Microwave Effect for Glycosylation Promoted by Solid Super Acid in Supercritical Carbon Dioxide

Abstract
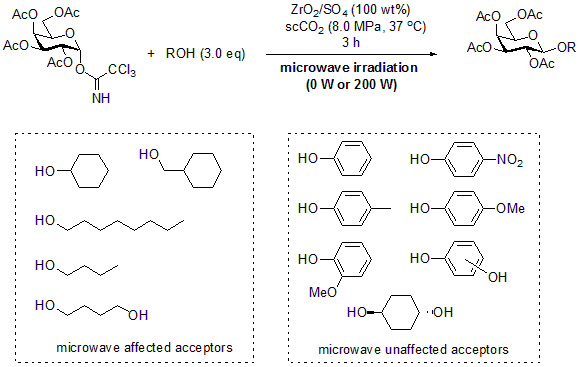
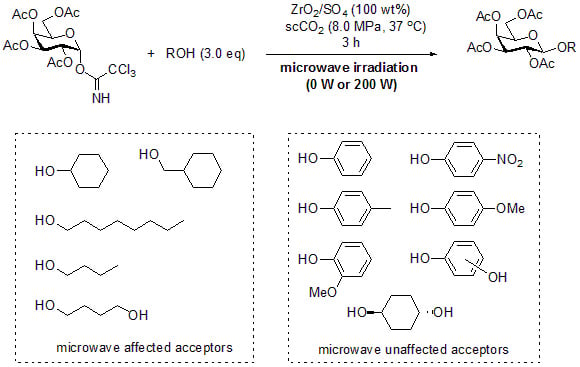
:
1. Introduction
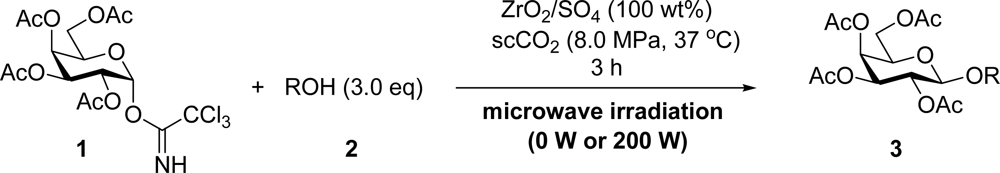
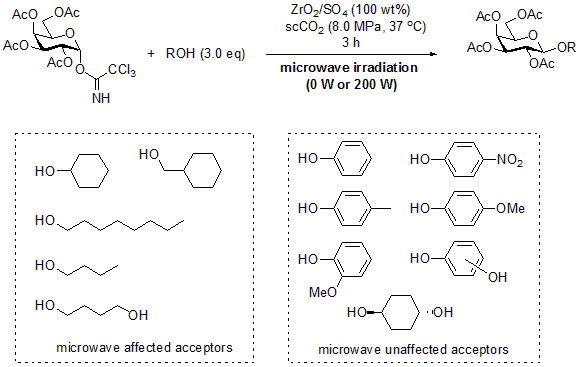
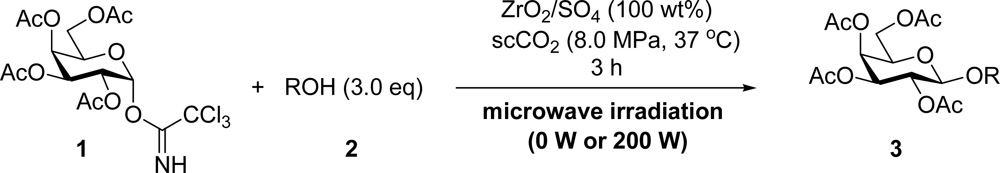
2. Results and Discussion
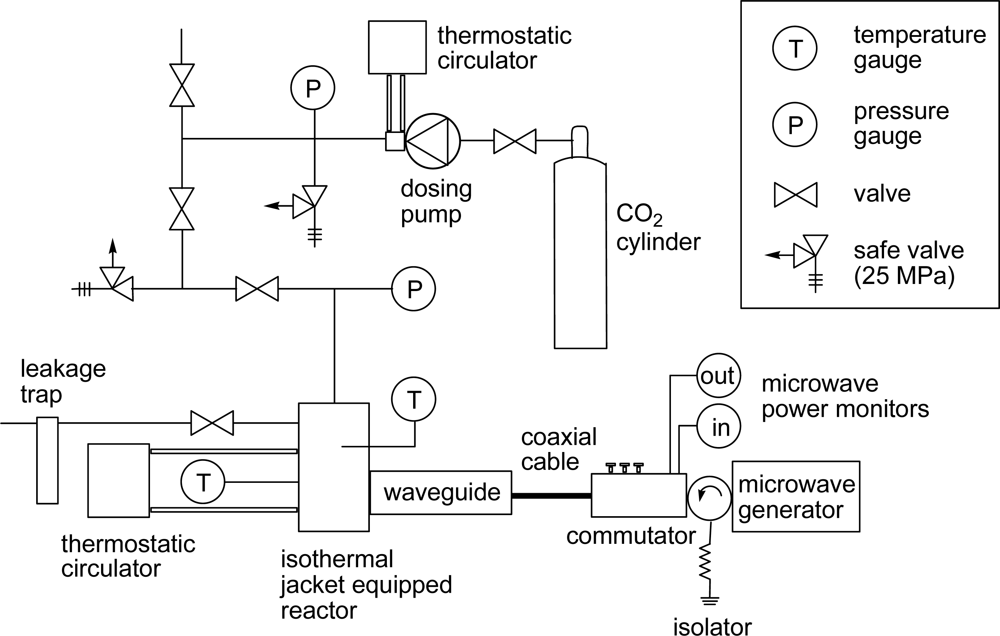
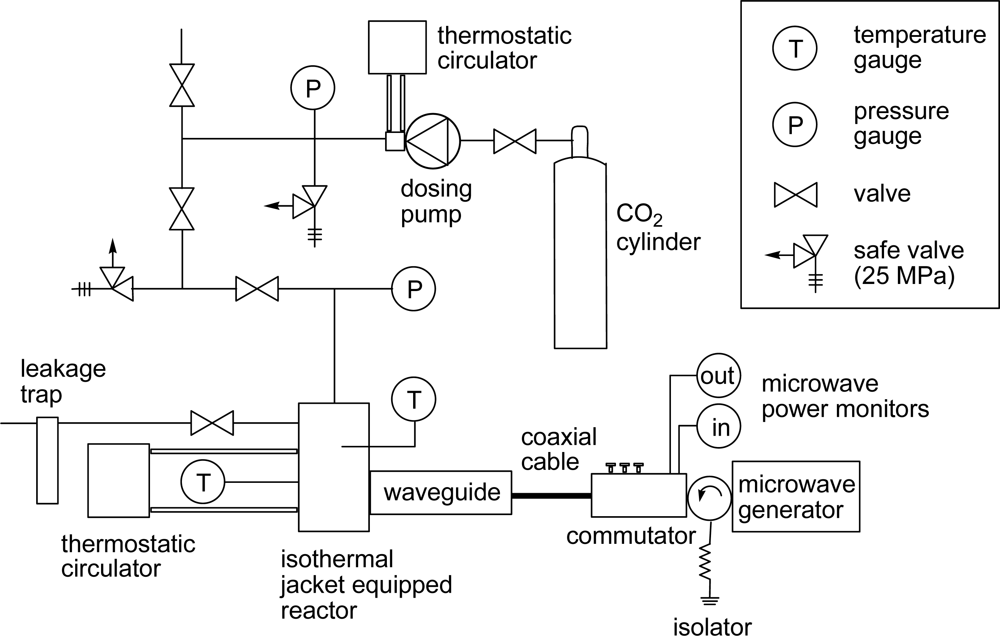
3. Experimental Section
3.1. Materials
3.2. General Method for Glycosylation Reactions
4. Conclusions
Acknowledgments
References and Notes
- Fischer, E. Über die Glucoside der Alkohole. Ber. Dtsch. Chem. Ges 1893, 26, 2400–2412. [Google Scholar]
- Toshima, K; Tatsuta, K. Recent progress in o-glycosylation methods and its application to natural product synthesis. Chem. Rev 1993, 93, 1503–1531. [Google Scholar]
- Toshima, K; Kasumi, K; Matsumura, S. Novel stereocontrolled glycosidations using a solid acid, SO4/ZrO2, for direct synthesis of α- and β-mannopyranosides. Synlett 1998, 643–645. [Google Scholar]
- Nagai, H; Sasaki, K; Matsumura, S; Toshima, K. Environmental benign β–stereoselective glycosidations of glycosyl phosphites using a reusable heterogeneous solid acid, nomtmorillonite K-10. Carbohydr. Res 2005, 340, 337–353. [Google Scholar]
