
Prenylation of Flavanones by an Aromatic Prenyltransferase from Fusarium globosum

 ,
,
Abstract

1. Introduction
2. Results and Discussion
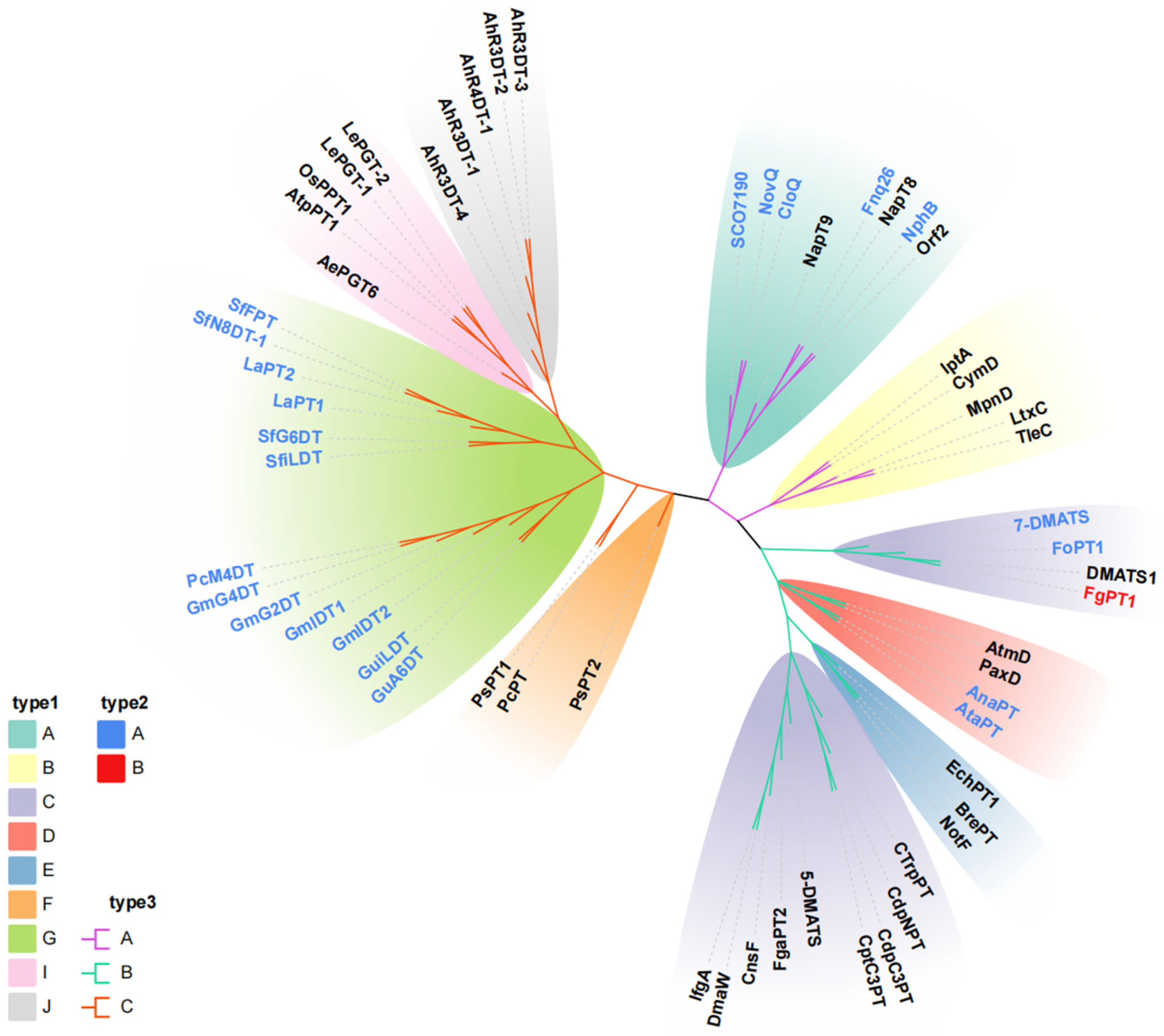
2.1. Sequence Analysis of FgPT1
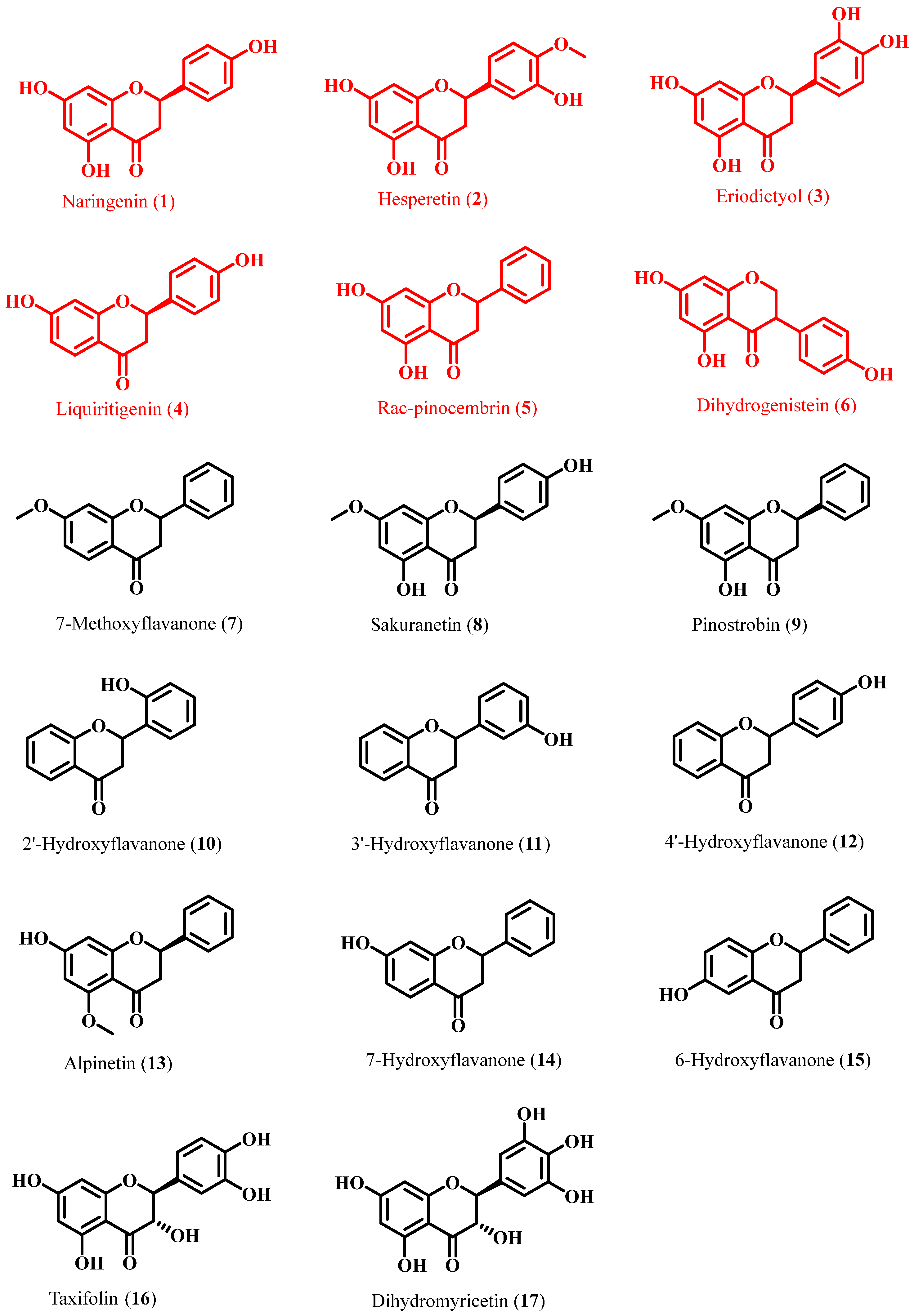
2.2. Preparation of FgPT1 and Functional Characterization of Its Prenylated Flavonoids
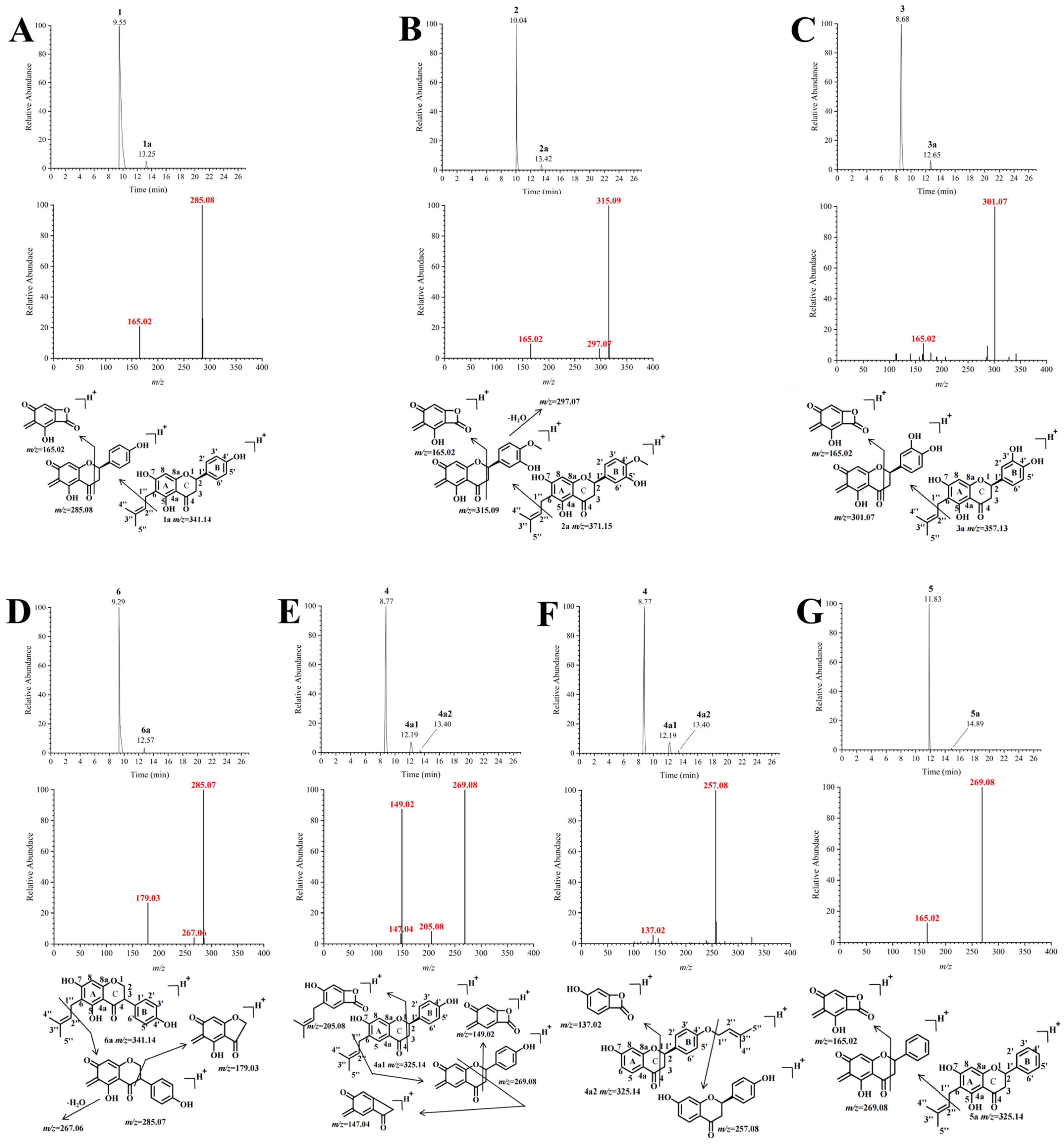
2.3. Isolated Identification and Structural Analysis of Prenylation Products
2.4. Characterization of the Catalytic Properties of FgPT1
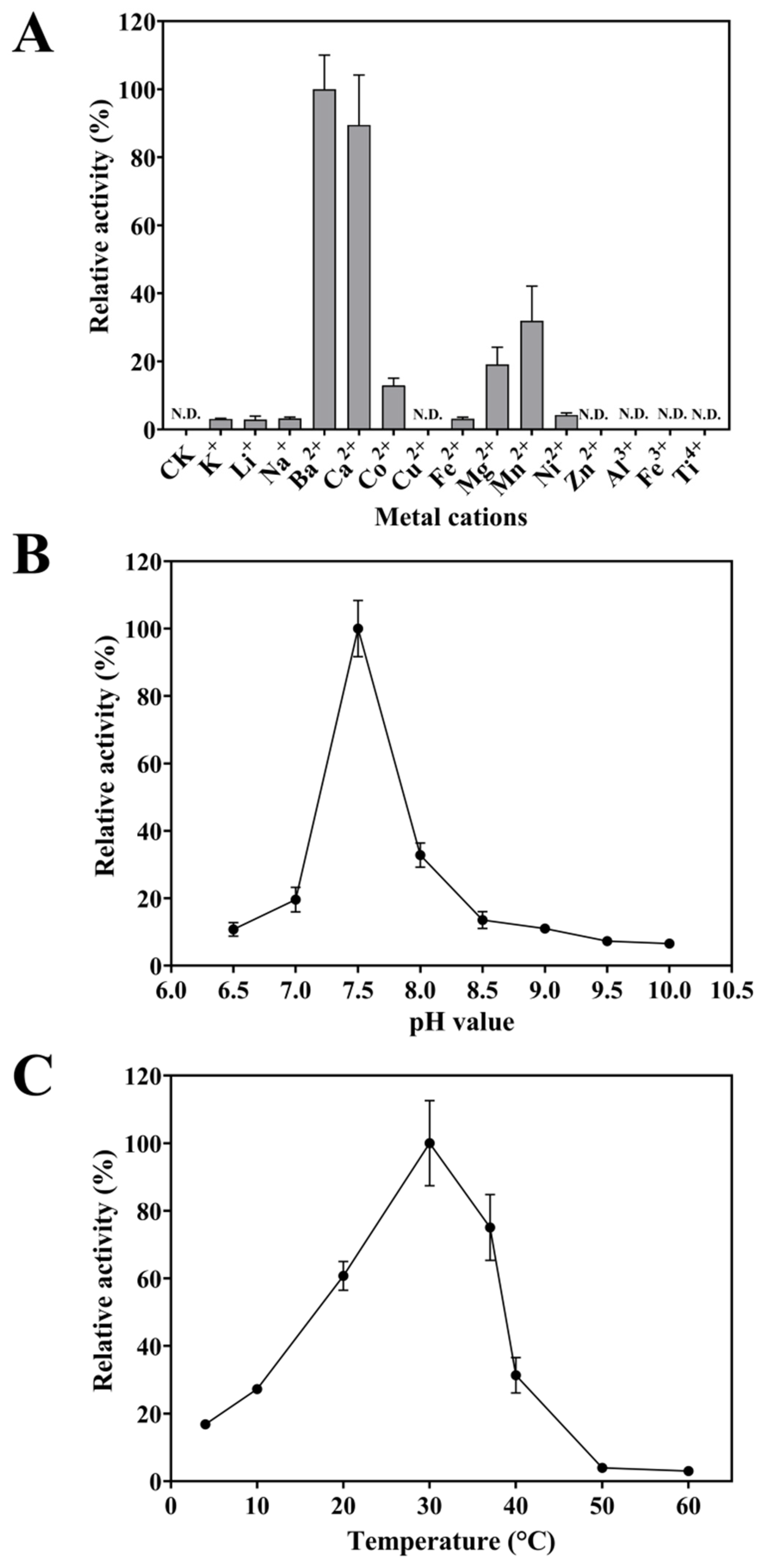
2.4.1. Metal-Cation Dependence of FgPT1
2.4.2. Optimum Reaction PH of FgPT1
2.4.3. Optimum Reaction Temperatures of FgPT1
2.5. Kinetic Parameters of the FgPT1 Reaction of Substrates in the Presence of DMAPP
2.6. Structural Requirements of FgPT1 for Flavanone Substrates
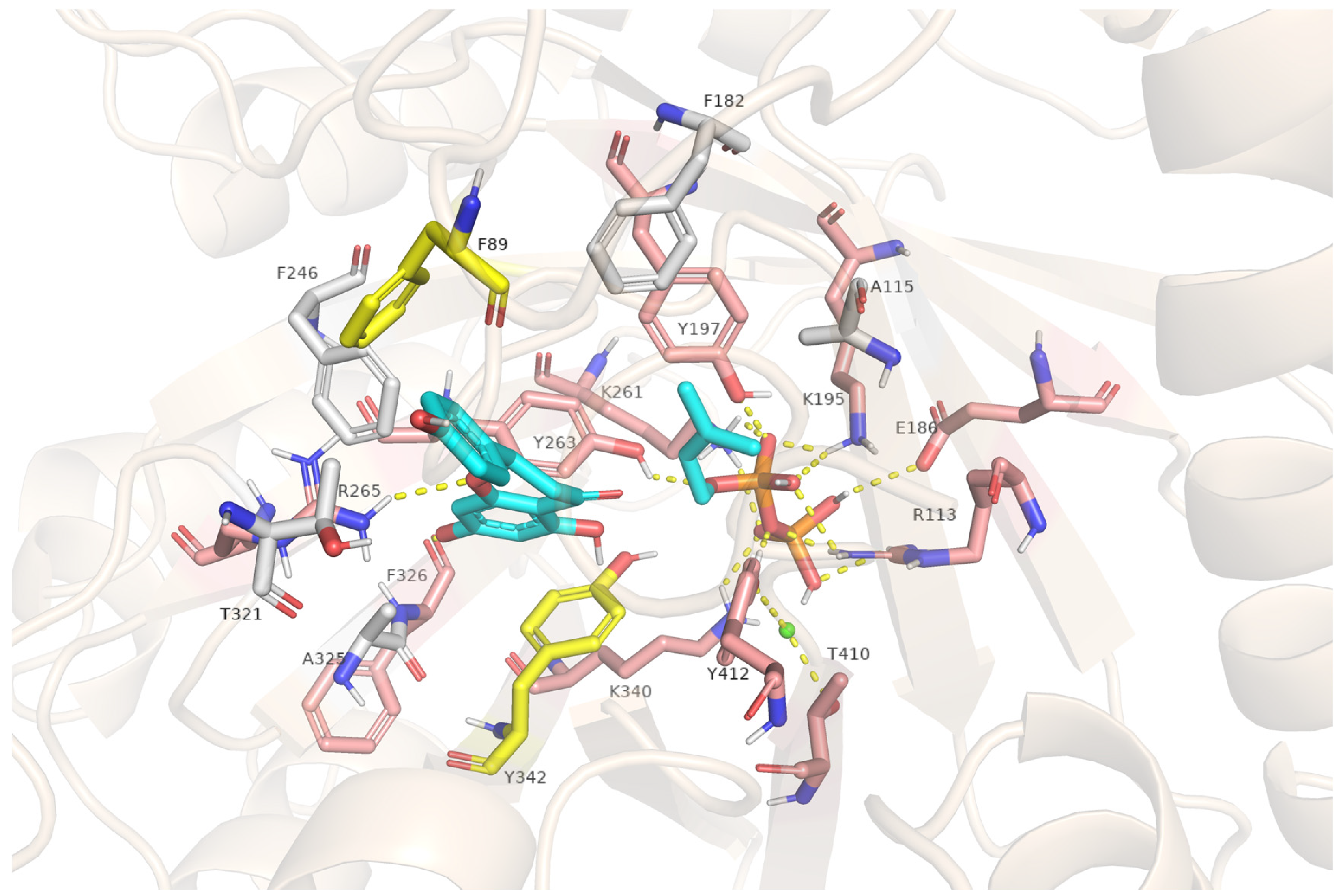
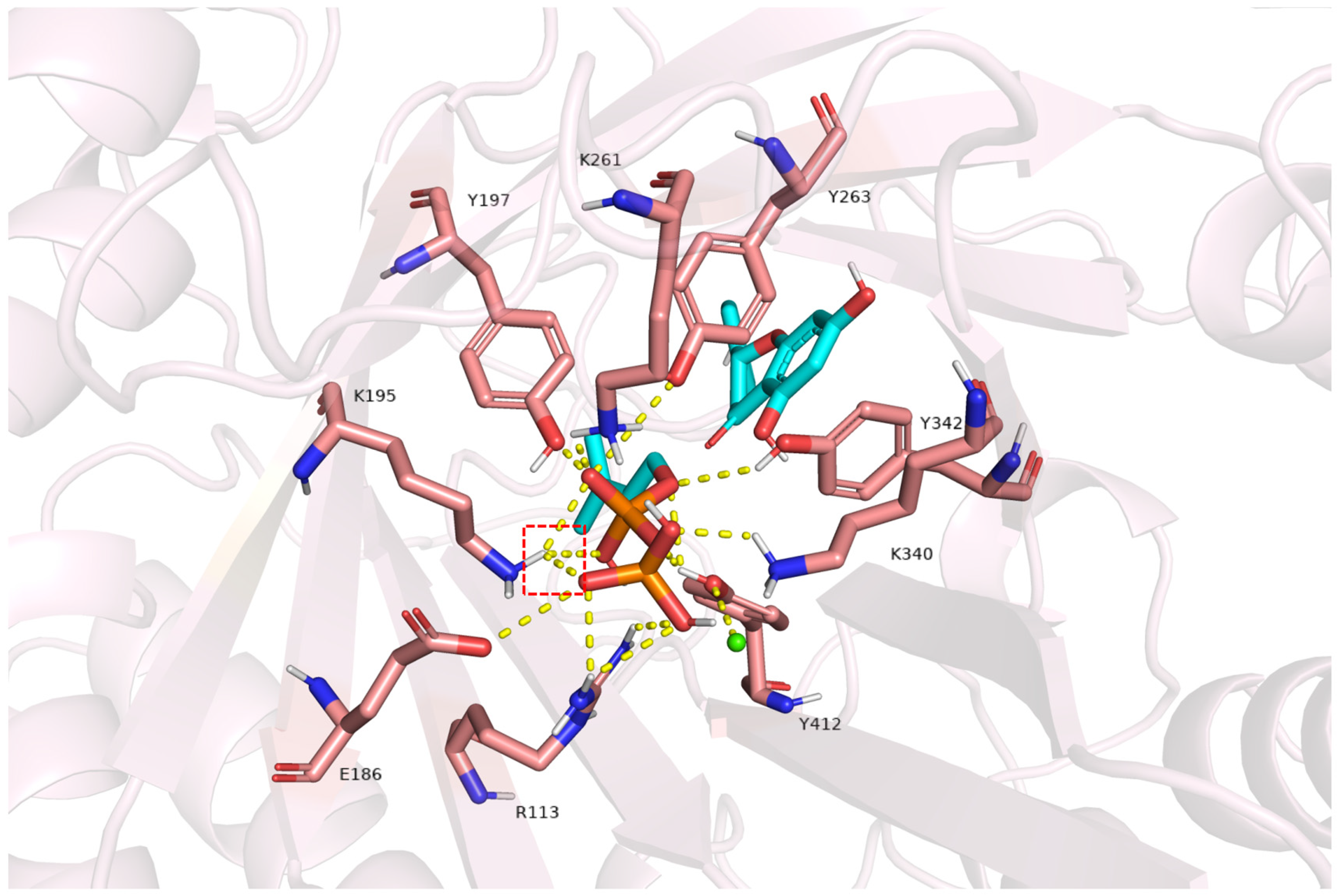
2.7. Structural Insights into FgPT1-Substrate Interactions from Homology Modeling and Docking
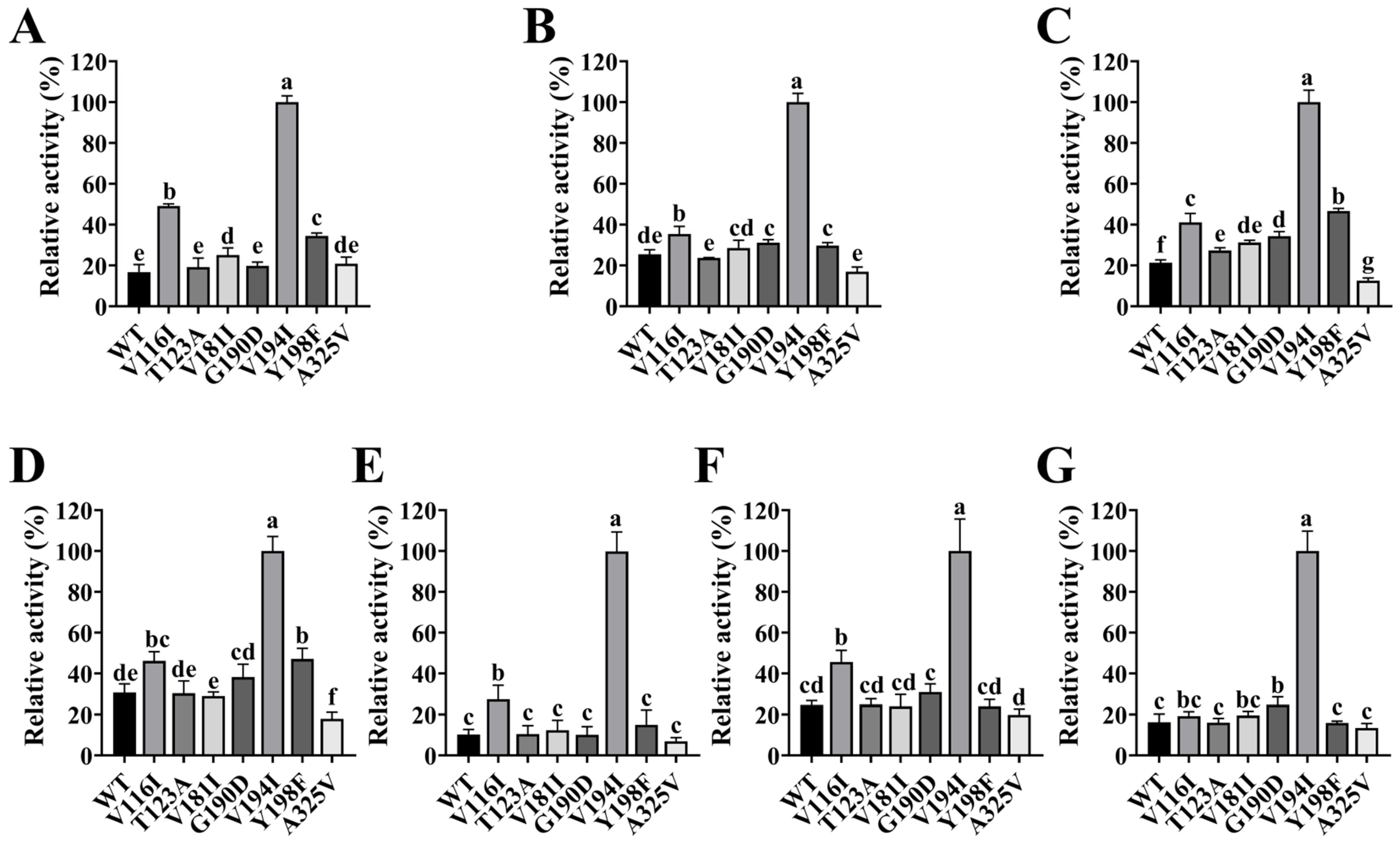
2.8. Impact of Site-Directed Mutagenesis on FgPT1 Catalytic Activity
3. Materials and Methods
3.1. Chemical Reagents
3.2. Synthesis of the FgPT1 Gene
3.3. Expression and Purification of the FgPT1 Gene
3.4. Measurement of FgPT1 Activity
3.5. Identification and Preparation of Products
3.6. Determination of Apparent KM Value
3.7. Homology Modeling and Molecular Docking Method for FgPT1
3.8. Site-Directed Mutagenesis Method for FgPT1
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jeong, H.; Lee, J.; Kim, S.; Yeo, Y.Y.; So, H.; Wu, H.; Song, Y.S.; Jang, C.Y.; Kim, H.D.; Kim, M.J.; et al. Hepatic metabolism of sakuranetin and its modulating effects on cytochrome P450s and UDP-glucuronosyltransferases. Molecules 2018, 23, 1542. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.B.; Li, Y.H.; Shu, X.C.; Pu, Y.T.; Wang, X.J.; Wang, T.; Wang, Z. The classification, molecular structure and biological biosynthesis of flavonoids, and their roles in biotic and abiotic stresses. Molecules 2023, 28, 3599. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Saini, S.; Gangwar, A.; Sharma, R.; Anal, J.M.H. Antibacterial activity of structurally diverse natural prenylated isobavachalcone derivatives. RSC Adv. 2024, 14, 32771–32785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Goto, Y.; Suga, H. Discovery, biochemical characterization, and bioengineering of cyanobactin prenyltransferases. Trends Biochem. Sci. 2023, 48, 360–374. [Google Scholar] [CrossRef]
