
Porphyrins as Chiroptical Conformational Probes for Biomolecules





Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
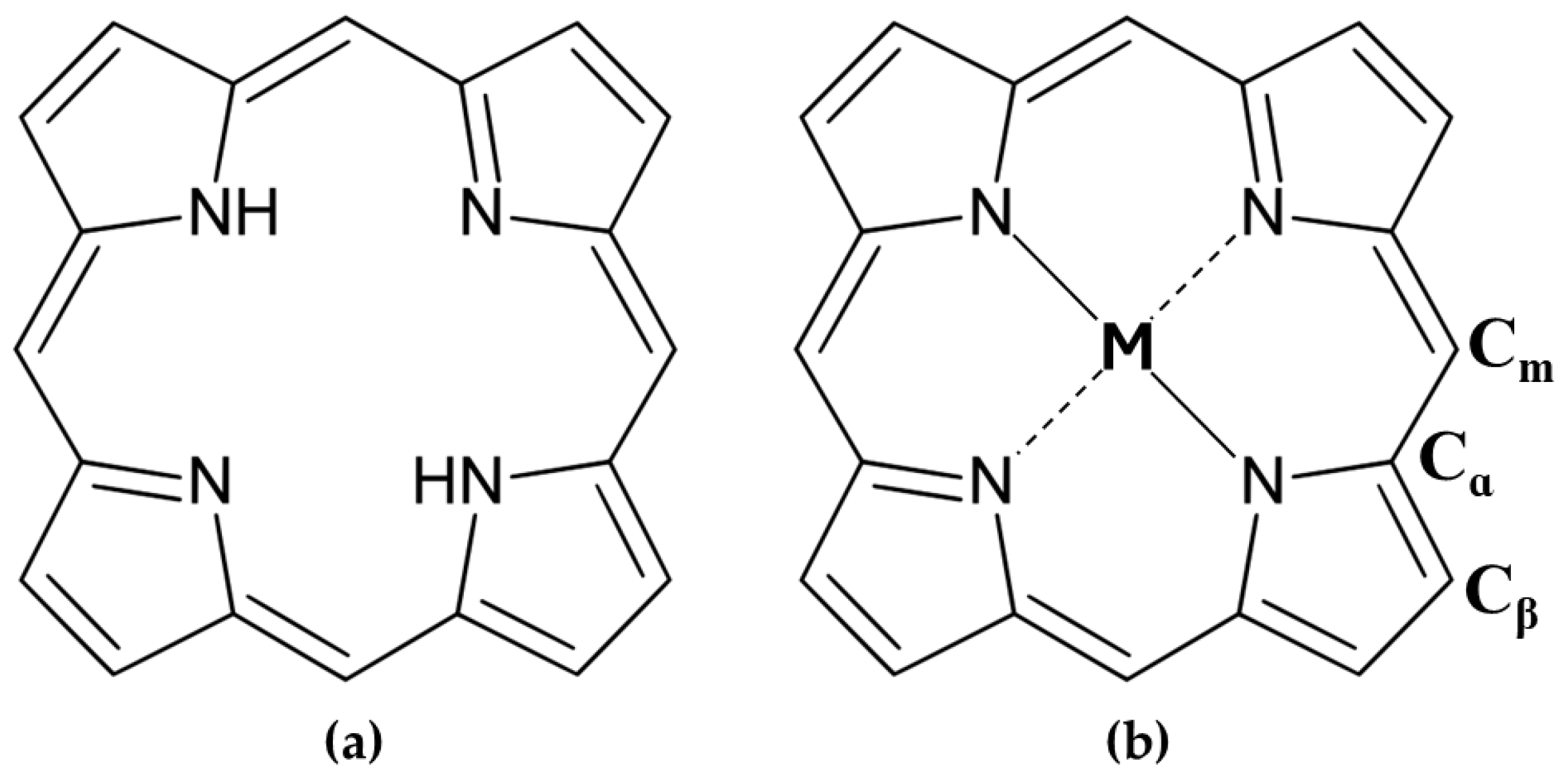
1. Introduction
- Their highly conjugated electronic system, which produces intense absorption bands in the 380–450 nm region (Soret region), allows their use in the micromolar range, far from the UV region, where most of biomolecules absorb, avoiding spectroscopic interferences.
- Their structural versatility, allowing functionalization at the meso positions with specific groups or charged moieties to tailor properties such as water solubility.
- Their ability to coordinate metal ions in the central core, such as zinc or magnesium, which provide additional stereochemical differentiation through Lewis acid sites for binding functional groups like OH, NH2, and COO− [42].
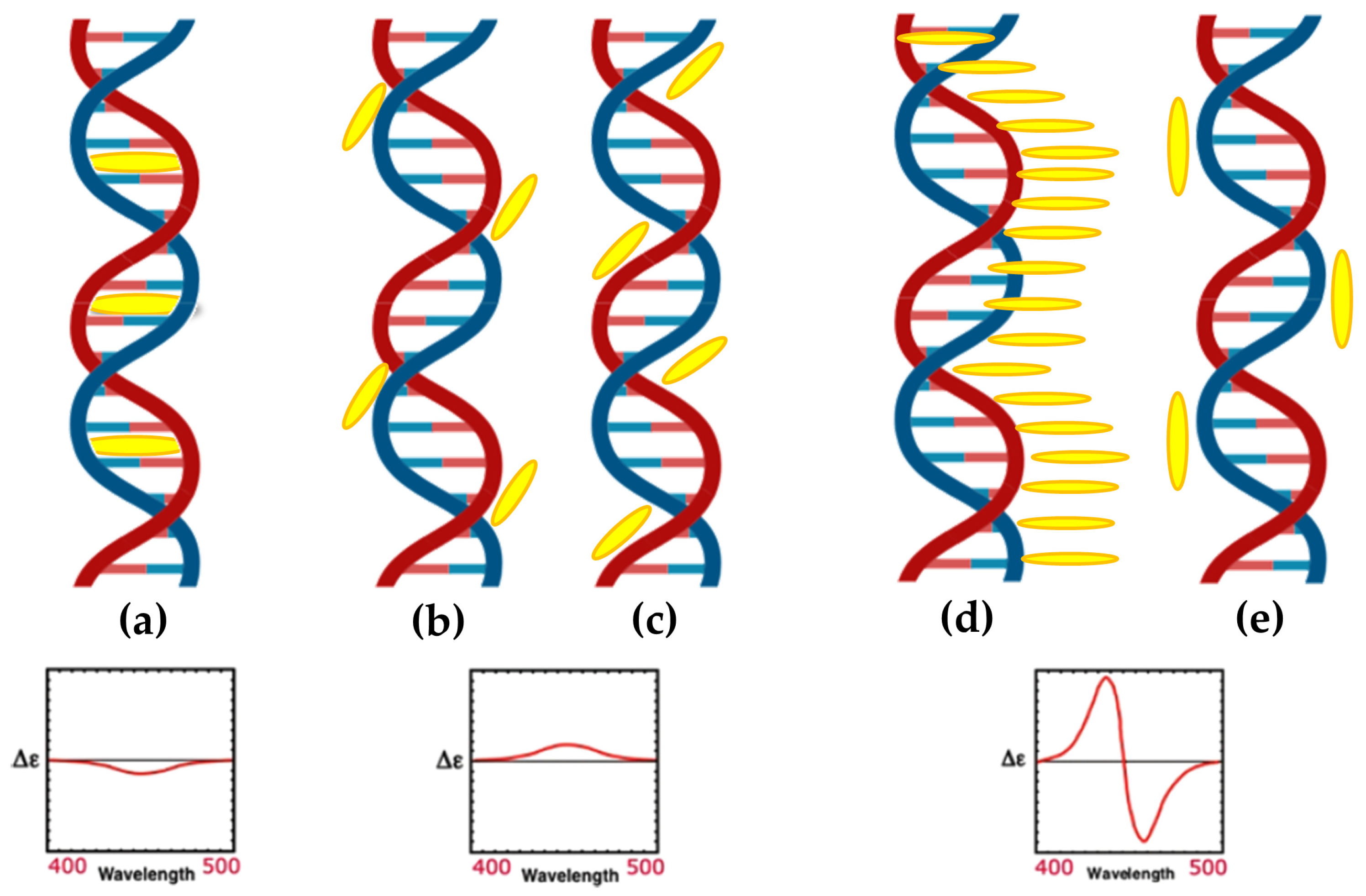
- When non-chiral substituents are present, these molecules are achiral and do not exhibit intrinsic chiroptical signals. However, interactions with chiral molecules could induce CD signals in the Soret absorption band region, which are highly indicative of the binding mode and are far from the UV region where most biomolecules, such as DNA and RNA, absorb [43].