- Sasaki, K; Matsumura, S; Toshima, K. A novel glycosidation of glycosyl fluoride using a designed ionic liquid and its effect on the stereoselectivity. Tetrahedron Lett 2004, 45, 7043–7047. [Google Scholar]
- Seko, A; Koketsu, M; Enoki, Y; Ibrahim, HR; Juneja, LR; Kim, M; Yamamoto, T. Occurrence of a sialylglycopeptide and free sialylglycans in hen’s egg yolk. Biochim Biophys Acta 1997, 1335, 23–32. [Google Scholar]
- Endo, T; Koizumi, S. Large-scale production of oligosaccharides using engineered bacteria. Curr. Opin. Struct. Biol 2000, 10, 536–541. [Google Scholar]
- Chen, X; Kowal, P; Wang, PG. Large-scale enzymatic synthesis of oligosaccharides. Curr. Opin. Drug Discov. Devel 2000, 3, 756–763. [Google Scholar]
- Fumoto, M; Hinou, H; Ohta, T; Ito, T; Yamada, K; Takimoto, A; Kondo, H; Shimizu, H; Inazu, T; Nakahara, Y; Nishimura, S-I. Combinatorial synthesis MUC1 glycopeptides polymer blotting facilitates chemical enzymatic synthesis highly complicated mucin glycopeptides. J. Am. Che. Soc 2005, 127, 11804–11818. [Google Scholar]
- Hanashima, S; Manabe, S; Ito, Y. Divergent synthesis of sialylated glycan chains: Combined use of polymer support, resin capture-release, and chemoenzymatic strategies. Angew. Chem. Int. Ed 2005, 44, 4218–4224. [Google Scholar]
