Manganese Superoxide Dismutase: Guardian of the Powerhouse

{kind=link}
Abstract
:1. Introduction
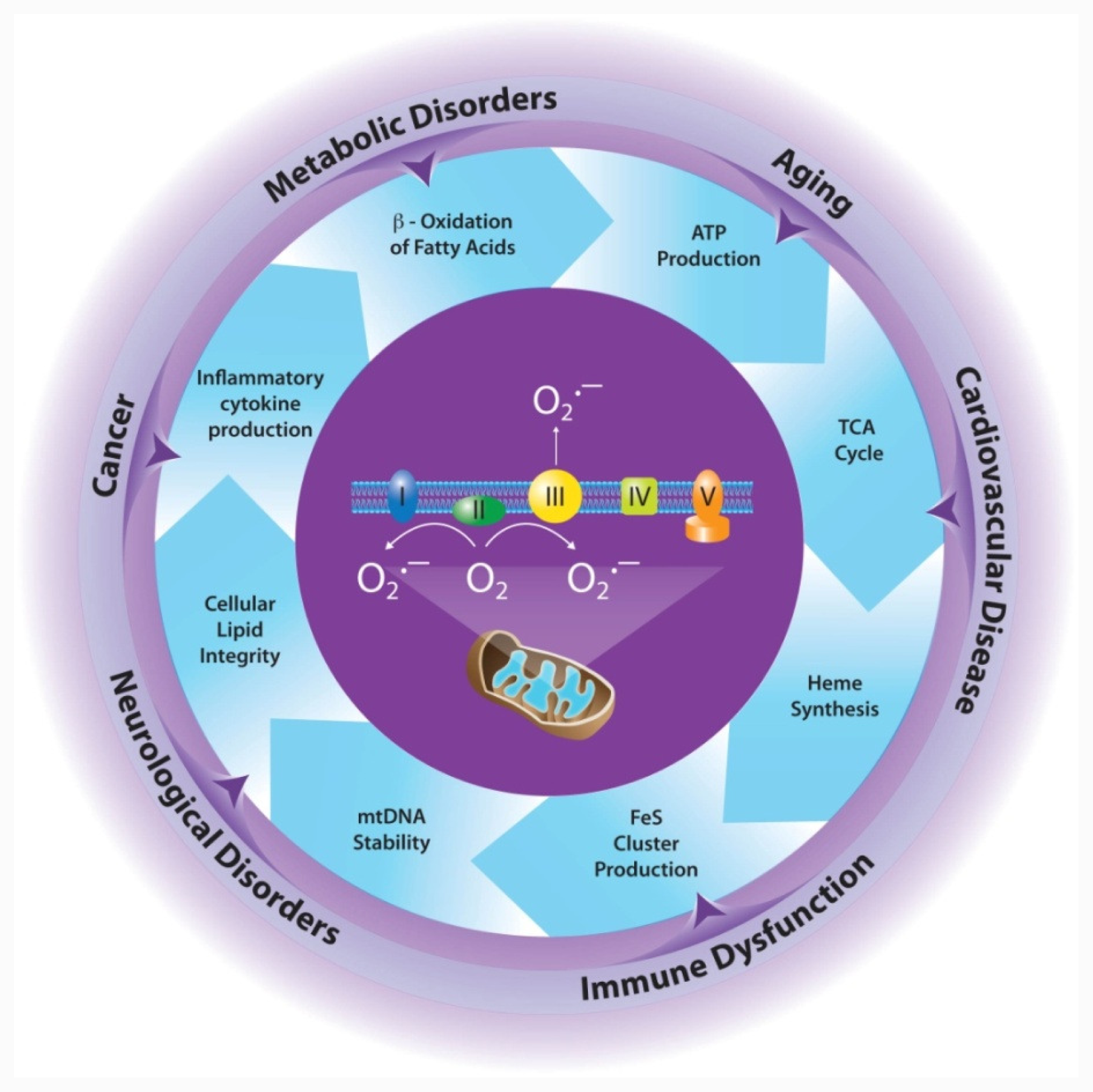
2. Mitochondrial Production of ROS
2.1. Mitochondria is a Major Source of ROS
2.2. Ways to Scavenge Mitochondrial ROS
3. MnSOD is Essential for Aerobic Life
4. Effects of MnSOD on Mitochondrial Integrity/Function
4.1. Electron Transport Chain
4.2. Tricarboxylic Acid (TCA) Cycle
4.3. Iron Metabolism
4.4. Apoptosis
4.5. Mitochondrial Control of Innate and Adaptive Immunity
4.6. Mitochondrial DNA (mtDNA) Stability
4.7. Cellular Lipid Integrity
5. Implications for Disease
5.1. Cancer
5.2. Cardiovascular Disease
5.3. Neurological Disorders
6. Conclusions
Acknowledgments
References
- Fridovich, I. The biology of oxygen radicals. Science 1978, 201, 875–880. [Google Scholar]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev 2002, 82, 47–95. [Google Scholar]
- Boonstra, J; Post, JA. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar]
- Forman, HJ; Fukuto, JM; Torres, M. Redox signaling: Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am. J. Physiol. Cell Physiol 2004, 287, C246–C256. [Google Scholar]
- Rhee, SG; Chang, T-S; Bae, YS; Lee, S-R; Kang, SW. Cellular regulation by hydrogen peroxide. J. Am. Soc. Nephrol 2003, 14, S211–S215. [Google Scholar]
- Waris, G; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog 2006, 5, 14–21. [Google Scholar]
- Ajioka, RS; Phillips, JD; Kushner, JP. Biosynthesis of heme in mammals. Biochim. Biophys. Acta 2006, 1763, 723–736. [Google Scholar]
- Houten, SM; Wanders, RJA. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis 2010, 33, 469–477. [Google Scholar]
- Briere, J-J; Favier, J; Gimenez-Roqueplo, A-P; Rustin, P. Tricarboxylic acid cycle dysfunction as a cause of human diseases and tumor formation. Am. J. Physiol. Cell Physiol 2006, 291, C1114–C1120. [Google Scholar]
- Hutson, SM; Fenstermacher, D; Mahar, C. Role of mitochondrial transamination in branched chain amino acid metabolism. J. Biol. Chem 1988, 263, 3618–3625. [Google Scholar]
- Guda, P; Guda, C; Subramaniam, S. Reconstruction of pathways associated with amino acid metabolism in human mitochondria. Genomics Proteomics Bioinform 2007, 5, 166–176. [Google Scholar]
- Walker, V. Ammonia toxicity and its prevention in inherited defects of the urea cycle. Diabetes Obes. Metab 2009, 11, 823–835. [Google Scholar]
- Lemarie, A; Grimm, S. Mitochondrial respiratory chain complexes: apoptosis sensors mutated in cancer. Oncogene 2011, 30, 3985–4003. [Google Scholar]
- Adam-Vizi, V; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci 2006, 27, 639–645. [Google Scholar]
- Hoye, AT; Davoren, JE; Wipf, P; Fink, MP; Kagan, VE. Targeting mitochondria. Acc. Chem. Res 2008, 41, 87–97. [Google Scholar]
- Huie, RE; Padmaja, S. The reaction of no with superoxide. Free Radic. Res. Commun 1993, 18, 195–199. [Google Scholar]
- Wang, X; Martindale, JL; Liu, Y; Holbrook, NJ. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signaling pathways on cell survival. Biochem. J 1998, 333, 291–300. [Google Scholar]
- Schafer, M; Schafer, C; Ewald, N; Piper, HM; Noll, T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ. Res 2003, 92, 1010–1015. [Google Scholar]
- Poli, G; Leonarduzzi, G; Biasi, F; Chiarpotto, E. Oxidative stress and cell signaling. Curr. Med. Chem 2004, 11, 1163–1182. [Google Scholar]
- Wright, VP; Reiser, PJ; Clanton, TL. Redox modulation of global phosphatase activity and protein phosphorylation in intact skeletal muscle. J. Physiol 2009, 587, 5767–5781. [Google Scholar]
- Abate, C; Patel, L; Rauscher, FJ, III; Curran, T. Redox regulation of fos and jun DNA-bindign activity in vitro. Science 1990, 249, 1157–1161. [Google Scholar]
- Kabe, Y; Ando, K; Hirao, S; Yoshida, M; Handa, H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox Signal 2005, 7, 395–403. [Google Scholar]
- Galanis, A; Pappa, A; Giannakakis, A; Lanitis, E; Dangaj, D; Sandaltzopoulos, R. Reactive oxygen species and HIF-1 signaling in cancer. Cancer Lett 2008, 266, 12–20. [Google Scholar]
- Sun, XZ; Vinci, C; Makmura, L; Han, S; Tran, D; Nguyen, J; Hamann, M; Grazziani, S; Sheppard, S; Gutova, M; et al. Formation of disulfide bond in p53 correlates with inhibition of DNA binding and tetramerization. Antioxid. Redox Signal 2003, 5, 655–665. [Google Scholar]
- Fojta, M; Kubicarova, T; Vojtesek, B; Palecek, E. Effect of p53 protein redox states on binding to supercoiled and linear DNA. J. Biol. Chem 1999, 274, 25749–25755. [Google Scholar]
- Hainaut, P; Milner, J. Redox modulation of p53 conformation and sequence-specific DNA binding in vitro. Cancer Res 1993, 53, 4469–4473. [Google Scholar]
- Miao, L; St Clair, DK. Regulation of superoxide dismutase genes: Implications in disease. Free Radic. Biol. Med 2009, 47, 344–356. [Google Scholar]
- Oberley, LW; Buettner, GR. Role of superoxide dismutase in cancer: a review. Cancer Res 1979, 39, 1141–1149. [Google Scholar]
- Lenaz, G. The mitochondrial production of reactive oxygen species: Mechanisms and implications in human pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar]
- Murphy, MP. How mitochondria produce reactive oxygen species. Biochem. J 2009, 417, 1–13. [Google Scholar]
- Brand, MD. The sites and topology of mitochondrial superoxide production. Exp. Gerontol 2010, 45, 466–472. [Google Scholar]
- Takeshige, K; Minakami, S. NADH- and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NADH-ubiquinone reductase preparation. Biochem. J 1979, 180, 129–135. [Google Scholar]
- Grivennikova, VG; Vinogradov, AD. Generation of superoxide by the mitochondrial complex I. Biochim. Biophys. Acta 2006, 1757, 553–561. [Google Scholar]
- Trumpower, BL. The protonmotive q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J. Biol. Chem 1990, 265, 11409–11412. [Google Scholar]
- Herrero, A; Barja, G. Localization of the site of oxygen radical generation inside the complex I of heart and nonsynaptic brain mammalian mitochondria. J. Bioenerg. Biomembr 2000, 32, 609–615. [Google Scholar]
- Kushnareva, Y; Murphy, AN; Andreyev, A. Complex i-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J 2002, 368, 545–553. [Google Scholar]
- Genova, ML; Ventura, B; Giuliano, G; Bovina, C; Formiggini, G; Castelli, GP; Lenaz, G. The site of production of superoxide radical in mitochondrial complex I is not a bound ubisemiquinone but presumably iron-sulfur cluster N2. FEBS Lett 2001, 505, 364–368. [Google Scholar]
- Dlaskova, A; Hlavata, L; Jezek, P. Oxidative stress caused by blocking of mitochondrial complex I H+ pumping as a link in aging/disease vicious cycle. Int. J. Biochem. Cell Biol 2008, 40, 1792–1805. [Google Scholar]
- Muller, FL; Liu, Y; van Remmen, H. Complex iii releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem 2004, 279, 49064–49073. [Google Scholar]
- Chen, Q; Vazquez, EJ; Moghaddas, S; Hoppel, CL; Lesnefsky, EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem 2003, 278, 36027–36031. [Google Scholar]
- Han, D; Williams, E; Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J 2001, 353, 411–416. [Google Scholar]
- McLennan, HR; Degli Esposti, M. The contribution fo mitochondrial respiratory complexes to the production of reactive oxygen species. J. Bioenerg. Biomembr 2000, 32, 153–162. [Google Scholar]
- Zhang, L; Yu, L; Yu, C-A. Generation of superoxide anion by succinate-cytochrome c reductase from bovine heart mitochondria. J. Biol. Chem 1998, 273, 33972–33976. [Google Scholar]
- Tretter, L; Adam-Vizi, V. Generation of reactive oxygen species in the reaction catalyzed by α-ketoglutarate dehydrogenase. J. Neurosci 2004, 24, 7771–7778. [Google Scholar]
- Starkov, AA; Fiskum, G; Chinopoulos, C; Lorenzo, BJ; Browne, SE; Patel, MS; Beal, MF. Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci 2004, 24, 7779–7788. [Google Scholar]
- Forman, HJ; Kennedy, J. Superoxide production and electron transport in mitochondrial oxidation of dihydroorotic acid. J. Biol. Chem 1975, 250, 4322–4326. [Google Scholar]
- Forman, HJ; Kennedy, J. Dihydroorotate-dependent superoxide production in rat brain and liver: a function of the primary dehydrogenase. Arch. Biochem. Biophys 1976, 173, 219–224. [Google Scholar]
- Drahota, Z; Chowdhury, SKR; Floryk, D; Mracek, T; Wilhelm, J; Rauchova, H; Lenaz, G; Houstek, J. Glycerophosphate-dependent hydrogen peroxide production by brown adipose tissue mitochondria and its activation by ferricyanide. J. Bioenerg. Biomembr 2002, 34, 105–113. [Google Scholar]
- Miwa, S; St-Pierre, J; Partridge, L; Brand, MD. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic. Biol. Med 2003, 35, 938–948. [Google Scholar]
- Hanukoglu, I. Antioxidant protective mechanisms against reactive oxygen species (ROS) generated by mitochondrial P450 systems in steroidogenic cells. Drug Metab. Rev 2006, 38, 171–196. [Google Scholar]
- Hanukoglu, I; Rapoport, R; Weiner, L; Sklan, D. Electron leakage from the mitochondrial NADPH-adrenodoxin reductase-adrenodoxin-P450scc (cholesterol side chain cleavage) system. Arch. Biochem. Biophys 1993, 305, 489–498. [Google Scholar]
- Brookes, PS; Yoon, Y; Robotham, JL; Anders, MW; Sheu, S-S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol 2004, 287, 817–833. [Google Scholar]
- Raha, S; Robinson, BH. Mitochondria, oxygen free radicals, disease, and ageing. Trends Biochem. Sci 2000, 25, 502–508. [Google Scholar]
- Raha, S; Robinson, BH. Mitochondria, oxygen free radicals, and apoptosis. Am. J. Med. Genet 2001, 106, 62–70. [Google Scholar]
- Fong, K-L; McCay, PB; Poyer, JL. Evidence for superoxide-dependent reduction of Fe3+ and its role in enzyme-generated hydroxyl radical formation. Chem. Biol. Interact 1976, 15, 77–89. [Google Scholar]
- Squadrito, GL; Pryor, WA. The formation of peroxynitrite in vivo from nitric oxide and superoxide. Chem. Biol. Interact 1995, 96, 203–206. [Google Scholar]
- Radi, R; Beckman, JS; Bush, KM; Freeman, BA. Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J. Biol. Chem 1991, 266, 4244–4250. [Google Scholar]
- Abello, N; Kerstjens, HAM; Postma, DS; Bischoff, R. Protein tyrosine nitration: Selectivity, physicochemical and biological consequences, denitration, and proteomics methods for the identification of tyrosine-nitrated proteins. J. Proteome Res 2009, 8, 3222–3238. [Google Scholar]
