
Anticancer Activity and Safety Profile of Novel 1-(4-Fluorophenoxyacetyl)-4-substituted Thio/Semicarbazide Derivatives

 , , , , and
, , , , and
Abstract
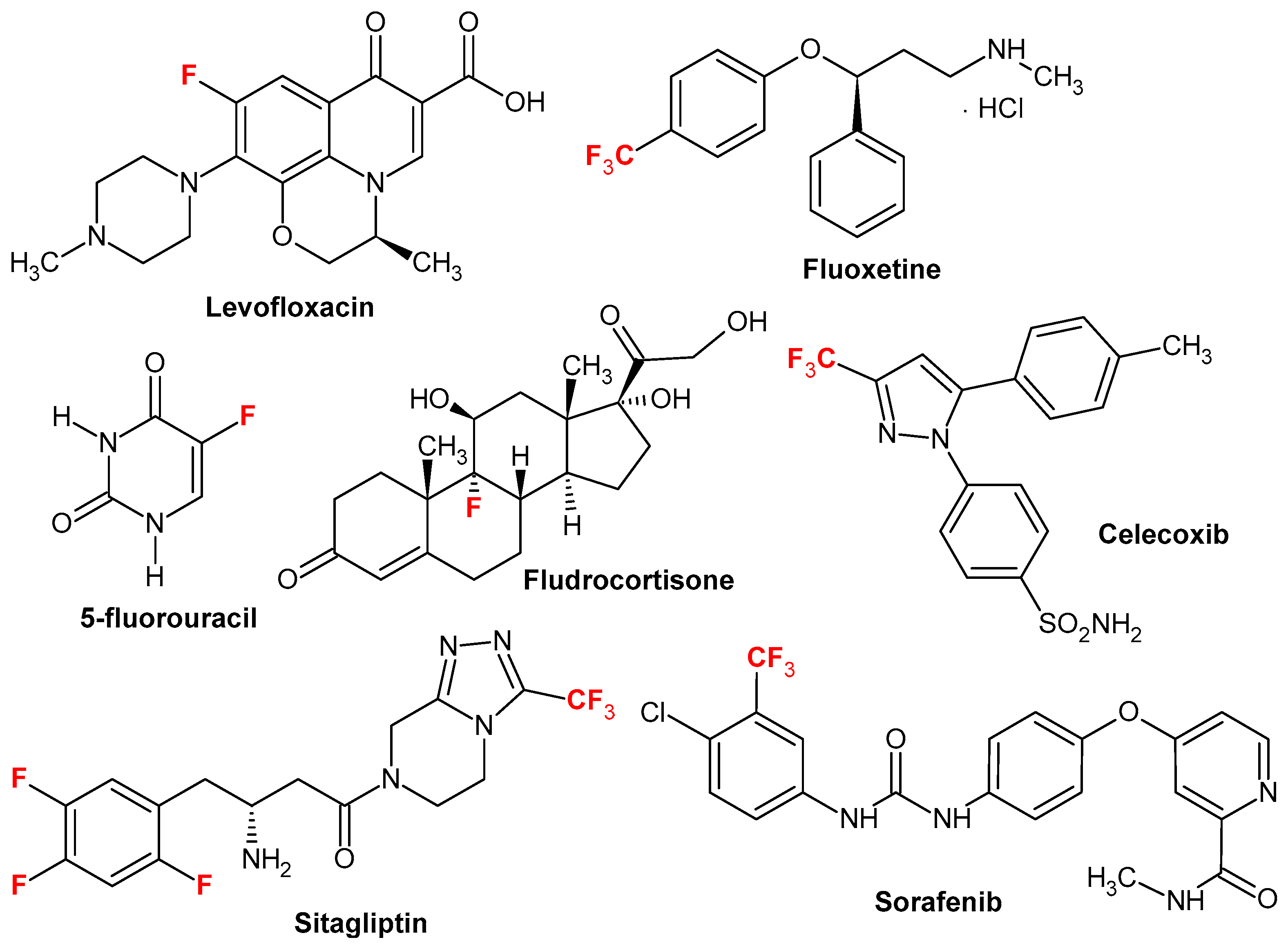
1. Introduction
2. Results and Discussion
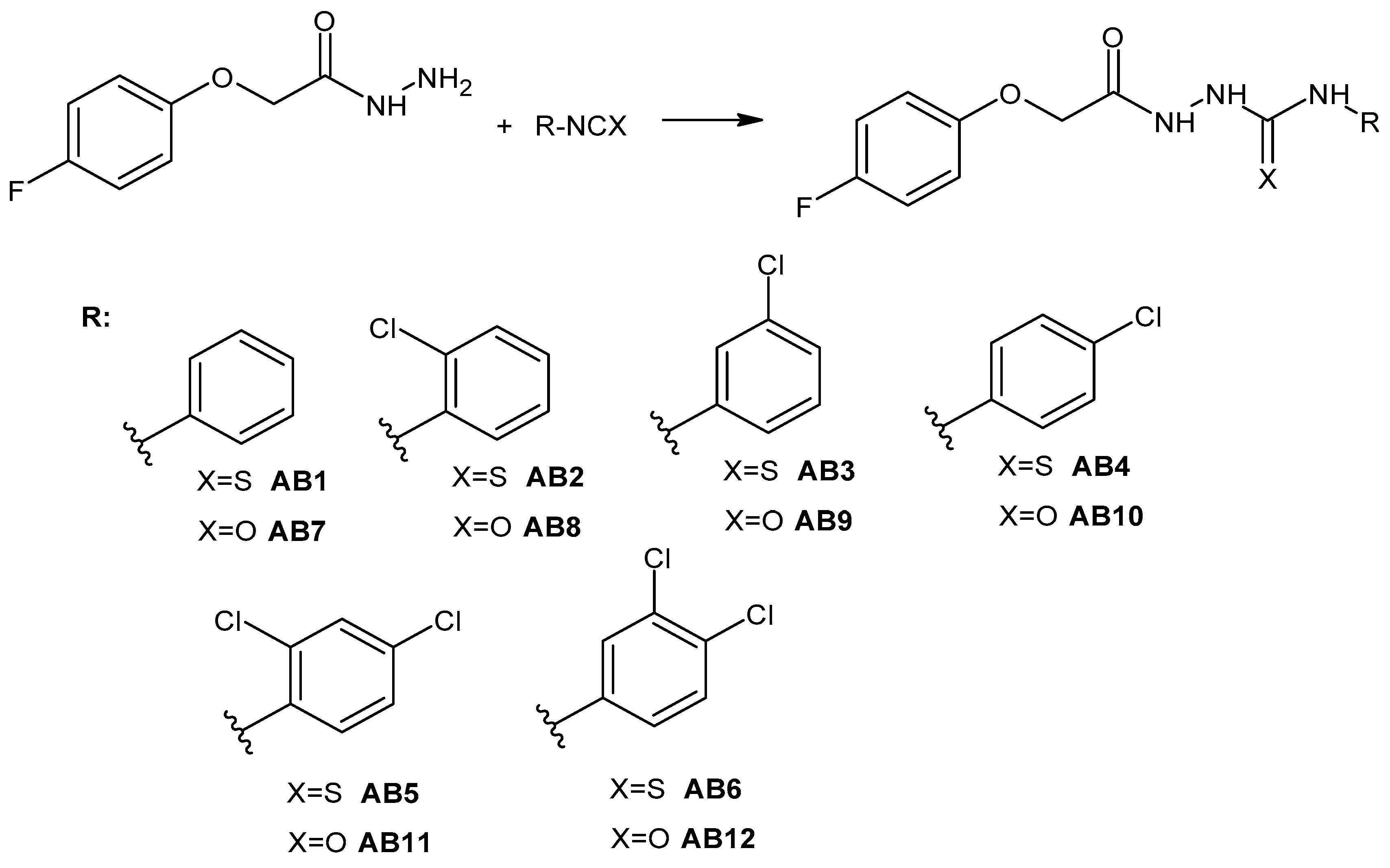
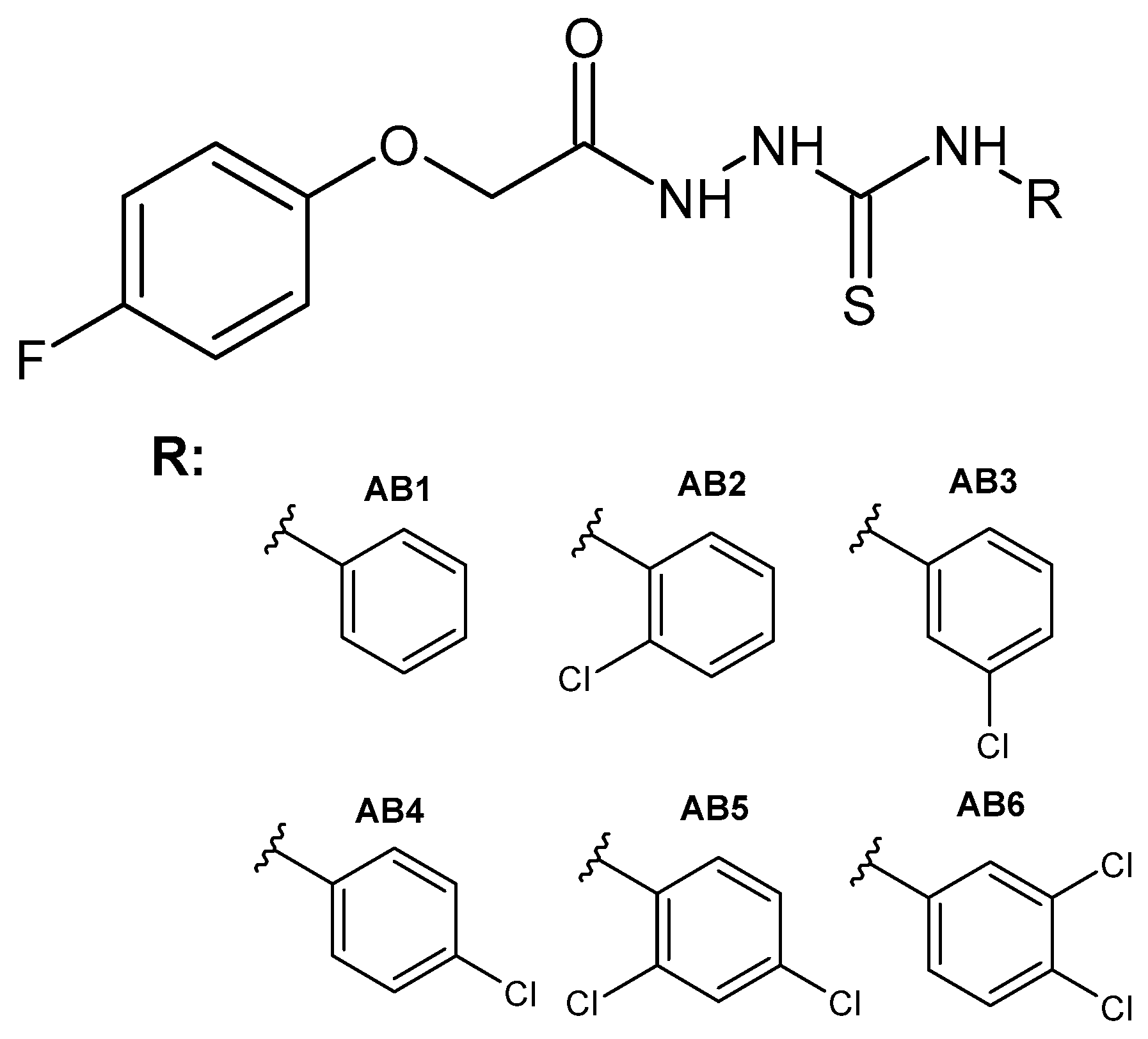
2.1. Chemistry
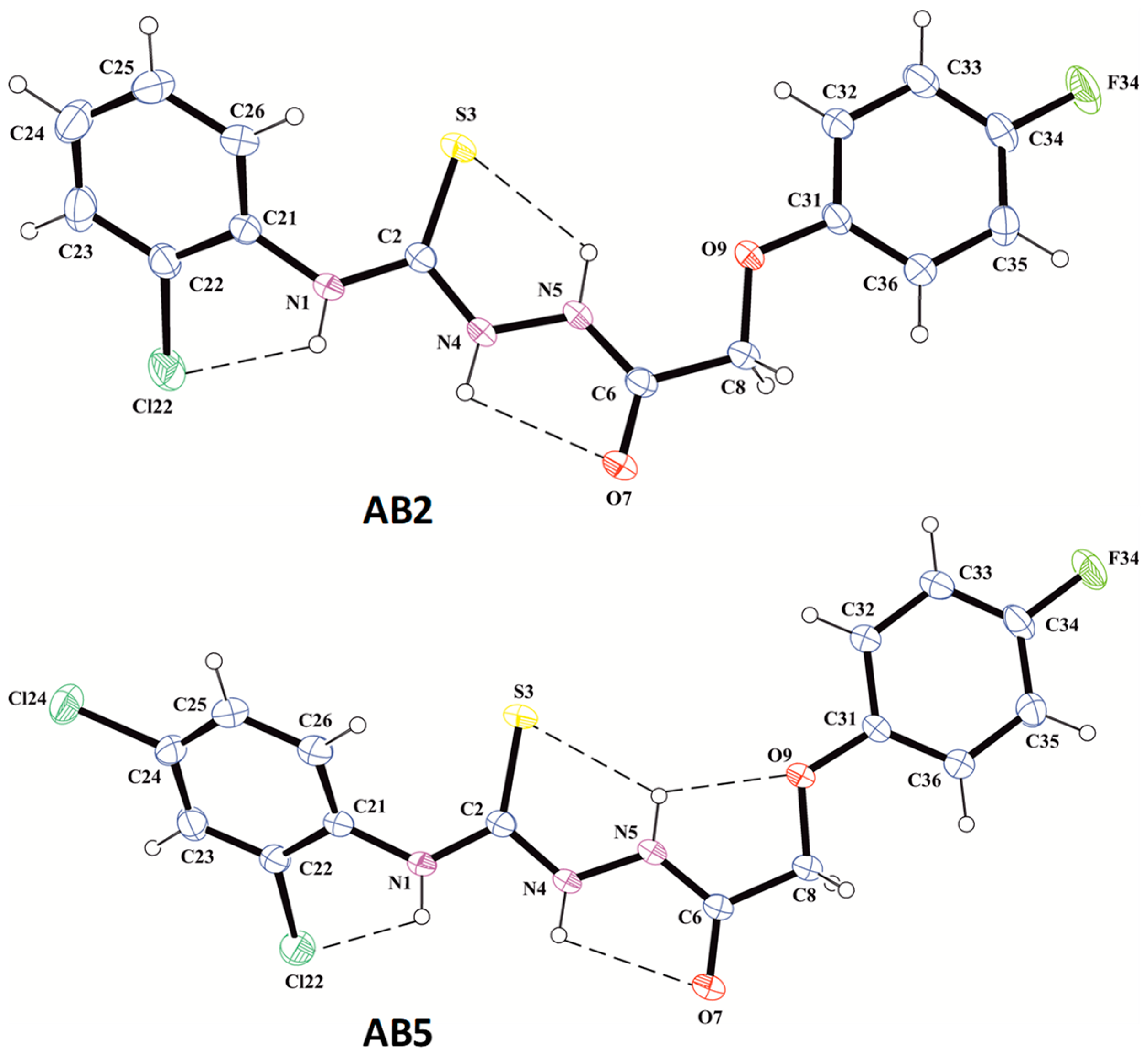
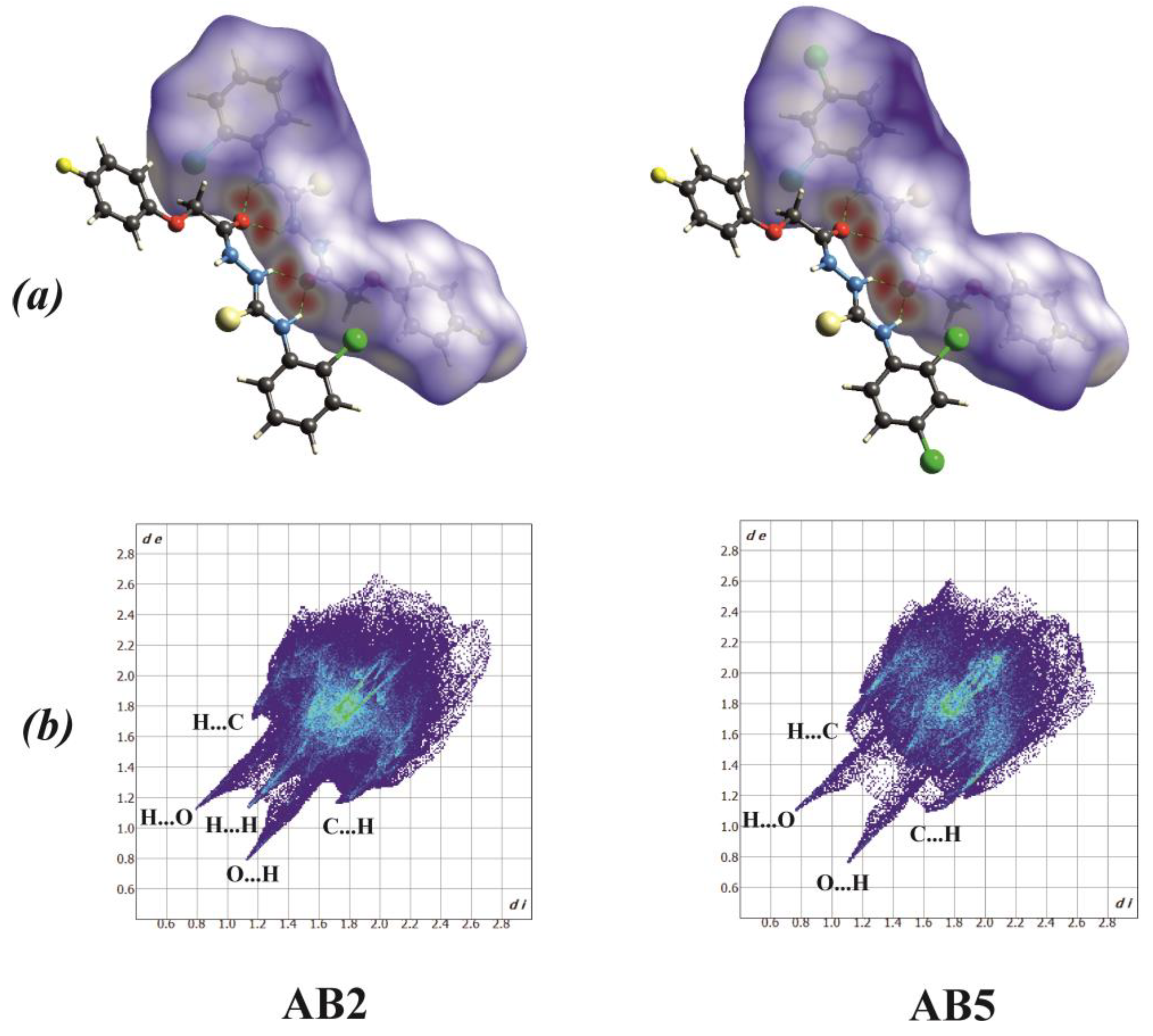
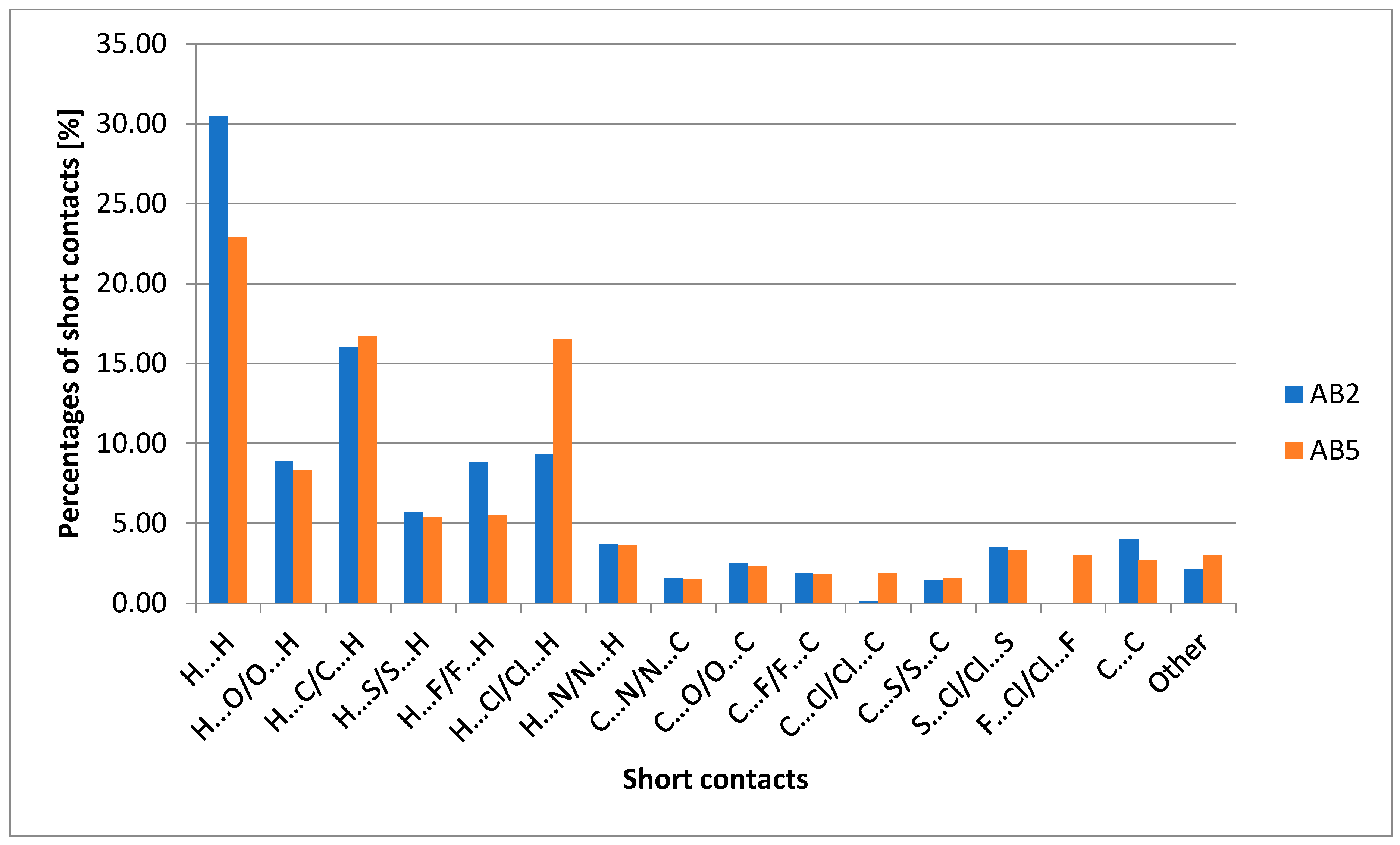
2.2. X-Ray Structure Determination
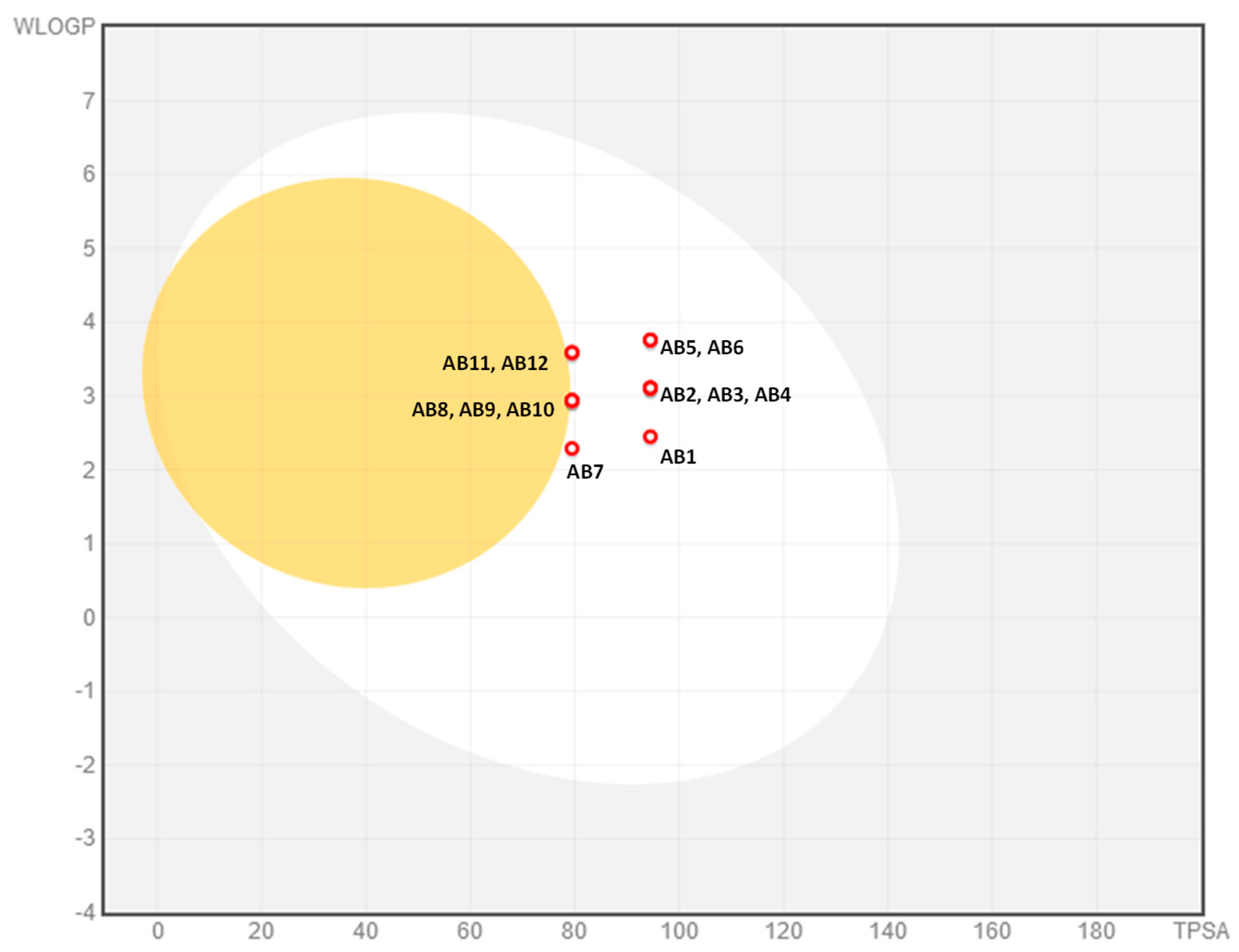
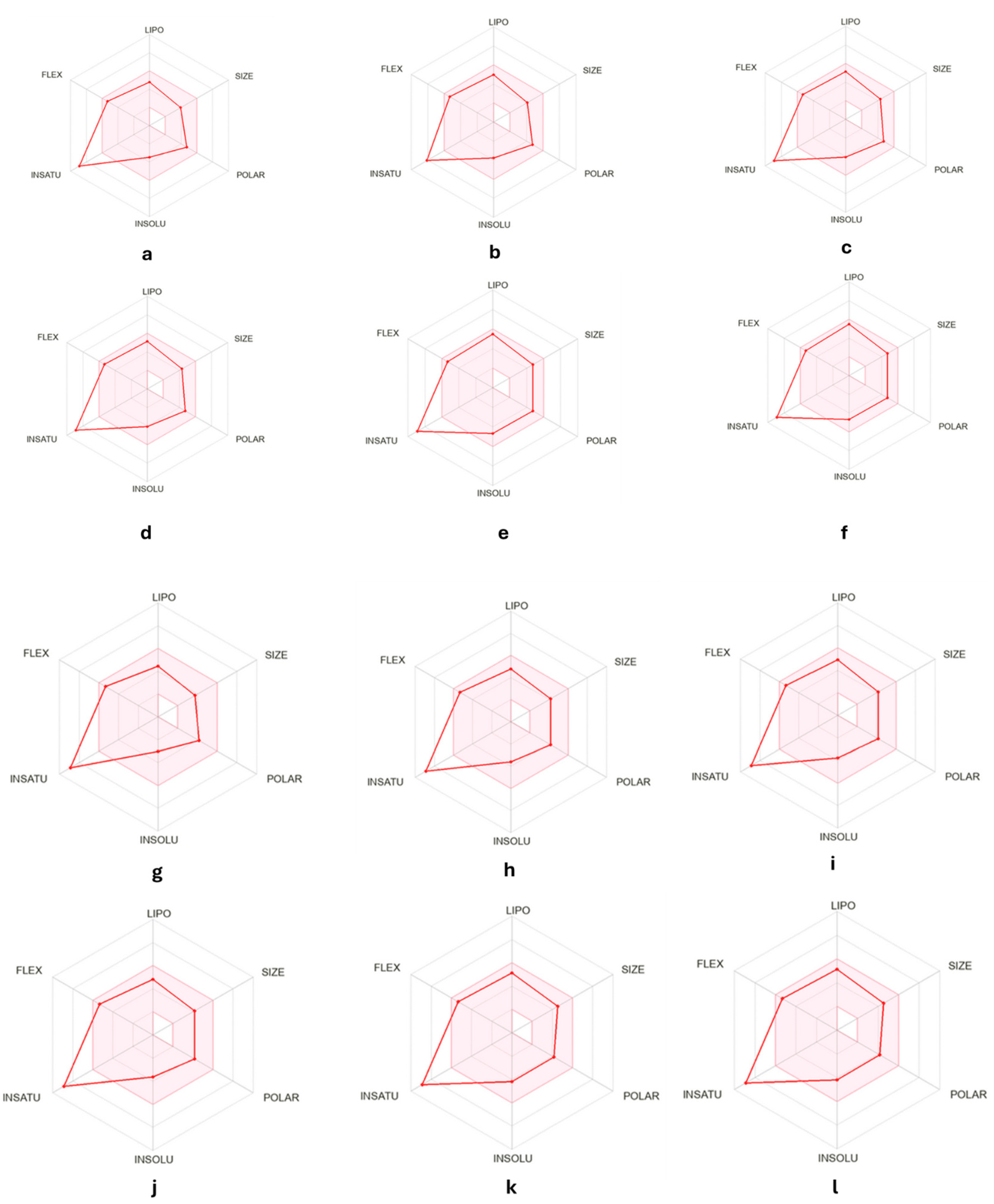
2.3. ADMET Analysis
2.4. Biological Activity
2.4.1. Cytotoxicity Assessment
2.4.2. Cell Cycle Analysis and Apoptosis Detection
2.4.3. Analysis of Gene Expression Related to Antioxidant Defense, Cell Cycle Regulation, DNA Damage Sensing and Repair and Apoptosis
2.5. Molecular Docking
3. Experimental
3.1. Chemical Reagents and General Method
3.2. General Procedure for the Synthesis of 1-(4-Fluorophenoxyacetyl)-4-substituted Thiosemicarbazide (AB1–AB6)
3.3. General Procedure for the Synthesis of 1-(4-Fluorophenoxyacetyl)-4-substituted Semicarbazide (AB7–AB12)
3.4. X-Ray Structure Determinations
3.5. ADMET Analysis
3.6. Biological Evaluation
3.6.1. Cell Culture and Treatment
3.6.2. MTT Assay
3.6.3. Cell Cycle Analysis
3.6.4. Apoptosis Detection
3.6.5. Quantitative Real-Time PCR Analysis (qRT-PCR)
3.6.6. Statistical Analysis
3.7. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patel, J.J.; Modh, R.P.; Asamdi, M.; Chikhalia, K.H. Comparative Biological Study between Quinazolinyl–Triazinyl Semicarbazide and Thiosemicarbazide Hybrid Derivatives. Mol. Divers. 2021, 25, 2271–2287. [Google Scholar] [CrossRef] [PubMed]
- Zarrinzadeh, G.; Tajbakhsh, M.; Hosseinzadeh, R.; Khalilzadeh, M.A.; Hosseinzadeh, M. Biological Evaluation and Molecular Docking Study of Euparin and Its Maleic Anhydride and Semicarbazide Derivatives. Polycycl. Aromat. Compd. 2023, 43, 409–420. [Google Scholar] [CrossRef]
- Çelik, B.; Buran Uğur, S.; Baran, M.; Gündüz, M.G.; Keskin, S.; Önder, G.Ö.; Bitgen, N.; Kaya, S.; Doğan, Ş.D. Semicarbazides Carrying Indole Core: Synthesis, Cytotoxicity Evaluation against Human Breast Cancer Cell Lines, and Molecular Modeling Studies. Chem. Biodivers. 2023, 20, e202300609. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Ling, Y.; Li, B.; Li, Y.; Rui, C.; Cui, J.; Shi, Y.; Yang, X. Synthesis and Bioactivity of N-Benzoyl-N’-[5-(2’-Substituted Phenyl)-2-Furoyl] Semicarbazide Derivatives. Molecules 2010, 15, 4267–4282. [Google Scholar] [CrossRef]
