3. Materials and Methods
General: All the chemicals and solvents used for these syntheses were obtained from commercial sources and were used without modification Nuclear magnetic resonance (NMR) spectra (
1H and
13C) were recorded using a Bruker AX-400 spectrometer (Bruker, Bremerhaven, Germany). Chemical shifts in
1H NMR spectra recorded in CDCl
3 are referenced to δ = 7.26 (Data are reported according to the following format: chemical shift (ppm) [multiplicity (e.g., s, d, t, q, etc.), coupling constant(s) (in Hz), integral value (to the nearest whole integer), and structural assignment of the proton]. Chemical shifts in
13C NMR spectra are referenced to the carbon atom of CDCl
3 (δ 77.16). Infrared (IR) spectra were measured on a FT-IR spectrophotometer (Thermo Fisher, Waltham, MA, USA). The samples were placed on a diamond window as thin films (solids by evaporation from a CH
2Cl
2 solution and liquids by direct deposition) and recorded in the attenuated total reflectance (ATR) mode. The absorption peak maxima are given in cm
−1. High-resolution mass spectrometry (HRMS) measurements were obtained on a Waters Q-TOF Premier Spectrometer (ESI or EI Source) (Waters Corporation, Milford, MA, USA). Reactions above the ambient laboratory temperature were performed in silicone oil baths that had been pre-equilibrated to the temperature of choice before