- Radi, R; Cassina, A; Hodara, R; Quijano, C; Castro, L. Peroxynitrite reactions and formation in mitochondria. Free Radic. Biol. Med 2002, 33, 1451–1464. [Google Scholar]
- Riobo, NA; Clementi, E; Melani, M; Boveris, A; Cadenas, E; Moncada, S; Poderoso, JJ. Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem. J 2001, 359, 139–145. [Google Scholar]
- Radi, R; Rodriquez, M; Castro, L; Telleri, R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch. Biochem. Biophys 1994, 308, 89–95. [Google Scholar]
- Murray, J; Taylors, SW; Zhang, B; Ghosh, SS; Capaldi, RA. Oxidative damage to mitochondrial complex I due to peroxynitrite. Identification of reactive tyrosines by mass spectrometry. J. Biol. Chem 2003, 278, 37223–37230. [Google Scholar]
- Cassina, A; Radi, R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch. Biochem. Biophys 1996, 328, 309–316. [Google Scholar]
- Padmaja, S; Squadrito, GL; Pryor, WA. Inactivation of glutathione peroxidase by peroxynitrite. Arch. Biochem. Biophys 1998, 349, 1–6. [Google Scholar]
- Hausladen, A; Fridovich, I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J. Biol. Chem 1994, 269, 29405–29408. [Google Scholar]
- Castro, L; Rodriquez, M; Radi, R. Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. J. Biol. Chem 1994, 269, 29409–29415. [Google Scholar]
- Andreyev, AY; Kushnareva, YE; Starkov, AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc. ) 2005, 70, 246–264. [Google Scholar]
- Koehler, CM; Beverley, KN; Leverich, EP. Redox pathways of the mitochondrion. Antioxid. Redox Signal 2006, 8, 813–822. [Google Scholar]
- Fridovich, I. Superoxide dismutases. An adaptation to a paramagnetic gas. J. Biol. Chem 1989, 264, 7761–7764. [Google Scholar]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem 1995, 64, 97–112. [Google Scholar]
- Esworthy, RS; Ho, Y-S; Chu, F-F. The gpx1 gene encodes mitochondrial glutathione peroxidase in the mouse liver. Arch. Biochem. Biophys 1997, 340, 59–63. [Google Scholar]
- Maiorino, M; Scapin, M; Ursini, F; Biasolo, M; Bosello, V; Flohe, L. Distinct promoters determine alternative transcription of GPX-4 into phospholipid-hydroperoxide glutathione peroxidase variants. J. Biol. Chem 2003, 278, 34286–34290. [Google Scholar]
- Oberley, TD; Verwiebe, E; Zhong, W; Kang, SW; Rhee, SG. Localization of the thioredoxin system in normal rat kidney. Free Radic. Biol. Med 2001, 30, 412–424. [Google Scholar]
- Nohl, H; Jordan, W. The metabolic fate of mitochondrial hydrogen peroxide. Eur. J. Biochem 1980, 111, 203–210. [Google Scholar]
- Zelko, IN; Mariani, TJ; Felz, RJ. Superoxide dismutase multigene family: a comparison of CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structure, evolution, and expression. Free Radic. Biol. Med 2002, 33, 337–349. [Google Scholar]
- Slot, JW; Geuze, HJ; Freeman, BA; Crapo, JD. Intracellular localization of the copper-zinc and manganese superoxide dismutases in rat liver parenchymal cells. Lab. Invest 1986, 55, 363–371. [Google Scholar]
- Okado-Matsumoto, A; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem 2001, 276, 38388–38393. [Google Scholar]
- Weisiger, RA; Fridovich, I. Mitochondrial superoxide dismutase. Site of synthesis and intramitochondrial localization. J. Biol. Chem 1973, 248, 4793–4796. [Google Scholar]
- Hjalmarsson, K; Marklund, SL; Engstrom, A; Edlund, T. Isolation and sequence of complimentary DNA encoding human extracellular superoxide dismutase. Proc. Natl. Acad. Sci. USA 1987, 84, 6340–6344. [Google Scholar]
- Folz, RJ; Crapo, JD. Extracellular superoxide dismutase (SOD3): Tissue-specific expression, genomic characterization, and computer-assisted sequence analysis of the human EC SOD gene. Genomics 1994, 22, 162–171. [Google Scholar]
- Borgstahl, GEO; Parge, HE; Hickey, MJ; Beyer, WF, Jr; Hallewell, RA; Tainer, JA. The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell 1992, 71, 107–118. [Google Scholar]
- Ravindranath, SD; Fridovich, I. Isolation and characterization of a manganese-containing superoxide dismutase from yeast. J. Biol. Chem 1975, 250, 6107–6112. [Google Scholar]
- Wispe, JR; Clark, JC; Burhans, MS; Kropp, KE; Korfhagen, TR; Whitsett, JA. Synthesis and processing of the precursor for human mangano-superoxide dismutase. Biochim. Biophys. Acta 1989, 994, 30–36. [Google Scholar]
- Misra, HP; Fridovich, I. Purification and properties of superoxide dismutase from a red alga, Porphyridium cruentum. J. Biol. Chem 1977, 252, 6421–6423. [Google Scholar]
- Keele, BB, Jr; McCord, JM; Fridovich, I. Superoxide dismutase from Escherichia coli B: a new manganese-containing enzyme. J. Biol. Chem 1970, 245, 6176–6181. [Google Scholar]
- Chang, T-S; Cho, C-S; Park, S; Yu, S; Kang, SW. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulate apoptotic signaling by mitochondria. J. Biol. Chem 2004, 279, 41975–41984. [Google Scholar]
- Shibata, E; Nanri, H; Ejima, K; Araki, M; Fukuda, J; Yoshimura, K; Toki, N; Ikeda, M; Kashimura, M. Enhancement of mitochondrial oxidative stress and up-regulation of antioxidant protein peroxiredoxin III/SP-22 in the mitochondria of human pre-eclamptic placentae. Placenta 2003, 24, 698–705. [Google Scholar]
- Seo, MS; Kang, SW; Kim, K; Baines, IC; Lee, TH; Rhee, SG. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem 2000, 275, 20346–20354. [Google Scholar]
- Lee, S-R; Kim, J-R; Kwon, K-S; Yoon, HW; Levine, RL; Ginsburg, A; Rhee, SG. Molecular cloning and characterization of a mitochondrial selenocysteine-containing thioredoxin reductase from rat liver. J. Biol. Chem 1999, 274, 4722–4734. [Google Scholar]
- Choi, JH; Kim, TN; Kim, S; Baek, SH; Kim, JH; Lee, SR; Kim, JR. Overexpression of mitochondrial thioredoxin reductase and peroxiredoxin III in hepatocellular carcinoma. Anticancer Res 2002, 22, 3331–3335. [Google Scholar]
- Panfili, E; Sandri, G; Ernster, L. Distribution of glutathione peroxidases and glutathione reductase in rat brain mitochondria. FEBS Lett 1991, 290, 35–37. [Google Scholar]
- Kelner, MJ; Montoya, MA. Structural organization of the human glutathione reductase gene: Determination of correct cDNA sequence and identification of a mitochondrial leader sequence. Biochem. Biophys. Res. Commun 2000, 269, 366–368. [Google Scholar]
- Chelikani, P; Fita, I; Loewen, PC. Diversity of structures and properties among catalases. Cell Mol. Life Sci 2004, 61, 192–208. [Google Scholar]
- Zamocky, M; Furtmuller, PG; Obinger, C. Evolution of catalases from bacteria to humans. Antioxid. Redox Signal 2008, 10, 1527–1547. [Google Scholar]
- Zhou, Z; Kang, YJ. Cellular and subcellular localization of catalase in the heart of transgenic mice. J. Histochem. Cytochem 2000, 48, 585–594. [Google Scholar]
- Salvi, M; Battaglia, V; Brunati, AM; La Rocca, N; Tibaldi, E; Pietrangeli, P; Marcocci, L; Mondovi, B; Rossi, CA; Toninello, A. Catalase takes part in rat liver mitochondria oxidative stress defense. J. Biol. Chem 2007, 282, 24407–24415. [Google Scholar]
- Radi, R; Turrens, JF; Chang, LY; Bush, KM; Crapo, JD; Freeman, BA. Detection of catalase in rat heart mitochondria. J. Biol. Chem 1991, 266, 22028–22034. [Google Scholar]
- Gregory, EM; Fridovich, I. Oxygen toxicity and the superoxide dismutase. J. Bacteriol 1973, 114, 1193–1197. [Google Scholar]
- Gregory, EM; Goscin, SA; Fridovich, I. Superoxide dismutase and oxygen toxicity in a eukaryote. J. Bacteriol 1974, 117, 456–460. [Google Scholar]
- Li, Y; Huang, T-T; Carlson, EJ; Melov, S; Ursell, PC; Olson, JL; Noble, LJ; Yoshimura, MP; Berger, C; Chan, PH; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet 1995, 11, 376–381. [Google Scholar]
- Duttaroy, A; Paul, A; Kundu, M; Belton, A. A sod2 null mutation confers severely reduced adult life span in Drosophila. Genetics 2003, 165, 2295–2299. [Google Scholar]
- Paul, A; Belton, A; Nag, S; Martin, I; Grotewiel, MS; Duttaroy, A. Reduced mitochondrial SOD displays mortality characteristics reminiscent of natural aging. Mech. Ageing Dev 2007, 128, 706–716. [Google Scholar]
- van Remmen, H; Ikeno, Y; Hamilton, M; Pahlavani, M; Wolf, N; Thorpe, SR; Alderson, NL; Baynes, JW; Epstein, CJ; Huang, T-T; et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics 2003, 16, 29–37. [Google Scholar]
- Copin, J-C; Gasche, Y; Chan, PH. Overexpression of copper/zinc superoxide dismutase does not prevent neonatal lethality in mutant mice that lack manganese superoxide dismutase. Free Radic. Biol. Med 2000, 28, 1571–1576. [Google Scholar]
- Mukherjee, S; Forde, R; Belton, A; Duttaroy, A. SOD2, the principal scavenger of mitochondrial superoxide, is dispensable for embryogenesis and imaginal tissue development but essential for adult survival. Fly 2011, 5, 39–46. [Google Scholar]
- Lebovitz, RM; Zhang, H; Vogel, H; Cartwright, J, Jr; Dionne, L; Lu, N; Huang, S; Matzuk, MM. Neurodegeneration, mycardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar]
- Ikegami, T; Suzuki, Y-I; Shimizu, T; Isono, K-I; Koseki, H; Shirasawa, T. Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene. Biochem. Biophys. Res. Commun 2002, 296, 729–736. [Google Scholar]
- Parajuli, N; Marine, A; Simmons, S; Saba, H; Mitchell, T; Shimizu, T; Shirasawa, T; MacMillan-Crow, LA. Generation and characterization of a novel kidney-specific manganese superoxide dismutase knockout mouse. Free Radic. Biol. Med 2011, 51, 406–416. [Google Scholar]
- Lustgarten, MS; Jang, YC; Liu, Y; Muller, FL; Qi, W; Steinhelper, M; Brooks, SV; Larkin, L; Shimizu, T; Shirasawa, T; et al. Conditional knockout of Mn-SOD targeted to type IIB skeletal muscle fibers increases oxidative stress and is sufficient to alter aerobic exercise capacity. Am. J. Physiol. Cell Physiol 2009, 297, C1520–C1532. [Google Scholar]
- Lustgarten, MS; Jang, YC; Liu, Y; Qi, W; Qin, Y; Dahia, PL; Shi, Y; Bhattacharya, A; Muller, FL; Shimizu, T; et al. MnSOD deficiency results in elevated oxidative stress and decreased mitochondrial function but does not lead to muscle atrophy during aging. Aging Cell 2011, 10, 493–505. [Google Scholar]
- Misawa, H; Nakata, K; Matsuura, J; Moriwaki, Y; Kawashima, K; Shimizu, T; Shirawawa, T; Takahashi, R. Conditional knockout of mn superoxide dismutase in postnatal motor neurons reveals resistance to mitochondrial generated superoxide radicals. Neurobiol. Dis 2006, 23, 169–177. [Google Scholar]