- Pavić, K.; Perković, I.; Cindrić, M.; Pranjić, M.; Martin-Kleiner, I.; Kralj, M.; Schols, D.; Hadjipavlou-Litina, D.; Katsori, A.-M.; Zorc, B. Novel Semicarbazides and Ureas of Primaquine with Bulky Aryl or Hydroxyalkyl Substituents: Synthesis, Cytostatic and Antioxidative Activity. Eur. J. Med. Chem. 2014, 86, 502–514. [Google Scholar] [CrossRef]
- Perković, I.; Tršinar, S.; Žanetić, J.; Kralj, M.; Martin-Kleiner, I.; Balzarini, J.; Hadjipavlou-Litina, D.; Katsori, A.M.; Zorc, B. Novel 1-Acyl-4-Substituted Semicarbazide Derivatives of Primaquine-Synthesis, Cytostatic, Antiviral and Antioxidative Studies. J. Enzym. Inhib. Med. Chem. 2013, 28, 601–610. [Google Scholar] [CrossRef]
- Perković, I.; Butula, I.; Kralj, M.; Martin-Kleiner, I.; Balzarini, J.; Hadjipavlou-Litina, D.; Katsori, A.-M.; Zorc, B. Novel NSAID 1-Acyl-4-Cycloalkyl/Arylsemicarbazides and 1-Acyl-5-Benzyloxy/Hydroxy Carbamoylcarbazides as Potential Anticancer Agents and Antioxidants. Eur. J. Med. Chem. 2012, 51, 227–238. [Google Scholar] [CrossRef]
- Szopa, A.; Herbet, M.; Pachuta-Stec, A.; Lachowicz, J.; Pawłowski, K.; Iwan, M.; Jarecka-Florek, D.; Krasińska, O.; Serefko, A.; Poleszak, E.; et al. Evaluation of Developmental Toxicity in Zebrafish Embryos and Antiproliferative Potential against Human Tumor Cell Lines of New Derivatives Containing 4-Nitrophenyl Group. Toxicol. Appl. Pharmacol. 2023, 458, 116325. [Google Scholar] [CrossRef]
- Rane, R.A.; Naphade, S.S.; Bangalore, P.K.; Palkar, M.B.; Shaikh, M.S.; Karpoormath, R. Synthesis of Novel 4-Nitropyrrole-Based Semicarbazide and Thiosemicarbazide Hybrids with Antimicrobial and Anti-Tubercular Activity. Bioorg. Med. Chem. Lett. 2014, 24, 3079–3083. [Google Scholar] [CrossRef]
- Song, M.; Wang, S.; Wang, Z.; Fu, Z.; Zhou, S.; Cheng, H.; Liang, Z.; Deng, X. Synthesis, Antimicrobial and Cytotoxic Activities, and Molecular Docking Studies of N-Arylsulfonylindoles Containing an Aminoguanidine, a Semicarbazide, and a Thiosemicarbazide Moiety. Eur. J. Med. Chem. 2019, 166, 108–118. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Liang, W.-L.; Lee, C.-C.; Tsai, Y.-F.; Hou, W.-C. Antioxidant and Semicarbazide-Sensitive Amine Oxidase Inhibitory Activities of Glucuronic Acid Hydroxamate. Food Chem. 2011, 129, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Manaf, A.; Khan, M.; Zaman, K.; Ali, M.; Alam, F.; Ali, K.M.K.; Ali, B. Synthesis, Characterization and Antioxidant Activities of Semicarbazide and Thiosemicarbazide Derivatives. JCSP 2021, 43, 475. [Google Scholar]