- Huang, J.B.; Chen, Y.S.; Wang, M.R.; Li, R.S.; Zhao, X.R.; Kaunda, J.S.; Zhang, R.H.; Zhang, X.J.; Xiao, W.L.; Li, H.L.; et al. Ten new prenylated flavonoids from Macaranga denticulata and their antitumor activities. Fitoterapia 2022, 162, 105302. [Google Scholar] [CrossRef]
- Brunelli, E.; Minassi, A.; Appendino, G.; Moro, L. 8-Prenylnaringenin, inhibits estrogen receptor-α mediated cell growth and induces apoptosis in MCF-7 breast cancer cells. J. Steroid Biochem. Mol. Biol. 2007, 107, 140–148. [Google Scholar] [CrossRef]
- Potaniec, B.; Grabarczyk, M.; Stompor, M.; Szumny, A.; Zieliński, P.; Żołnierczyk, A.K.; Anioł, M. Antioxidant activity and spectroscopic data of isoxanthohomol oxime and related compounds. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 118, 716–723. [Google Scholar] [CrossRef]
- Feng, S.; Hao, J.; Xu, Z.; Chen, T.; Qiu, S.X. Polyprenylated isoflavanone and isoflavonoids from Ormosia henryi and their cytotoxicity and anti-oxidation activity. Fitoterapia 2012, 83, 161–165. [Google Scholar] [CrossRef]
- Sekiguchi, F.; Fujita, T.; Deguchi, T.; Yamaoka, S.; Tomochika, K.; Tsubota, M.; Ono, S.; Horaguchi, Y.; Ichii, M.; Ichikawa, M.; et al. Blockade of T-type calcium channels by 6-prenylnaringenin, a hop component, alleviates neuropathic and visceral pain in mice. Neuropharmacology 2018, 138, 232–244. [Google Scholar] [CrossRef]
- Štulíková, K.; Karabín, M.; Nešpor, J.; Dostálek, P. Therapeutic perspectives of 8-prenylnaringenin, a potent phytoestrogen from hops. Molecules 2018, 23, 660. [Google Scholar] [CrossRef]
- Trantas, E.A.; Koffas, M.A.; Xu, P.; Ververidis, F. When plants produce not enough or at all: Metabolic engineering of flavonoids in microbial hosts. Front. Plant Sci. 2015, 6, 7. [Google Scholar] [CrossRef]