- Blixt, O; Vasiliu, D; Allin, K; Jacobsen, N; Warnock, D; Razi, N; Paulson, JC; Bernatchez, S; Gilbert, M; Wakarchuk, W. Chemoenzymatic synthesis of 2-azidoethyl-ganglio-oligosaccharides GD3, GT3, GM2, GD2, GT2, GM1 and GD1a. Carbohydr. Res 2005, 340, 1963–1972. [Google Scholar]
- Nishimura, S-I. Combinatorial syntheses of sugar derivatives. Curr. Opin. Chem. Biol 2001, 5, 325–335. [Google Scholar]
- Li, X-B; Ogawa, M; Monden, T; Maeda, T; Yamashita, E; Naka, M; Matsuda, M; Hinou, H; Nishimura, S-I. Glycosidation promoted by a reusable solid superacid in supercritical carbon dioxide. Angew. Chem. Int. Ed 2006, 45, 5652–5655. [Google Scholar]
- Nuchter, M; Ondrusehka, B; Bonreth, W; Gum, A. Microwave assisted synthesis–A critical technology overview. Green Chem 2004, 6, 128–141. [Google Scholar]
- Bornaghi, LF; Poulsen, SA. Microwave-accelerated fisher glycosylation. Tetrahedr. Lett 2005, 46, 3485–3488. [Google Scholar]
- Yoshimura, Y; Shimizu, H; Hinou, H; Nishimura, SI. A novel glycosylation concept; microwave-assisted acetal-exchange type glycosylations from methyl glycosides as donors. Tetrahedr. Lett 2005, 46, 4701–4705. [Google Scholar]
- Shimizu, H; Yoshimura, Y; Hinou, H; Nishimura, SI. A new glycosylation method part 3: Study of microwave dffects at low temperatures to control reaction pathways and reduce byproducts. Tetrahedron 2008, 64, 10091–10096. [Google Scholar]