- Sazeli, S.; Nath, A.R.; Ahmad, M.H.; Zulkifli, N.W.M.; Johan, M.R.; Yehye, W.A.; Voon, L.H. Semicarbazide and Thiosemicarbazide Containing Butylated Hydroxytoluene Moiety: New Potential Antioxidant Additives for Synthetic Lubricating Oil. RSC Adv. 2021, 11, 7138–7145. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Alkskas, I.A.; Khokra, S.L.; Prakash, O. Synthesis of Some Novel N4-(Naphtha[1,2-d]Thiazol-2-Yl)Semicarbazides as Potential Anticonvulsants. Eur. J. Med. Chem. 2009, 44, 203–211. [Google Scholar] [CrossRef]
- Jain, J.; Kumar, Y.; Stables, J.; Sinha, R. Menthone Semicarbazides and Thiosemicarbazides as Anticonvulsant Agents. Med. Chem. 2010, 6, 44–50. [Google Scholar] [CrossRef]
- Nie, Y.; Zhong, M.; Jiang, Z.; Sun, J.; Gao, Y.; Ding, F.; Li, H.; Zhang, Y.; He, X. Synthesis and Potential Anticonvulsant Activity of New Aryl Sulfonyl Semicarbazide Derivatives. Med. Chem. Res. 2016, 25, 1425–1432. [Google Scholar] [CrossRef]
- Sharma, C.S.; Verma, T.; Singh, H.P.; Kumar, N. Synthesis, Characterization and Preliminary Anticonvulsant Evaluation of Some Flavanone Incorporated Semicarbazides. Med. Chem. Res. 2014, 23, 4814–4824. [Google Scholar] [CrossRef]
- Yogeeswari, P.; Sriram, D.; Saraswat, V.; Ragavendran, J.V.; Kumar, M.M.; Murugesan, S.; Thirumurugan, R.; Stables, J.P. Synthesis and Anticonvulsant and Neurotoxicity Evaluation of N4-Phthalimido Phenyl (Thio) Semicarbazides. Eur. J. Pharm. Sci. 2003, 20, 341–346. [Google Scholar] [CrossRef]
- Pavić, K.; Perković, I.; Gilja, P.; Kozlina, F.; Ester, K.; Kralj, M.; Schols, D.; Hadjipavlou-Litina, D.; Pontiki, E.; Zorc, B. Design, Synthesis and Biological Evaluation of Novel Primaquine-Cinnamic Acid Conjugates of the Amide and Acylsemicarbazide Type. Molecules 2016, 28, 1629. [Google Scholar] [CrossRef]
- Pitucha, M.; Karczmarzyk, Z.; Swatko-Ossor, M.; Wysocki, W.; Wos, M.; Chudzik, K.; Ginalska, G.; Fruzinski, A. Synthesis, In Vitro Screening and Docking Studies of New Thiosemicarbazide Derivatives as Antitubercular Agents. Molecules 2019, 24, 251. [Google Scholar] [CrossRef]