- Ji, W.; Sun, W.; Feng, J.; Song, T.; Zhang, D.; Ouyang, P.; Gu, Z.; Xie, J. Characterization of a novel N-acetylneuraminic acid lyase favoring industrial N-acetylneuraminic acid synthesis process. Sci. Rep. 2015, 5, 9341. [Google Scholar] [CrossRef]
- Sasaki, K.; Mito, K.; Ohara, K.; Yamamoto, H.; Yazaki, K. Cloning and characterization of naringenin 8-prenyltransferase, a flavonoid-specific prenyltransferase of Sophora flavescens. Plant Physiol. 2008, 146, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liu, X.; Zou, J.; Yin, Y.; Ou, B.; Li, J.; Wang, R.; Xie, D.; Zhang, P.; Dai, J. Regio- and stereospecific prenylation of flavonoids by Sophora flavescens prenyltransferase. Adv. Synth. Catal. 2013, 355, 1817–1828. [Google Scholar] [CrossRef]
- Liu, J.; Xia, Y.; Jiang, W.; Shen, G.; Pang, Y. LaPT2 gene encodes a flavonoid prenyltransferase in white lupin. Front. Plant Sci. 2021, 12, 673337. [Google Scholar] [CrossRef]
- Ohara, K.; Mito, K.; Yazaki, K. Homogeneous purification and characterization of LePGT1—A membrane-bound aromatic substrate prenyltransferase involved in secondary metabolism of Lithospermum erythrorhizon. FEBS J. 2013, 280, 2572–2580. [Google Scholar] [CrossRef]
- Yazaki, K.; Kunihisa, M.; Fujisaki, T.; Sato, F. Geranyl diphosphate:4-hydroxybenzoate geranyltransferase from Lithospermum erythrorhizon: Cloning and characterization of a ket enzyme in shikonin biosynthesis. J. Biol. Chem. 2002, 277, 6240–6246. [Google Scholar] [CrossRef]
- Cho, J.S.; Kim, G.B.; Eun, H.; Moon, C.W.; Lee, S.Y. Designing microbial cell factories for the production of chemicals. JACS Au 2022, 2, 1781–1799. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Y.; Zhang, J.; Sun, L.; Sheng, J.Z.; Tan, Z.; Qi, Q.; Hou, J. Metabolic engineering and strain mating of Yarrowia lipolytica for sustainable production of prenylated aromatic compounds. ACS Sustain. Chem. Eng. 2025, 13, 3149–3159. [Google Scholar] [CrossRef]
- Miller, E.T.; Tsodikov, O.V.; Garneau-Tsodikova, S. Structural insights into the diverse prenylating capabilities of DMATS prenyltransferases. Nat. Prod. Rep. 2024, 41, 113–147. [Google Scholar] [CrossRef]