- Jang, YC; Perez, VI; Song, W; Lustgarten, MS; Salmon, AB; Mele, J; Qi, W; Liu, Y; Liang, H; Chaudhuri, A; et al. Overexpression of mn superoxide dismutase does not increase life span in mice. J. Gerontol. A Biol. Sci. Med. Sci 2009, 64, 1114–1125. [Google Scholar]
- Perez, VI; van Remmen, H; Bokov, A; Epstein, CJ; Vijg, J; Richardson, A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 2009, 8, 73–75. [Google Scholar]
- Sun, J; Folk, D; Bradley, TJ; Tower, J. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics 2002, 161, 661–672. [Google Scholar]
- Albracht, SPJ. The prosthetic groups in succinate dehydrogenase number and stoichiometry. Biochim. Biophys. Acta 1980, 612, 11–28. [Google Scholar]
- Albracht, SPJ; Subramanian, J. The number of Fe atoms in the iron-sulfur centers of the respiratory chain. Biochim. Biophys. Acta 1977, 462, 36–48. [Google Scholar]
- Ohnishi, T. Iron-sulfur clusters/semiquinones in complex I. Biochim. Biophys. Acta 1998, 1364, 186–206. [Google Scholar]
- Ohnishi, T. Thermodynamic and EPR characterization of iron-sulfur centers in the NADH-ubiquinone segment of the mitochondrial respiratory chain in pigeon heart. Biochim. Biophys. Acta 1975, 387, 475–490. [Google Scholar]
- Teintze, M; Slaughter, M; Weiss, H; Neupert, W. Biogenesis of mitochondrial ubiquinol:Cytochrome c reductase (cytochrome bc1 complex). Precursor proteins and their transfer into mitochondria. J. Biol. Chem 1982, 257, 10364–10371. [Google Scholar]
- Roessler, MM; King, MS; Robinson, AJ; Armstrong, FA; Harmer, J; Hirst, J. Direct assignment of EPR spectra to structurally defined iron-sulfur clusters in complex I by double electron-electron resonance. Proc. Natl. Acad. Sci. USA 2010, 107, 1930–1935. [Google Scholar]
- Chen, Y-R; Chen, C-L; Zhang, L; Green-Church, KB; Zweier, JL. Superoxide generation from mitochondrial NADH dehydrogenase induces self-inactivation with specific protein radical formation. J. Biol. Chem 2005, 280, 37339–37348. [Google Scholar]
- Keeney, PM; Xie, J; Capaldi, RA; Bennett, JP, Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci 2006, 26, 5256–5264. [Google Scholar]
- Yamamoto, T; Maruyama, W; Kato, Y; Yi, H; Shamoto-Nagai, M; Tanaka, M; Sato, Y; Naoi, M. Selective nitration of mitochondrial complex I by peroxynitrite: Involvement of mitochondria dysfunction and cell death of dopaminergic SH-SY5Y cells. J. Neural Transm 2002, 109, 1–13. [Google Scholar]
- Brown, GC; Borutaite, V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. Biophys. Acta 2004, 1658, 44–49. [Google Scholar]
- Chinta, SJ; Andersen, JK. Nitrosylation and nitration of mitochondrial complex I in Parkinson’s disease. Free Radic. Res 2011, 45, 53–58. [Google Scholar]
- Pearce, LL; Epperly, MW; Greenberger, JS; Pitt, BR; Peterson, J. Identification of respiratory complexes I and III as mitochondrial sites of damage following exposure to ionizing radiation and nitric oxide. Nitric Oxide Biol. Chem 2001, 5, 128–136. [Google Scholar]
- Williams, MD; Van Remmen, H; Conrad, CC; Huang, T-T; Epstein, CJ; Richardson, A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J. Biol. Chem 1998, 273, 28510–28515. [Google Scholar]
- Martin, FM; Xu, X; von Lohneysen, K; Gilmartin, TJ; Friedman, J. SOD2 deficient erythroid cells up-regulate transferrin receptor and down-regulate mitochondrial biogenesis and metabolism. PLoS One 2011, 6, e16894. [Google Scholar]
- Larosche, I; Letteron, P; Berson, A; Fromenty, B; Huang, T-T; Moreau, R; Pessayre, D; Mansouri, A. Hepatic mitochondrial DNA depletion after an alcohol binge in mice: probable role of peroxynitrite and modulation by manganese superoxide dismutase. J. Pharmacol. Exp. Ther 2010, 332, 886–897. [Google Scholar]
- Simbre, VC, II; Duffy, SA; Dadlani, GH; Miller, TL; Lipshultz, SE. Cardiotoxicity of cancer chemotherapy: implications for children. Paediatr Drugs 2005, 7, 187–202. [Google Scholar]
- Minotti, G; Menna, P; Salvatorelli, E; Cairo, G; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev 2004, 56, 185–229. [Google Scholar]
- Sarvazyan, N. Visualization of doxorubicin-induced oxidative stress in isolated cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol 1996, 271, H2079–H2085. [Google Scholar]
- Kang, YJ; Sun, X; Chen, Y; Zhou, Z. Inhibition of doxorubicin chronic toxicity in catalase-overexpressing transgenic mouse hearts. Chem. Res. Toxicol 2002, 15, 1–6. [Google Scholar]
- Sun, X; Zhou, Z; Kang, YJ. Attenuation of doxorubicin chronic toxicity in metallothionein-overexpressing transgenic mouse heart. Cancer Res 2001, 61, 3382–3387. [Google Scholar]
- Shioji, K; Kishimoto, C; Nakamura, H; Masutani, H; Yuan, Z; Oka, S-I; Yodoi, J. Overexpression of thioredoxin-1 in transgenic mice attenuates adriamycin-induced cardiotoxicity. Circulation 2002, 106, 1403–1409. [Google Scholar]
- Yen, H-C; Oberley, TD; Vichitbandha, S; Ho, Y-S; St Clair, DK. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J. Clin. Invest 1996, 98, 1253–1260. [Google Scholar]
- Yen, H-C; Oberley, TD; Gairola, CG; Szweda, LI; St Clair, DK. Manganese superoxide dismutase protects mitochondrial complex I against adriamycin-induced cardiomyopathy in transgenic mice. Arch. Biochem. Biophys 1999, 362, 59–66. [Google Scholar]
- Tannock, IF; Ahles, TA; Ganz, PA; van Dam, FS. Cognitive impairment associated with chemotherapy for cancer: report of a workshop. J. Clin. Oncol 2004, 22, 2233–2239. [Google Scholar]
- Ahles, TA; Saykin, AJ. Candidate mechanisms for chemotherapy-induced cognitive changes. Nat. Rev. Cancer 2007, 7, 192–201. [Google Scholar]
- Wefel, JS; Lenzi, R; Theriault, R; Buzdar, AU; Cruickshank, S; Meyers, CA. “Chemobrain” in breast carcinoma?: a prologue”. Cancer 2004, 101, 466–475. [Google Scholar]
- Nelson, CJ; Nandy, N; Roth, AJ. Chemotherapy and cognitive deficits: Mechanisms, findings, and potential interventions. Palliat. Support. Care 2007, 5, 273–280. [Google Scholar]
- Inagaki, M; Yoshikawa, E; Matsuoka, Y; Sugawara, Y; Nakano, T; Akechi, T; Wada, N; Imoto, S; Murakami, K; Uchitomi, Y; et al. Smaller regional volumes of brain gray and white matter demonstrated in breast cancer survivors exposed to adjuvant chemotherapy. Cancer 2007, 109, 146–156. [Google Scholar]
- Brown, MS; Stemmer, SM; Simon, JH; Stears, JC; Jones, RB; Cagnoni, PJ; Sheeder, JL. White matter disease induced by high-dose chemotherapy: longitudinal study with MR imaging and proton spectroscopy. AJNR Am. J. Neuroradiol 1998, 19, 217–221. [Google Scholar]
- Silverman, DHS; Dy, CJ; Castellon, SA; Lai, J; Pio, BS; Abraham, L; Waddell, K; Petersen, L; Phelps, ME; Ganz, PA. Altered frontocortical, cerebellar, and basal ganglia activity in adjuvant-treated breast cancer survivors 5–10 years after chemotherapy. Breast Cancer Res. Treat 2007, 103, 303–311. [Google Scholar]
- Tangpong, J; Cole, MP; Sultana, R; Estus, S; Vore, M; St Clair, W; Ratanachaiyavong, S; St Clair, DK; Butterfield, DA. Adriamycin-mediated nitration of manganese superoxide dismutase in the central nervous system: insight into the mechanism of chemobrain. J. Neurochem 2007, 100, 191–201. [Google Scholar]
- Joshi, G; Sultana, R; Tangpong, J; Cole, MP; St Clair, DK; Vore, M; Estus, S; Butterfield, DA. Free radical mediated oxidative stress and toxic side effects in brain induced by the anticancer drug adriamycin: Insight into chemobrain. Free Radic. Res 2005, 29, 1147–1154. [Google Scholar]
- Ohnishi, T; Tamai, I; Sakanaka, K; Sakata, A; Yamashima, T; Yamashita, J; Tsuji, A. In vivo and in vitro evidence for ATP-dependency of P-glycoprotein-mediated efflux of doxorubicin at the blood-brain barrier. Biochem. Pharmacol 1995, 49, 1541–1544. [Google Scholar]
- Tangpong, J; Cole, MP; Sultana, R; Joshi, G; Estus, S; Vore, M; St Clair, W; Ratanachaiyavong, S; St Clair, DK; Butterfield, DA. Adriamycin-induced, TNF-α-mediated central nervous system toxicity. Neurobiol. Dis 2006, 23, 127–139. [Google Scholar]
- Rotig, A; de Lonlay, P; Chretien, D; Foury, F; Koenig, M; Sidi, D; Munnich, A; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in friedreich ataxia. Nat. Genet 1997, 17, 215–217. [Google Scholar]
- Gardner, PR; Raineri, I; Epstein, LB; White, CW. Superoxide radical and iron modulate aconitase activity in mammalian cells. J. Biol. Chem 1995, 270, 13399–13405. [Google Scholar]
- Cantu, D; Schaack, J; Patel, M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS One 2009, 4, e7095. [Google Scholar]
- Gardner, PR. Superoxide-driven aconitase FE-S center cycling. Biosci. Rep 1997, 17, 33–42. [Google Scholar]
- Tortora, V; Quijano, C; Freeman, B; Radi, R; Castro, L. Mitochondrial aconitase reaction with nitric oxide, s-nitrosoglutathione, and peroxynitrite: mechanisms and relative contributions to aconitase inactivation. Free Radic. Biol. Med 2007, 42, 1075–1088. [Google Scholar]
- Han, D; Canali, R; Garcia, J; Aguilera, R; Gallaher, TK; Cadenas, E. Sites and mechanisms of aconitase inactivation by peroxynitrite: Modulation by citrate and glutathione. Biochemistry 2005, 44, 11986–11996. [Google Scholar]
- Morgan, MJ; Lehmann, M; Schwarzlander, M; Baxter, CJ; Sienkiewicz-Porzucek, A; Williams, TCR; Schauer, N; Fernie, AR; Fricker, MD; Ratcliffe, RG; et al. Decrease in manganese superoxide dismutase leads to reduced root growth and affects tricarboxylic acid cycle flux and mitochondrial redox homeostasis. Plant Physiol 2008, 147, 101–114. [Google Scholar]
- Longo, VD; Liou, L-L; Valentine, JS; Gralla, EB. Mitochondrial superoxide decreases yeast survival in stationary phase. Arch. Biochem. Biophys 1999, 365, 131–142. [Google Scholar]
- Powell, CS; Jackson, RM. Mitochondrial complex I, aconitase, and succinate dehydrogenase during hypoxia-reoxygenation: modulation of enzyme activities by MnSOD. Am. J. Physiol. Lung Cell Mol. Physiol 2003, 285, L189–L198. [Google Scholar]
- Levi, S; Rovida, E. The role of iron in mitochondrial function. Biochim. Biophys. Acta 2009, 1790, 629–636. [Google Scholar]
- Rouault, TA; Tong, W-H. Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat. Rev. Mol. Cell Biol 2005, 6, 345–351. [Google Scholar]