- Kedzierska, E.; Orzelska, J.; Perković, I.; Knežević, D.; Fidecka, S.; Kaiser, M.; Zorc, B. Pharmacological Effects of Primaquine Ureas and Semicarbazides on the Central Nervous System in Mice and Antimalarial Activity in Vitro. Fundam. Clin. Pharmacol. 2016, 30, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Horváth, Á.; Menghis, A.; Botz, B.; Borbély, É.; Kemény, Á.; Tékus, V.; Csepregi, J.Z.; Mócsai, A.; Juhász, T.; Zákány, R.; et al. Analgesic and Anti-Inflammatory Effects of the Novel Semicarbazide-Sensitive Amine-Oxidase Inhibitor SzV-1287 in Chronic Arthritis Models of the Mouse. Sci. Rep. 2017, 7, 39863. [Google Scholar] [CrossRef]
- Wang, E.Y.; Gao, H.; Salter-Cid, L.; Zhang, J.; Huang, L.; Podar, E.M.; Miller, A.; Zhao, J.; O’Rourk, A.; Linnik, M.D. Design, Synthesis, and Biological Evaluation of Semicarbazide-Sensitive Amine Oxidase (SSAO) Inhibitors with Anti-Inflammatory Activity. J. Med. Chem. 2006, 49, 2166–2173. [Google Scholar] [CrossRef]
- Kozyra, P.; Adamczuk, G.; Karczmarzyk, Z.; Matysiak, J.; Podkościelna, B.; Humeniuk, E.; Wysocki, W.; Korga-Plewko, A.; Senczyna, B.; Pitucha, M. Novel Phenoxyacetylthiosemicarbazide Derivatives as Novel Ligands in Cancer Diseases. Toxicol. Appl. Pharmacol. 2023, 475, 116634. [Google Scholar] [CrossRef]
- Kozyra, P.; Korga-Plewko, A.; Karczmarzyk, Z.; Hawrył, A.; Wysocki, W.; Człapski, M.; Iwan, M.; Ostrowska-Leśko, M.; Fornal, E.; Pitucha, M. Potential Anticancer Agents against Melanoma Cells Based on an As-Synthesized Thiosemicarbazide Derivative. Biomolecules 2022, 12, 151. [Google Scholar] [CrossRef]
- Pitucha, M.; Korga-Plewko, A.; Kozyra, P.; Iwan, M.; Kaczor, A.A. 2,4-Dichlorophenoxyacetic Thiosemicarbazides as a New Class of Compounds against Stomach Cancer Potentially Intercalating with DNA. Biomolecules 2020, 10, 296. [Google Scholar] [CrossRef]
- Altalhi, A.A.; Hashem, H.E.; Negm, N.A.; Mohamed, E.A.; Azmy, E.M. Synthesis, Characterization, Computational Study, and Screening of Novel 1-Phenyl-4-(2-Phenylacetyl)-Thiosemicarbazide Derivatives for Their Antioxidant and Antimicrobial Activities. J. Mol. Liq. 2021, 333, 115977. [Google Scholar] [CrossRef]
- Lahari, K.; Sundararajan, R. Design and Synthesis of Novel Isatin Derivatives as Potent Analgesic, Anti-Inflammatory and Antimicrobial Agents. J. Chem. Sci. 2020, 132, 94. [Google Scholar] [CrossRef]