- Zhou, K.; Yu, X.; Xie, X.; Li, S.-M. Complementary flavonoid prenylations by fungal indole prenyltransferases. J. Nat. Prod. 2015, 78, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, X.; Li, S.M.; Zhang, Y.; Yuan, C.M.; He, S.; Yang, Z.; Yang, S.; Zhou, K. Increasing structural diversity of prenylated chalcones by two fungal prenyltransferases. J. Agric. Food Chem. 2022, 70, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Gao, B.; Liu, X.; Ruan, F.; Zhang, Y.; Lou, J.; Feng, K.; Wunsch, C.; Li, S.-M.; Dai, J.; et al. Molecular insights into the enzyme promiscuity of an aromatic prenyltransferase. Nat. Chem. Biol. 2016, 13, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, J.; Jiang, Y.; Yang, H.; Yun, Z.; Rong, W.; Yang, B. Regiospecific synthesis of prenylated flavonoids by a prenyltransferase cloned from Fusarium oxysporum. Sci. Rep. 2016, 6, 24819. [Google Scholar] [CrossRef]
- Xu, K.; Yang, C.; Xu, Y.; Li, D.; Bao, S.; Zou, Z.; Kang, F.; Tan, G.; Li, S.-M.; Yu, X. Selective geranylation of biflavonoids by Aspergillus terreus aromatic prenyltransferase (AtaPT). Org. Biomol. Chem. 2020, 18, 28–31. [Google Scholar] [CrossRef]
- Araya-Cloutier, C.; Martens, B.; Schaftenaar, G.; Leipoldt, F.; Gruppen, H.; Vincken, J.-P. Structural basis for non-genuine phenolic acceptor substrate specificity of Streptomyces roseochromogenes prenyltransferase CloQ from the ABBA/PT-barrel superfamily. PLoS ONE 2017, 12, e0174665. [Google Scholar] [CrossRef]
- Xu, M.J.; Wu, B.; Ding, T.; Chu, J.H.; Li, C.Y.; Zhang, J.; Wu, T.; Wu, J.; Liu, S.J.; Liu, S.L.; et al. Simultaneous characterization of prenylated flavonoids and isoflavonoids in Psoralea corylifolia L. by liquid chromatography with diode-array detection and quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2012, 26, 2343–2358. [Google Scholar] [CrossRef]
- Grundmann, A.; Kuznetsova, T.; Afiyatullov, S.S.; Li, S.M. FtmPT2, an N-prenyltransferase from Aspergillus fumigatus, catalyses the last step in the biosynthesis of fumitremorgin B. ChemBioChem 2008, 9, 2059–2063. [Google Scholar] [CrossRef]
- Grundmann, A.; Li, S.M. Overproduction, purification and characterization of FtmPT1, a brevianamide F prenyltransferase from Aspergillus fumigatus. Microbiology 2005, 151, 2199–2207. [Google Scholar] [CrossRef]