the reaction vessel was immersed. Column chromatography was generally performed on silica gel (300–400 mesh). Reactions were monitored by thin-layer chromatography (TLC) using UV light to visualize the course of the reactions and an ethanolic solution of phosphomolybdic acid and heat as developing agents. The spectroscopic data of
1–
4,
8a,
8b,
9–
11,
14–
18 are in
Supplementary Materials.
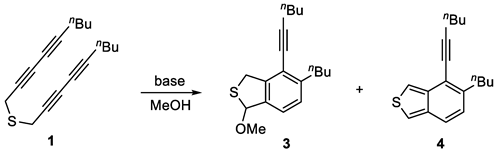
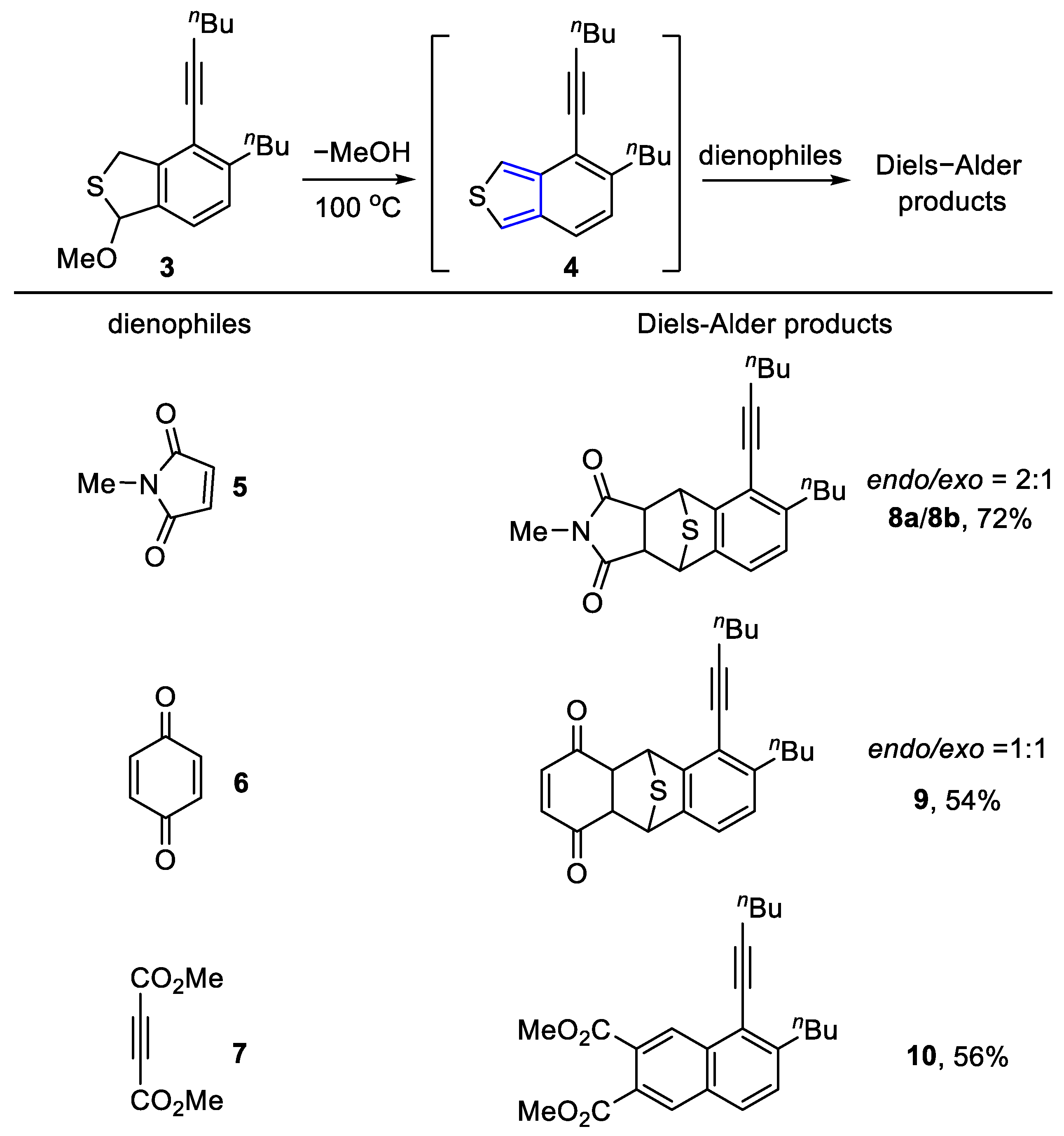
To a stirred solution of 1-bromonona-2,4-diyne (1.2 g, 6.03 mmol) in MeOH (8 mL) was added sodium sulfide nonahydrate (658 mg, 2.74 mmol). The resulting mixture was stirred at room temperature for 10 min before it was diluted by the addition of H2O (5 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (5 × 10 mL). The combined organic layers were washed with brine (20 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (aluminum oxide, hexane) afforded thioether-tethered tetryne 1 (519 mg, 70%) as a reddish brown oil. 1H NMR (400 MHz, CDCl3): δ 3.48 [s, 4H, S(CH2)2)], 2.27 [t, J = 7.4 Hz, 4H, (C≡CCH2)2], 1.57–1.47 [m, 4H, (C≡CCH2CH2)2], 1.45–1.36 [m, 4H, (CH2CH3)2], and 0.91 [t, J = 7.2 Hz, 6H, (CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 80.3 (2C), 71.0 (2C), 68.5 (2C), 64.8 (2C), 30.3 (2C), 22.0 (2C), 20.0 (2C), 19.0 (2C), and 13.6 (2C). HRMS (ESI) m/z: [M + H]+ Calcd for C18H23S+ 271.1515; Found 271.1519. IR (film): 2954, 2255 (C≡C), 1158, 1073, and 856 cm−1.