- Özcan, E.; Vagolu, S.K.; Gündüz, M.G.; Stevanovic, M.; Kökbudak, Z.; Tønjum, T.; Nikodinovic-Runic, J.; Çetinkaya, Y.; Doğan, Ş.D. Novel Quinoline-Based Thiosemicarbazide Derivatives: Synthesis, DFT Calculations, and Investigation of Antitubercular, Antibacterial, and Antifungal Activities. ACS Omega 2023, 8, 40140–40152. [Google Scholar] [CrossRef]
- Cihan-Üstündağ, G.; Gürsoy, E.; Naesens, L.; Ulusoy-Güzeldemirci, N.; Çapan, G. Synthesis and Antiviral Properties of Novel Indole-Based Thiosemicarbazides and 4-Thiazolidinones. Bioorg. Med. Chem. 2016, 24, 240–246. [Google Scholar] [CrossRef]
- Moharana, A.K.; Dash, R.N.; Subudhi, B.B. Thiosemicarbazides: Updates on Antivirals Strategy. Mini Rev. Med. Chem. 2020, 20, 2135–2152. [Google Scholar] [CrossRef] [PubMed]
- Bulut, N.; Kocyigit, U.M.; Gecibesler, I.H.; Dastan, T.; Karci, H.; Taslimi, P.; Durna Dastan, S.; Gulcin, I.; Cetin, A. Synthesis of Some Novel Pyridine Compounds Containing Bis-1,2,4-Triazole/Thiosemicarbazide Moiety and Investigation of Their Antioxidant Properties, Carbonic Anhydrase, and Acetylcholinesterase Enzymes Inhibition Profiles. J. Biochem. Mol. Toxicol. 2018, 32, e22006. [Google Scholar] [CrossRef]
- Kulandasamy, R.; Adhikari, A.V.; Taranalli, A.; Venkataswamy, T. New Hydrazides and Thiosemicarbazides Derived from Ethylenedioxythiophene as Potential Anticonvulsants. Phosphorus Sulfur Silicon Relat. Elem. 2010, 185, 1358–1368. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Dhake, A.S.; Narkhede, H.I.; Kaur, P. Design, Synthesis and Biological Evaluation of Novel Thiosemicarbazide Analogues as Potent Anticonvulsant Agents. Bioorg. Chem. 2014, 54, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Abhale, Y.K.; Shinde, A.; Deshmukh, K.K.; Nawale, L.; Sarkar, D.; Mhaske, P.C. Synthesis, Antitubercular and Antimicrobial Potential of Some New Thiazole Substituted Thiosemicarbazide Derivatives. Med. Chem. Res. 2017, 26, 2557–2567. [Google Scholar] [CrossRef]
- Parks, R.E.; Kidder, G.W.; Dewey, V.C. Thiosemicarbazide Toxicity in Mice. Proc. Soc. Exp. Biol. Med. 1952, 79, 287–289. [Google Scholar] [CrossRef]
- Chandra, G.; Singh, D.V.; Mahato, G.K.; Patel, S. Fluorine-a Small Magic Bullet Atom in the Drug Development: Perspective to FDA Approved and COVID-19 Recommended Drugs. Chem. Zvesti 2023, 77, 4085–4106. [Google Scholar] [CrossRef]