- Yin, W.B.; Ruan , H.L.; Westrich, L.; Grundmann, A.; Li, S.M. CdpNPT, an N-prenyltransferase from Aspergillus fumigatus: Overproduction, purification and biochemical characterisation. ChemBioChem 2007, 8, 1154–1161. [Google Scholar] [CrossRef]
- Kuzuyama, T.; Noel, J.P.; Richard, S.B. Structural basis for the promiscuous biosynthetic prenylation of aromatic natural products. Nature 2005, 435, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Liu, Y.; Wu, Y.; Zhao, L.; Pei, J. Biochemical characterization of a novel prenyltransferase from Streptomyces sp. NT11 and development of a recombinant strain for the production of 6-prenylnaringenin. J. Agric. Food Chem. 2021, 69, 14231–14240. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Bian, G.; Herbst-Gervasoni, C.J.; Mori, T.; Shinsky, S.A.; Hou, A.; Mu, X.; Huang, M.; Cheng, S.; Deng, Z. Discovery of the cryptic function of terpene cyclases as aromatic prenyltransferases. Nat. Commun. 2020, 11, 3958. [Google Scholar] [CrossRef]
- Tadera, K.; Minami, Y.; Takamatsu, K.; Matsuoka, T. Inhibition of .ALPHA.-glucosidase and .ALPHA.-amylase by flavonoids. J. Nutr. Sci. Vitaminol. 2006, 52, 149–153. [Google Scholar] [CrossRef]
- Shao, X.; Chen, H.; Zhu, Y.; Sedighi, R.; Ho, C.T.; Sang, S. Essential structural requirements and additive effects for flavonoids to scavenge methylglyoxal. J. Agric. Food Chem. 2014, 62, 3202–3210. [Google Scholar] [CrossRef]
- Qing, L.S.; Xue, Y.; Liu, Y.M.; Liang, J.; Xie, J.; Liao, X. Rapid magnetic solid-phase extraction for the selective determination of isoflavones in soymilk using baicalin-functionalized magnetic nanoparticles. J. Agric. Food Chem. 2013, 61, 8072–8078. [Google Scholar] [CrossRef]
- Eaton, S.A.; Ronnebaum, T.A.; Roose, B.W.; Christianson, D.W. Structural basis of substrate promiscuity and catalysis by the reverse prenyltransferase N-dimethylallyl-l-tryptophan synthase from Fusarium fujikuroi. Biochemistry 2022, 61, 2025–2035. [Google Scholar] [CrossRef]
- Christianson, D.W. Structural and chemical biology of terpenoid cyclases. Chem. Rev. 2017, 117, 11570–11648. [Google Scholar] [CrossRef]
- Tello, M.; Kuzuyama, T.; Heide, L.; Noel, J.P.; Richard, S.B. The ABBA family of aromatic prenyltransferases: Broadening natural product diversity. Cell. Mol. Life Sci. 2008, 65, 1459–1463. [Google Scholar] [CrossRef]