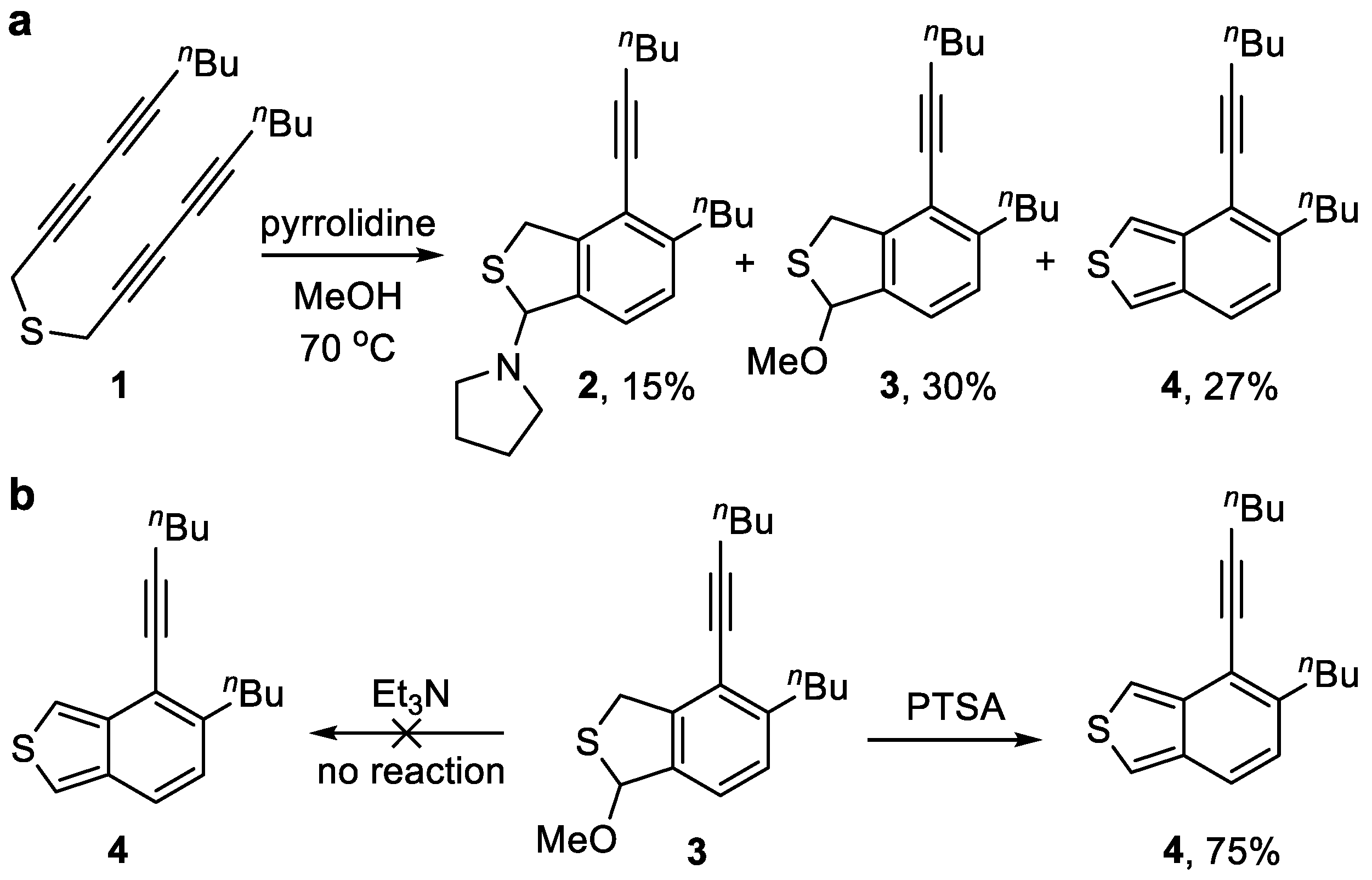
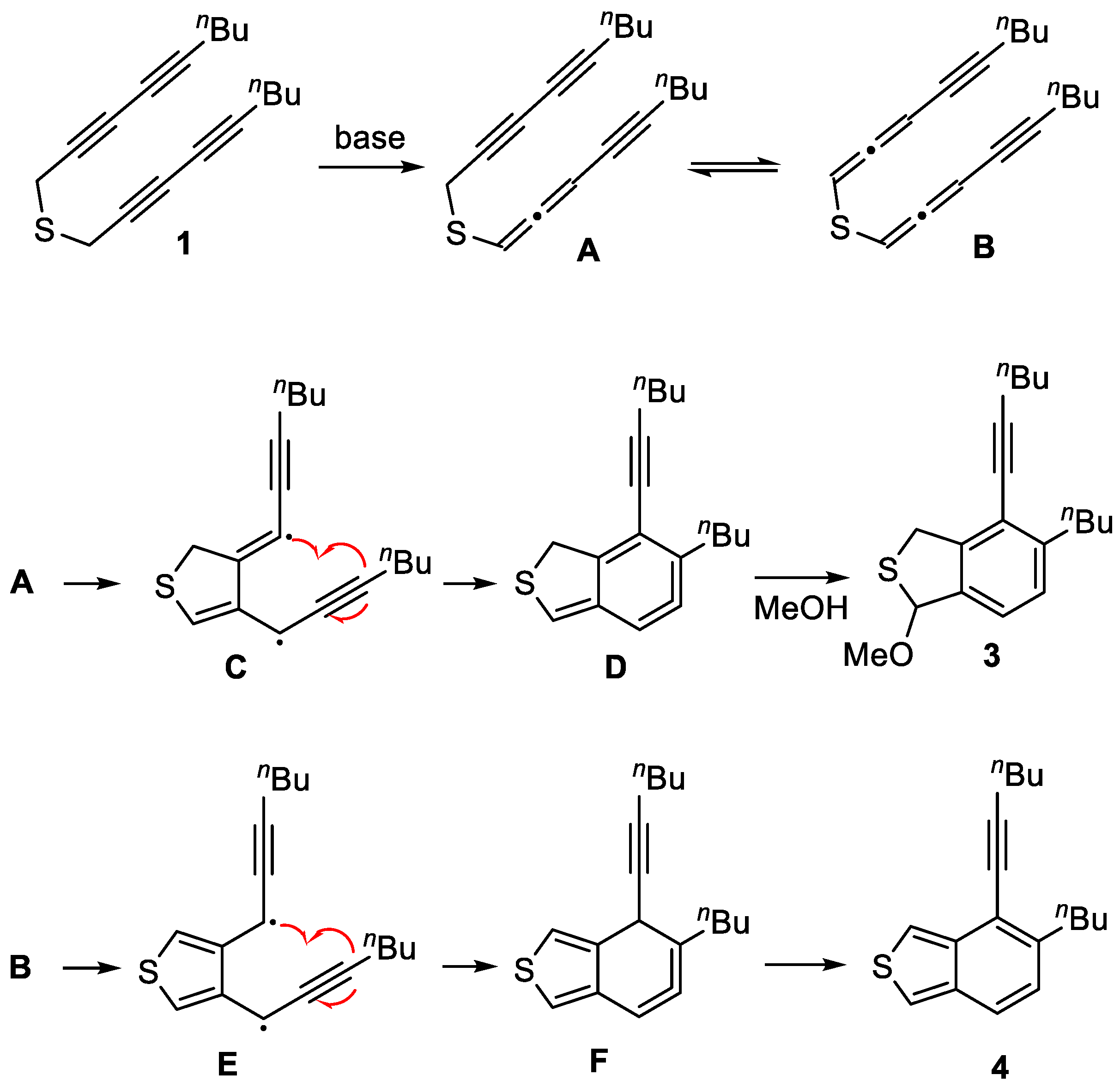
To a stirred solution of thioether 1 (386 mg, 1.43 mmol) in MeOH (5 mL) was added pyrrolidine (1.20 mL, 14.27 mmol). The resulting mixture was stirred at 70 °C for 4 h before it was quenched by the addition of NH4Cl (10 mL, satd aq). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc = 20:1) to afford benzo[c]thiophene 4 (104 mg, 27%) as a yellow oil, followed by the slower eluting 3 (130 mg, 30%) as a brown oil, and the pyrrolidine adduct 2 (74 mg, 15%) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.23 (d, J = 7.8 Hz, 1H, ArH), 7.08 (d, J = 7.9 Hz, 1H, ArH), 6.32 (d, J = 1.9 Hz, 1H, NCH), 4.23–4.12 (m, 2H, SCH2), 2.85–2.70 (m, 2H, ArCH2), 2.70–2.65 (m, H, NCH2), 2.48 (t, J = 6.8 Hz, 2H, C≡CCH2), 2.36–2.26 (m, 2H NCH2), 1.78–1.72 [m, 4H, N(CH2CH2)2], 1.63–1.56 [m, 4H, (CH2CH2CH3)2], 1.54–1.47 (m, 2H, (CH2CH2CH3), 1.40–1.33 (m, 2H, CH2CH2CH3), and 0.95 [q, J = 7.4 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 144.7, 143.2, 138.9, 127.6, 124.6, 120.2, 98.8, 80.9, 77.2, 48.8, 36.9, 34.1, 33.0, 31.1, 23.7, 22.7, 22.1, 19.5, 14.1, and 13.8. HRMS (ESI) m/z: [M + H]+ Calcd for C22H32NS+ 342.2250; Found 342.2247. IR (film): 3449, 2261 (C≡C), 1506 (Ar), 1227, and 1159 cm−1.