- Seibel, J; Hillringhaus, L; Moraru, R. Microwave-assisted glycosylation for the synthesis of glycopeptides. Carbohydr. Res 2005, 340, 507–511. [Google Scholar]
- Larsen, K; Worm-Leonhard, K; Olsen, P; Hoel, A; Jensen, KJ. Reconsidering glycosylations at high temperature: precise microwave heating. Org. Biomol. Chem 2005, 3, 3966–3970. [Google Scholar]
- Hosseini, M; Stiasni, N; Barbieri, V; Kappe, CO. Microwave-assisted asymmetric organocatalysis. A probe for nonthermal microwave effects and the concept of simultaneous cooling. J. Org. Chem 2007, 72, 1417–1424. [Google Scholar]
- Moriyoshi, T; Kita, T; Usaki, Y. Static relative permittivity of carbon-dioxide and nitrous-oxide up to 30 Mpa. Ber. Bunsenges. Phys. Chem 1993, 97, 589–596. [Google Scholar]
- Michels, A; Kleerekoper, L. Measurements on the dielectric constant of CO2 at 25 °C, 50 °C and 100 °C up to 1700 atmospheres. Physical 1939, 7, 586–590. [Google Scholar]
- Kremsner, JM; Kappe, CO. Silicon carbide passive heating elements in microwave-assisted organic synthesis. J. Org. Chem 2006, 71, 4651–4658. [Google Scholar]
- Staudt, R; Nitzsche, J; Harting, P. Supercritical fluid extraction using microwave heating. Proceedings 6th International Symposium on Supercritical Fluids, Versailles, France, April 2003; Brunner, G, Ed.;