2. Porphyrins as Chiroptical Probes in the Structural Investigation of Nucleic Acids
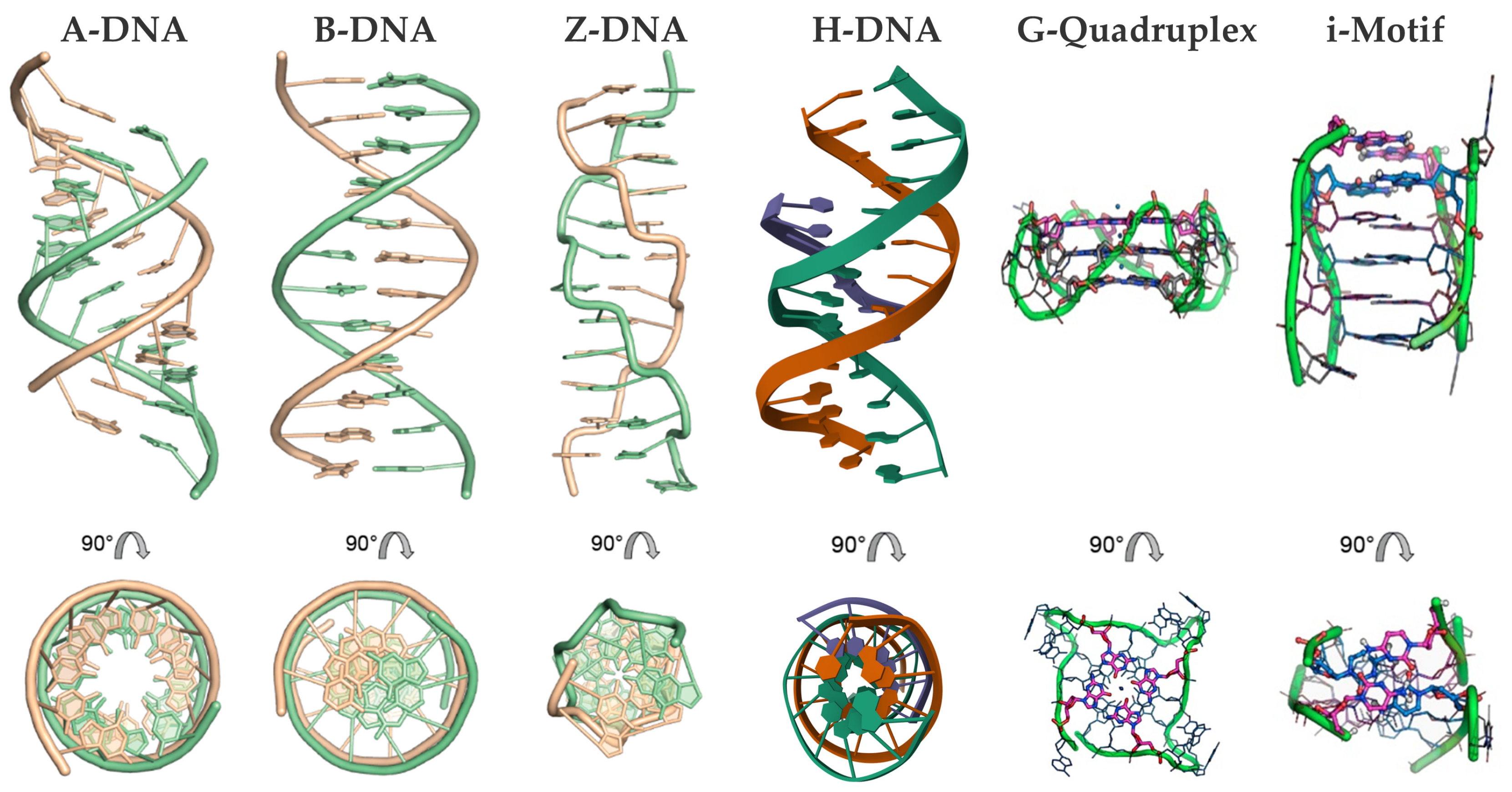
2.1. DNA Polymorphism and Types of Porphyrin Interactions
2.1.1. Chiroptical and Structural Insights into Porphyrin Interactions with B-DNA
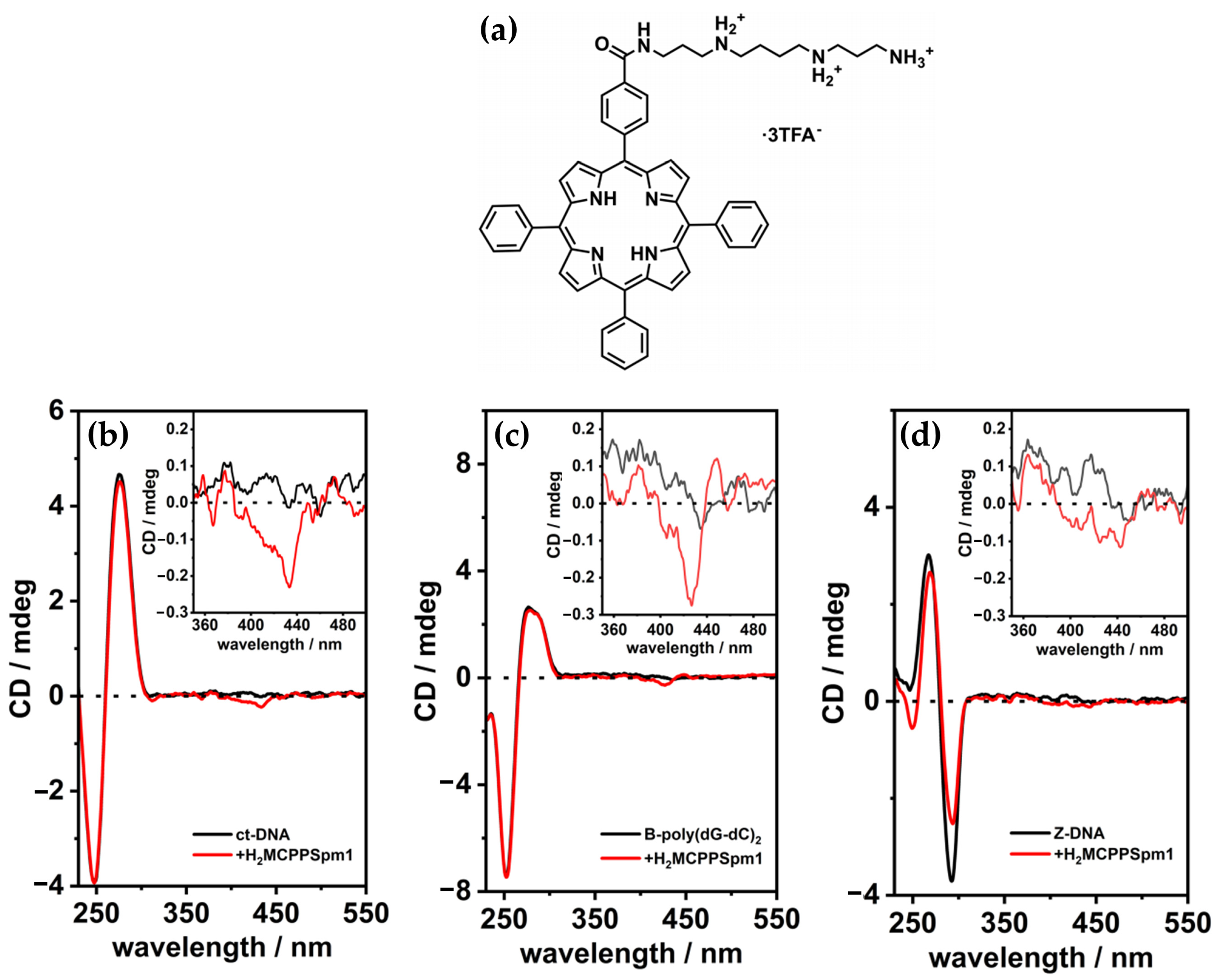
2.1.2. Porphyrin Derivatives as Probes for the Z-DNA
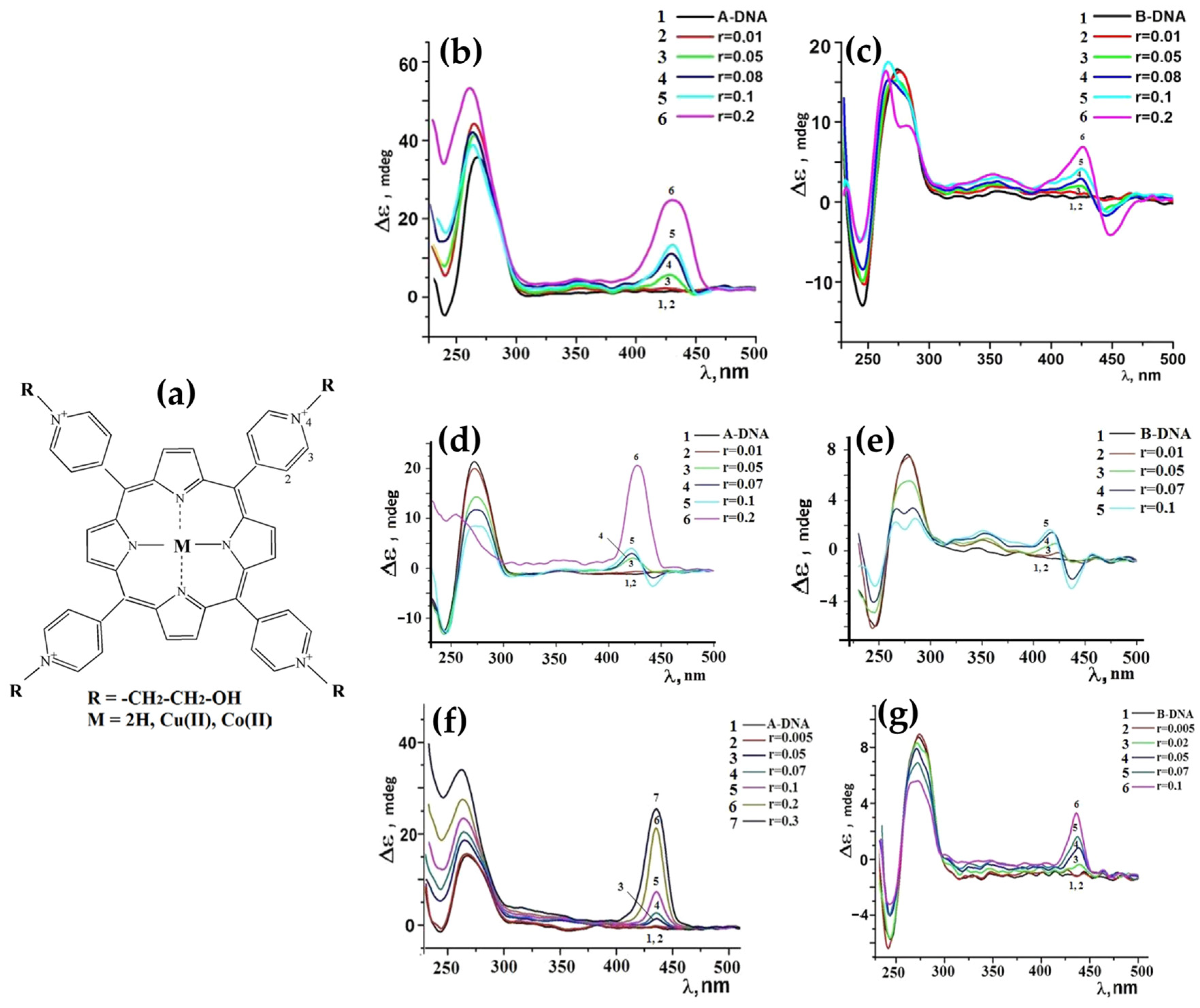
2.1.3. Porphyrin Derivatives as Probes for the A-DNA
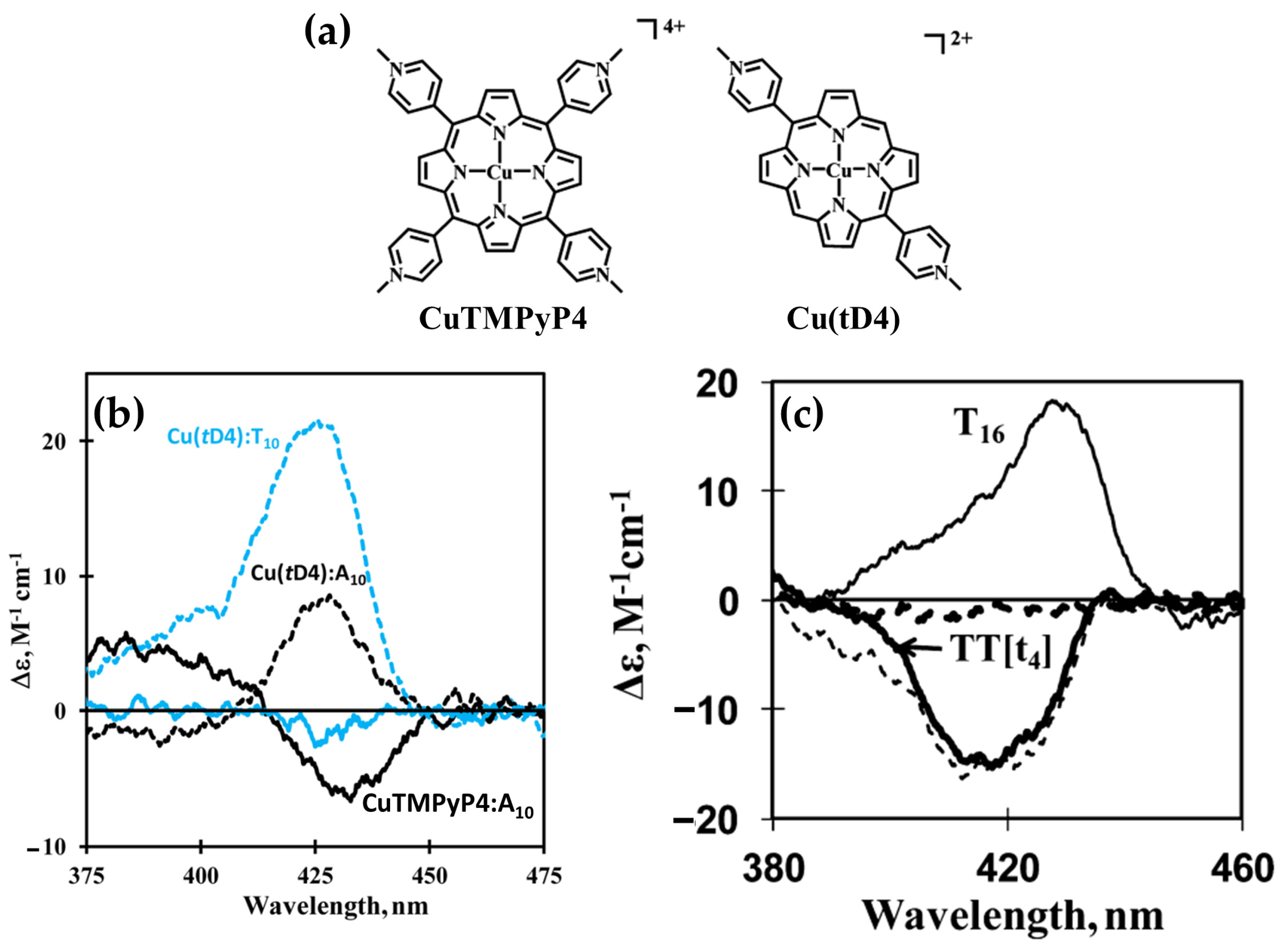
2.1.4. Chiroptical and Structural Insights into Porphyrin Interactions with Single-Stranded DNA
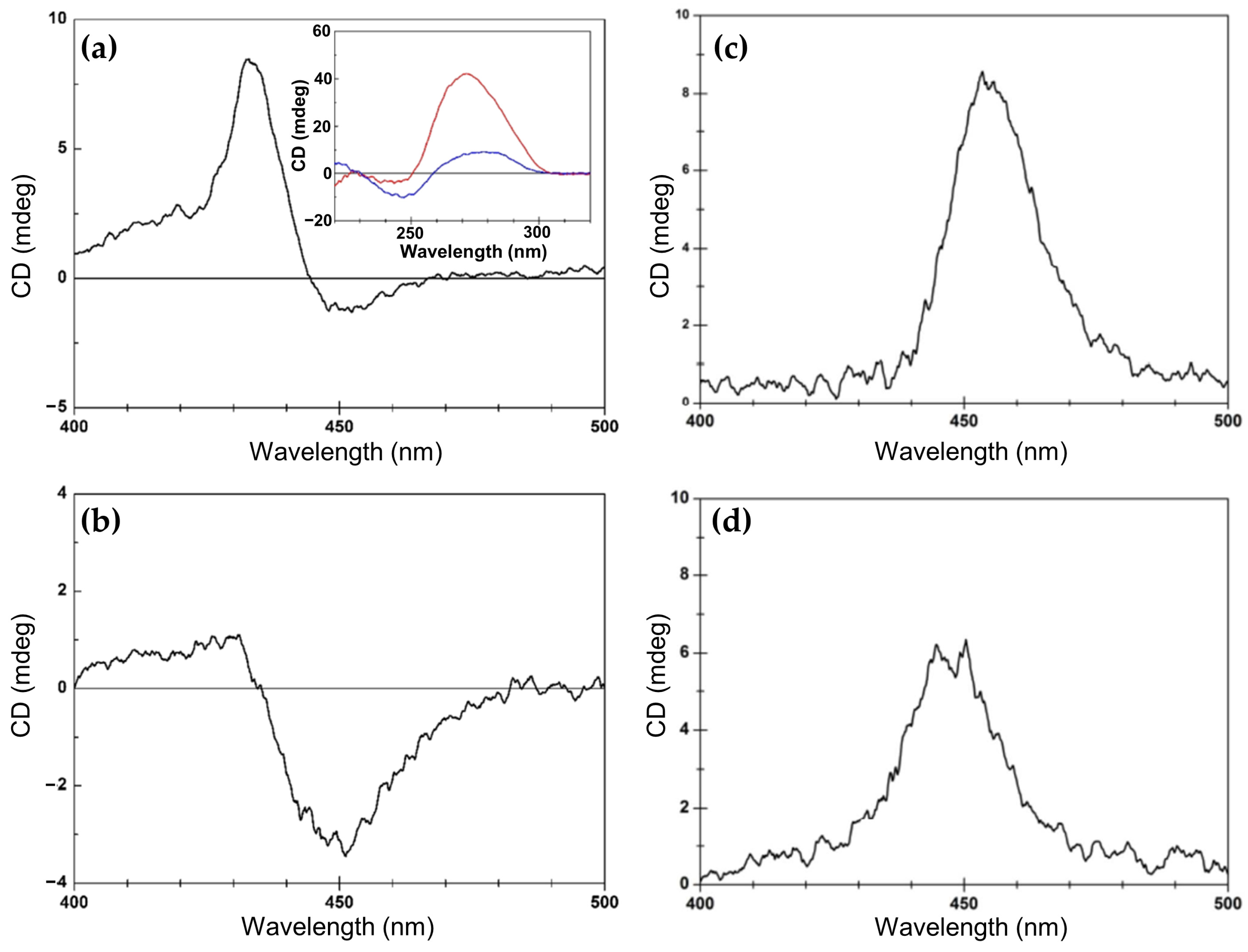
2.1.5. Chiroptical and Structural Insights into Porphyrin Interactions with G-Quadruplex DNA
2.1.6. Chiroptical Signals and Interactions of Porphyrin Derivatives with I-Motif and E-Motif Structures
2.2. RNA Structural Diversity
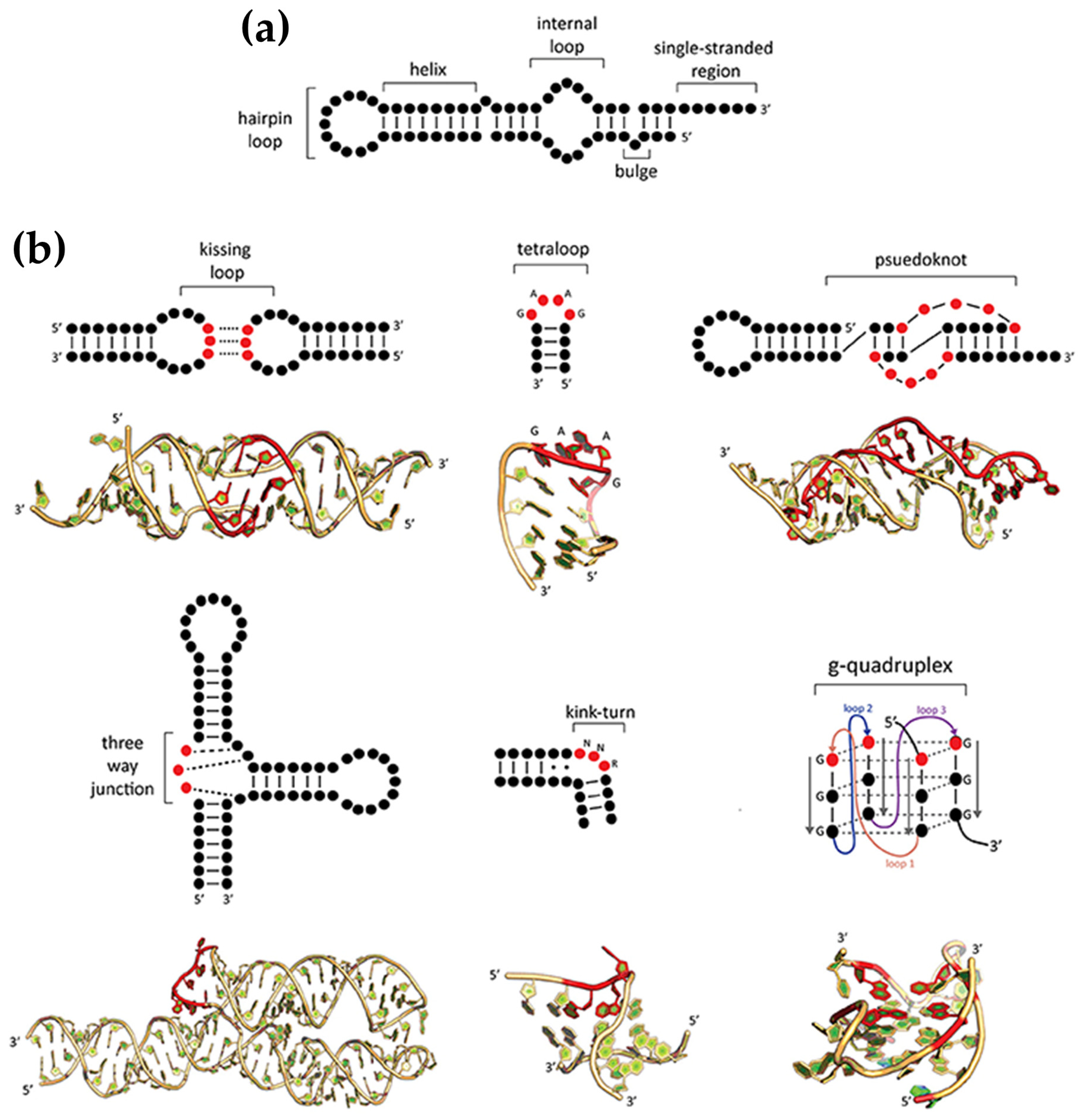
Chiroptical and Structural Insights into Porphyrin Interactions with RNA Structures
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Auwärter, W.; Écija, D.; Klappenberger, F.; Barth, J.V. Porphyrins at Interfaces. Nat. Chem. 2015, 7, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Battersby, A.R. Tetrapyrroles: The Pigments of Life. Nat. Prod. Rep. 2000, 17, 507–526. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Osuka, A.; Vogel, E.; Osuka, A.; Saito, S. Expanded Porphyrins: Intriguing Structures, Electronic Properties, and Reactivities. Angew. Chem. Int. Ed. 2011, 50, 4342–4373. [Google Scholar] [CrossRef]
- Tran-Thi, T.H. Assemblies of Phthalocyanines with Porphyrins and Porphyrazines: Ground and Excited State Optical Properties. Coord. Chem. Rev. 1997, 160, 53–91. [Google Scholar] [CrossRef]
- Senge, M.O.; Fazekas, M.; Notaras, E.G.A.; Blau, W.J.; Zawadzka, M.; Locos, O.B.; Mhuircheartaigh, E.M.N. Nonlinear Optical Properties of Porphyrins. Adv. Mater. 2007, 19, 2737–2774. [Google Scholar] [CrossRef]
- Adinehnia, M.; Eskelsen, J.R.; Hipps, K.W.; Mazur, U. Mechanical Behavior of Crystalline Ionic Porphyrins. J. Porphyr. Phthalocyanines 2019, 23, 855–866. [Google Scholar] [CrossRef]
- Phillips, J.N. Physico-Chemical Properties of Porphyrins. Compr. Biochem. 1963, 9, 34–72. [Google Scholar] [CrossRef]
- Biesaga, M.; Pyrzyńska, K.; Trojanowicz, M. Porphyrins in Analytical Chemistry. A Review. Talanta 2000, 51, 209–224. [Google Scholar] [CrossRef]
- Brothers, P.J.; Collman, J.P. The Organometallic Chemistry of Transition-Metal Porphyrin. Acc. Chem. Res. 1986, 19, 209–215. [Google Scholar] [CrossRef]
- Chilukuri, B.; Mazur, U.; Hipps, K.W. Structure, Properties, and Reactivity of Porphyrins on Surfaces and Nanostructures with Periodic DFT Calculations. Appl. Sci. 2020, 10, 740. [Google Scholar] [CrossRef]
- Huang, H.; Song, W.; Rieffel, J.; Lovell, J.F. Emerging Applications of Porphyrins in Photomedicine. Front. Phys. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Ramzan, M.; Qureshi, A.K.; Azhar Khan, M.; Tariq, M. Emerging Applications of Porphyrins and Metalloporphyrins in Biomedicine and Diagnostic Magnetic Resonance Imaging. Biosensors 2018, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Tsolekile, N.; Nelana, S.; Oluwafemi, O.S. Porphyrin as Diagnostic and Therapeutic Agent. Molecules 2019, 24, 2669. [Google Scholar] [CrossRef] [PubMed]
- Barona-Castaño, J.C.; Carmona-Vargas, C.C.; Brocksom, T.J.; de Oliveira, K.T.; Graça, M.; Neves, P.M.S.; Amparo, M.; Faustino, F. Porphyrins as Catalysts in Scalable Organic Reactions. Molecules 2016, 21, 310. [Google Scholar] [CrossRef]
- Li, L.L.; Diau, E.W.G. Porphyrin -Sensitized Solar Cells. Chem. Soc. Rev. 2012, 42, 291–304. [Google Scholar] [CrossRef]
- Villari, V.; Gaeta, M.; D’Urso, A.; Micali, N. Porphyrin/Carbon Nanodot Supramolecular Complexes and Their Optical Properties. Colloids Surf. A Physicochem. Eng. Asp. 2022, 648, 129436. [Google Scholar] [CrossRef]