1H NMR (400 MHz, CDCl3): δ 7.26 (d, J = 7.7 Hz, 1H, ArH), 7.16 (d, J = 7.5 Hz, 1H, ArH), 6.44 (q, J = 2.3 Hz, 1H, CH3OCH), 4.38 (d, J = 15.1 Hz, 1H, SCHaHb), 4.21 (d, J = 15.6 Hz, 1H, SCHaHb), 3.30 (s, 3H, OCH3), 2.81 (t, J = 7.5 Hz, 2H, ArCH2), 2.52 (td, J = 7.1, 2.6 Hz, 2H, C≡CCH2), 1.65–1.60 [m, 4H, (CH2CH2CH3)2], 1.57–1.51 (m, 2H, CH2CH2CH3), 1.43–1.36 (m, 2H, CH2CH2CH3), and 0.98 [q, J = 7.7 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 145.6, 143.7, 137.5, 128.1, 124.1, 120.4, 99.1, 94.4, 77.0, 53.9, 37.4, 34.1, 33.0, 31.0, 22.7, 22.1, 19.4, 14.1, and 13.7. HRMS (ESI) m/z: [M + H]+ Calcd for C19H27OS+ 303.1777; Found 303.1780. IR (film): 3473, 2090 (C≡C), 1640, 1465 (Ar), and 1186 cm−1.
1H NMR (400 MHz, CDCl3): δ 7.75 (dd, J = 3.4, 1.1 Hz, 1H, SCH=C), 7.61 (d, J = 3.4 Hz, 1H, SCH=C), 7.45 (dd, J = 8.9, 1.1 Hz, 1H, ArH), 6.93 (d, J = 8.9 Hz, 1H, ArH), 2.85–2.81 (m, 2H, ArCH2), 2.58 (t, J = 6.9 Hz, 2H, C≡CCH2), 1.72–1.65 (m, 2H, CH2CH2CH3), 1.61–1.55 [m, 4H, (CH2CH2CH3)2], 1.42–1.37 (m, 2H, CH2CH2CH3), and 1.01–0.93 [m, 6H, (CH2CH2CH3)2].
(3aS,4R,9S,9aR)-6-Butyl-5-(hex-1-yn-1-yl)-2-methyl-3a,4,9,9a-tetrahydro-1H-4,9-epithiobenzo[f]isoindole-1,3(2H)-dione (8a)
To a stirred solution of thioether 3 (120 mg, 0.40 mmol) in chlorobenzene (3 mL) was added N-methylmaleimide 5 (220 mg, 1.98 mmol). The resulting mixture was stirred at 70 °C for 1 h before it was quenched by the addition of NaHCO3 (10 mL, satd aq). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc = 10:1) to afford compound 8a (72 mg, 48%) as a light yellow oil and followed by the slower eluting 8b (36 mg, 24%) as a light yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.00 (d, J = 7.4 Hz, 1H, ArH), 6.85 (d, J = 7.5 Hz, 1H, ArH), 5.11 (t, J = 1.4 Hz, 1H, SHa), 4.82 (t, J = 1.3 Hz, 1H, SHb), 3.28 (dd, J = 6.5, 1.4 Hz, 1H, COCHa), 3.24 (dd, J = 6.6, 1.4 Hz, 1H, COCHb), 3.03 (s, 3H, NCH3), 2.76–2.59 (m, 2H, ArCH2), 2.52 (t, J = 6.9 Hz, 2H, C≡CCH2), 1.68–1.60 (m, 2H, CH2CH2CH3), 1.59–1.49 [m, 4H, (CH2CH2CH3)2], 1.41–1.30 (m, 2H, CH2CH2CH3), and 0.95 [dt, J = 23.1, 7.3 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 176.2, 176.0, 148.2, 143.5, 143.3, 126.6, 118.8, 116.7, 98.4, 75.8, 55.9, 55.1, 51.7, 51.0, 34.3, 32.9, 30.9, 25.1, 22.7, 22.1, 19.5, 14.0, and 13.7. HRMS (ESI) m/z: [M + H]+ Calcd for C23H28NO2S+ 382.1835; Found 382.1836. IR (film): 3450, 1705 (C=O), 1643, 1456 (Ar), and 1109 cm−1.