- Kappe, CO; Dallinger, D. Controlled microwave heating in modern organic synthesis: Highlights from the 2004–2008 literature. Mol. Divers 2009, 13, 71–193. [Google Scholar]
- Schmidt, RR; Stumpp, M. Glycosylimidates. 8. Synthesis of 1-thioglycosides. Liebigs Ann. Chem 1983, 7, 1249–1256. [Google Scholar]
- Schroeder, LR; Counts, KM; Haigh, FC. Stereoselectivity of Koenigs-Knorr syntheses of alkyl β-d-galactopyranoside and β-d-xylopyranoside peracetates promoted by mercuric bromide and mercuric oxide. Carbohydr. Res 1974, 37, 368–372. [Google Scholar]
- Tsui, DSK; Gorin, PAJ. Methods for the preparation of alkyl 1,2-orthoacetates of d-glucopyranose and d-galactopyranose in high yield. Carbohydr. Res 1985, 144, 137–147. [Google Scholar]
- Dess, D; Kleine, HP; Weinberg, DV; Kaufmann, RJ; Sidhu, RS. Phase-transfer catalyzed synthesis of acetylated aryl β-d-glucopyranosides and aryl β-d-galactopyranosides. Synthesis 1981, 883–885. [Google Scholar]
- de Bruyne, CK; Wouters-Leysen, J. Synthesis of substituted phenyl β-d-galactopyranosides. Carbohyd. Res 1971, 18, 124–126. [Google Scholar]