- Richardson, DR; Lane, DJR; Becker, EM; Huang, ML-H; Witnall, M; Rahmanto, YS; Sheftel, AD; Ponka, P. Mitochondrial iron trafficking and the integration of metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar]
- Eaton, JW; Qian, M. Molecular bases of cellular iron toxicity. Free Radic. Biol. Med 2002, 32, 833–840. [Google Scholar]
- Napier, I; Ponka, P; Richardson, DR. Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood 2005, 105, 1867–1874. [Google Scholar]
- Ye, H; Rouault, TA. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 2010, 49, 4945–4956. [Google Scholar]
- Srinivasan, C; Liba, A; Imlay, JA; Valentine, JS; Gralla, EB. Yeast lacking superoxide dismutase(s) show elevated levels of “Free iron” As measured by whole cell electron paramagnetic resonance. J. Biol. Chem 2000, 275, 29187–29192. [Google Scholar]
- Nahon, P; Charnaux, N; Friand, V; Prost-Squarcioni, C; Zoil, M; Lievre, N; Trinchet, J-C; Beaugrand, M; Gattegno, L; Pessayre, D; et al. The manganese superoxide dismutase Ala16Val dimorphism modulates iron accumulation in human hepatoma cells. Free Radic. Biol. Med 2008, 45, 1308–1317. [Google Scholar]
- Shimada, Y; Okuno, S; Kawai, A; Shinomiya, H; Saito, A; Suzuki, M; Omori, Y; Nishino, N; Kanemoto, N; Fujiwara, T; et al. Cloning and chromosomal mapping of a novel ABC transporter gene (hABC7), a candidate for X-linked sideroblastic anemia with spinocerebellar ataxia. J. Hum. Genet 1998, 43, 115–122. [Google Scholar]
- Pondarre, C; Antiochos, BB; Campagna, DR; Clarke, SL; Greer, EL; Deck, KM; McDonald, A; Han, A-P; Medlock, A; Kutok, JL; et al. The mitochondrial ATP-binding cassette transporter ABCB7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum. Mol. Genet 2006, 15, 953–964. [Google Scholar]
- Bekri, S; Kispal, G; Lange, H; Fitzsimons, E; Tolmie, J; Lill, R; Bishop, DF. Human ABC7 transporter: Gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood 2000, 96, 3256–3264. [Google Scholar]
- Allikmets, R; Raskind, WH; Hutchinson, A; Scheuck, ND; Dean, M; Koeller, DM. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum. Mol. Genet 1999, 8, 743–749. [Google Scholar]
- Sato, K; Torimoto, Y; Hosoki, T; Ikuta, K; Takahashi, H; Yamamoto, M; Ito, S; Okamura, N; Ichiki, K; Tanaka, H; et al. Loss of ABCB7 gene: pathogenesis of mitochondrial iron accumulation in erythroblasts in refractory anemia with ringed siderblast with isodicentric (X)(q13). Int. J. Hematol 2011, 93, 311–318. [Google Scholar]
- Cavadini, P; Biasiotto, G; Poli, M; Levi, S; Verardi, R; Zanella, I; Derosas, M; Ingrassia, R; Corrado, M; Arosio, P. RNA silencing of the mitochondrial ABCB7 transporter in hela cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood 2007, 109, 3552–3559. [Google Scholar]
- Miyake, A; Higashijima, S-I; Kobayashi, D; Narita, T; Jindo, T; Setiamarga, DHE; Ohisa, S; Orihara, N; hibiya, K; Konno, S; et al. Mutation in the abcb7 gene causes abnormal iron and fatty acid metabolism in developing medaka fish. Dev. Growth Differ 2008, 50, 703–716. [Google Scholar]
- Pinkham, JL; Wang, Z; Alsina, J. Heme regulates sod2 transcription by activation and repression in Saccharomyces cerevisiae. Curr. Genet 1997, 31, 281–291. [Google Scholar]
- Converso, DP; Taille, C; Carreras, C; Jaitovich, A; Poderoso, JJ; Boczkowski, J. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J 2006, 20, E482–E492. [Google Scholar]
- Frankel, D; Mehindate, K; Schipper, HM. Role of heme oxygenase-1 in the regulation of manganese superoxide dismutase gene expression in oxidatively-challenged astroglia. J. Cell Physiol 2000, 185, 80–86. [Google Scholar]
- Jiralerspong, S; Ge, B; Hudson, TJ; Pandolfo, M. Manganese superoxide dismutase induction by iron is impaired in friedreich ataxia cells. FEBS Lett 2001, 509, 101–105. [Google Scholar]
- Majno, G; Joris, I. Apoptosis, oncosis, and necrosis: an overview of cell death. Am. J. Pathol 1995, 146, 3–15. [Google Scholar]
- Afford, S; Randhawa, S. Demystified apoptosis. J. Clin. Pathol. Mol. Pathol 2000, 53, 55–63. [Google Scholar]
- Schwartzman, RA; Cidlowski, JA. Apoptosis: The biochemistry and molecular biology of programmed cell death. Endocr. Rev 1993, 14, 133–151. [Google Scholar]
- Reed, JC. Mechanisms of apoptosis. Am. J. Pathol 2000, 157, 1415–1430. [Google Scholar]
- Roy, S; Nicholson, DW. Cross-talk in cell death signaling. J. Exp. Med 2000, 192, F21–F25. [Google Scholar]
- Basu, A; Castle, VP; Bouziane, M; Bhalla, K; Haldar, S. Crosstalk between extrinsic and intrainsic cell death pathways in pancreatic cancer: Synergistic action of estrogen metabolite and ligands of death receptor family. Cancer Res 2006, 66, 4309–4318. [Google Scholar]
- Spierings, D; McStay, G; Saleh, M; Bender, C; Chipuk, J; Maurer, U; Green, DR. Connected to death: the (unexpurgated) mitochondrial pathway of apoptosis. Science 2005, 310, 66–67. [Google Scholar]
- Gulbins, E; Dreschers, S; Bock, J. Role of mitochondria in apoptosis. Exp. Physiol 2003, 88, 85–90. [Google Scholar]
- Green, DR; Reed, JC. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar]
- Adams, JM. Ways of dying: Multiple pathways to apoptosis. Genes Dev 2003, 17, 2481–2495. [Google Scholar]
- Epperly, MW; Sikora, CA; DeFilippi, SJ; Gretton, JE; Zhan, Q; Kufe, DW; Greenberger, JS. Manganese superoxide dismutase (SOD2) inhibits radiation-induced apoptosis by stabilization of the mitochondrial membrane. Radiat. Res 2002, 157, 568–577. [Google Scholar]
- Keller, JN; Kindy, MS; Holtsberg, FW; St Clair, DK; Yen, H-C; Germeyer, A; Steiner, SM; Bruce-Keller, AJ; Hutchins, JB; Mattson, MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: Suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci 1998, 18, 687–697. [Google Scholar]
- Mohr, A; Buneker, C; Gough, RP; Zwacka, RM. MnSOD protects colorectal cancer cells from TRAIL-induced apoptosis by inhibition of Smac/DIABLO release. Oncogene 2007, 27, 763–774. [Google Scholar]
- Kiningham, KK; Oberley, TD; Lin, S-M; Mattingly, CA; St Clair, DK. Overexpression of manganese superoxide dismutase protects against mitochondrial-initiated poly(ADP-ribose) polymerase-mediated cell death. FASEB J 1999, 13, 1601–1610. [Google Scholar]
- Hirose, K; Longo, DI; Oppenheim, JJ; Matsushima, K. Overexpression of mitochondrial manganese superoxide dismutase promotes the survival of tumor cells exposed to interleukin-1, tumor necrosis factor, selected anticancer drugs, and ionizing radiation. FASEB J 1993, 7, 361–368. [Google Scholar]
- Goossens, V; Grooten, J; De Vos, K; Fiers, W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 8115–8119. [Google Scholar]
- Wong, GHW; Goeddel, DV. Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective role. Science 1988, 242, 941–944. [Google Scholar]
- Wong, GHW; Elwell, JH; Oberley, LW; Goeddel, DV. Manganese superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell 1989, 58, 923–931. [Google Scholar]
- Mattson, MP; Goodman, Y; Luo, H; Fu, W; Furukawa, K. Activation of NF-κB protects hippocampal neurons against oxidative stress-induced apoptosis: Evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J. Neurosci. Res 1997, 49, 681–697. [Google Scholar]
- Daosukho, C; Kiningham, K; Kasarskis, EJ; Ittarat, W; St Clair, DK. Tamoxifen enhancement of TNF-α induced MnSOD expression: Modulation of NF-κB dimerization. Oncogene 2002, 21, 3603–3610. [Google Scholar]
- Daosukho, C; Ittarat, W; Lin, S-M; Sawyer, DB; Kiningham, K; Lien, Y-C; St Clair, DK. Induction of manganese superoxide dismutase (MnSOD) mediates cardioprotective effect of tamoxifen (TAM). J. Mol. Cell. Cardiol 2005, 39, 792–803. [Google Scholar]
- Pardo, M; Melendez, JA; Tirosh, O. Manganese superoxide dismutase inactivation during Fas (CD95)-mediated apoptosis in Jurkat T cells. Free Radic. Biol. Med 2006, 41, 1795–1806. [Google Scholar]
- Dasgupta, J; Subbaram, S; Connor, KM; Rodriguez, AM; Tirosh, O; Beckman, JS; Jourd’Heuil, D; Melendez, JA. Manganese superoxide dismutase protects from TNF-α-induced apoptosis by increasing the steady-state production of H2O2. Antioxid. Redox Signal 2006, 8, 1295–1305. [Google Scholar]
- Kawai, T; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol 2009, 21, 317–337. [Google Scholar]
- Cinel, I; Opal, SM. Molecular biology of inflammation and sepsis: A primer. Crit. Care Med 2009, 37, 291–304. [Google Scholar]
- Martinon, F; Mayor, A; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol 2009, 27, 229–265. [Google Scholar]
- Sidiropoulos, PI; Goulielmos, G; Voloudakis, GK; Petraki, E; Boumpas, DT. Inflammasomes and rheumatic diseases: Evolving concepts. Ann. Rheum. Dis 2008, 67, 1382–1389. [Google Scholar]
- Seth, RB; Sun, L; Ea, C-K; Chen, ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar]
- Zhang, Q; Raoof, M; Chen, Y; Sumi, Y; Sursal, T; Junger, W; Brohi, K; Itagaki, K; Hauser, CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar]
- Nakahira, K; Hapsel, JA; Rathinam, VAK; Lee, S-J; Dolinay, T; Lam, HC; Englert, JA; Rabinovitch, M; Cernadas, M; Kim, HP; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the Nalp3 inflammasome. Nat. Immunol 2011, 8, 222–230. [Google Scholar]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol 2010, 40, 616–619. [Google Scholar]
- Dostert, C; Petrilli, V; van Bruggen, R; Steele, C; Mossman, BT; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar]
- Meissner, F; Seger, RA; Moshous, S; Fischer, A; Reichenbach, J; Zychlinsky, A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood 2010, 116, 1570–1573. [Google Scholar] [Green Version]
- Zhou, R; Yazdi, AS; Menu, P; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–226. [Google Scholar]
- Bulua, AC; Simon, A; Maddipati, R; Pelletier, M; Park, H; Kim, K-Y; Sack, MN; Kastner, DL; Siegel, RM. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med 2011, 208, 519–533. [Google Scholar]
- Lu, S-P; Feng, M-HL; Huang, H-L; Huang, Y-C; Tsou, W-I; Lai, M-Z. Reactive oxygen species promote raft formation in T lymphocytes. Free Radic. Biol. Med 2007, 42, 936–944. [Google Scholar]
- Moulian, N; Truffault, F; Gaudry-Talarmain, YM; Serraf, A; Berrih-Aknin, S. In vivo and in vitro apoptosis of human thymocytes are associated with nitrotyrosine formation. Blood 2008, 97, 3521–3530. [Google Scholar]