- Morgenthaler, M.; Schweizer, E.; Hoffmann-Röder, A.; Benini, F.; Martin, R.E.; Jaeschke, G.; Wagner, B.; Fischer, H.; Bendels, S.; Zimmerli, D.; et al. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amine Basicities. ChemMedChem 2007, 2, 1100–1115. [Google Scholar] [CrossRef]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef]
- Pal, S.; Chandra, G.; Patel, S.; Singh, S. Fluorinated Nucleosides: Synthesis, Modulation in Conformation and Therapeutic Application. Chem. Rec. 2022, 22, e202100335. [Google Scholar] [CrossRef]
- Shet, H.; Sahu, R.; Sanghvi, Y.S.; Kapdi, A.R. Strategies for the Synthesis of Fluorinated Nucleosides, Nucleotides and Oligonucleotides. Chem. Rec. 2022, 22, e202200066. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Shu, Y.-Z.; Zhuo, X.; Meanwell, N.A. Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; Amata, S.; Pibiri, I.; Pace, A.; Buscemi, S.; Palumbo Piccionello, A. FDA-Approved Fluorinated Heterocyclic Drugs from 2016 to 2022. Int. J. Mol. Sci. 2023, 24, 7728. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Zhang, C. Fluorine in Medicinal Chemistry: In Perspective to COVID-19. ACS Omega 2022, 7, 18206–18212. [Google Scholar] [CrossRef]
- Kozyra, P.; Pitucha, M. Terminal Phenoxy Group as a Privileged Moiety of the Drug Scaffold—A Short Review of Most Recent Studies 2013–2022. Int. J. Mol. Sci. 2022, 23, 8874. [Google Scholar] [CrossRef]
- Kozyra, P.; Wysocki, W.; Walczak, Ł.J.; Herbet, M.; Karczmarzyk, Z.; Pitucha, M. Application of in Silico Methods to Assess the Anticancer Potential of New Derivatives of 4-Fluorophenoxythiosemicarbazide Derivatives. J. Mol. Struct. 2025, 1326, 141090. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of Bond Lengths Determined by X-Ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds. J. Chem. Soc. Perkin Trans. 2 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Turner, M.J.; Mckinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer; Version 17; University of Western Australia: Crawley, Australia, 2017; Available online: https://scholar.google.com/scholar_lookup?hl=en&publication_year=2017&author=M.+J.+Turner&author=J.+J.+McKinnon&author=S.+K.+Wolff&author=D.+J.+Grimwood&author=P.+R.+Spackman&author=D.+Jayatilaka&author=M.+A.+Spackman&title=CrystalExplorer17 (accessed on 27 March 2025).