- Paolesse, R.; Nardis, S.; Monti, D.; Stefanelli, M.; di Natale, C. Porphyrinoids for Chemical Sensor Applications. Chem. Rev. 2017, 117, 2517–2583. [Google Scholar] [CrossRef]
- Jurow, M.; Schuckman, A.E.; Batteas, J.D.; Drain, C.M. Porphyrins as Molecular Electronic Components of Functional Devices. Coord. Chem. Rev. 2010, 254, 2297–2310. [Google Scholar] [CrossRef]
- Travagliante, G.; Gaeta, M.; Purrello, R.; D’Urso, A. Recognition and Sensing of Chiral Organic Molecules by Chiral Porphyrinoids: A Review. Chemosensors 2021, 9, 204. [Google Scholar] [CrossRef]
- D’Urso, A.; Marino, N.; Gaeta, M.; Rizzo, M.S.; Cristaldi, D.A.; Fragalà, M.E.; Pappalardo, S.; Gattuso, G.; Notti, A.; Parisi, M.F.; et al. Porphyrin Stacks as an Efficient Molecular Glue to Induce Chirality in Hetero-Component Calixarene–Porphyrin Assemblies. New J. Chem. 2017, 41, 8078–8083. [Google Scholar] [CrossRef]
- Gaeta, M.; Rodolico, E.; Fragalà, M.E.; Pappalardo, A.; Pisagatti, I.; Gattuso, G.; Notti, A.; Parisi, M.F.; Purrello, R.; D’Urso, A. Self-Assembly of Discrete Porphyrin/Calix[4]Tube Complexes Promoted by Potassium Ion Encapsulation. Molecules 2021, 26, 704. [Google Scholar] [CrossRef] [PubMed]
- Bergin, E. Asymmetric Catalysis. Annu. Rep. Prog. Chem. Sect. B 2012, 108, 353–371. [Google Scholar] [CrossRef]
- Gaeta, M.; Randazzo, R.; Villari, V.; Micali, N.; Pezzella, A.; Purrello, R.; d’Ischia, M.; D’Urso, A. En Route to a Chiral Melanin: The Dynamic “From-Imprinted-to-Template” Supramolecular Role of Porphyrin Hetero-Aggregates During the Oxidative Polymerization of L-DOPA. Front. Chem. 2020, 8, 616961. [Google Scholar] [CrossRef]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of Electronic Circular Dichroism in Configurational and Conformational Analysis of Organic Compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef]
- Sato, H.; Yajima, T.; Yamagishi, A. Chiroptical Studies on Supramolecular Chirality of Molecular Aggregates. Chirality 2015, 27, 659–666. [Google Scholar] [CrossRef]
- David, A.H.G.; Stoddart, J.F. Chiroptical Properties of Mechanically Interlocked Molecules. Isr. J. Chem. 2021, 61, 608–621. [Google Scholar] [CrossRef]
- D’Urso, A.; Choi, J.K.; Shabbir-Hussain, M.; Ngwa, F.N.; Lambousis, M.I.; Purrello, R.; Balaz, M. Recognition of Left-Handed Z-DNA of Short Unmodified Oligonucleotides under Physiological Ionic Strength Conditions. Biochem. Biophys. Res. Commun. 2010, 397, 329–332. [Google Scholar] [CrossRef]
- Gjuroski, I.; Furrer, J.; Vermathen, M.; Zacharis, C.K.; Tzanavaras, P.D. Probing the Interactions of Porphyrins with Macromolecules Using NMR Spectroscopy Techniques. Molecules 2021, 26, 1942. [Google Scholar] [CrossRef]
- Mihara, H.; Haruta, Y.; Sakamoto, S.; Nishino, N.; Aoyagi, H. Chiral Assembly of Porphyrins Regulated by Amphiphilic α-Helix Peptides. Chem. Lett. 1996, 25, 1–2. [Google Scholar] [CrossRef]
- Fukushima, Y. Interaction of Porphyrin Derivatives with a β-Sheet Structure of a Zwitterionic Polypeptide in Aqueous Solution. Polym. Bull. 2001, 45, 479–485. [Google Scholar] [CrossRef]
- Urbanova, M.; Urbanova, U. Bioinspired Interactions Studied by Vibrational Circular Dichroism. Chirality 2009, 21, E215–E230. [Google Scholar] [CrossRef] [PubMed]
- Palivec, L.; Urbanová, M.; Volka, K. Circular Dichroism Spectroscopic Study of Non-Covalent Interactions of Poly-L-Glutamic Acid with a Porphyrin Derivative in Aqueous Solutions. J. Pept. Sci. 2005, 11, 536–545. [Google Scholar] [CrossRef]
- Occhiuto, I.G.; Samperi, M.; Trapani, M.; de Luca, G.; Romeo, A.; Pasternack, R.F.; Scolaro, L.M. Aggregates of a Cationic Porphyrin as Supramolecular Probes for Biopolymers. J. Inorg. Biochem. 2015, 153, 361–366. [Google Scholar] [CrossRef]
- Thorpe, S.L.; Snyder, G.N.; Mammana, A. Spectroscopic Study of Porphyrin Self-Assembly: Role of PH, Time, and Chiral Template. Chirality 2020, 32, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Charvátová, J.; Kaika, V.; Král, V.; Kdeyl, Z. Capillary Electrochromatographic Study of the Interactions of Porphyrin Derivatives with Amino Acids and Oligopeptides. J. Chromatogr. B 2002, 770, 165–175. [Google Scholar] [CrossRef]