(3aR,4R,9S,9aS)-6-Butyl-5-(hex-1-yn-1-yl)-2-methyl-3a,4,9,9a-tetrahydro-1H-4,9-epithiobenzo[f]isoindole-1,3(2H)-dione (8b)
1H NMR (400 MHz, CDCl3): δ 6.93 (d, J = 7.5 Hz, 1H, ArH), 6.84 (d, J = 7.6 Hz, 1H, ArH), 5.16 (dd, J = 4.2, 1.4 Hz, 1H, SHa), 4.82 (dd, J = 4.1, 1.4 Hz, 1H, SHb), 4.09–3.95 [m, 2H, (COCH)2], 2.70–2.60 (m, 2H, ArCH2), 2.52 (t, J = 7.0 Hz, 2H, C≡CCH2), 2.37 (s, 3H, NCH3), 1.70–1.61 (m, 2H, CH2CH2CH3), 1.57–1.46 [m, 4H (CH2CH2CH3)2], 1.34–1.24 (m, 2H, CH2CH2CH3), and 0.93 [dt, J = 32.6, 7.4 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 174.8, 173.7, 144.9, 143.9, 139.8, 127.2, 120.1, 117.6, 98.1, 75.8, 55.3, 54.6, 53.2, 52.9, 34.2, 33.0, 31.1, 24.1, 22.5, 22.2, 19.6, 14.0, and 13.8. HRMS (ESI) m/z: [M + H]+ Calcd for C23H28NO2S+ 382.1835; Found 382.1834. IR (film): 3461, 2114 (C≡C), 1706 (C=O), 1644, and 1113 cm−1.
6-Butyl-5-(hex-1-yn-1-yl)-4a,9,9a,10-tetrahydro-9,10-epithioanthracene-1,4-dione (9)
To a stirred solution of thioether 3 (128 mg, 0.42 mmol) in chlorobenzene (3 mL) was added 1,4-benzoquinone 6 (137 mg, 1.27 mmol). The resulting mixture was stirred at 100 °C for 5 h before it was quenched by the addition of NaHCO3 (10 mL, satd aq). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc = 10:1) afforded 9 (86 mg, endo and exo 1:1, 54%) as a bright yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.05 (d, J = 7.6 Hz, 1H, COCH=CH), 6.86 (m, 2H, ArH), 6.85 (d, J = 7.8 Hz, 1H, COCH=CH), 6.79 (d, J = 7.5 Hz, 2H, ArH), 6.07 [q, J = 9.7 Hz, 2H, (COCH=CH)2], 5.32–5.30 (m, 1H, SH), 5.29 (dd, J = 3.6, 1.6 Hz, 1H, SH), 5.01–4.99 (m, 1H, SH), 4.95 (dd, J = 3.6, 1.6 Hz, 1H, SH), 3.82 (dd, J = 7.5, 3.6 Hz, 2H, COCH), 3.25 (dd, J = 4.4, 1.0 Hz, 2H, COCH), 2.75–2.58 [m, 4H, (ArCH2)2], 2.57–2.48 [m, 4H, (C≡CCH2)2], 1.69–1.64 [m, 4H, (CH2CH2CH3)2], 1.59–1.52 [m, 4H, (CH2CH2CH3)2], 1.49–1.44 [m, 4H, (CH2CH2CH3)2], 1.38–1.24 [m, 4H, (CH2CH2CH3)2], 0.99 [td, J = 7.3, 3.7 Hz, 6H, CH2CH2CH3)2], and 0.91 [dt, J = 15.9, 7.2 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 197.4, 196.4, 195.9, 195.5, 148.5, 145.9, 143.8, 143.7, 143.1, 142.6, 142.2, 140.9, 139.6, 139.0, 127.0, 126.5, 120.0, 118.5, 117.5, 116.3, 98.4, 98.3, 75.9, 75.5, 58.9, 58.7, 58.0, 57.9, 54.0, 53.4, 53.2, 53.0, 34.3, 34.2, 33.03, 32.97,31.00, 30.97, 22.7, 22.5, 22.20, 22.16, 19.50, 19.45, 14.1, 14.0, and 13.78. HRMS (ESI) m/z: [M + H]+ Calcd for C24H27O2S+ 379.1726; Found 379.1728. IR (film): 3446, 2089 (C≡C), 1672 (C=O), 1457 (Ar), and 1054 cm−1.
To a stirred solution of thioether 3 (135 mg, 0.45 mmol) in chlorobenzene (3 mL) was added dimethyl acetylenedicarboxylate 7 (1.10 mL, 8.93 mmol). The resulting mixture was stirred at 100 °C for 2 h before it was quenched by the addition of NaHCO3 (10 mL, satd aq). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc = 10:1) afforded 10 (95 mg, 56%) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ 8.69 (s, 1H, ArH), 8.21 (s, 1H, ArH), 7.75 (d, J = 8.4 Hz, 1H, ArH), 7.46 (d, J = 8.5 Hz, 1H, ArH), 3.97 (s, 3H, CO2CH3), 3.95 (s, 3H, CO2CH3), 3.01–2.93 (m, 2H, ArCH2), 2.63 (t, J = 6.9 Hz, 2H, C≡CCH2), 1.75–1.65 [m, 4H, (CH2CH2CH3)2], 1.62–1.56 (m, 2H, CH2CH2CH3), 1.44–1.37 (m, 2H, CH2CH2CH3), and 0.98 [dt, J = 18.0, 7.4 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 168.8, 168.2, 146.5, 134.3, 131.8, 130.5, 130.1, 129.2, 128.6, 127.8, 127.7, 121.3, 101.1, 76.4, 52.8 (2C), 35.2, 32.9, 31.0, 22.8, 22.2, 19.7, 14.1, and 13.8. HRMS (ESI) m/z: [M + H]+ Calcd for C24H29O4+ 381.2060; Found 381.2063. IR (film): 3443, 2080 (C≡C), 1742 (C=O), 1464 (Ar), and 1122 cm−1.