- Lee, S-M; Lee, Y-S; Choi, J-H; Park, S-G; Choi, I-W; Joo, Y-D; Lee, W-S; Lee, J-N; Choi, I; Seo, S-K. Tryptophan metabolite 3-hydroxyanthranilic acid selectively induces activated T cell death via intracellular GSH depletion. Immunol. Lett 2010, 132, 53–60. [Google Scholar]
- Owusu-Ansah, E; Banerjee, U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 2009, 461, 537–541. [Google Scholar]
- Case, AJ; McGill, JL; Tygrett, LT; Shirasawa, T; Spitz, DR; Waldschmidt, TJ; Legge, KL; Domann, FE. Elevated mitochondrial superoxide disrupts normal T cell development, impairing adaptive immune response to an influenza challenge. Free Radic. Biol. Med 2011, 50, 448–458. [Google Scholar]
- Anderson, S; Bankier, AT; Barrell, BG; de Bruijn, MHL; Coulson, AR; Drouin, J; Eperon, IC; Nierlich, DP; Roe, BA; Sanger, F; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar]
- Iborra, FJ; Kimura, H; Cook, PR. The functional organization of mitochondrial genomes in human cells. BMB Biol 2004, 2, 9–22. [Google Scholar]
- Legros, F; Malka, F; Frachon, P; Lombes, A; Rojo, M. Organization and dynamics of human mitochondrial DNA. J. Cell Sci 2004, 117, 2653–2662. [Google Scholar]
- Chen, XJ; Butow, RA. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet 2005, 6, 815–825. [Google Scholar]
- Garrido, N; Griparic, L; Jokitalo, E; Wartiovaara, J; van der Bliek, AM; Spelbrink, JN. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell 2003, 14, 1583–1596. [Google Scholar]
- Boffoli, D; Scacco, SC; Vergari, R; Solarino, G; Santacroce, G; Papa, S. Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta 1994, 1226, 73–82. [Google Scholar]
- Short, KR; Bigelow, ML; Kahl, J; Singh, R; Coenen-Schimke, J; Raghavakaimal, S; Nair, KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar]
- Tanaka, M; Kovalenko, SA; Gong, J-S; Borgeld, H-JW; Katsumata, K; Hayakawa, M; Yoneda, M; Ozawa, T. Accumulation of deletions and point mutations in mitochondrial genome in degenerative diseases. Ann. N. Y. Acad. Sci 1996, 786, 102–111. [Google Scholar]
- Kang, D; Hamasaki, N. Alterations of mitochondrial DNA in common diseases and disease states: aging, neurodegeneration, heart failure, diabetes, and cancer. Curr. Med. Chem 2005, 12, 429–441. [Google Scholar]
- Brandon, M; Baldi, P; Wallace, DC. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar]
- Lu, J; Sharma, LK; Bai, Y. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res 2009, 19, 802–815. [Google Scholar]
- Takai, D; Park, S-H; Takada, Y; Ichinose, S; Kitagawa, M; Akashi, M. UV-irradiation induces oxidative damage to mitochondrial DNA primarily through hydrogen peroxide: analysis of 8-oxodguo by HPLC. Free Radic. Res 2006, 40, 1138–1148. [Google Scholar]
- Richter, C; Park, J-W; Ames, BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar]
- Mambo, E; Gao, X; Cohen, Y; Guo, Z; Talalay, P; Sidransky, D. Electrophile and oxidant damage of mitochondrial DNA leading to rapid evolution of homplasmic mutations. Proc. Natl. Acad. Sci. USA 2003, 100, 1838–1843. [Google Scholar]
- Cortopassi, G; Wang, E. Modelling the effects of age-related mtdna mutation accumulation; complex I deficiency, superoxide and cell death. Biochim. Biophys. Acta 1995, 1271, 171–176. [Google Scholar]
- Indo, HP; Davidson, M; Yen, H-C; Suenaga, S; Tomita, K; Nishii, T; Higuchi, M; Koga, Y; Ozawa, T; Majima, HJ. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar]
- Park, SY; Chang, I; Kim, J-Y; Kang, SW; Park, S-H; Singh, K; Lee, M-S. Resistance of mitochondrial DNA-depleted cells against cell death: Role of mitochondrial superoxide dismutase. J. Biol. Chem 2004, 279, 7512–7520. [Google Scholar]
- Birch-Machin, MA; Swalwell, H. How mitochondria record the effects of UV exposure and oxidative stress using human skin as a model tissue. Mutagenesis 2010, 25, 101–107. [Google Scholar]
- Yakes, FM; van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar]
- Steinman, HM; Weinstein, L; Brenowitz, M. The manganese superoxide dismutase of Escherichia coli K-12 associates with DNA. J. Biol. Chem 1994, 269, 28629–28634. [Google Scholar]
- Garcia-Ramirez, M; Francisco, G; Garcia-Arumi, E; Hernandez, C; Martinez, R; Andreu, AL; Simo, R. Mitochondrial DNA oxidation and manganese superoxide dismutase activity in peripheral blood mononuclear cells fro type 2 diabetic patients. Diabetes Metab 2008, 34, 117–124. [Google Scholar]
- Madsen-Bouterse, SA; Zhong, Q; Mohammad, G; Ho, Y-S; Kowluru, RA. Oxidative damage of mitochondrial DNA in diabetes and its protection by manganese superoxide dismutase. Free Radic. Res 2010, 44, 313–321. [Google Scholar]
- Mansouri, A; Tarhuni, A; Larosche, I; Reyl-Desmars, F; Demeilliers, C; Degoul, F; Nahon, P; Sutton, A; Moreau, R; Fromenty, B; et al. MnSOD overexpression prevents liver mitochondrial DNA depletion after an alcohol binge but worsens this effect after prolonged alcohol consumption in mice. Dig. Dis 2010, 28, 756–775. [Google Scholar]
- Kienhofer, J; Haussler, DJF; Ruckelshausen, F; Muessig, E; Weber, K; Pimentel, D; Ullrich, V; Burkle, A; Bachschmid, MM. Association of mitochondrial antioxidant enzymes with mitochondrial DNA as integral nucleoid constitutents. FASEB J 2009, 23, 2034–2044. [Google Scholar]
- Bakthavatchalu, V; Dey, S; St Clair, D. Manganese superoxide dismutase protects Pol γ against UV-induced inactivation: implication for DNA repair. Free Radic. Biol. Med 2010, 49, S55. [Google Scholar]
- Spickett, CM; Wiswedel, I; Siems, W; Zarkovic, K; Zarkovic, N. Advances in methods for the determination of biologically relevant lipid peroxidation products. Free Radic. Res 2010, 44, 1172–1202. [Google Scholar]
- Baker, PRS; Schopfer, FJ; O’Donnell, VB; Freeman, BA. Convergence of nitric oxid and lipid signaling: Anti-inflammatory nitro-fatty acids. Free Radic. Biol. Med 2009, 46, 989–1003. [Google Scholar]
- O’Donnell, VB; Eiserich, JP; Bloodsworth, A; Chumley, PH; Kirk, M; Barnes, S; Darley-Usmar, VM; Freeman, BA. Nitration of unsaturated fatty acids by nitric oxide-derived reactive species. Methods Enzymol 1999, 301, 454–470. [Google Scholar]
- Rubbo, H; Trostchansky, A; O’Donnell, VB. Peroxynitrite-mediated lipid oxidation and nitration: Mechanisms and consequences. Arch. Biochem. Biophys 2009, 484, 167–172. [Google Scholar]
- Radi, R; Beckman, JS; Bush, KM; Freeman, BA. Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Arch. Biochem. Biophys 1991, 288, 481–487. [Google Scholar]
- Nadtochiy, SM; Baker, PRS; Freeman, BA; Brooks, PS. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: Implications for cardioprotection. Cardiovasc. Res 2009, 82, 333–340. [Google Scholar]
- Strassburger, M; Bloch, W; Sulyok, S; Schuller, J; Keist, AF; Schmidt, A; Wenk, J; Peters, T; Wlaschek, M; Krieg, T; et al. Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic. Biol. Med 2005, 38, 1458–1470. [Google Scholar]
- Epperly, MW; Tyurina, YY; Nie, S; Niu, YY; Zhang, X; Kagan, VE; Greenberger, JS. MnSOD-plasmid liposome gene therapy decreases ionizing irradiation-induced lipid peroxidation of the esophagus. Vivo 2005, 19, 997–1004. [Google Scholar]
- Ohtsuki, T; Matsumoto, M; Suzuki, K; Taniguchi, N; Kamada, T. Mitochondrial lipid peroxidation and superoxide dismutase in rat hypertensive target organs. Am. J. Physiol. Heart Circ. Physiol 1995, 37, H1418–H1421. [Google Scholar]
- Zidenberg-Cherr, S; Keen, CL; Lonnerdal, B; Hurley, LS. Superoxide dismutase activity and lipid peroxidation in the rat: Developmental correlations affected by manganese deficiency. J. Nutr 1983, 113, 2498–2504. [Google Scholar]
- Malecki, EA; Greger, JL. Manganese protects against heart mitochondrial lipid peroxidation in rats fed high levels of polyunsaturated fatty acids. J. Nutr 1996, 126, 27–33. [Google Scholar]
- Carl, GF; Keen, CL; Gallagher, BB; Clegg, MS; Littleton, WH; Flannery, DB; Hurley, LS. Association of low blood manganese concentrations with epilepsy. Neurology 1986, 36, 1584–1587. [Google Scholar]
- Kazi, TG; Afridi, HI; Kazi, N; Jamali, MK; Arain, MB; Jalbani, N; Kandhro, GA. Copper, chromium, manganese, iron, nickel, and zinc levels in biological samples of diabetes mellitus patients. Biol. Trace Elem. Res 2008, 122, 1–18. [Google Scholar]
- Tonelli, M; Wiebe, N; Hemmelgarn, B; Klarenbach, S; Field, C; Manns, B; Thadhani, R; Gill, J. Trace elements in hemodialysis patients: A systematic review and meta-analysis. BMC Med 2009, 7, 25. [Google Scholar]
- Garrett, TA; Kordestani, R; Raetz, CRH. Quantification of Cardiolipin by Liquid Chromatography-Electrospray Ionization Mass Spectrometry. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2007; Volume 433. [Google Scholar]
- Claypool, SM. Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim. Biophys. Acta 2009, 1788, 2059–2068. [Google Scholar]
- Paradies, G; Petrosillo, G; Paradies, V; Ruggiero, FM. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 2009, 45, 643–650. [Google Scholar]
- Sparagna, GC; Johnson, CA; McCune, SA; Moore, RL; Murphy, RC. Quantitation of cardiolipin molecular species in spontaneously hypertensive heart failure rats using electrospray ionization mass spectrometry. J. Lipid Res 2005, 46, 1196–1204. [Google Scholar]
- Paradies, G; Petrosillo, G; Paradies, V; Ruggiero, FM. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic. Biol. Med 2010, 48, 1286–1295. [Google Scholar]
- Lesnefsky, EJ; Minkler, P; Hoppel, CL. Enhanced modification of cardiolipin during ischemia in the aged heart. J. Mol. Cell. Cardiol 2009, 46, 1008–1015. [Google Scholar]
- Canuto, RA; Biocca, ME; Muzio, G; Dianzani, MU. Fatty acid composition of phospholipids in mitochondria and microsomes during diethylnitrosamine carcinogenesis in rat liver. Cell Biochem. Funct 1989, 7, 11–19. [Google Scholar]
- Reynier, M; Sari, H; d’Anglebermes, M; Kye, EA; Pasero, L. Differences in lipid characteristics of undifferentiated and enterocytic-differentiated HT29 human colonic cells. Cancer Res 1991, 51, 1270–1277. [Google Scholar]
- Jahnke, VE; Sabido, O; Defour, A; Castells, J; Lefai, E; Roussel, D; Freyssenet, D. Evidence for mitochondrial respiratory deficiency in rat rhabdomyosarcoma cells. PLoS One 2010, 5, e8637. [Google Scholar]
- Hauff, KD; Hatch, GM. Cardiolipin metabolism and barth syndrome. Prog. Lipid Res 2006, 45, 91–101. [Google Scholar]