- Gibbs, E.J.; Tinoco, I.; Maestre, M.F.; Ellinas, P.A.; Pasternack, R.F. Self-Assembly of Porphyrins on Nucleic Acid Templates. Biochem. Biophys. Res. Commun. 1988, 157, 350–358. [Google Scholar] [CrossRef]
- Pasternack, R.F. Circular Dichroism and the Interactions of Water Soluble Porphyrins with DNA—A Minireview. Chirality 2003, 15, 329–332. [Google Scholar] [CrossRef]
- Qin, T.; Liu, K.; Song, D.; Yang, C.; Zhao, H.; Su, H. Binding Interactions of Zinc Cationic Porphyrin with Duplex DNA: From B-DNA to Z-DNA. Int. J. Mol. Sci. 2018, 19, 1071. [Google Scholar] [CrossRef]
- Gangemi, C.M.A.; D’Agostino, B.; Randazzo, R.; Gaeta, M.; Fragalà, M.E.; Purrello, R.; D’Urso, A. Interaction of Spermine Derivative Porphyrin with DNA. J. Porphyr. Phthalocyanines 2018, 22, 581–587. [Google Scholar] [CrossRef]
- Sabharwal, N.C.; Chen, J.; Lee, J.H.J.; Gangemi, C.M.A.; D’urso, A.; Yatsunyk, L.A. Interactions Between Spermine-Derivatized Tentacle Porphyrins and The Human Telomeric DNA G-Quadruplex. Int. J. Mol. Sci. 2018, 19, 3686. [Google Scholar] [CrossRef]
- Zhang, L.M.; Cui, Y.X.; Zhu, L.N.; Chu, J.Q.; Kong, D.M. Cationic Porphyrins with Large Side Arm Substituents as Resonance Light Scattering Ratiometric Probes for Specific Recognition of Nucleic Acid G-Quadruplexes. Nucleic Acids Res. 2019, 47, 2727–2738. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Nakanishi, K.; Berova, N. Porphyrins and Metalloporphyrins: Versatile Circular Dichroic Reporter Groups for Structural Studies. Chirality 2000, 12, 237–255. [Google Scholar] [CrossRef]
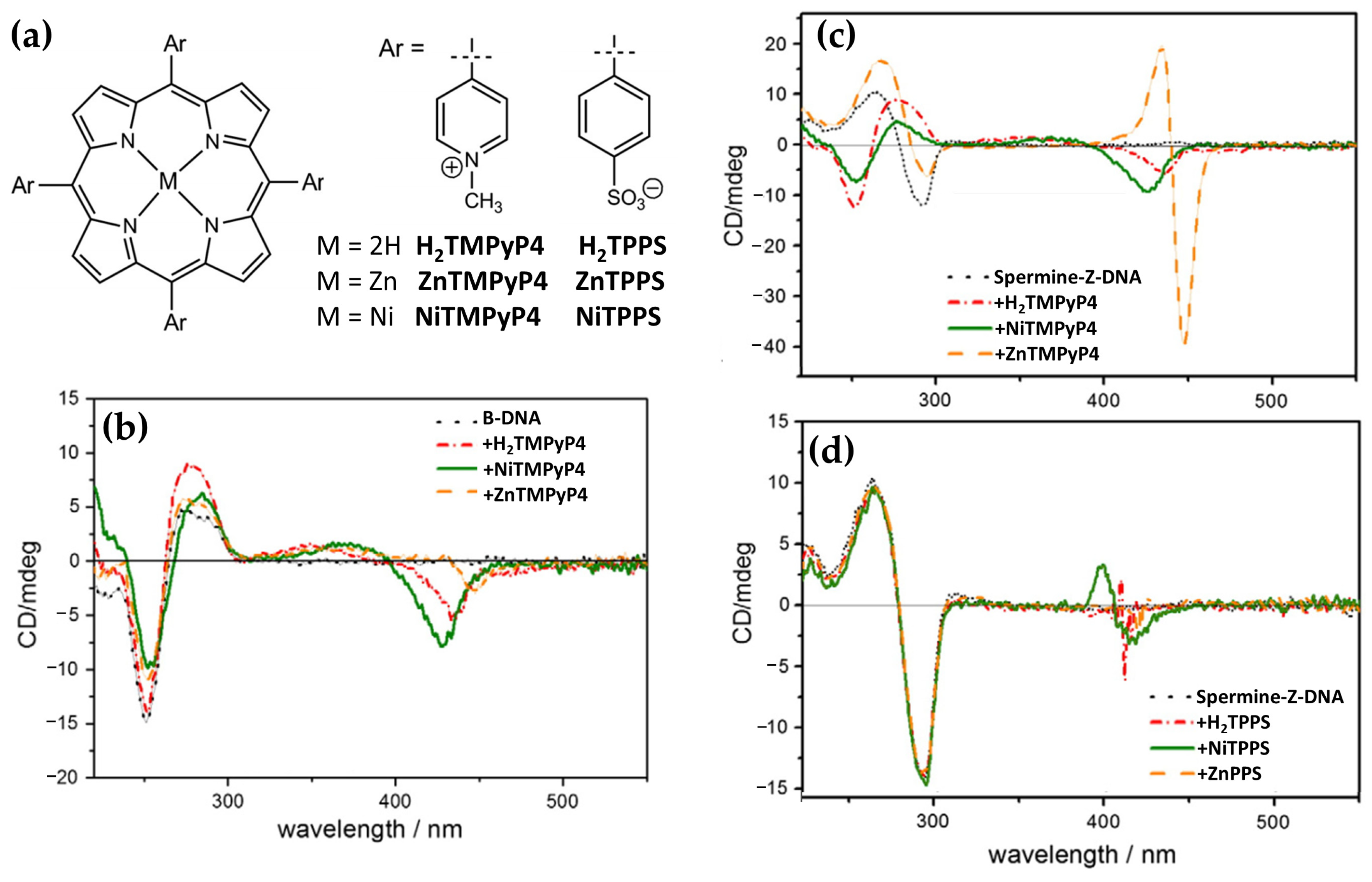
- Choi, J.K.; D’Urso, A.; Balaz, M. Chiroptical Properties of Anionic and Cationic Porphyrins and Metalloporphyrins in Complex with Left-Handed Z-DNA and Right-Handed B-DNA. J. Inorg. Biochem. 2013, 127, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Spikes, J.D.; Jori, G. Photodynamic Therapy of Tumours and Other Diseases Using Porphyrins. Lasers Med. Sci. 1987, 2, 3–15. [Google Scholar] [CrossRef]
- Tian, J.; Huang, B.; Nawaz, M.H.; Zhang, W. Recent Advances of Multi-Dimensional Porphyrin-Based Functional Materials in Photodynamic Therapy. Coord. Chem. Rev. 2020, 420, 213410. [Google Scholar] [CrossRef]
- D’Urso, A.; Mammana, A.; Balaz, M.; Holmes, A.E.; Berova, N.; Lauceri, R.; Purrello, R. Interactions of a Tetraanionic Porphyrin with DNA: From a Z-DNA Sensor to a Versatile Supramolecular Device. J. Am. Chem. Soc. 2009, 131, 2046–2047. [Google Scholar] [CrossRef]
- Wahl, M.C.; Sundaralingam, M. Crystal Structures of A-DNA. Biopolymers 1997, 44, 45–63. [Google Scholar] [CrossRef]
- Dickerson, R.E.; Ng, H.L. DNA Structure from A to B. Proc. Natl. Acad. Sci. USA 2001, 98, 6986–6988. [Google Scholar] [CrossRef]
- Herbert, A.; Rich, A. Left-Handed Z-DNA: Structure and Function. Genetica 1999, 106, 37–47. [Google Scholar] [CrossRef]
- Rich, A.; Zhang, S. Z-DNA: The Long Road to Biological Function. Nat. Rev. Genet. 2003, 4, 566–572. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA Secondary Structures: Stability and Function of G-Quadruplex Structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M.; Frank-Kamenetskii, M.D. H-DNA and related structures. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 541–576. [Google Scholar] [CrossRef] [PubMed]
- Schroth, G.P.; Ho, P.S. Occurrence of Potential Cruciform and H-DNA Forming Sequences in Genomic DNA. Nucleic Acids Res. 1995, 23, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Lipps, H.J.; Rhodes, D. G-Quadruplex Structures: In Vivo Evidence and Function. Trends Cell Biol. 2009, 19, 414–422. [Google Scholar] [CrossRef]
- Travers, A.; Muskhelishvili, G. DNA Structure and Function. FEBS J. 2015, 282, 2279–2295. [Google Scholar] [CrossRef]