- Bonitz, T.; Alva, V.; Saleh, O.; Lupas, A.N.; Heide, L. Evolutionary relationships of microbial aromatic prenyltransferases. PLoS ONE 2011, 6, e27336. [Google Scholar] [CrossRef]
- Biagioli, M.; Marchianò, S.; Roselli, R.; Di Giorgio, C.; Bellini, R.; Bordoni, M.; Gidari, A.; Sabbatini, S.; Francisci, D.; Fiorillo, B.; et al. Discovery of a AHR pelargonidin agonist that counter-regulates Ace2 expression and attenuates ACE2-SARS-CoV-2 interaction. Biochem. Pharmacol. 2021, 188, 114564. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, J.; Zhan, C.; Qiao, J.; Caiyin, Q.; Huang, M. Fine-tuning the function of farnesene synthases for selective synthesis of farnesene Stereoisomers. J. Agric. Food Chem. 2024, 72, 27355–27364. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Levin, E.J.; Liu, S.; Bai, Y.; Lockless, S.W.; Zhou, M. Structure of a membrane-embedded prenyltransferase homologous to UBIAD1. PLoS Biol. 2014, 12, e1001911. [Google Scholar] [CrossRef]
- Eklund, B.I.; Mannervik, B. Importance of a hypervariable active-site residue in human Mu class glutathione transferases catalyzing the bioactivation of chemotherapeutic thiopurine prodrugs. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2007, 1770, 1098–1103. [Google Scholar] [CrossRef]
- Wen, K.; Wang, S.; Sun, Y.; Wang, M.; Zhang, Y.; Zhu, J.; Li, Q. Mechanistic insights into the conversion of flavin adenine dinucleotide (FAD) to 8-formyl FAD in formate oxidase: A combined experimental and in-silico study. Bioresour. Bioprocess. 2024, 11, 67. [Google Scholar] [CrossRef]
- Bertsch, U.; Deschermeier, C.; Fanick, W.; Girkontaite, I.; Hillemeier, K.; Johnen, H.; Weglöhner, W.; Emmrich, F.; Mayr, G.W. The second messenger binding site of inositol 1,4,5-trisphosphate 3-kinase is centered in the catalytic domain and related to the Inositol trisphosphate receptor site. J. Biol. Chem. 2000, 275, 1557–1564. [Google Scholar] [CrossRef]
- Köllner, T.G.; Degenhardt, J.; Gershenzon, J. The product specificities of maize terpene synthases TPS4 and TPS10 are determined both by active site amino acids and residues adjacent to the active site. Plants 2020, 9, 552. [Google Scholar] [CrossRef]