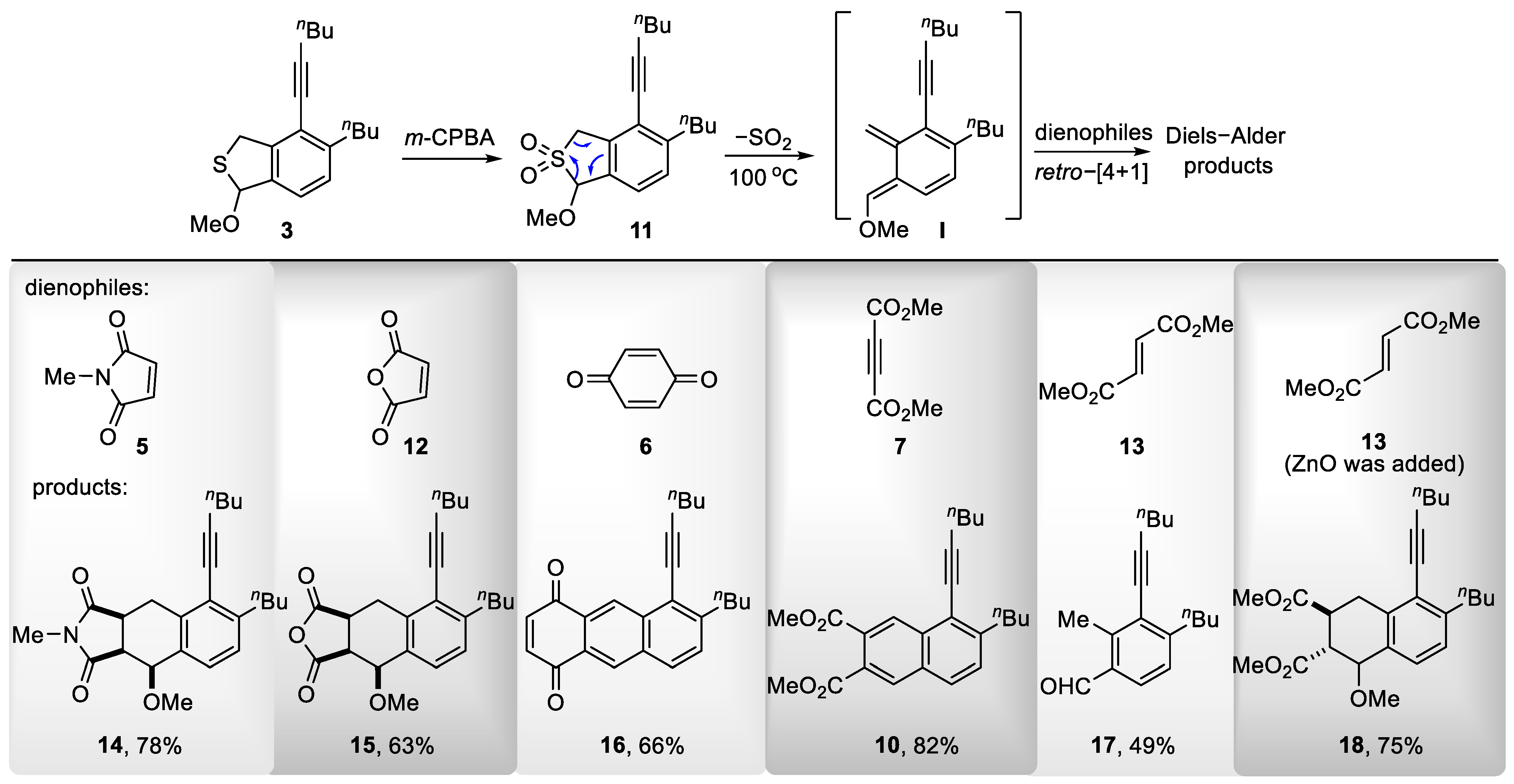
To a stirred solution of thioether 3 (500 mg, 1.65 mmol) in CH2Cl2 (8 mL) at 0 °C was added m-CPBA (761 mg, 3.31 mmol). The resulting mixture was stirred at room temperature for 30 min before it was quenched by the addition of NaHCO3 (10 mL, satd aq). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc = 10:1) afforded sulfone 11 (425 mg, 77%) as a light yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.28 (d, J = 8.0 Hz, 1H, ArH), 7.23 (d, J = 8.0 Hz, 1H, ArH), 5.26 (s, 1H, CH3OCH), 4.40 (d, J = 16.3 Hz, 1H, SO2CH), 4.33 (d, J = 16.3 Hz, 1H, SO2CH), 3.82 (s, 3H, OCH3), 2.80–2.73 (m, 2H, ArCH2), 2.47 (t, J = 6.9 Hz, 2H, C≡CCH2), 1.61–1.56 [m, 4H, (CH2CH2CH3)2], 1.51–1.46 (m, 2H, CH2CH2CH3), 1.38–1.32 (m, 2H, CH2CH2CH3), and 0.94 [dt, J = 11.0, 7.3 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 147.7, 133.5, 130.1, 129.5, 126.0, 121.8, 100.6, 96.3, 76.2, 59.9, 55.1, 34.6, 32.7, 30.9, 22.6, 22.1, 19.4, 14.0, and 13.7. HRMS (ESI) m/z: [M + H]+ Calcd for C19H27O3S+ 335.1675; Found 335.1675. IR (film): 3293, 2090 (C≡C), 1640, 1317, and 1208 cm−1.