- Day, H.A.; Pavlou, P.; Waller, Z.A.E. I-Motif DNA: Structure, Stability and Targeting with Ligands. Bioorg. Med. Chem. 2014, 22, 4407–4418. [Google Scholar] [CrossRef]
- Yoga, Y.M.K.; Traore, D.A.K.; Sidiqi, M.; Szeto, C.; Pendini, N.R.; Barker, A.; Leedman, P.J.; Wilce, J.A.; Wilce, M.C.J. Contribution of the First K-Homology Domain of Poly(C)-Binding Protein 1 to Its Affinity and Specificity for C-Rich Oligonucleotides. Nucleic Acids Res. 2012, 40, 5101–5114. [Google Scholar] [CrossRef]
- Bansal, A.; Kaushik, S.; Kukreti, S. Non-Canonical DNA Structures: Diversity and Disease Association. Front. Genet. 2022, 13, 959258. [Google Scholar] [CrossRef]
- Chen, L.; Dickerhoff, J.; Sakai, S.; Yang, D. DNA G-Quadruplex in Human Telomeres and Oncogene Promoters: Structures, Functions, and Small Molecule Targeting. Acc. Chem. Res. 2022, 55, 2628–2646. [Google Scholar] [CrossRef]
- Xu, Y. Chemistry in Human Telomere Biology: Structure, Function and Targeting of Telomere DNA/RNA. Chem. Soc. Rev. 2011, 40, 2719–2740. [Google Scholar] [CrossRef]
- Sun, D.; Hurley, L.H. The Importance of Negative Superhelicity in Inducing the Formation of G-Quadruplex and i-Motif Structures in the c-Myc Promoter: Implications for Drug Targeting and Control of Gene Expression. J. Med. Chem. 2009, 52, 2863–2874. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Gan, J.; Huang, Z. Structure-Based DNA-Targeting Strategies with Small Molecule Ligands for Drug Discovery. Med. Res. Rev. 2013, 33, 1119–1173. [Google Scholar] [CrossRef] [PubMed]
- Greco, F.; Musumeci, D.; Borbone, N.; Falanga, A.P.; D’errico, S.; Terracciano, M.; Piccialli, I.; Roviello, G.N.; Oliviero, G. Exploring the Parallel G-Quadruplex Nucleic Acid World: A Spectroscopic and Computational Investigation on the Binding of the c-Myc Oncogene NHE III1 Region by the Phytochemical Polydatin. Molecules 2022, 27, 2997. [Google Scholar] [CrossRef] [PubMed]
- Fiel, R.J.; Howard, J.C.; Mark, E.H.; Gupta, N.D. Interaction of DNA with a Porphyrin Ligand: Evidence for Intercalation. Nucleic Acids Res. 1979, 6, 3093. [Google Scholar] [CrossRef]
- Fiel, R.J.; Munson, B.R. Binding of Meso-Tetra (4-N-Methylpyridyl) Porphine to DNA. Nucleic Acids Res. 1980, 8, 2835. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Gibbs, E.J.; Villafranca, J.J. Interactions of Porphyrins with Nucleic Acids. Biochemistry 1983, 22, 2406–2414. [Google Scholar] [CrossRef]
- Scolaro, L.M.; Romeo, A.; Pasternack, R.F. Tuning Porphyrin/DNA Supramolecular Assemblies by Competitive Binding. J. Am. Chem. Soc. 2004, 126, 7178–7179. [Google Scholar] [CrossRef]
- Urso, A.D.; Fragalà, M.E.; Purrello, R. Non-Covalent Interactions of Porphyrinoids with Duplex DNA. Top. Heterocycl. Chem. 2013, 34, 139–174. [Google Scholar] [CrossRef]
- Fiel, R.J. Porphyrin-Nucleic Acid Interactions: A Review. J. Biomol. Struct. Dyn. 1989, 6, 1259–1274. [Google Scholar] [CrossRef]
- Mathew, D.; Sujatha, S. Interactions of Porphyrins with DNA: A Review Focusing Recent Advances in Chemical Modifications on Porphyrins as Artificial Nucleases. J. Inorg. Biochem. 2021, 219, 111434. [Google Scholar] [CrossRef]
- D’Urso, A.; Holmes, A.E.; Berova, N.; Balaz, M.; Purrello, R. Z-DNA Recognition in B-Z-B Sequences by a Cationic Zinc Porphyrin. Chem. Asian J. 2011, 6, 3104–3109. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, A.; Petrovic, A.G.; Fragalà, M.E.; Tamargo, M.A.; Ellestad, G.A.; Purrello, R.; Berova, N. Exploring Nucleic Acid Conformations by Employment of Porphyrin Non-covalent and Covalent Probes and Chiroptical Analysis. In DNA in Supramolecular Chemistry and Nanotechnology, 1st ed.; Stulz, E., Clever, G.H., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; pp. 172–208. [Google Scholar] [CrossRef]
- Dixon, I.M.; Lopez, F.; Tejera, A.M.; Estève, J.P.; Blasco, M.A.; Pratviel, G.; Meunier, B. A G-Quadruplex Ligand with 10000-Fold Selectivity over Duplex DNA. J. Am. Chem. Soc. 2007, 129, 1502–1503. [Google Scholar] [CrossRef] [PubMed]