- Xu, Y; Condell, M; Plesken, H; Edelman-Novemsky, I; Ma, J; Ren, M; Schlame, M. A Drosophila model of barth syndrome. Proc. Natl. Acad. Sci. USA 2006, 103, 11584–11588. [Google Scholar]
- Bione, S; D’Adamo, P; Maestrini, E; Gedeon, AK; Bolhuis, PA; Toniolo, D. A novel x-linked gene, G4.5. is responsible for barth syndrome. Nat. Genet 1996, 12, 385–389. [Google Scholar]
- Houtkooper, RH; Turkenburg, M; Poll-The, BT; Karall, D; Perez-Cerda, C; Morrone, A; Malvagia, S; Wanders, RJ; Kulik, W; Vaz, FM. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta 2009, 1788, 2003–2014. [Google Scholar]
- Petrosillo, G; Casanova, G; Matera, M; Ruggiero, FM; Paradies, G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS Lett 2006, 580, 6311–6316. [Google Scholar]
- Nakagawa, Y. Initiation of apoptotic signal by the peroxidation of cardiolipin of mitochondria. Ann. N. Y. Acad. Sci 2004, 1011, 177–184. [Google Scholar]
- Sorice, M; Manganelli, V; Matarrese, P; Tinari, A; Misasi, R; Malorni, W; Garofalo, T. Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett 2009, 583, 2447–2450. [Google Scholar]
- Fernandez, MG; Troiano, L; Moretti, L; Nasi, M; Pinti, M; Salvioli, S; Dobrucki, J; Cossarizza, A. Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ 2002, 13, 449–455. [Google Scholar]
- Gonzalez, F; Schug, ZT; Houtkooper, RH; MacKenzie, ED; Brooks, DG; Wanders, RJA; Petit, PX; Vaz, FM; Gottlieb, E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol 2008, 183, 681–696. [Google Scholar]
- Schug, ZT; Gottlieb, E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta 2009, 1788, 2022–2031. [Google Scholar]
- McMillin, JB; Dowhan, W. Cardiolipin and apoptosis. Biochim. Biophys. Acta 2002, 1585, 97–107. [Google Scholar]
- Petrosillo, G; Ruggiero, FM; Pistolese, M; Paradies, G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett 2001, 509, 435–438. [Google Scholar]
- Fry, M; Green, DE. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J. Biol. Chem 1981, 256, 1874–1880. [Google Scholar]
- Pfeiffer, K; Gohil, V; Stuart, RA; Hunte, C; Brandt, U; Greenberg, ML; Schagger, H. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem 2003, 278, 52873–52880. [Google Scholar]
- Wittig, I; Schagger, H. Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim. Biophys. Acta 2009, 1787, 672–680. [Google Scholar]
- Tyurina, YY; Tyurin, VA; Kaynar, AM; Kapralova, VI; Wasserloos, K; Li, J; Mosher, M; Wright, L; Wipf, P; Watkins, S; et al. Oxidative lipidomics of hyperoxic acute lung injury: Mass spectrometric characterization of cardiolipin and phosphatidylserine. Am. J. Physiol. Lung Cell. Mol. Physiol 2010, 299, L73–L85. [Google Scholar]
- Wiswedel, I; Gardemann, A; Storch, A; Peter, D; Schild, L. Degradation of phospholipids by oxidative stress—exceptional significance of cardiolipin. Free Radic. Res 2010, 44, 135–145. [Google Scholar]
- Paradies, G; Petrosillo, G; Pistolese, M; Ruggiero, FM. The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett 2000, 466, 323–326. [Google Scholar]
- Paradies, G; Petrosillo, G; Pistolese, M; Di Venosa, N; Federici, A; Ruggiero, FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ. Res 2004, 94, 53–59. [Google Scholar]
- Kiebish, MA; Han, X; Cheng, H; Chuang, JH; Seyfried, TN. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: Lipidomic evidence supporting the warburg theory of cancer. J. Lipid Res 2008, 49, 2545–2556. [Google Scholar]
- Paradies, G; Petrosillo, G; Pistolese, M; Ruggiero, FM. Reactive oxygen species generated by the mitochondrial respiratory chain affect the complex III activity via cardiolipin peroxidation in beef-heart submitochondrial particles. Mitochondrion 2001, 1, 151–159. [Google Scholar]
- Paradies, G; Petrosillo, G; Pistolese, M; Ruggiero, FM. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 2002, 286, 135–141. [Google Scholar]
- Wiswedel, I; Keilhoff, G; Dorner, L; Navarro, A; Bockelmann, R; Bonnekoh, B; Gardemann, A; Gollnick, H. UVB irradiation-induced impairment of keratinocytes and adaptive responses to oxidative stress. Free Radic. Res 2007, 41, 1017–1027. [Google Scholar]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci 2005, 10, 1881–1896. [Google Scholar]
- Behrend, I; Henderson, G; Zwacka, RM. Reactive oxygen species in oncogenic transformation. Biochem. Soc. Trans 2003, 31, 1441–1444. [Google Scholar]
- Gius, D; Spitz, DR. Redox signaling in cancer biology. Antioxid. Redox Signal 2006, 8, 1249–1252. [Google Scholar]
- Hempel, N; Carrico, PM; Melendez, JA. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anticancer Agents Med. Chem 2011, 11, 191–201. [Google Scholar]
- Malafa, M; Margenthaler, J; Webb, B; Neitzel, L; Christophersen, M. MnSOD expression is increased in metastatic gastric cancer. J. Surg. Res 2000, 88, 130–134. [Google Scholar]
- Izutani, R; Asano, S; Imano, M; Kuroda, D; Kato, M; Ohyanagi, H. Expression of manganese superoxide dismutase in esophageal and gastric cancers. J. Gastroenterol 1998, 33, 816–822. [Google Scholar]
- Janssen, AML; Bosman, CB; van Duijn, W; Oostendorp-van de Ruit, MM; Kubben, FJGM; Griffioen, G; Lamers, BBHW; van Krieken, JHJM; van de Velde, CJH; Verspaget, HW. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clin. Cancer Res 2000, 6, 3183–3192. [Google Scholar]
- Toh, Y; Kuninaka, S; Oshiro, T; Ikeda, Y; Nakashima, H; Baba, H; Kohnoe, S; Okamura, T; Mori, M; Sugimachi, K. Overexpression of manganese superoxide dismutase mRNA may correlate with aggressiveness in gastric an colorectal adenocarcinomas. Int. J. Oncol 2000, 17, 107–112. [Google Scholar]
- Hu, H; Luo, M-L; Du, X-L; Feng, Y-B; Zhang, Y; Shen, X-M; Xu, X; Cai, Y; Han, Y-L; Wang, M-R. Up-regulated manganese superoxide dismutase expression increases apoptosis resistance in human esophageal squamous cell carcinomas. Chin. Med. J 2007, 120, 2092–2098. [Google Scholar]
- Ho, JC-M; Zheng, S; Comhair, SAA; Farver, C; Erzurum, SC. Differential expression of manganese superoxide dismutase and catalase in lung cancer. Cancer Res 2001, 61, 8578–8585. [Google Scholar]
- Tsanou, E; Ioachim, E; Briasoulis, E; Damala, K; Charchanti, A; Karavasilis, V; Pavlidis, N; Agnantis, NJ. Immunohistochemical expression of superoxide dismutase (MnSOD) anti-oxidant enzyme in invasive breast carcinoma. Histol. Histopathol 2004, 19, 807–813. [Google Scholar]
- Soini, Y; Vakkala, M; Kahlos, K; Paakko, P; Kinnla, V. MnSOD expression is less frequent in tumour cells of invasive breast carcinomas than in in situ carcinomas or non-neoplastic breast epithelial cells. J. Pathol 2001, 195, 156–162. [Google Scholar]
- Chuang, T-C; Liu, J-Y; Lin, C-T; Tang, Y-T; Yeh, M-H; Chang, S-C; Li, J-W; Kao, M-C. Human manganese superoxide dismutase suppresses HER2/neu-mediated breast cancer malignancy. FEBS Lett 2007, 581, 4443–4449. [Google Scholar]
- Cullen, JJ; Weydert, C; Hinkhouse, MM; ritchie, J; Domann, FE; Spitz, D; Oberley, LW. The role of manganese superoxide dismutase in the growth of pancreatic adenocarcinoma. Cancer Res 2003, 63, 1297–1303. [Google Scholar]
- Hu, Y; Rosen, DG; Zhou, Y; Feng, L; Yang, G; Liu, J; Huang, P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: Role in cell proliferation and response to oxidative stress. J. Biol. Chem 2005, 280, 39485–39492. [Google Scholar]
- Nozoe, T; Honda, M; Inutsuka, S; Yasuda, M; Korenaga, D. Significance of immunohistochemical expression of manganese superoxide dismutase as a marker of malignant potential in colorectal carcinoma. Oncol. Rep 2003, 10, 39–43. [Google Scholar]
- Kattan, Z; Minig, V; Leroy, P; Dauca, M; Becuwe, P. Role of manganese superoxide dismtuase on growth and invasive properties of human estrogen-independent breast cancer cells. Breast Cancer Res. Treat 2008, 108, 203–215. [Google Scholar]
- Holley, AK; Kiningham, KK; Spitz, DR; Edwards, DP; Jenkins, JT; Moore, MR. Progestin stimulation of manganese superoxide dismutase and invasive properties in T47D human breast cancer cells. J. Steroid Biochem. Mol. Biol 2009, 117, 23–30. [Google Scholar]
- Nelson, KK; Ranganathan, AC; Mansouri, J; Rodriguez, AM; Providence, KM; Rutter, JL; Pumiglia, K; Bennett, JA; Melendez, JA. Elevated Sod2 activity augments matrix metalloproteinase expression: Evidence for the involvement of endogenous hydrogen peroxide in regulating metastasis. Clin. Cancer Res 2003, 9, 424–432. [Google Scholar]
- Ranganathan, AC; Nelson, KK; Rodriguez, AM; Kim, K-H; Tower, GB; Rutter, JL; Brinckerhoff, CE; Huang, T-T; Epstein, CJ; Jeffrey, JJ; et al. Manganese superoxide dismutase signals matrix metalloproteinase expression via H2O2-dependent ERK1/2 activation. J. Biol. Chem 2001, 276, 14264–14270. [Google Scholar]
- Liu, R; Oberley, TD; Oberley, LW. Transfection and expression of MnSOD cDNA decreases tumor malignancy of human oral sqamous carcinoma SCC-25 cells. Hum. Gene Ther 1997, 8, 585–595. [Google Scholar]
- Venkataraman, S; Jiang, X; Weydert, C; Zhang, Y; Zhang, HJ; Goswami, PC; Ritchie, JM; Oberley, LW; Buettner, GR. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene 2005, 24, 77–89. [Google Scholar]
- Weydert, C; Roling, B; Liu, J; Hinkhouse, MM; Ritchie, JM; Oberley, LW; Cullen, JJ. Suppression of the malignant phenotype in human pancreatic cancer cells by the overexpression of manganese superoxide dismutase. Mol. Cancer Ther 2003, 2, 361–369. [Google Scholar]
- Behrend, L; Mohr, A; Dick, T; Zwacka, RM. Manganese superoxide dismutase induces p53-dependent senescence in colorectal cancer cells. Mol. Cell. Biol 2005, 25, 7758–7769. [Google Scholar]
- Ridnour, LA; Oberley, TD; Oberley, LW. Tumor suppressive effects of MnSOD overexpression may involve imbalance in peroxide generation versus peroxide removal. Antioxid. Redox Signal 2004, 6, 501–512. [Google Scholar]
- Oberley, LW. Mechanism of the tumor suppressive effect of MnSOD overexpression. Biomed. Pharmacother 2005, 59, 143–148. [Google Scholar]
- Zhang, Y; Smith, BJ; Oberley, LW. Enzymatic activity is necessary for the tumor-suppressive effects of MnSOD. Antioxid. Redox Signal 2006, 8, 1283–1293. [Google Scholar]
- Li, N; Oberley, TD; Oberley, LW; Zhong, W. Overexpression of manganese superoxide dismutase in DU145 human prostate carcinoma cells has mutliple effects on cell phenotype. Prostate 1998, 35, 221–233. [Google Scholar]
