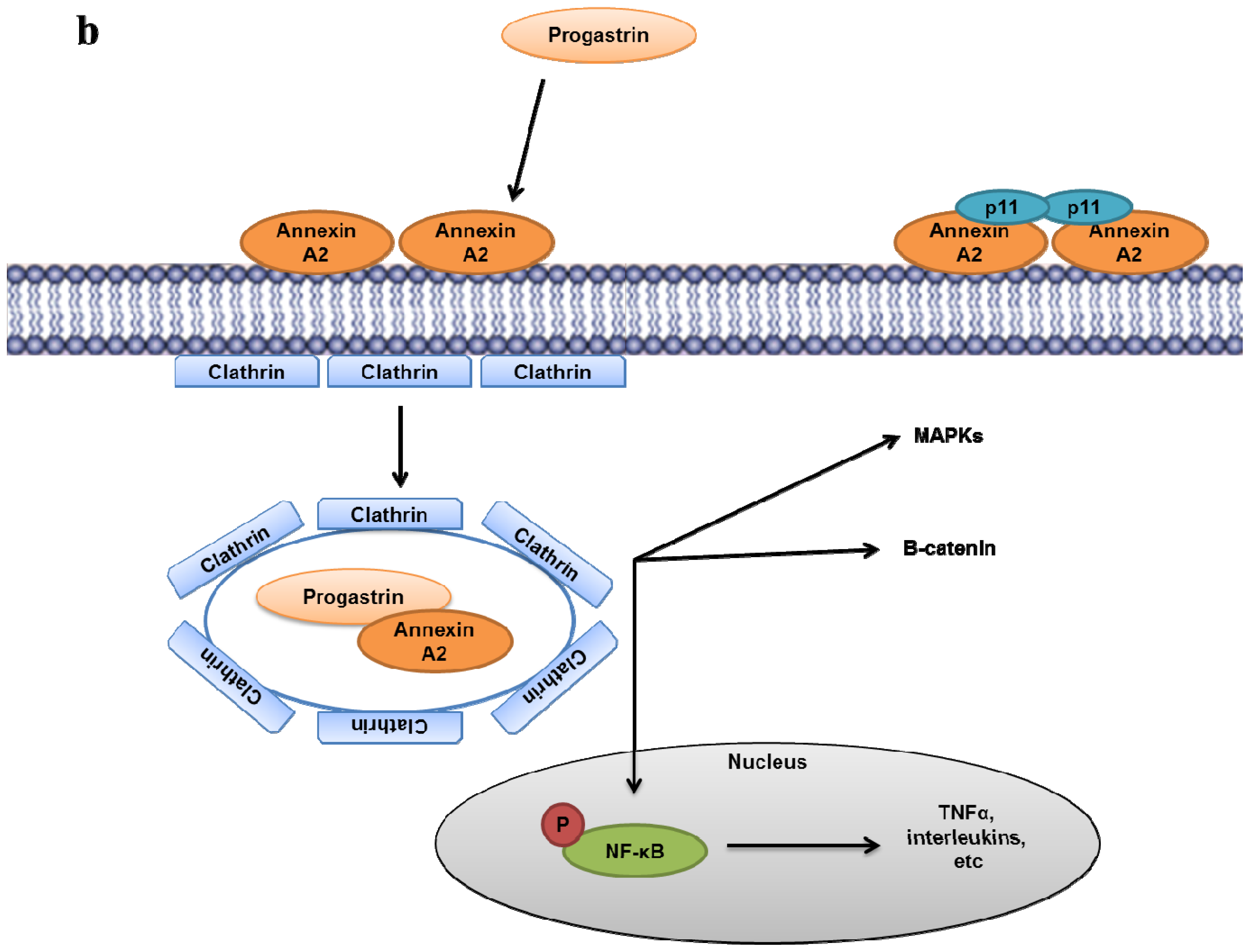
- Singh, P.; Wu, H.; Clark, C.; Owlia, A. Annexin II binds progastrin and gastrin-like peptides, and mediates growth factor effects of autocrine and exogenous gastrins on colon cancer and intestinal epithelial cells. Oncogene 2007, 26, 425–440. [Google Scholar]
- Sarkar, S.; Swiercz, R.; Kantara, C.; Hajjar, K.A.; Singh, P. Annexin A2 mediates up-regulation of NF-kappaB, beta-catenin, and stem cell in response to progastrin in mice and HEK-293 cells. Gastroenterology 2011, 140. [Google Scholar]
- Sarkar, S.; Kantara, C.; Singh, P. Clathrin mediates endocytosis of progastrin and activates MAPKs: Role of cell surface annexin A2. Am. J. Physiol. Gastrointest Liver Physiol 2012, 302, G712–722. [Google Scholar]
- Swisher, J.F.; Burton, N.; Bacot, S.M.; Vogel, S.N.; Feldman, G.M. Annexin A2 tetramer activates human and murine macrophages through TLR4. Blood 2010, 115, 549–558. [Google Scholar]
- Li, F.; Chung, H.; Reddy, S.V.; Lu, G.; Kurihara, N.; Zhao, A.Z.; Roodman, G.D. Annexin II stimulates RANKL expression through MAPK. J. Bone Miner. Res 2005, 20, 1161–1167. [Google Scholar]
- Menaa, C.; Devlin, R.D.; Reddy, S.V.; Gazitt, Y.; Choi, S.J.; Roodman, G.D. Annexin II increases osteoclast formation by stimulating the proliferation of osteoclast precursors in human marrow cultures. J. Clin. Invest 1999, 103, 1605–1613. [Google Scholar]
- Bouma, B.; de Groot, P.G.; van den Elsen, J.M.; Ravelli, R.B.; Schouten, A.; Simmelink, M.J.; Derksen, R.H.; Kroon, J.; Gros, P. Adhesion mechanism of human beta(2)-glycoprotein I to phospholipids based on its crystal structure. EMBO J 1999, 18, 5166–5174. [Google Scholar]
- Schwarzenbacher, R.; Zeth, K.; Diederichs, K.; Gries, A.; Kostner, G.M.; Laggner, P.; Prassl, R. Crystal structure of human beta2-glycoprotein I: Implications for phospholipid binding and the antiphospholipid syndrome. EMBO J 1999, 18, 6228–6239. [Google Scholar]
- Ma, K.; Simantov, R.; Zhang, J.C.; Silverstein, R.; Hajjar, K.A.; McCrae, K.R. High affinity binding of beta 2-glycoprotein I to human endothelial cells is mediated by annexin II. J. Biol. Chem 2000, 275, 15541–15548. [Google Scholar]
- Zhang, J.; McCrae, K.R. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood 2005, 105, 1964–1969. [Google Scholar]
- Cesarman-Maus, G.; Rios-Luna, N.P.; Deora, A.B.; Huang, B.; Villa, R.; del Cravioto, M.C.; Alarcon-Segovia, D.; Sanchez-Guerrero, J.; Hajjar, K.A. Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood 2006, 107, 4375–4382. [Google Scholar]
- Cockrell, E.; Espinola, R.G.; McCrae, K.R. Annexin A2: Biology and relevance to the antiphospholipid syndrome. Lupus 2008, 17, 943–951. [Google Scholar]
- Allen, K.L.; Fonseca, F.V.; Betapudi, V.; Willard, B.; Zhang, J.; McCrae, K.R. A novel pathway for human endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood 2012, 119, 884–893. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Characteristics/Mechanisms | References |
|---|---|---|