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-Score—A Comprehensive Scoring Function for Evaluation of Chemical Drug-Likeness. Medchemcomm 2018, 10, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Czylkowska, A.; Lanka, S.; Szczesio, M.; Czarnecka, K.; Szymański, P.; Pitucha, M.; Drabińska, A.; Camargo, B.C.; Szczytko, J. New Derivatives of 5-((1-Methyl-Pyrrol-2-Yl) Methyl)-4-(Naphthalen-1-Yl)-1,2,4-Triazoline-3-Thione and Its Coordination Compounds with Anticancer Activity. Int. J. Mol. Sci. 2022, 23, 9162. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Malki, A.; Elbayaa, R.Y.; Ashour, H.M.A.; Loffredo, C.A.; Youssef, A.M. Novel Thiosemicarbazides Induced Apoptosis in Human MCF-7 Breast Cancer Cells via JNK Signaling. J. Enzyme Inhib. Med. Chem. 2015, 30, 786–795. [Google Scholar] [CrossRef]
- Chen, R.; Huo, L.; Jaiswal, Y.; Huang, J.; Zhong, Z.; Zhong, J.; Williams, L.; Xia, X.; Liang, Y.; Yan, Z. Design, Synthesis, Antimicrobial, and Anticancer Activities of Acridine Thiosemicarbazides Derivatives. Molecules 2019, 24, 2065. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Qian, Y.; Zhu, D.-D.; Yang, X.-G.; Zhu, H.-L. Synthesis, Molecular Modeling and Biological Evaluation of Chalcone Thiosemicarbazide Derivatives as Novel Anticancer Agents. Eur. J. Med. Chem. 2011, 46, 4702–4708. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First Line Defence Antioxidants-Superoxide Dismutase (SOD), Catalase (CAT) and Glutathione Peroxidase (GPX): Their Fundamental Role in the Entire Antioxidant Defence Grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Abuetabh, Y.; Wu, H.H.; Chai, C.; Al Yousef, H.; Persad, S.; Sergi, C.M.; Leng, R. DNA Damage Response Revisited: The P53 Family and Its Regulators Provide Endless Cancer Therapy Opportunities. Exp. Mol. Med. 2022, 54, 1658–1669. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. P21 in Cancer: Intricate Networks and Multiple Activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Foo, T.K.; Vincelli, G.; Huselid, E.; Her, J.; Zheng, H.; Simhadri, S.; Wang, M.; Huo, Y.; Li, T.; Yu, X.; et al. ATR/ATM-Mediated Phosphorylation of BRCA1 T1394 Promotes Homologous Recombinational Repair and G2-M Checkpoint Maintenance. Cancer Res. 2021, 81, 4676–4684. [Google Scholar] [CrossRef]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The Role of BCL-2 Family Proteins in Regulating Apoptosis and Cancer Therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Davalli, P.; Marverti, G.; Lauriola, A.; D’Arca, D. Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies. Oxidative Med. Cell. Longev. 2018, 2018, 2389523. [Google Scholar] [CrossRef]
- Barzilai, A.; Yamamoto, K.-I. DNA Damage Responses to Oxidative Stress. DNA Repair 2004, 3, 1109–1115. [Google Scholar] [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base Excision Repair of Oxidative DNA Damage and Association with Cancer and Aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef]
- Hongo, H.; Kosaka, T.; Suzuki, Y.; Mikami, S.; Fukada, J.; Oya, M. Topoisomerase II Alpha Inhibition Can Overcome Taxane-Resistant Prostate Cancer through DNA Repair Pathways. Sci. Rep. 2021, 11, 22284. [Google Scholar] [CrossRef]
- Linka, R.M.; Porter, A.C.G.; Volkov, A.; Mielke, C.; Boege, F.; Christensen, M.O. C-Terminal Regions of Topoisomerase II α and II β Determine Isoform-Specific Functioning of the Enzymes in Vivo. Nucleic Acids Res. 2007, 35, 3810–3822. [Google Scholar] [CrossRef] [PubMed]
- Schaefer-Klein, J.L.; Murphy, S.J.; Johnson, S.H.; Vasmatzis, G.; Kovtun, I.V. Topoisomerase 2 Alpha Cooperates with Androgen Receptor to Contribute to Prostate Cancer Progression. PLoS ONE 2015, 10, e0142327. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Chen, S.-F.; Wu, C.-C.; Liao, Y.-W.; Lin, T.-S.; Liu, K.-T.; Chen, Y.-S.; Li, T.-K.; Chien, T.-C.; Chan, N.-L. Producing Irreversible Topoisomerase II-Mediated DNA Breaks by Site-Specific Pt(II)-Methionine Coordination Chemistry. Nucleic Acids Res. 2017, 45, 10861–10871. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro. Agilent Technologies. Version 1.171.37.35h, 2015 (Release 09-02-2015 CrysAlis171.NET), (Compiled Feb 9 2015, 16: 28: 20); Agilent Technologies: Oxfordshire, UK, 2015. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking1. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- BIOVIA. Dassault Systèmes, Discovery Studio Visualizer, V21. 1.0.20298; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 |
|---|---|---|---|---|---|
| Inhibitor | Inhibitor | Inhibitor | Inhibitor | Inhibitor | |
| AB1 | No | Yes | Yes | No | No |
| AB2 | No | Yes | Yes | No | No |
| AB3 | Yes | Yes | Yes | No | No |
| AB4 | Yes | Yes | Yes | Yes | Yes |
| AB5 | Yes | Yes | Yes | No | Yes |
| AB6 | Yes | Yes | Yes | No | Yes |
| AB7 | No | No | No | No | No |
| AB8 | No | Yes | Yes | No | No |
| AB9 | No | Yes | No | No | No |
| AB10 | No | Yes | No | No | No |
| AB11 | No | Yes | Yes | No | No |
| AB12 | No | Yes | Yes | No | No |