- Davis, CA; Hearn, AS; Fletcher, B; Bickford, J; Garcia, JE; Leveque, V; Melendez, JA; Silverman, DN; Zucali, J; Agarwal, A; et al. Potent anti-tumor effects of an active site mutant of human manganese-superoxide dismutase. Evolutionary conservation of product inhibition. J. Biol. Chem 2004, 279, 12769–12776. [Google Scholar]
- Kiningham, KK; St Clair, DK. Overexpression of manganese superoxide dismutase selectively modulates the activity of Jun-associated transcription factors in fibrosarcoma cells. Cancer Res 1997, 57, 5265–5271. [Google Scholar]
- Zhao, Y; Kiningham, KK; Lin, S-M; St Clair, DK. Overexpression of MnSOD protects murine fibrosarcoma cells (FSa-II) from apoptosis and promotes a differentiation program upon treatment with 5-azacytidine: Involvement of MAPK and NF-κB pathways. Antioxid. Redox Signal 2001, 3, 375–386. [Google Scholar]
- Zhao, Y; Xue, Y; Oberley, TD; Kiningham, KK; Lin, S-M; Yen, H-C; Majima, H; Hines, J; St Clair, DK. Overexpression of manganese superoxide dismutase suppresses tumor formation by modulation of activator protein-1 signaling in a multistage skin carcinogenesis model. Cancer Res 2001, 61, 6082–6088. [Google Scholar]
- Zhao, Y; Oberley, TD; Chaiswing, L; Lin, S-M; Epstein, CJ; Huang, T-T; St Clair, DK. Manganese superoxide dismutase deficiency enhances cell turnover via tumor promoter-induced alterations in AP-1 and p53-mediated pathways in a skin cancer model. Oncogene 2002, 21, 3836–3846. [Google Scholar]
- Zhao, Y; Chaiswing, L; Oberley, TD; Batinic-Haberle, I; St Clair, W; Epstein, CJ; St Clair, DK. A mechanism-based antioxidant approach for the reduction of skin carcinogenesis. Cancer Res 2005, 65, 1401–1405. [Google Scholar]
- Rosenblum, JS; Gilula, NB; Lerner, RA. On signal sequence polymorphisms and diseases of distribution. Proc. Natl. Acad. Sci. USA 1996, 93, 4471–4473. [Google Scholar]
- Sutton, A; Khoury, H; Prip-Buus, C; Cepanec, C; Pessayre, D; Degoul, F. The Ala16Val genetic dimorphism modulates the import of human manganese superoxide dismutase into rat liver mitochondria. Pharmacogenetics 2003, 13, 145–157. [Google Scholar]
- Shimoda-Matsubayashi, S; Matsumine, H; Kobayashi, T; Nakagawa-Hattori, Y; Shimizu, Y; Mizuno, Y. Structural dimorphism in the mitochondrial targeting sequence in the human manganese superoxide dismutase gene. Biochem. Biophys. Res. Commun 1996, 226, 561–565. [Google Scholar]
- Bag, A; Bag, N. Target sequence polymorphism of human manganese superoxide dismutase gene and its association with cancer risk: A review. Cancer Epidemiol. Biomark. Prev 2008, 17, 3298–3305. [Google Scholar]
- Johnatty, SE; Nagle, CM; Spurdle, AB; Chen, X; Study, ABCF; Webb, PM; Chenevix-Trench, G. The MnSOD Val9Ala polymorphism, dietary antioxidant intake, risk and survival in ovarian cancer (Australia). Gynecol. Oncol 2007, 107, 388–391. [Google Scholar]
- Dalan, AB; Ergen, A; Yilmaz, H; Karateke, A; Isbar, T. Manganese superoxide dismutase gene polymorphism, MnSOD plasma levels and risk of epithelial ovarian cancer. J. Obstet. Gynaecol. Res 2008, 34, 878–884. [Google Scholar]
- Sun, L; König, IR; Homann, N. Manganese superoxide dismutase (MnSOD) polymorphism, alcohol, cigarette smoking and risk of oesophageal cancer. Alcohol Alcohol 2009, 44, 353–357. [Google Scholar]
- Zejnilovic, J; Akev, N; Yilmaz, H; Isbir, T. Association between manganese superoxide dismutase polymorphism and risk of lung cancer. Cancer Genet. Cytogenet 2009, 189, 1–4. [Google Scholar]
- Wang, LI; Miller, DP; Sai, Y; Liu, G; Su, L; Wain, JC; Lynch, TJ; Christiani, DC. Manganese superoxide dismutase alanine-to-valine polymorphism at codon 16 and lung cancer risk. J. Natl. Cancer Inst 2001, 93, 1818–1821. [Google Scholar]
- Woodson, K; Tangrea, JA; Lehman, TA; Modali, R; Taylor, KM; Snyder, K; Taylor, PR; Virtamo, J; Albanes, D. Manganese superoxide dismutase polymorphism, α-tocopherol supplementation and prostate cancer risk in the alpha-tocopherol, beta-carotene cancer prevention study (Finland). Cancer Causes Control 2003, 14, 513–518. [Google Scholar]
- Arsova-Sarafinovska, Z; Matevska, N; Petrovski, D; Banev, S; Dzikova, S; Georgiev, V; Sikole, A; Sayal, A; Aydin, A; Suturkova, L; et al. Manganese superoxide dismutase (MnSOD) genetic polymorphism is associated with risk of early-onset prostate cancer. Cell Biochem. Funct 2008, 26, 771–777. [Google Scholar]
- Borgstahl, GEO; Parge, HE; Hickey, MJ; Johnson, MJ; Boissinot, M; Hallewell, RA; Lepock, JR; Cabelli, DE; Tainer, JA. Human mitochondrial manganese superoxide dismutase polymorphic variant Ile58Thr reduces activity by destabilizing the tetrameric interface. Biochemistry 1996, 35, 4287–4297. [Google Scholar]
- Zhang, HJ; Yan, T; Oberley, TD; Oberley, LW. Comparison of effects of two polymorphic variants of manganese superoxide dismutase on human breast MCF-7 cancer cell phenotype. Cancer Res 1999, 59, 6276–6283. [Google Scholar]
- Hernandez-Saavedra, D; McCord, JM. Paradoxical effects of thiol reagents on jurkat cells and a new thiol-sensitive mutant form of human mitochondrial superoxide dismutase. Cancer Res 2003, 63, 159–163. [Google Scholar]
- Hernandez-Saavedra, D; McCord, JM. Association of a new intronic polymorphism of the SOD2 gene (G1677T) with cancer. Cell Biochem. Funct 2009, 27, 223–227. [Google Scholar]
- Glynn, SA; Boersma, BJ; Howe, TM; Edvardsen, H; Geisler, SB; Goodman, JE; Ridnour, LA; Lonning, PE; Borresen-Dale, A-L; Naume, B; et al. A mitochondrial target sequence polymorphism in MnSOD predicts inferior survival in breast cancer patients treted with cyclophosphamide. Clin. Cancer Res 2009, 15, 4165–4173. [Google Scholar]
- Yao, S; Barlow, WE; Albain, KS; Choi, J-Y; Zhao, H; Livingston, RB; Davis, W; Rae, JM; Yeh, I-T; Hutchins, LF; et al. Manganese superoxide dismutase polymorphism, treatment-related toxicity and disease-free survival in SWOG 8897 clinical trial for breast cancer. Breast Cancer Res. Treat 2010, 124, 433–439. [Google Scholar]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Solaini, G; Sgarbi, G; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys. Acta 2011, 1807, 534–542. [Google Scholar]
- Chen, Z; Lu, W; Garcia-Prieto, C; Huang, P. The warburg effect and its cancer therapeutic implications. J. Bioenerg. Biomembr 2007, 39, 267–274. [Google Scholar]
- Altenberg, B; Greulich, KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar]
- Lopez-Lazaro, M. The warburg effect: Why and how do cancer cells activate glycolysis in the presence of oxygen? Anticancer Agents Med. Chem 2008, 8, 305–312. [Google Scholar]
- Ruckenstuhl, C; Buttner, S; Carmona-Gutierrez, D; Eisenberg, T; Kroemer, G; Sigrist, SJ; Frohlich, K-U; Madeo, F. The warburg effect suppresses oxidative stress induced apoptosis in a yeast model for cancer. PLoS One 2009, 4, e4592. [Google Scholar]
- Xu, R-H; Pelicano, H; Zhou, Y; Carew, JS; Feng, L; Bhalla, KN; Keating, MJ; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res 2005, 65, 613–621. [Google Scholar]
- Michishita, E; Park, JY; Burneskis, JM; Barrett, JC; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar]
- Kim, H-S; Patel, K; Muldoon-Jacobs, K; Bisht, KS; Aykin-Burns, N; Pennington, JD; van der Meer, R; Nguyen, P; Savage, J; Owens, KM; et al. SirT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar]
- Bell, EL; Emerling, BM; Ricoult, SJH; Guarente, L. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar]
- Finley, LWS; Carracedo, A; Lee, J; Souza, A; Egia, A; Zhang, J; Teruya-Feldstein, J; Moreira, PI; Cardoso, SM; Clish, CB; et al. Sirt3 opposes reprograming of cancer cell metabolism through hif1α destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar]
- Yamakura, F; Kawasaki, H. Post-translational modifications of superoxide dismutase. Biochim. Biophys. Acta 2010, 1804, 318–325. [Google Scholar]
- Tao, R; Coleman, MC; Pennington, JD; Ozden, O; Park, S-H; Jiang, H; Kim, H-S; Flynn, CR; Hill, S; McDonald, WH; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar]
- Ozden, O; Park, S-H; Kim, H-S; Jiang, H; Coleman, MC; Spitz, DR; Gius, D. Acetylation of MnSOD directs enzymatic activity responding to cellular nutrient status or oxidative stress. Aging 2011, 3, 102–107. [Google Scholar]
- Sam, F; Kerstetter, DL; Pimental, DR; Mulukutla, S; Tabaee, A; Bristow, MR; Colucci, WS; Sawyer, DB. Increased reactive oxygen species production and functional alterations in antioxidant enzymes in human failing myocardium. J. Card. Fail 2005, 11, 473–480. [Google Scholar]
- Assem, M; Teyssier, J-R; Benderitter, M; Terrand, J; Laubriet, A; Javouhey, A; David, M; Rochette, L. Pattern of superoxide dismutase enzymatic activity and rna changes in rat heart ventricles after myocardial infarction. Am. J. Pathol 1997, 151, 549–555. [Google Scholar]
- Csonka, C; Pataki, T; Kovacs, P; Muller, SL; Schroeter, ML; Tosaki, A; Blasig, IE. Effects of oxidative stress on the expression of antioxidative defense enzymes in spontaneously hypertensive rat hearts. Free Radic. Biol. Med 2000, 29, 612–619. [Google Scholar]
- Khaper, N; Kaur, K; Li, T; Farahmand, F; Singal, PK. Antioxidant enzyme gene expression in congestive heart failure following myocardial infarction. Mol. Cell. Biochem 2003, 251, 9–15. [Google Scholar]
- Loch, T; Vakhrusheva, O; Piotrowska, I; Ziolkowski, W; Ebelt, H; Braun, T; Bober, E. Different extent of cardiac malfunction and resistance to oxidative stress in heterozygous and homozygous manganese-dependent superoxide dismutase-mutant mice. Cardiovasc. Res 2009, 82, 448–457. [Google Scholar]
- Van Remmen, H; Williams, MD; Guo, Z; Estlack, L; Yang, H; Carlson, EJ; Epstein, CJ; Huang, T-T; Richardson, A. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am. J. Physiol. Heart Circ. Physiol 2001, 281, H1422–H1432. [Google Scholar]
- Wenzel, P; Schuhmacher, S; Kienhofer, J; Muller, J; Hortmann, M; Oelze, M; Schulz, E; Treiber, N; Kawamoto, T; Scharffetter-Kochanek, K; et al. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc. Res 2008, 80, 280–289. [Google Scholar]
- Brown, KA; Didion, SP; Andresen, JJ; Faraci, FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler. Thromb. Vasc. Biol 2007, 27, 1941–1946. [Google Scholar]