| Breast cancer | -over-expression in invasive breast cancer and DCIS; absent in normal breast epithelium; predictor of response to neoadjuvant chemotherapy; tPA-mediated invasiveness and angiogenesis (through S100A10); metastatic cell proliferation (interaction with TNC); resistance to anthracyclines and taxanes. | [20,100,182–184] |
| PDAC | -over-expression in invasive lesions and PDAC; PDAC invasiveness and metastasis (probably through S100A10); activation of EMT; interaction with TNC in advanced pancreatic cancers; increased recurrence in post-operative patients pre-treated with gemcitabine; correlation with patient survival. | [83,87,185,190] |
| RCC | -over-expression in RCC patients (along with S100A10); low expression in normal renal tubules; AIIt as a potential diagnostic marker; -decreased metastasis-free survival of annexin A2-positive patients. | [191,192] |
| CRC | -correlation with increased tumor size, advanced histology and pTNM; plasmin-mediated invasiveness (through S100A10); correlation with TNC expression. | [193–195] |
| HCC | -over-expression in HCC tissues; activation of pro-inflammatory responses (through NF-κB); indicator of histological grade and improved reliability in diagnosis in HCC patients. | [197–200,202] |
| APL | -Activation of fibrinolysis (through S100A10); -ATRA and ATO treatments alleviate APL-associated bleeding by inhibiting annexin A2 and S100A10. | [204,205,207,208] |
| Prostate cancer | -annexin A2 and S100A10 expression lost especially in androgen-independent prostate cancer; prostate cancer cell migration; -annexin A2 down-regulation by DNA hypermethylation; Conflicting results: annexin A2 expression promotes cell proliferation, bone metastasis and tumor relapse | [84,168,210–212] |
| Head and neck cancers | ESCC -decreased expression in ESCC tissues; correlation with less differentiated ESCC tumors. | [210,214] |
| NPC -reduced expression in NPC patients and cell lines; -implication with higher incidence of lymph node metastasis. | [215] | |
| HNSCC -down-regulation of annexin A2 in HNSCC patients; correlation with less differentiated tumors and lymph node metastasis; Conflicting results: annexin A2 down-regulation present in metastatic tumors and not primary tumors | [215–217] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bharadwaj, A.; Bydoun, M.; Holloway, R.; Waisman, D. Annexin A2 Heterotetramer: Structure and Function. Int. J. Mol. Sci. 2013, 14, 6259-6305. https://doi.org/10.3390/ijms14036259
Bharadwaj A, Bydoun M, Holloway R, Waisman D. Annexin A2 Heterotetramer: Structure and Function. International Journal of Molecular Sciences. 2013; 14(3):6259-6305. https://doi.org/10.3390/ijms14036259
Chicago/Turabian StyleBharadwaj, Alamelu, Moamen Bydoun, Ryan Holloway, and David Waisman. 2013. "Annexin A2 Heterotetramer: Structure and Function" International Journal of Molecular Sciences 14, no. 3: 6259-6305. https://doi.org/10.3390/ijms14036259
APA StyleBharadwaj, A., Bydoun, M., Holloway, R., & Waisman, D. (2013). Annexin A2 Heterotetramer: Structure and Function. International Journal of Molecular Sciences, 14(3), 6259-6305. https://doi.org/10.3390/ijms14036259