- Nicoludis, J.M.; Barrett, S.P.; Mergny, J.L.; Yatsunyk, L.A. Interaction of Human Telomeric DNA with N-Methyl Mesoporphyrin IX. Nucleic Acids Res. 2012, 40, 5432. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, I.; Borovok, N.; Kotlyar, A. Interaction of Monomolecular G4-DNA Nanowires with TMPyP: Evidence for Intercalation. Biochemistry 2007, 46, 12925–12929. [Google Scholar] [CrossRef]
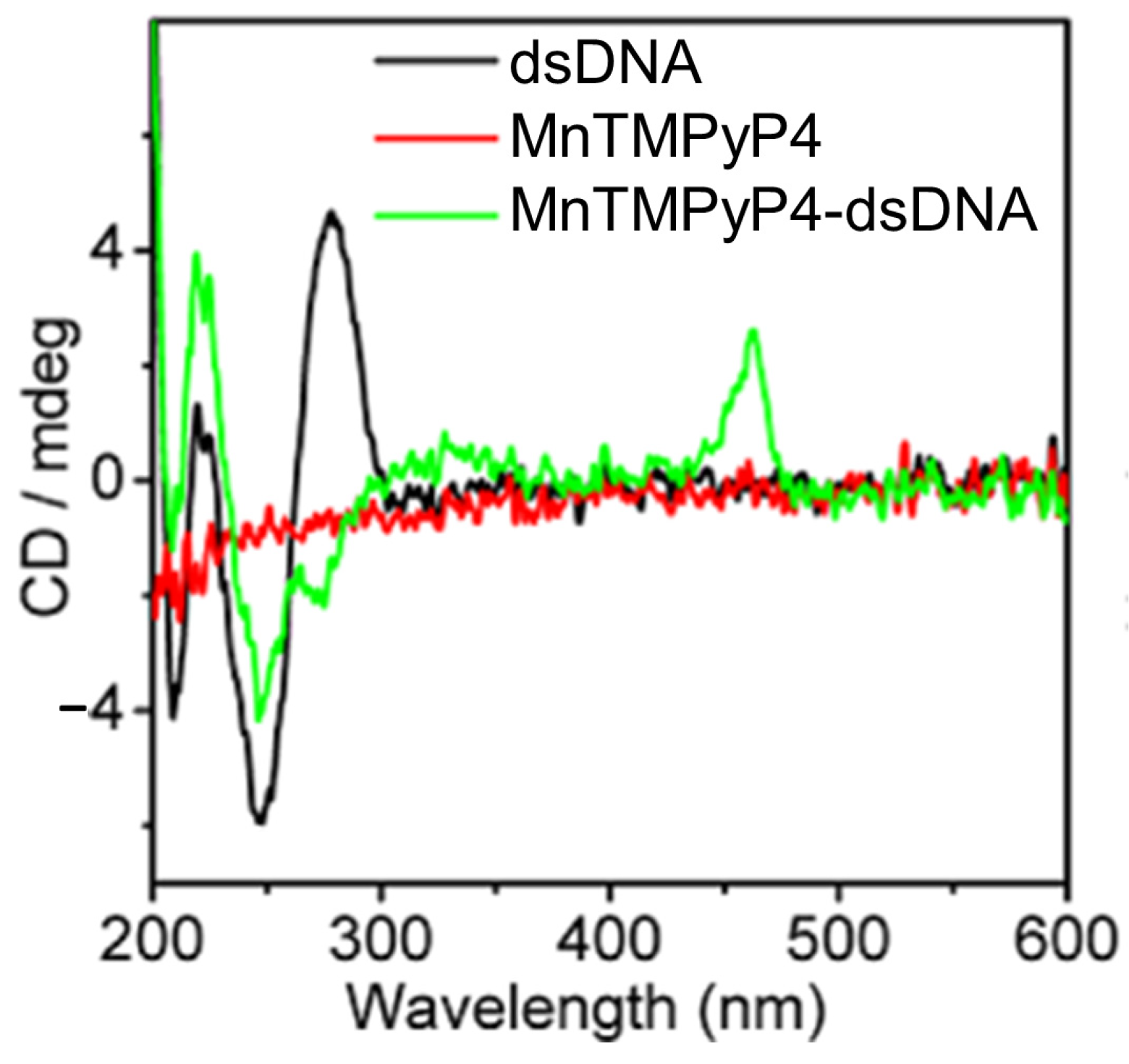
- Xu, J.; Wu, J.; Zong, C.; Ju, H.; Yan, F. Manganese Porphyrin-DsDNA Complex: A Mimicking Enzyme for Highly Efficient Bioanalysis. Anal. Chem. 2013, 85, 3374–3379. [Google Scholar] [CrossRef]
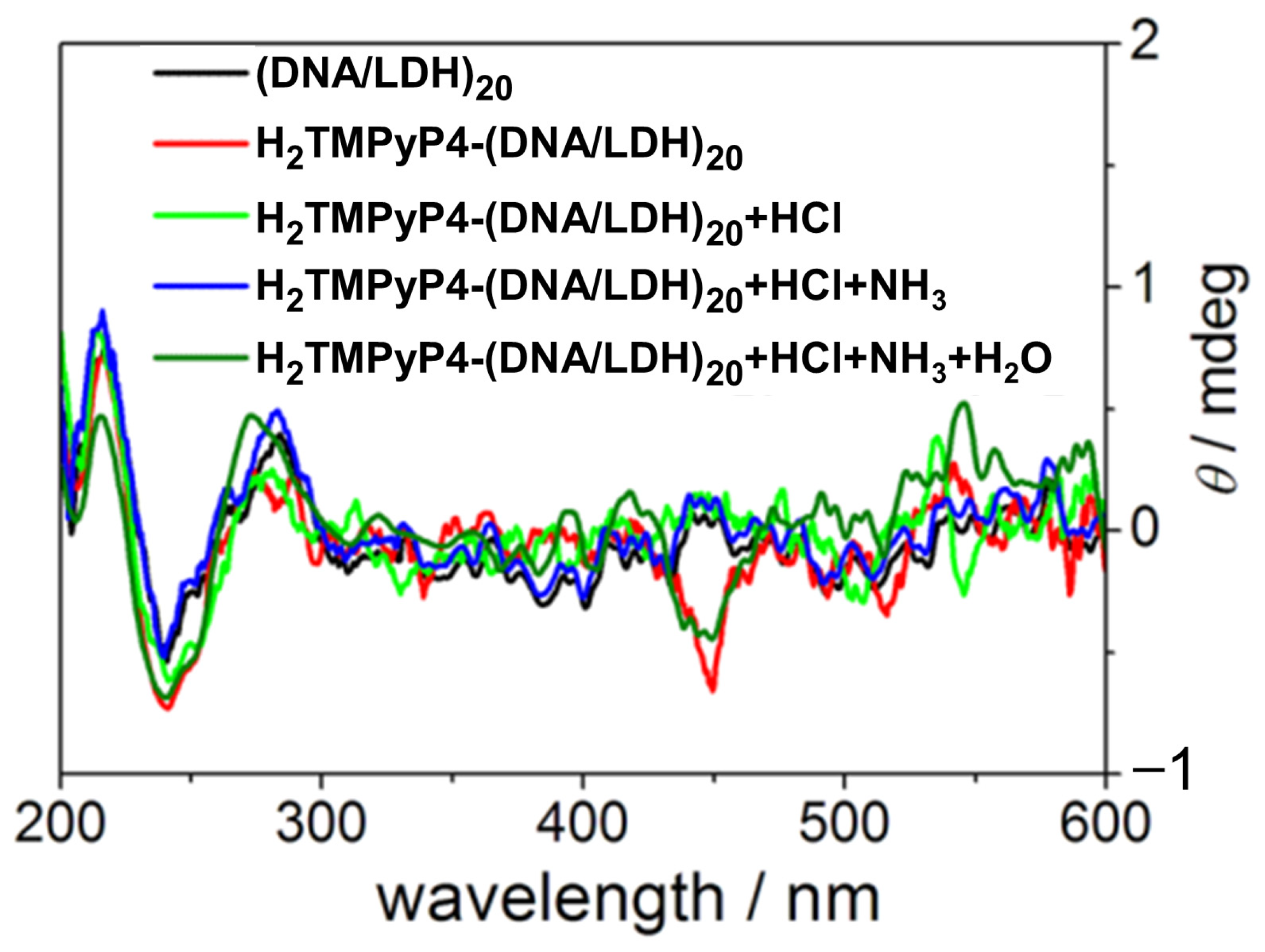
- Shi, W.; Jia, Y.; Xu, S.; Li, Z.; Fu, Y.; Wei, M.; Shi, S. A Chiroptical Switch Based on DNA/Layered Double Hydroxide Ultrathin Films. Langmuir 2014, 30, 12916–12922. [Google Scholar] [CrossRef]
- Bork, M.A.; Gianopoulos, C.G.; Zhang, H.; Fanwick, P.E.; Choi, J.H.; McMillin, D.R. Accessibility and External versus Intercalative Binding to DNA as Assessed by Oxygen-Induced Quenching of the Palladium(II)-Containing Cationic Porphyrins Pd(T4) and Pd(t D4). Biochemistry 2014, 53, 714–724. [Google Scholar] [CrossRef]
- Ghimire, S.; Fanwick, P.E.; McMillin, D.R. DNA-Binding Studies of a Tetraalkyl-Substituted Porphyrin and the Mutually Adaptive Distortion Principle. Inorg. Chem. 2014, 53, 11108–11118. [Google Scholar] [CrossRef]
- Kovaleva, O.A.; Tsvetkov, V.B.; Mamaeva, O.K.; Ol’shevskaya, V.A.; Makarenkov, A.V.; Dezhenkova, L.G.; Semeikin, A.S.; Borisova, O.F.; Shtil, A.A.; Shchyolkina, A.K.; et al. Preferential DNA Photocleavage Potency of Zn(II) over Ni(II) Derivatives of Carboxymethyl Tetracationic Porphyrin: The Role of the Mode of Binding to DNA. Eur. Biophys. J. 2014, 43, 545–554. [Google Scholar] [CrossRef]
- Mandoj, F.; D’Urso, A.; Nardis, S.; Monti, D.; Stefanelli, M.; Gangemi, C.M.A.; Randazzo, R.; Fronczek, F.R.; Smith, K.M.; Paolesse, R. The Interaction of a β-Fused Isoindoline–Porphyrin Conjugate with Nucleic Acids. New J. Chem. 2016, 40, 5662–5665. [Google Scholar] [CrossRef]
- Lee, H.S.; Han, J.H.; Park, J.H.; Heo, M.E.; Hirakawa, K.; Kim, S.K.; Cho, D.W. Relationship between the Photoinduced Electron Transfer and Binding Modes of a Pyrene–Porphyrin Dyad to DNA. Phys. Chem. Chem. Phys. 2017, 19, 27123–27131. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Liu, D.; Zhao, Y.; Wu, F.; Yang, K.; Wang, K. Synthesis, DNA Binding Mode, Singlet Oxygen Photogeneration and DNA Photocleavage Activity of Ruthenium Compounds with Porphyrin–Imidazo[4,5-f]Phenanthroline Conjugated Ligand. Appl. Organomet. Chem. 2018, 32, e4468. [Google Scholar] [CrossRef]
- Cho, S.Y.; Han, J.H.; Jang, Y.J.; Kim, S.K.; Lee, Y.A. Binding Properties of Various Cationic Porphyrins to DNA in the Molecular Crowding Condition Induced by Poly(Ethylene Glycol). ACS Omega 2020, 5, 10459–10465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, W.; Liu, Z.; Li, H.; Liu, Y.; Liu, X.; Han, Z.; He, J.; Zeng, Y.; Guo, Y.; et al. Design, Synthesis, Antitumor Activity and Ct-DNA Binding Study of Photosensitive Drugs Based on Porphyrin Framework. Int. J. Biol. Macromol. 2023, 230, 123147. [Google Scholar] [CrossRef]
- Dröge, P. Protein Tracking-Induced Supercoiling of DNA: A Tool to Regulate DNA Transactions in Vivo? BioEssays 1994, 16, 91–99. [Google Scholar] [CrossRef]
- Kim, Y.G.; Lowenhaupt, K.; Oh, D.B.; Kim, K.K.; Rich, A. Evidence That Vaccinia Virulence Factor E3L Binds to Z-DNA in Vivo: Implications for Development of a Therapy for Poxvirus Infection. Proc. Natl. Acad. Sci. USA 2004, 101, 1514–1518. [Google Scholar] [CrossRef]
- Ditlevson, J.V.; Tornaletti, S.; Belotserkovskii, B.P.; Teijeiro, V.; Wang, G.; Vasquez, K.M.; Hanawalt, P.C. Inhibitory Effect of a Short Z-DNA Forming Sequence on Transcription Elongation by T7 RNA Polymerase. Nucleic Acids Res. 2008, 36, 3163–3170. [Google Scholar] [CrossRef]