- Mora-Gamboa, M.P.C.; Ferrucho-Calle, M.C.; Ardila-Leal, L.D.; Rojas-Ojeda, L.M.; Galindo, J.F.; Poutou-Piñales, R.A.; Pedroza-Rodríguez, A.M.; Quevedo-Hidalgo, B.E. Statistical improvement of rGILCC 1 and rPOXA 1B laccases activity assay conditions supported by molecular dynamics. Molecules 2023, 28, 7263. [Google Scholar] [CrossRef]
- Hao, M.; Huang, A.; Li, B.; Xin, Y.; Zhang, L.; Gu, Z.; Sun, H.; Li, Y.; Shi, G. Preparation and characterization of a iaccase-like enzyme from Thermomicrobium roseum. Int. J. Biol. Macromol. 2023, 242, 124992. [Google Scholar] [CrossRef]
- Lv, K.; Shao, W.; Pedroso, M.M.; Peng, J.; Wu, B.; Li, J.; He, B.; Schenk, G. Enhancing the catalytic activity of a GH5 processive endoglucanase from Bacillus subtilis BS-5 by site-directed mutagenesis. Int. J. Biol. Macromol. 2021, 168, 442–452. [Google Scholar] [CrossRef]
- Zheng, F.; Tu, T.; Wang, X.; Wang, Y.; Ma, R.; Su, X.; Xie, X.; Yao, B.; Luo, H. Enhancing the catalytic activity of a novel GH5 cellulase Gt Cel5 from Gloeophyllum trabeum CBS 900.73 by site-directed mutagenesis on loop 6. Biotechnol. Biofuels 2018, 11, 76. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Substrate | FgPT1 | Substrate | FgPT1 |
|---|---|---|---|---|
| Flavanones | Naringenin (1) | + | Hesperetin (2) | + |
| Eriodictyol (3) | + | Liquiritigenin (4) | + | |
| Rac-pinocembrin (5) | + | Dihydrogenistein (6) | + | |
| 7-Methoxyflavanone (7) | − | Sakuranetin (8) | − | |
| Pinostrobin (9) | − | 2′-Hydroxyflavanone (10) | − | |
| 3′-Hydroxyflavanone (11) | − | 4′-Hydroxyflavanone (12) | − | |
| Alpinetin (13) | − | 7-Hydroxyflavanone (14) | − | |
| 6-Hydroxyflavanone (15) | − | |||
| Flavanonols | Taxifolin (16) | − | Dihydromyricetin (17) | − |
| Chalcones | 2′-Hydroxychalcone (18) | − | 2′,4′-Dihydroxychalcone (19) | − |
| 2′,4,4′-Trihydroxychalcone (20) | − | 4-Hydroxychalcone (21) | − | |
| 4′-Hydroxychalcone (22) | − | 2′,5′-Dihydroxychalcone (23) | − | |
| Naringenin chalcone (24) | − | |||
| Flavones | Butein (25) | − | 6-Hydroxyflavone (26) | − |
| 6,2′-Dihydroxyflavone (27) | − | 6,3′-Dihydroxyflavone (28) | − | |
| 6,4′-Dihydroxyflavone (29) | − | 5,6-Dihydroxyflavone (30) | − | |