- Yamago, S; Hashidume, M; Yoshida, JI. A new synthetic route to substituted quinones by radical-mediated coupling of organotellurium compounds with quinines. Tetrahedron 2002, 58, 6805–6813. [Google Scholar]
- Peng, SQ; Li, L; Wu, JW; An, Y; Cheng, TM; Cai, MS. Studies on glycosides IV. Synthesis of arbutin analogs. Acta. Chim. Sin 1989, 47, 512–515. [Google Scholar]
- DeFrees, SA; Kosch, W; Way, W; Paulson, JC; Sabesan, S; Halcomb, RL; Huang, DH; Ichikawa, Y; Wong, CH. Ligand recognition by e-selectin: Synthesis, inhibitory activity and conformational analysis of bivalent sialyl lewis x analogs. J. Am. Chem. Soc 1995, 117, 66–79. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Acceptor 2 | MW (W) | Glycoside 3 | Yield [1st, 2nd] (%) |
|---|---|---|---|---|
| 1 |  | 0 | 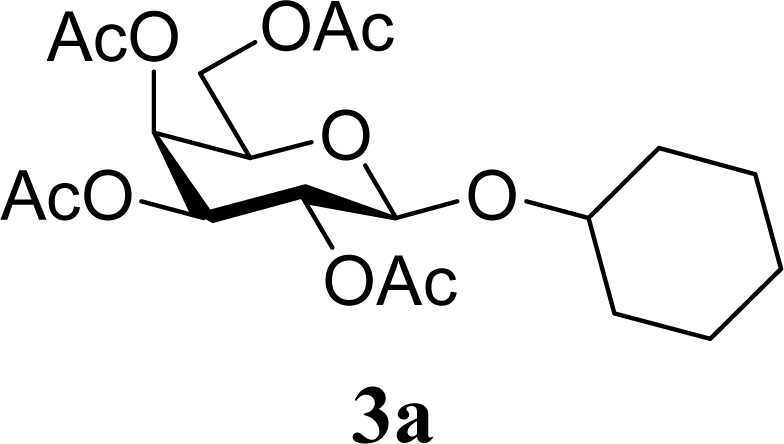 | 53 [51,55] |
| 2 | 2a | 200 | 3a | 71 [69,72] |
| 3 | 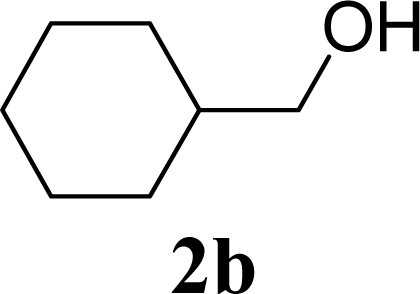 | 0 | 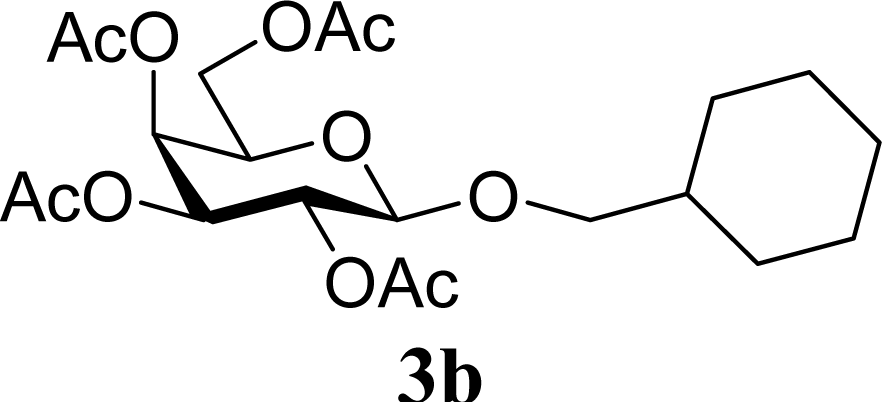 | 54 [52,55] |
| 4 | 2b | 200 | 3b | 68 [68,67] |
| 5 | 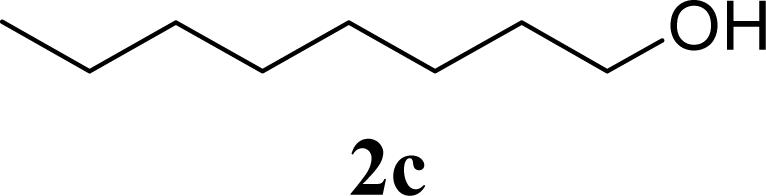 | 0 | 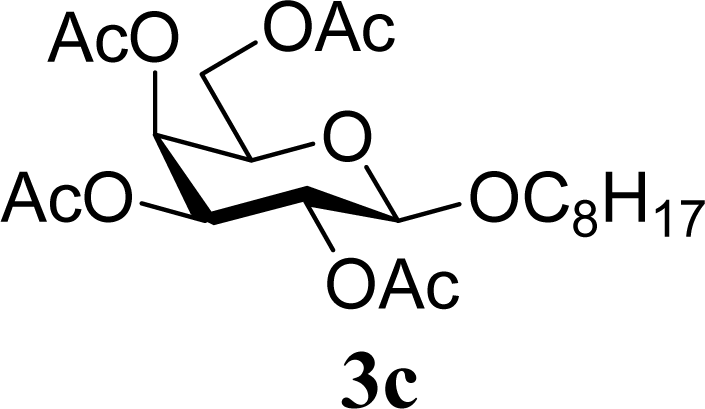 | 65 [68,62] |
| 6 | 2c | 200 | 3c | 77 [80,74] |
| 7 | 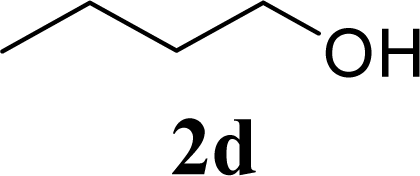 | 0 | 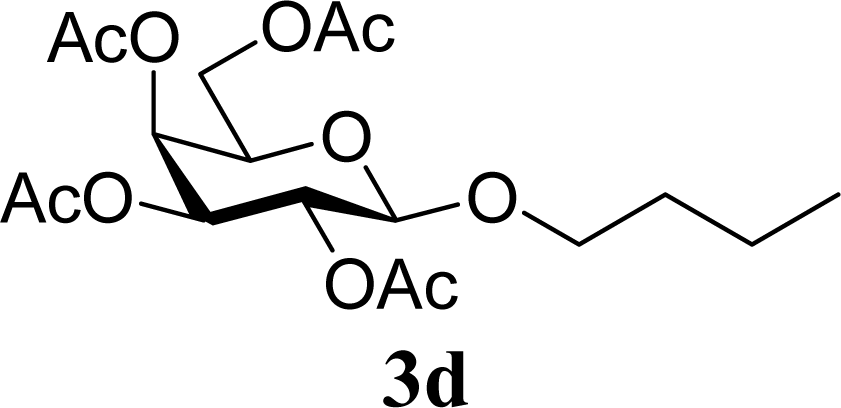 | 60 [58,61] |
| 8 | 2d | 200 | 3d | 86 [84,87] |
| Entry | Acceptor 2 | MW (W) | Glycoside 3 | Yield [1st, 2nd] (%) |
|---|---|---|---|---|