- Belmont, P.; Constant, J.F.; Demeunynck, M. Nucleic Acid Conformation Diversity: From Structure to Function and Regulation. Chem. Soc. Rev. 2001, 30, 70–81. [Google Scholar] [CrossRef]
- Krzyźaniak, A.; Salański, P.; Jurczak, J.; Barciszewski, J. B–Z DNA Reversible Conformation Changes Effected by High Pressure. FEBS Lett. 1991, 279, 1–4. [Google Scholar] [CrossRef]
- Ivanov, V.I.; Minyat, E.E. The Transitions between Left- and Right-Handed Forms of Poly(DG-DC). Nucleic Acids Res. 1981, 9, 4783–4798. [Google Scholar] [CrossRef]
- Rich, A.; Nordheim, A.; Wang, A.H.J. The Chemistry and Biology of Left-Handed Z-DNA. Annu. Rev. Biochem. 1983, 53, 791–846. [Google Scholar] [CrossRef]
- Wittig, B.; Dorbic, T.; Rich, A. Transcription Is Associated with Z-DNA Formation in Metabolically Active Permeabilized Mammalian Cell Nuclei. Proc. Natl. Acad. Sci. USA 1991, 88, 2259. [Google Scholar] [CrossRef] [PubMed]
- Latha, K.S.; Anitha, S.; Rao, K.S.J.; Viswamitra, M.A. Molecular Understanding of Aluminum-Induced Topological Changes in (CCG)12 Triplet Repeats: Relevance to Neurological Disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2002, 1588, 56–64. [Google Scholar] [CrossRef]
- Wang, A.H.J.; Quigley, G.J.; Kolpak, F.J.; Crawford, J.L.; Van Boom, J.H.; Van Der Marel, G.; Rich, A. Molecular Structure of a Left-Handed Double Helical DNA Fragment at Atomic Resolution. Nature 1979, 282, 680–686. [Google Scholar] [CrossRef]
- Oh, D.B.; Kim, Y.G.; Rich, A. Z-DNA-Binding Proteins Can Act as Potent Effectors of Gene Expression in Vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 16666–16671. [Google Scholar] [CrossRef]
- Liu, H.; Mulholland, N.; Fu, H.; Zhao, K. Cooperative Activity of BRG1 and Z-DNA Formation in Chromatin Remodeling. Mol. Cell. Biol. 2006, 26, 2550–2559. [Google Scholar] [CrossRef]
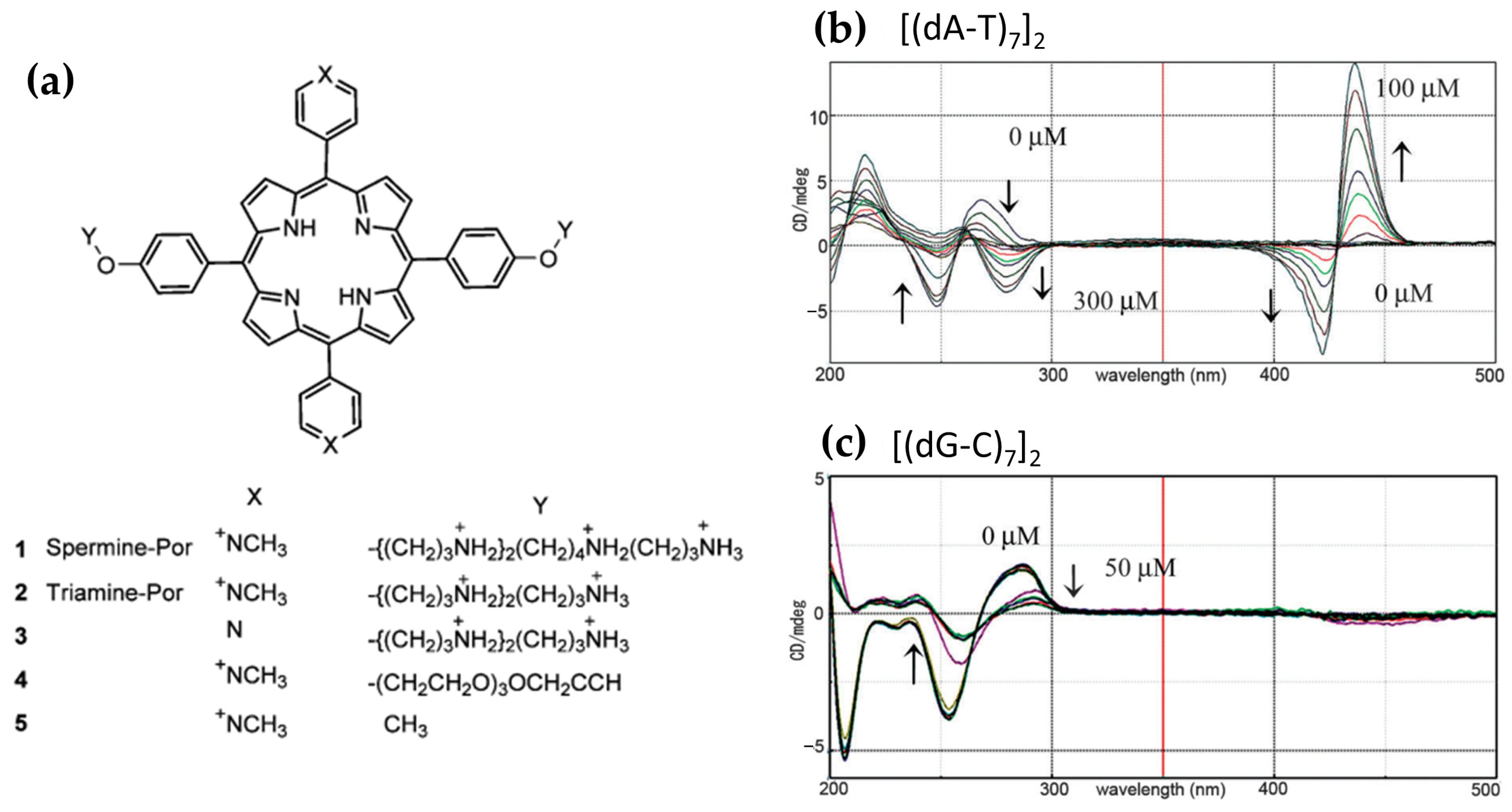
- Sasaki, H.; Sasaki, S. B–Z Transition of (DA-T)n Duplexes Induced by a Spermine Porphyrin-Conjugate via an Intermediate DNA Conformation. Chem. Commun. 2013, 49, 9024–9026. [Google Scholar] [CrossRef]
- Choi, J.K.; Reed, A.; Balaz, M. Chiroptical Properties, Binding Affinity, and Photostability of a Conjugated Zinc Porphyrin Dimer Complexed with Left-Handed Z-DNA and Right-Handed B-DNA. Dalton Trans. 2013, 43, 563–567. [Google Scholar] [CrossRef]
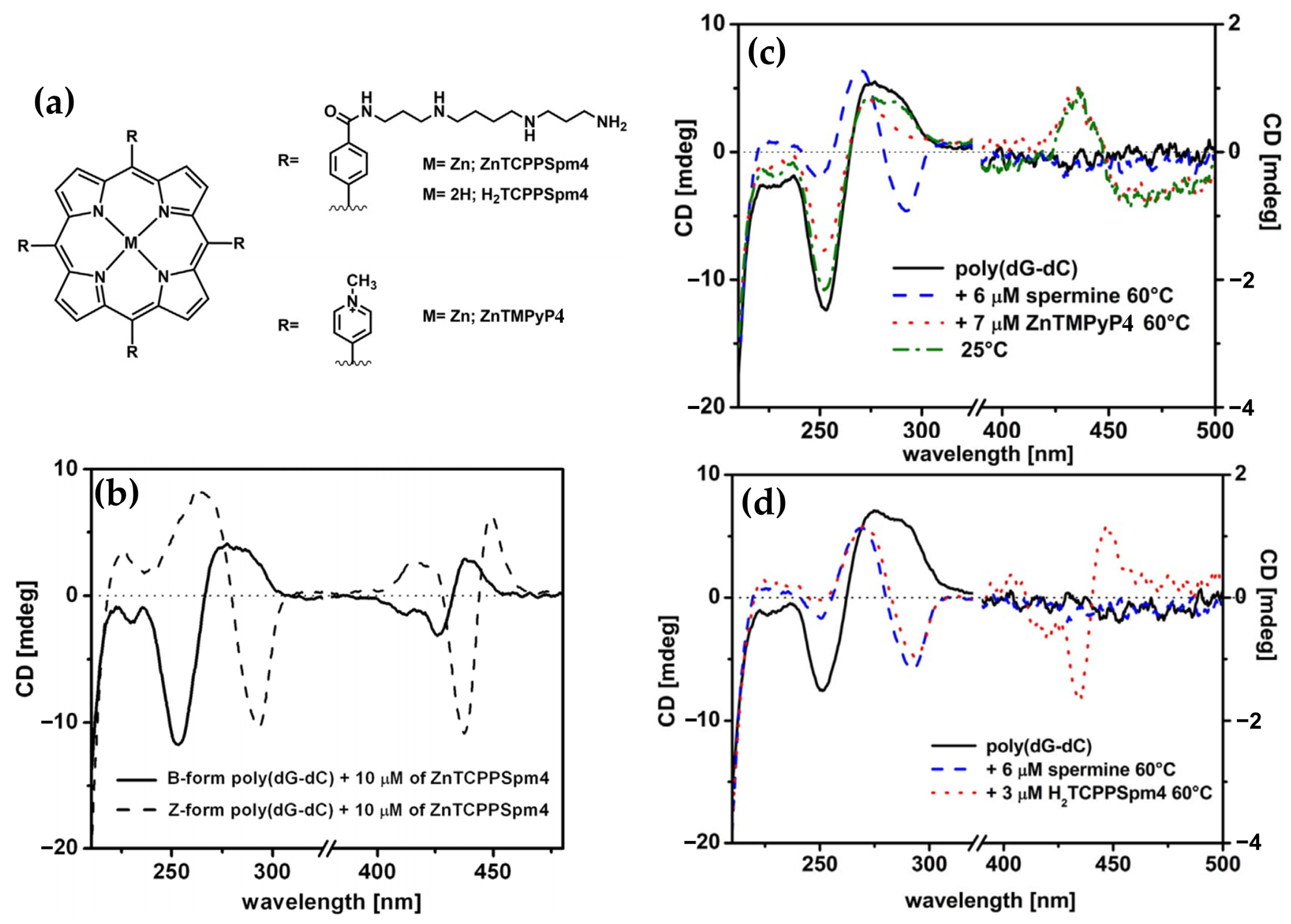
- Gangemi, C.M.A.; D’Urso, A.; Tomaselli, G.A.; Berova, N.; Purrello, R. A Novel Porphyrin-Based Molecular Probe ZnTCPPSpm4 with Catalytic, Stabilizing and Chiroptical Diagnostic Power towards DNA B-Z Transition. J. Inorg. Biochem. 2017, 173, 141–143. [Google Scholar] [CrossRef]
- Gaeta, M.; Farini, S.; Gangemi, C.M.A.; Purrello, R.; D’Urso, A. Interactions of Mono Spermine Porphyrin Derivative with DNAs. Chirality 2020, 32, 1243–1249. [Google Scholar] [CrossRef]
- Franklin, R.E.; Goslino, X.R.G. The Structure of Sodium Thymonucleate Fibres. I. The Influence of Water Content. Acta Crystallogr. 1953, 6, 673–677. [Google Scholar] [CrossRef]
- Neidle, S. New Insights into Sequence-Dependent DNA Structure. Nat. Struct. Biol. 1998, 5, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, V.I.; Minchenkova, L.E.; Minyat, E.E.; Frank-Kamenetskii, M.D.; Schyolkina, A.K. The B to Ā Transition of DNA in Solution. J. Mol. Biol. 1974, 87, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Trantírek, L.; Štefl, R.; Vorlíčková, M.; Koča, J.; Sklenářář, V.; Kypr, J. An A-Type Double Helix of DNA Having B-Type Puckering of the Deoxyribose Rings. J. Mol. Biol. 2000, 297, 907–922. [Google Scholar] [CrossRef]
- Štefl, R.; Trantírek, L.; Vorlíčková, M.; Koča, J.; Sklenář, V.; Kypr, J. A-like Guanine-Guanine Stacking in the Aqueous DNA Duplex of d(GGGGCCCC). J. Mol. Biol. 2001, 307, 513–524. [Google Scholar] [CrossRef]