| 6,7-Dihydroxyflavone (31) | − | 5,6,7-Trihydroxyflavone (32) | − | |
| Flavone (33) | − | 6-Aminoflavone (34) | − | |
| 6-Methoxyflavone (35) | − | 5-Hydroxyflavone (36) | − | |
| 7-Hydroxyflavone (37) | − | Apigenin (38) | − | |
| Chrysin (39) | − | Luteolin (40) | − | |
| Flavonols | 3-Hydroxyflavone (41) | − | 3,6-Dihydroxyflavone (42) | − |
| Kaempfenrol (43) | − | Fisetin (44) | − | |
| Kaempferde (45) | − | Morin hydrate (46) | − | |
| Quercetin (47) | − | Myricetin (48) | − | |
| Isoflavonoids | Genistein (49) | − | Formononetin (50) | − |
| Daidzein (51) | − | |||
| Stibenoid | Resveratrol (53) | − | ||
| Prenyl flavonoid | Icaritin (54) | − | ||
| Glycosilated flavonoid | Neohesperidin Dihydrochalcone (55) | − | ||
| Flavan-3-ol | Catechin (56) | − |
| Substrates | kcat (s−1) | KM (mM) | kcat/KM (s−1 M−1) |
|---|---|---|---|
| Hesperetin | 0.0032 ± (0.00023) | 0.94 ± (0.075) | 3.40 |
| Liquiritigenin | 0.0011 ± (0.00008) | 0.93 ± (0.161) | 1.18 |
| Naringenin | 0.0161 ± (0.00033) | 0.26 ± (0.077) | 61.92 |
| Eriodictyol | 0.0014 ± (0.00015) | 0.23 ± (0.026) | 6.09 |
| Dihydrogenistein | 0.0007 ± (0.00008) | 0.14 ± (0.026) | 5.00 |
| Pinocembrin | 0.0003 ± (0.00002) | 0.14 ± (0.037) | 2.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, D.; Quan, J.; Gao, Z.; He, B.; Hou, Y.; Fan, P.; Pan, M.; Yang, J. Prenylation of Flavanones by an Aromatic Prenyltransferase from Fusarium globosum. Molecules 2025, 30, 1558. https://doi.org/10.3390/molecules30071558
Tang D, Quan J, Gao Z, He B, Hou Y, Fan P, Pan M, Yang J. Prenylation of Flavanones by an Aromatic Prenyltransferase from Fusarium globosum. Molecules. 2025; 30(7):1558. https://doi.org/10.3390/molecules30071558
Chicago/Turabian StyleTang, Dingtao, Jiajie Quan, Zhengjiao Gao, Bingfeng He, Yu Hou, Peipei Fan, Meidong Pan, and Jiali Yang. 2025. "Prenylation of Flavanones by an Aromatic Prenyltransferase from Fusarium globosum" Molecules 30, no. 7: 1558. https://doi.org/10.3390/molecules30071558
APA StyleTang, D., Quan, J., Gao, Z., He, B., Hou, Y., Fan, P., Pan, M., & Yang, J. (2025). Prenylation of Flavanones by an Aromatic Prenyltransferase from Fusarium globosum. Molecules, 30(7), 1558. https://doi.org/10.3390/molecules30071558