7-Butyl-8-(hex-1-yn-1-yl)-4-methoxy-2-methyl-3a,4,9,9a-tetrahydro-1H-cyclopenta[b]naphthalene-1,3(2H)-dione (14)
To a stirred solution of sulfone 11 (150 mg, 0.45 mmol) in chlorobenzene (4 mL) was added N-methylmaleimide 5 (150 mg, 1.35 mmol). The resulting mixture was stirred at 100 °C for 6 h before it was quenched by the addition of NaHCO3 (10 mL, satd aq). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc = 5:1) afforded 14 (132 mg, 78%) as a light yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.05 (d, J = 2.7 Hz, 2H, ArH), 4.66 (d, J = 3.8 Hz, 1H, CH3OCH), 3.81 (dd, J = 14.9, 8.0 Hz, 1H, COCHa), 3.10 (m, 1H, ArHaHb), 3.05 (s, 3H, OCH3), 3.03 (m, 1H, COCHb), 2.99 (s, 3H, CO2CH3), 2.93 (dd, J = 14.9, 10.1 Hz, 1H, ArHaHb), 2.77 (td, J = 7.5, 3.8 Hz, 2H, ArCH2), 2.52 (t, J = 7.0 Hz, 2H, C≡CCH2), 1.67–1.58 [m, 4H, (CH2CH2CH3)2], 1.56–1.49 (m, 2H, CH2CH2CH3), 1.44–1.33 [m, 2H, CH2CH2CH3), and 0.96 [dt, J = 10.7, 7.3 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 180.2, 177.2, 146.3, 138.5, 132.2, 127.5, 126.3, 123.4, 99.5, 77.8, 76.7, 56.0, 46.7, 38.3, 35.0, 32.9, 31.1, 25.5, 25.0, 22.9, 22.2, 19.6, 14.1, and 13.8. HRMS (ESI) m/z: [M + H]+ Calcd for C24H32NO3+ 382.2377; Found 382.2377. IR (neat): 3447 2115 (C≡C), 1704 (C=O), 1644, and 1434 (Ar) cm−1.
7-Butyl-8-(hex-1-yn-1-yl)-4-methoxy-3a,4,9,9a-tetrahydronaphtho [2,3-c]furan-1,3-dione (15)
To a stirred solution of sulfone 11 (140 mg, 0.42 mmol) in chlorobenzene (3 mL) was added maleic anhydride 12 (205 mg, 2.09 mmol). The resulting mixture was stirred at 100 °C for 5 h before it was quenched by the addition of NaHCO3 (10 mL, satd aq). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc = 10:1) afforded 15 (95 mg, 63%) as a bright yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.07 (s, 2H, ArH), 4.67 (d, J = 3.6 Hz, 1H, CH3OCH), 3.83 (dd, J = 15.5, 9.3 Hz, 1H, COCHa), 3.44 (dt, J = 10.7, 9.1 Hz, 1H, ArHaHb)), 3.24 (dd, J = 10.7, 3.7 Hz, 1H, ArHaHb)), 3.11 (dd, J = 15.9, 7.0 Hz, 1H, ArHaHb)), 3.05 (s, 3H, OCH3), 2.78 (td, J = 7.7, 5.5 Hz, 2H, ArCH2), 2.52 (t, J = 7.0 Hz, 2H, C≡CCH2), 1.67–1.57 [m, 4H, (CH2CH2CH3)2], 1.55–1.49 (m, 2H, CH2CH2CH3), 1.43–1.35 (m, 2H, CH2CH2CH3), and 0.97 [dt, J = 10.9, 7.3 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 174.2, 171.3, 146.9, 137.1, 130.8, 127.7, 126.6, 123.6, 100.1, 77.5, 76.5, 56.1, 47.6, 38.1, 35.0, 32.8, 31.0, 25.0, 22.8, 22.2, 19.5, 14.1, and 13.8. HRMS (ESI) m/z: [M + H]+ Calcd for C23H29O4+ 369.2060; Found 369.2059. IR (film): 3443, 2079 (C≡C), 1641 (C=O), 1454 (Ar), and 1016 cm−1.