| Compound | Cell Lines | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fibroblast | Prostate Cancer | Melanoma | Breast Cancer | Lung Cancer | ||||||
| BJ | PC-3 | DU-145 | LNCaP | SK-MEL28 | A375 | G-361 | MCF-7 | MDA-MB-231 | NCI-H1563 | |
| IC50 (μM) | ||||||||||
| AB1 | 497.71 | >500 | >500 | 252.14 | >500 | >500 | 369.37 | >500 | >500 | >500 |
| AB2 | 453.31 | >500 | >500 | 108.14 | 469.96 | 428.8 | 222.74 | >500 | >500 | >500 |
| AB3 | 421.67 | 467.13 | 267.67 | 175.42 | 302.11 | 400.17 | 247.55 | 455.76 | 495.32 | 437.25 |
| AB4 | 312.55 | 355.29 | 297.31 | 257.12 | 321.71 | 398.54 | 265.32 | 342.92 | 421.27 | 293.01 |
| AB5 | 302.68 | 395.24 | 401.2 | 154.31 | 297.33 | 383.66 | 276.39 | 421.31 | 444.78 | 371.59 |
| AB6 | <100 | 387.79 | 342.67 | 121.11 | 195.28 | 257.48 | 210.24 | 239.77 | 334.12 | 241.58 |
| Cisplatin (reference) | - | 52.24 | 64.13 | 31.52 | 25.79 | 18.22 | 12.45 | 22.64 | 38.66 | 2.78 |
| Compounds | ChemScore Value | Ligand–Amino Acids Interactions |
|---|---|---|
| AB1 | 70.91 | (C=)O6…H-N(Gly488)(B) N1-H…O(=C)(Arg487)(B) |
| AB2 | 75.42 | C33-H…N(DA12)(D) |
| AB3 | 74.18 | (C=)S3…H-C(DC8)(E) C33-H…N(DA12)(D) |
| AB4 | 73.96 | (C=)34…H-C(ARG487)(B) N1-H…H-C(DC8)(E) C33-H…N(DG13)(D) C33-H…N(DA12)(D) |
| AB5 | 74.55 | C22-Cl…H-N(DG13)(D) C24-Cl…H-O(ASP463)(B) |
| AB6 | 72.16 | (C=)S3…H-C(DC8)(E) C33-H…N(DA12)(D) |
| Data Collection/Crystal Data | AB2 | AB5 |
|---|---|---|
| Diffractometer | KM4 CCD four-circle diffractometer | |
| Crystal size (mm) | 0.40 × 0.30 × 0.20 | 0.30 × 0.20 × 0.10 |
| Radiation type | Mo Kα (λ = 0.71073 Å) | Mo Kα (λ = 0.71073 Å) |
| Collected data scan | ω | ω |
| Absorption correction | multi-scan CrysAlisPro [75] | multi-scan CrysAlisPro [75] |
| Transmission factors, Tmin, Tmax | 0.609/1.000 | 0.730/1.000 |
| Chemical formula | C15H13ClFN3O2S | C15H12Cl2FN3O2S, |
| Mr (g mol−1) | 353.79 | 388.24 |
| Crystal system, space group | ||
| Lattice constant, a, b, c (Å), α, β, γ (°) | 8.6773 (15), 9.3488 (16), 10.6905 (15), 89.835 (13), 70.387 (14), 76.972 (15) | 8.7626 (8), 9.5122 (6), 10.7018 (9), 86.446 (6), 72.915 (7), 77.803 (6) |
| V (Å3) | 793.4 (2) | 833.40 (12) |
| Z | 2 | 2 |
| Calculated density, Dcalc. (g cm−1) | 1.481 | 1.547 |
| F(000) | 364 | 396 |
| Absorption coefficient, µ (mm−1) | 0.395 | 0.538 |
| Temperature (K) | 293 (2) | 293 (2) |
| No. of measured reflections | 6495 | 5484 |
| θ range (°) | 2.029–28.744 | 2.190–28.517 |
| No. of unique reflections | 6495 | 3654 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.052, 0.119, 0.861 | 0.050, 0.117, 1.041 |
| No. of observed reflections with I > 2σ(I), | 3395 | 2650 |
| Δρmax, Δρmin (eÅ−3) | +0.290, –0.062 | +0.316, –0.069 |
| Gene Symbol | Gene Name | Forward Sequence (3′→5′) | Reverse Sequence (3′→5′) |
|---|---|---|---|
| ATM | Ataxia telangiectasia mutated protein kinase | GCCGCGGTTGATACTACTTTG | GCAGCAGGGTGACAATAAACA |
| ATR | ATR serine/threonine kinase | AATGGTTGGAGAATGCTGGC | ACATCACCCTTGGACCAGAG |
| BAX | Bcl-2-like protein 4 | TCAGGATGCGTCCACCAAGAAG | TGTGTCCACGGCGGCAATCATC |
| BCL-2 | BCL-2 apoptosis regulator | ACTGAGTACCTGAACCGGCA | TACAGTTCCACAAAGGCATCCCAG |
| SOD2 | Superoxide dismutase 2 | CTTCAGGGTGGTATGGCTGT | TGGCCAGACCTTAATGTTCC |
| CAT | Catalase | GCTCCGCAATCCTACACCAT | GGACATCGGGTTTCTGAGGG |
| GPX1 | Glutathione peroxidase 1 | CAATCAGTTCGGACATCAGGAGA | TAAAGAGCGGGTGAGCCTTC |
| TP53 | Tumor protein p53 | CCTCAGCATCTTATCCGAGTGG | TGGATGGTGGTACAGTCAGAGC |
| CDKN1A | Cyclin dependent kinase inhibitor 1A/CDKN1A (p21) | CCTCATCCCGTGTTCTCCTTT | GTACCACCCAGCGGACAAGT |
| CYCD | D-type cyclins | CCGTCCATGCGGAAGATC | GAAGACCTCCTCCTCGCACT |
| RNA18SN5 | 18S ribosomal N5 | GAAACTGCGAATGGCTCATTAAA | CACAGTTATCCAAGTGGGAGAGG |
| BACT | Beta-actin | AGAGCTACGAGCTGCCTGAC | AGCACTGTGTTGGCGTACAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozyra, P.; Humeniuk, E.; Karczmarzyk, Z.; Borzęcki, A.; Adamczuk, G.; Korga-Plewko, A.; Wysocki, W.; Pitucha, M. Anticancer Activity and Safety Profile of Novel 1-(4-Fluorophenoxyacetyl)-4-substituted Thio/Semicarbazide Derivatives. Molecules 2025, 30, 1576. https://doi.org/10.3390/molecules30071576
Kozyra P, Humeniuk E, Karczmarzyk Z, Borzęcki A, Adamczuk G, Korga-Plewko A, Wysocki W, Pitucha M. Anticancer Activity and Safety Profile of Novel 1-(4-Fluorophenoxyacetyl)-4-substituted Thio/Semicarbazide Derivatives. Molecules. 2025; 30(7):1576. https://doi.org/10.3390/molecules30071576
Chicago/Turabian StyleKozyra, Paweł, Ewelina Humeniuk, Zbigniew Karczmarzyk, Adrian Borzęcki, Grzegorz Adamczuk, Agnieszka Korga-Plewko, Waldemar Wysocki, and Monika Pitucha. 2025. "Anticancer Activity and Safety Profile of Novel 1-(4-Fluorophenoxyacetyl)-4-substituted Thio/Semicarbazide Derivatives" Molecules 30, no. 7: 1576. https://doi.org/10.3390/molecules30071576
APA StyleKozyra, P., Humeniuk, E., Karczmarzyk, Z., Borzęcki, A., Adamczuk, G., Korga-Plewko, A., Wysocki, W., & Pitucha, M. (2025). Anticancer Activity and Safety Profile of Novel 1-(4-Fluorophenoxyacetyl)-4-substituted Thio/Semicarbazide Derivatives. Molecules, 30(7), 1576. https://doi.org/10.3390/molecules30071576