| 9 |  | 0 |  | 14 [12,15] |
| 10 | 2e | 200 | 3e | 20 [17,23] |
| 11 | 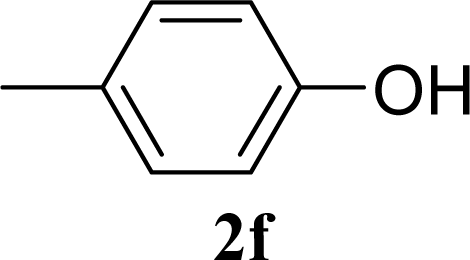 | 0 |  | 41 [49,33] |
| 12 | 2f | 200 | 3f | 39 [39,38] |
| 13 | 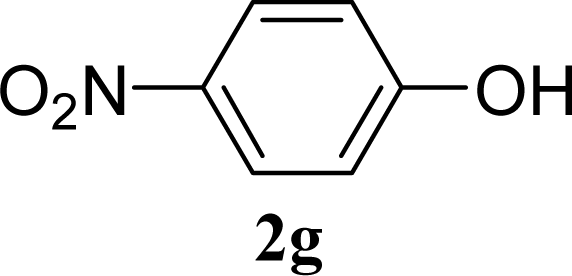 | 0 |  | 5 [6,4] |
| 14 | 2g | 200 | 3g | 3 [5,1] |
| 15 | 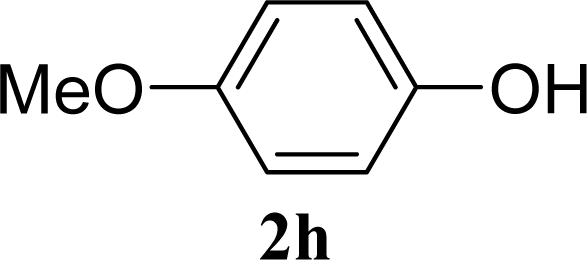 | 0 | 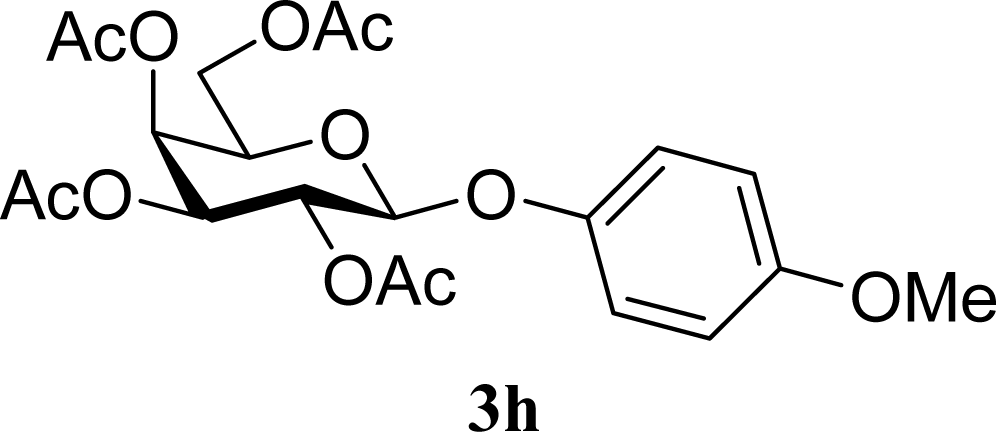 | 43 [42,44] |
| 16 | 2h | 200 | 3h | 40 [42,38] |
| 17 | 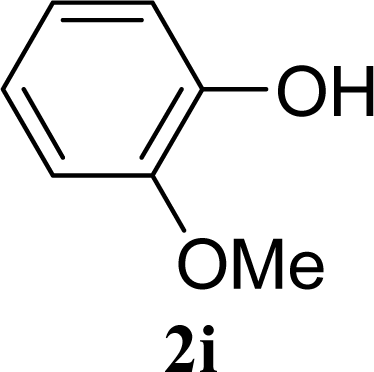 | 0 | 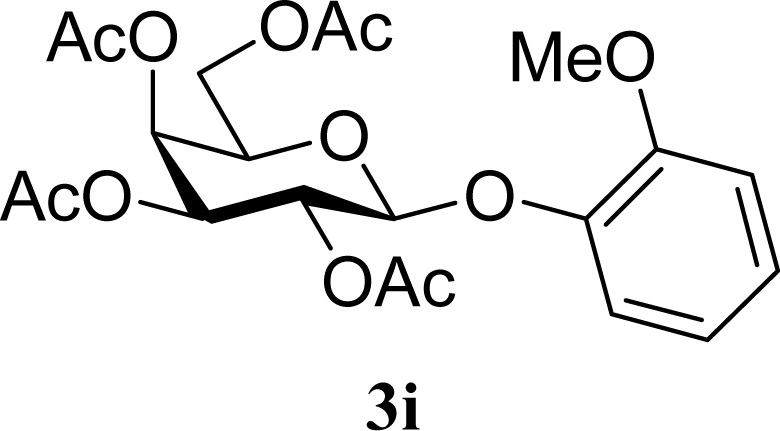 | 24 [26,22] |
| 18 | 2i | 200 | 3i | 22 [24,20] |
| Entry | Acceptor 2 | MW (W) | Monoglycosylated product 3 | Yield of 3 [1st, 2nd] (%) | Yield of diglycosylated product 4 (%) |
|---|---|---|---|---|---|
| 19 | 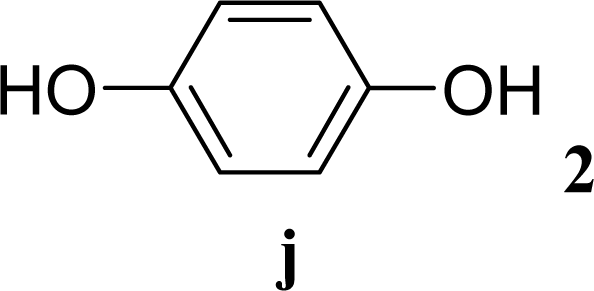 | 0 | 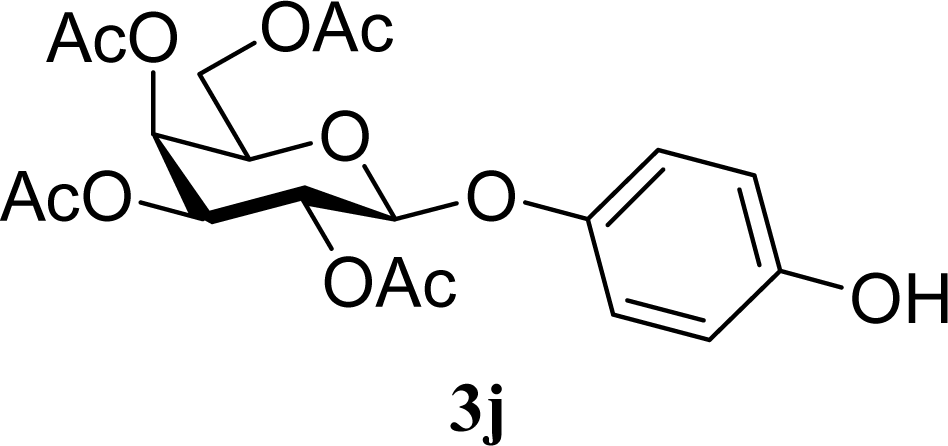 | 6 [7,4] | n. d. |
| 20 | 2j | 200 | 3j | 5 [4,6] | n. d. |
| 21 | 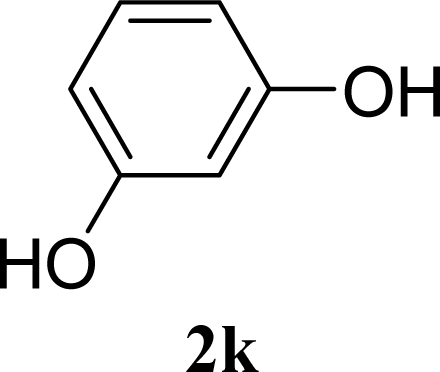 | 0 |  | 14 [8,19] | n. d. |
| 22 | 2k | 200 | 3k | 19 [10,28] | n. d. |
| 25 |  | 0 |  | 20 [19,21] | n. d. |
| 26 | 2l | 200 | 3l | 17 [16,18] | n. d. |
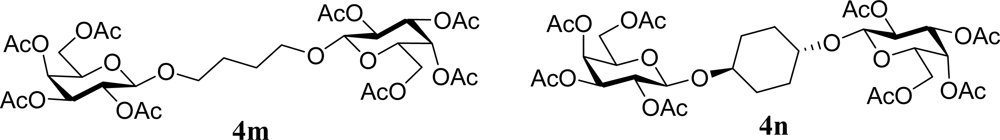
| 27 | 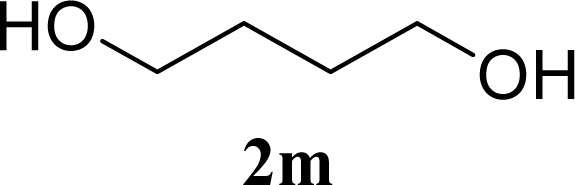 | 0 |  | 12 [12,12] | 4ma,b 12 [11,12] |
| 28 | 2m | 200 | 3m | 30 [33,26] | 4ma,b |