- Kakko, S; Paivansalo, M; Koistinen, P; Kesaniemi, YA; Kinnula, VL; Savolainen, MJ. The signal sequence polymorphism of the MnSOD gene is associated with the degree of carotid atherosclerosis. Atherosclerosis 2003, 168, 147–152. [Google Scholar]
- Fujimoto, H; Taguchi, J-I; Imai, Y; Ayabe, S; Hashimoto, H; Kobayashi, H; Ogasawara, K; Aizawa, T; Yamakado, M; Nagai, R; et al. Manganese superoxide dismutase polymorphism affects the oxidized low-density lipoprotein-induced apoptosis of macrophages and coronary artery disease. Eur. Heart J 2008, 29, 1267–1274. [Google Scholar]
- Fujimoto, H; Kobayashi, H; Ogasawara, K; Yamakado, M; Ohno, M. Association of the manganese superoxide dismutase polymorphism with vasospastic angina pectoris. J. Cardiol 2010, 55, 205–210. [Google Scholar]
- Miller, JD; Peotta, VA; Chu, Y; Weiss, RM; Zimmerman, K; Brooks, RM; Heistad, DD. MnSOD protects against COX1-mediated endothelial dysfunction in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol 2010, 298, H1600–H1607. [Google Scholar]
- Negoro, S; Kunisada, K; Fujio, Y; Funamoto, M; Darville, MI; Eizirik, DL; Osugi, T; Izumi, M; Oshima, Y; Nakaoka, Y; et al. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation 2001, 104, 979–981. [Google Scholar]
- Jin, Z-Q; Zhou, H-Z; Cecchini, G; Gray, MO; Karliner, JS. MnSOD in mouse heart: Acute responses to ischemic preconditioning and ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol 2005, 288, H2986–H2994. [Google Scholar]
- MacMillan-Crow, LA; Crow, JP; Kerby, JD; Beckman, JS; Thompson, JA. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc. Natl. Acad. Sci. USA 1996, 93, 11853–11858. [Google Scholar]
- Yamakura, F; Taka, H; Fujimura, T; Murayama, K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J. Biol. Chem 1998, 273, 14085–14089. [Google Scholar]
- MacMillan-Crow, LA; Crow, JP; Thompson, JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar]
- Quint, P; Reutzel, R; Mikulski, R; McKenna, R; Silverman, DN. Crystal structure of nitrated human manganese superoxide dismutase: Mechanism of inactivation. Free Radic. Biol. Med 2006, 40, 453–458. [Google Scholar]
- Xu, S; Ying, J; Jiang, B; Guo, W; Adachi, T; Sharov, V; Lazar, H; Menzoian, J; Knyushko, TV; Bigelow, D; et al. Detection of sequence-specific tyrosine nitration of manganese sod and serca in cardiovascular disease and aging. Am. J. Physiol. Heart Circ. Physiol 2006, 290, H2220–H2227. [Google Scholar]
- Miller, LW. Cardiovascular toxicities of immunosuppressive agents. Am. J. Transplant 2002, 2, 807–818. [Google Scholar]
- Lamas, S. Cellular mechanisms of vascular injury mediated by calcineurin inhibitors. Kidney Int 2005, 68, 898–907. [Google Scholar]
- Navarro-Antolin, J; Redondo-Horcajo, M; Zaragoza, C; Alvarez-Barrientos, A; Fernandez, AP; Leon-Gomez, E; Rodrigo, J; Lamas, S. Role of peroxynitrite in endothelial damage mediated by cyclosporine a. Free Radic. Biol. Med 2007, 42, 394–403. [Google Scholar]
- Redondo-Horcajo, M; Romero, N; Martinez-Acedo, P; Martinez-Ruiz, A; Quijano, C; Lourenco, CF; Movilla, N; Enriquez, JA; Rodriguez-Pascual, F; Rial, E; et al. Cyclosporine A-induced nitration of tyrosine 34 MnSOD in endothelial cells: Role of mitochondrial superoxide. Cardiovasc. Res 2010, 87, 356–365. [Google Scholar]
- Kim, GW; Kondo, T; Noshita, N; Chan, PH. Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice: Implications for the production and role of superoxide radicals. Stroke 2002, 33, 809–815. [Google Scholar]
- Shan, X; Chi, L; Ke, Y; Luo, C; Qian, S; Gozal, D; Liu, R. Manganese superoxide dismutase protects mouse cortical neurons from chronic intermittent hypoxia-mediated oxidative damage. Neurobiol. Dis 2007, 28, 206–215. [Google Scholar]
- Saha, R; Pahan, K. Differential regulation of mn-superoxide dismutase in neurons and astroglia by HIV-1 gp120: Implications for hiv-associted dementia. Free Radic. Biol. Med 2007, 42, 1866–1878. [Google Scholar]
- Xiong, Y; Shie, F-S; Zhang, J; Lee, C-P; Ho, Y-S. Prevention of mitochondrial dysfunction in post-traumatic mouse brain by superoxide dismutase. J. Neurochem 2005, 95, 732–744. [Google Scholar]
- Bayir, H; Kagan, VE; Clark, RSB; Janesko-Feldman, K; Rafikov, R; Huang, Z; Zhang, X; Vagni, V; Billiar, TR; Kochanek, PM. Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J. Neurochem 2007, 101, 168–181. [Google Scholar]
- Li, Y; Copin, JC; Reola, LF; Calagui, G; Gobbel, GT; Chen, SF; Sato, S; Epstein, CJ; Chan, PH. Reduced mitochondrial manganese-superoxide dismutase activity exacerbates glutamate toxicity in cultured mouse cortical neurons. Brain Res 1998, 814, 164–170. [Google Scholar]
- Gonzalez-Zulueta, M; Ensz, LM; Mukhina, G; Lebovitz, RM; Zwacka, RM; Engelhardt, JF; Oberley, LW; Dawson, VL; Dawson, TM. Manganese superoxide dismutase protects nNOS neurons from nmda and nitric oxide-mediated neurotoxicity. J. Neurosci 1998, 18, 2040–2055. [Google Scholar]
- Sullivan, PG; Bruce-Keller, AJ; Rabchevsky, AG; Cristakos, S; Clair, DK; Mattson, MP; Scheff, SW. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J. Neurosci 1999, 19, 6248–6256. [Google Scholar]
- Lynn, S; Huang, EJ; Elchuri, S; Naeemuddin, M; Nishinaka, Y; Yodoi, J; Ferriero, DM; Epstein, CJ; Huang, T-T. Selective neuronal vulnerability and inadequate stress response in superoxide dismutase mutant mice. Free Radic. Biol. Med 2005, 38, 817–828. [Google Scholar]
- Marcus, DL; Strafaci, JA; Freedman, ML. Differential neuronal expression of manganese superoxide dismutase in Alzheimer’s disease. Med Sci Monit 2006, 12, BR8–14. [Google Scholar]
- Dumont, M; Wille, E; Stack, C; Calingasan, NY; Beal, MF; Lin, MT. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J 2009, 23, 2459–2466. [Google Scholar]
- Li, F; Calingasan, NY; Yu, F; Mauck, WM; Toidze, M; Almeida, CG; Takahashi, RH; Carlson, GA; Beal, MF; Lin, MT; et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J. Neurochem 2004, 89, 1308–1312. [Google Scholar]
- Sompol, P; Ittarat, W; Tangpong, J; Chen, Y; Doubinskaia, I; Batinic-Haberle, I; Abdul, HM; Butterfield, DA; St Clair, DK. A neuronal model of Alzheimer’s disease: An insight into the mechanisms of oxidative stress-mediated mitochondrial injury. Neuroscience 2008, 153, 120–130. [Google Scholar]
- Anantharaman, M; Tangpong, J; Keller, JN; Murphy, MP; Markesbery, WR; Kiningham, KK; St Clair, DK. β-amyloid mediated nitration of manganese superoxide dismutase: Implication for oxidative stress in a APPNLH/NLH × PS-1P264L/P264L double knock-in mouse model of Alzheimer’s disease. Am. J. Pathol 2006, 168, 1608–1618. [Google Scholar]
- Eldridge, A; Fan, M; Candas, D; Chromy, B; Li, JJ. Profiling of MnSOD Interaction Proteins in Low-Dose Radiation Induced Adaptive Response. Proceedings of the Low Dose Radiation Research Investigators’ Workshop, Washington, DC, USA, 9–11 May 2011.
- Jin, C; Fan, M; Liu, R; Nantajit, D; Candas, D; Vaughan, ATM; Murley, JS; Woloschak, G; Grdina, DJ; Li, JJ. Cyclin D1 and CDK4 Translocate into Mitochondria and Interact with MnSOD in Low-Dose Radiation-Induced Adaptive Radioprotection. Proceedings of the Low Dose Radiation Research Investigators’ Workshop, Washington, DC, USA, 9–11 May 2011.
- Holley, AK; Velez, JM; St Clair, DK. Mechanisms of p53-mediated paclitaxel-induced neurotoxicity. Free Radic. Biol. Med 2009, 47, S110–S111. [Google Scholar]
- Shadrina, MI; Slominsky, PA; Limborska, SA. Molecular mechanisms of pathogenesis of Parkinson’s disease. Int. Rev. Cell Mol. Biol 2010, 281, 229–266. [Google Scholar]
- Lim, K-L; Ng, X-H; Grace, LG-Y; Yao, T-P. Mitochondrial dynamics and Parkinson’s disease: Focus on parkin. Antioxid Redox Signal 2011. [Google Scholar] [CrossRef]
- Zhu, J; Chu, CT. Mitochondrial dysfunction in Parkinson’s disease. J. Alzheimers Dis 2010, 20, S325–S334. [Google Scholar]
- Schapira, AHV; Gegg, M. Mitochondrial contribution to Parkinson’s disease pathogenesis. Parkinsons Dis 2011, 2011, 159–160. [Google Scholar]
- Tsang, AHK; Chung, KKK. Oxidative and nitrosative stress in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 643–650. [Google Scholar]
- Fong, C-S; Wu, R-M; Shieh, J-C; Chao, Y-T; Fu, Y-P; Kuao, C-L; Cheng, C-W. Pesticide exposure on southwestern taiwanese with MnSOD and NQO1 polymorphisms is associated with increased risk of Parkinson’s disease. Clin. Chim. Acta 2007, 378, 136–141. [Google Scholar]
- Grasbon-Frodl, EM; Kosel, S; Riess, O; Muller, U; Mehraein, P; Graeber, MB. Analysis of mitochondrial targeting sequence and coding region polymorphisms of the manganese superoxide dismutase gene in german Parkinson disease patients. Biochem. Biophys. Res. Commun 1999, 255, 749–752. [Google Scholar]
- Wang, V; Chen, S-Y; Chuang, T-C; Shan, D-E; Soong, B-W; Kao, M-C. Val-9Ala and Ile+58Thr polymorphism of MnSOD in Parkinson’s disease. Clin. Biochem 2010, 43, 979–982. [Google Scholar]
- Andreassen, OA; Ferrante, RJ; Dedeoglu, A; Albers, DW; Klivenyl, P; Carlson, EJ; Epstein, CJ; Beal, MF. Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and mptp. Exp. Neurol 2001, 167, 189–195. [Google Scholar]
- Liang, L-P; Patel, M. Iron-sulfur enzyme mediated mitochondrial superoxide toxicity in experimental Parkinson’s disease. J. Neurochem 2004, 90, 1076–1084. [Google Scholar]
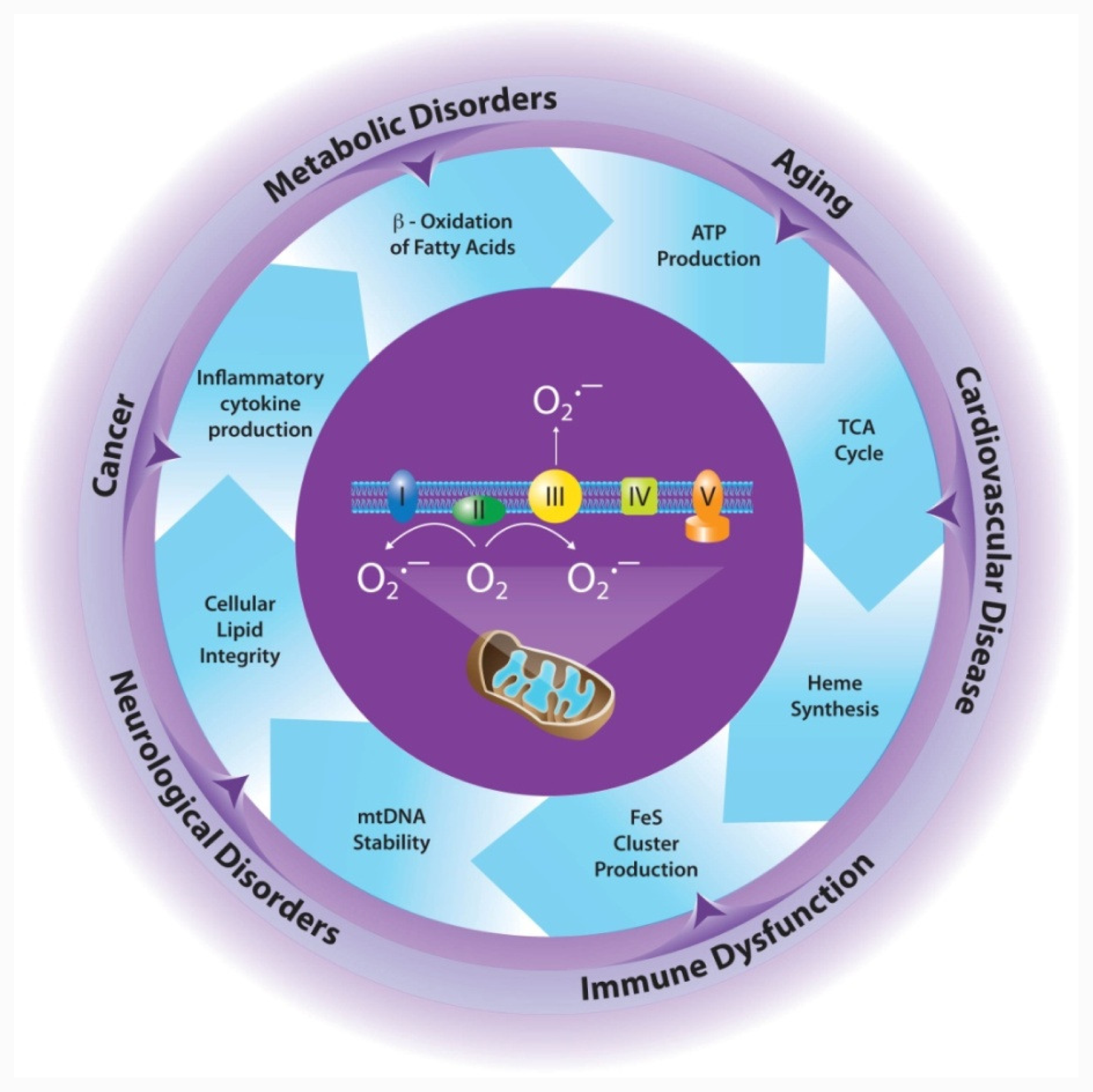
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St. Clair, D.K. Manganese Superoxide Dismutase: Guardian of the Powerhouse. Int. J. Mol. Sci. 2011, 12, 7114-7162. https://doi.org/10.3390/ijms12107114
Holley AK, Bakthavatchalu V, Velez-Roman JM, St. Clair DK. Manganese Superoxide Dismutase: Guardian of the Powerhouse. International Journal of Molecular Sciences. 2011; 12(10):7114-7162. https://doi.org/10.3390/ijms12107114
Chicago/Turabian StyleHolley, Aaron K., Vasudevan Bakthavatchalu, Joyce M. Velez-Roman, and Daret K. St. Clair. 2011. "Manganese Superoxide Dismutase: Guardian of the Powerhouse" International Journal of Molecular Sciences 12, no. 10: 7114-7162. https://doi.org/10.3390/ijms12107114
APA StyleHolley, A. K., Bakthavatchalu, V., Velez-Roman, J. M., & St. Clair, D. K. (2011). Manganese Superoxide Dismutase: Guardian of the Powerhouse. International Journal of Molecular Sciences, 12(10), 7114-7162. https://doi.org/10.3390/ijms12107114