To a stirred solution of sulfone 11 (145 mg, 0.43 mmol) in chlorobenzene (3 mL) was added 1,4-benzoquinone 6 (141 mg, 1.30 mmol). The resulting mixture was stirred at 100 °C for 6 h before it was quenched by the addition of NaHCO3 (10 mL, satd aq). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc = 10:1) afforded 16 (100 mg, 66%) as a bright yellow oil. 1H NMR (400 MHz, CDCl3): δ 9.06 (s, 1H, COCH=CH), 8.51 (s, 1H, COCH=CH), 7.85 (d, J = 8.4 Hz, 1H, ArH), 7.51 (d, J = 8.4 Hz, 1H, ArH), 7.04 (q, J = 1.7 Hz, 2H, ArH), 3.03–2.91 (m, 2H, ArCH2), 2.67 (t, J = 6.9 Hz, 2H, C≡CCH2), 1.77–1.66 [m, 4H, (CH2CH2CH3)2], 1.64–1.58 (m, 2H, CH2CH2CH3), 1.47–1.37 (m, 2H, CH2CH2CH3), and 1.00 [dt, J = 24.9, 7.7 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 184.9 (2C), 147.6, 140.3, 140.0, 135.6, 133.3, 131.1, 129.2, 128.9, 128.7, 128.0, 127.6, 123.0, 102.0, 76.2, 35.3, 32.8, 31.0, 22.8, 22.3, 19.8, 14.1, and 13.8. HRMS (ESI) m/z: [M + H]+ Calcd for C24H25O2+ 345.1849; Found 345.1845. IR (film): 2957, 2089 (C≡C), 1667 (C=O), 1457 (Ar), and 1056 cm−1.
To a stirred solution of sulfone 11 (115 mg, 0.34 mmol) in chlorobenzene (3 mL) was added dimethyl fumarate 13 (145 mg, 1.0 mmol). The resulting mixture was stirred at 100 °C for 4 h before it was quenched by the addition of NaHCO3 (10 mL, satd aq). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc = 20:1) afforded 17 (80 mg, 49%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 10.25 (s, 1H, CHO), 7.64 (d, J = 7.9 Hz, 1H, ArH), 7.17 (d, J = 7.9 Hz, 1H, ArH), 2.86–2.80 (m, 2H, ArCH2), 2.78 (s, 3H, ArCH3), 2.53 (t, J = 6.9 Hz, 2H, C≡CCH2), 1.69–1.59 [m, 4H, (CH2CH2CH3)2], 1.55–1.48 (m, 2H, CH2CH2CH3), 1.43–1.36 (m, 2H, CH2CH2CH3), and 0.96 [q, J = 7.6 Hz, 6H, (CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 192.4, 151.5, 143.0, 132.2, 130.2, 126.5, 125.7, 100.3, 77.0, 35.5, 32.4, 31.0, 22.8, 22.2, 19.5, 16.9, 14.1, and 13.8. HRMS (ESI) m/z: [M + H]+ Calcd for C18H25O+ 257.1900; Found 257.1903. IR (film): 2827, 2098 (C≡C), 1715 (C=O), 1455 (Ar), and 1054 cm−1.
Dimethyl (2R,3S)-6-butyl-5-(hex-1-yn-1-yl)-1-methoxy-1,2,3,4-tetrahydronaphthalene-2,3-dicarboxylate (18)
To a stirred solution of sulfone 11 (85 mg, 0.25 mmol) in chlorobenzene (2 mL) were added dimethyl fumarate 13 (108 mg, 0.75 mmol) and ZnO (61 mg, 0.75 mmol). The resulting mixture was stirred at 100 °C for 6 h before it was quenched by the addition of NaHCO3 (10 mL, satd aq). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4), and concentrated in vacuo. Flash column chromatography (silica gel, hexanes:EtOAc = 10:1) afforded 18 (80 mg, 75%) as a light yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.22 (d, J = 8.0 Hz, 0.35H, ArH, minor), 7.07 (t, J = 7.7 Hz, 1.25H, ArH, major), 7.04 (d, J = 8.0 Hz, 0.35H, ArH, minor), 4.78 (d, J = 8.0 Hz, 0.35H, CH3OCH, minor), 4.56 (t, J = 2.6 Hz, 0.63H, CH3OCH, major), 3.77 (s, 3.8H, CO2CH3, major), 3.75 (s, 1H, CO2CH3, minor), 3,73 (s, 1H, CO2CH3, minor), 3.51–3.33 (m, 2H, ArCH2CH), 3.3 (s, 1H, ArCH2CH minor), 3.26 (s, 1.9H, ArCH2CH, major), 3.18–2.76 (m, 2H, ArCH2CH), 2.74 (m, 2H, ArCH2), 2.48 (m, 2H, C≡CCH2), 11.66–1.12 [m, 8H, (CH2CH2CH3)2], and 0.94 [m, 6H, CH2CH2CH3)2]. 13C NMR (101 MHz, CDCl3): δ 176.4, 174.05, 174.03 172.7, 145.9, 145.0, 136.7, 136.5, 132.2, 130.5, 128.8, 126.9, 126.3, 126.0, 123.5, 122.8, 100.6, 99.9, 79.0, 77.7, 57.0, 55.2, 52.4, 52.3, 52.2, 47.3, 46.4, 41.4, 36.9, 34.7, 34.6, 32.8, 32.8, 31.4, 31.1, 31.0, 30.0, 29.8, 22.8, 22.09, 22.07, 19.5, 14.1, 13.75, and 13.73. HRMS (ESI) m/z: [M + H]+ Calcd for C25H35O5+ 415.2479; Found 415.2475. IR (film): 2850, 2085 (C≡C), 1732 (C=O), 1483 (Ar), and 1030 cm−1.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}