| 29 | 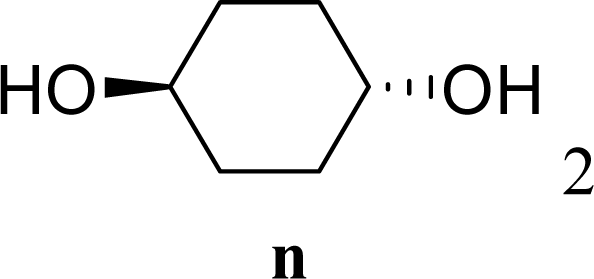 | 0 | 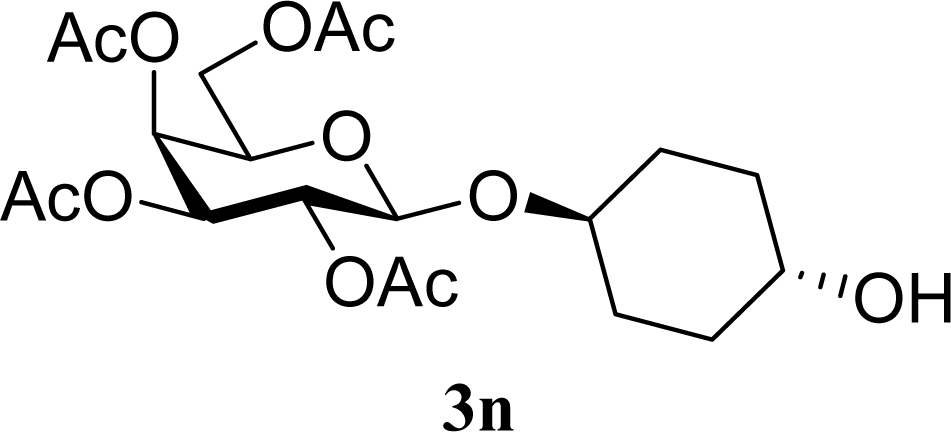 | 17 [14,20] | 18 [16,20] 4na,b |
| 30 | 2n | 200 | 3n | 21 [23,19] | 17 [15,18] 4na,b 14 [10,17] |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hinou, H.; Saito, N.; Ogawa, M.; Maeda, T.; Nishimura, S.-I. Microwave Effect for Glycosylation Promoted by Solid Super Acid in Supercritical Carbon Dioxide. Int. J. Mol. Sci. 2009, 10, 5285-5295. https://doi.org/10.3390/ijms10125285
Hinou H, Saito N, Ogawa M, Maeda T, Nishimura S-I. Microwave Effect for Glycosylation Promoted by Solid Super Acid in Supercritical Carbon Dioxide. International Journal of Molecular Sciences. 2009; 10(12):5285-5295. https://doi.org/10.3390/ijms10125285
Chicago/Turabian StyleHinou, Hiroshi, Naohiro Saito, Masato Ogawa, Takahiko Maeda, and Shin-Ichiro Nishimura. 2009. "Microwave Effect for Glycosylation Promoted by Solid Super Acid in Supercritical Carbon Dioxide" International Journal of Molecular Sciences 10, no. 12: 5285-5295. https://doi.org/10.3390/ijms10125285
APA StyleHinou, H., Saito, N., Ogawa, M., Maeda, T., & Nishimura, S.-I. (2009). Microwave Effect for Glycosylation Promoted by Solid Super Acid in Supercritical Carbon Dioxide. International Journal of Molecular Sciences, 10(12), 5285-5295. https://doi.org/10.3390/ijms10125285