- Ki, S.L.; Bumbaca, D.; Kosman, J.; Setlow, P.; Jedrzejas, M.J. Structure of a Protein-DNA Complex Essential for DNA Protection in Spores of Bacillus Species. Proc. Natl. Acad. Sci. USA 2008, 105, 2806–2811. [Google Scholar] [CrossRef]
- Mohr, S.C.; Sokolov, N.V.H.A.; He, C.; Setlow, P. Binding of Small Acid-Soluble Spore Proteins from Bacillus Subtilis Changes the Conformation of DNA from B to A. Proc. Natl. Acad. Sci. USA 1991, 88, 77–81. [Google Scholar] [CrossRef]
- DiMaio, F.; Yu, X.; Rensen, E.; Krupovic, M.; Prangishvili, D.; Egelman, E.H. A Virus That Infects a Hyperthermophile Encapsidates A-Form DNA. Science 2015, 348, 914–917. [Google Scholar] [CrossRef]
- Whelan, D.R.; Hiscox, T.J.; Rood, J.I.; Bambery, K.R.; McNaughton, D.; Wood, B.R. Detection of an En Masse and Reversible B- to A-DNA Conformational Transition in Prokaryotes in Response to Desiccation. J. R. Soc. Interface 2014, 11, 20140454. [Google Scholar] [CrossRef]
- Whitley, D.C.; Runfola, V.; Cary, P.; Nazlamova, L.; Guille, M.; Scarlett, G. APTE: Identification of Indirect Read-out A-DNA Promoter Elements in Genomes. BMC Bioinform. 2014, 15, 288. [Google Scholar] [CrossRef]
- Kawasaki, A.M.; Casper, M.D.; Freier, S.M.; Lesnik, E.A.; Zounes, M.C.; Cummins, L.L.; Gonzalez, C.; Dan Cook, P. Uniformly Modified 2′-Deoxy-2′-Fluoro Phosphorothioate Oligonucleotides as Nuclease-Resistant Antisense Compounds with High Affinity and Specificity for RNA Targets. J. Med. Chem. 1993, 36, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Sundaralingam, M. Crystal Structure and Conformation of a DNA–RNA Hybrid Duplex with a Polypurine RNA Strand: D(TTCTTBr5CTTC)–r(GAAGAAGAA). Structure 1998, 6, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Avetisyan, A.A.; Vardanyan, I.V.; Dalyan, Y.B. Thermodynamics of Interaction of Meso -Tetra-(4N-Oxyethylpyridyl) Porphyrin and Its Cu(II)- and Co(II)- Containing Derivatives with A and B Forms of DNA. J. Porphyr. Phthalocyanines 2017, 21, 731–738. [Google Scholar] [CrossRef]
- Sol Oh, Y.; Jung, M.J.; Kim, S.K.; Lee, Y.A. Comparison of the Binding Geometry of Free-Base and Hexacoordinated Cationic Porphyrins to A- and B-form DNA. ACS Omega 2018, 3, 1315–1321. [Google Scholar] [CrossRef]
- Ha, T.; Kozlov, A.G.; Lohman, T.M. Single-Molecule Views of Protein Movement on Single-Stranded DNA. Annu. Rev. Biophys. 2012, 41, 295–319. [Google Scholar] [CrossRef]
- Wold, M.S. Replication Protein A: A Heterotrimeric, Single-Stranded DNA-Binding Protein Required for Eukaryotic DNA Metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef]
- Shereda, R.D.; Kozlov, A.G.; Lohman, T.M.; Cox, M.M.; Keck, J.L. SSB as an Organizer/Mobilizer of Genome Maintenance Complexes. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 289. [Google Scholar] [CrossRef]
- Kowalczykowski, S.C. Initiation of Genetic Recombination and Recombination-Dependent Replication. Trends Biochem. Sci. 2000, 25, 156–165. [Google Scholar] [CrossRef]
- Bell, C.E. Structure and Mechanism of Escherichia Coli RecA ATPase. Mol. Microbiol. 2005, 58, 358–366. [Google Scholar] [CrossRef]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and Mechanism of Helicases and Nucleic Acid Translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef]
- Gaier, A.J.; Ghimire, S.; Fix, S.E.; McMillin, D.R. Internal versus External Binding of Cationic Porphyrins to Single-Stranded DNA. Inorg. Chem. 2014, 53, 5467–5473. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, G.; Leonard, B.M.; Kubelka, J.; Balaz, M. Supramolecular SsDNA Templated Porphyrin and Metalloporphyrin Nanoassemblies with Tunable Helicity. Chem. Eur. J. 2014, 20, 1878–1892. [Google Scholar] [CrossRef] [PubMed]
- Tannir, S.; Levintov, L.; Townley, M.A.; Leonard, B.M.; Kubelka, J.; Vashisth, H.; Varga, K.; Balaz, M. Functional Nanoassemblies with Mirror-Image Chiroptical Properties Templated by a Single Homochiral DNA Strand. Chem. Mater. 2020, 32, 2272–2281. [Google Scholar] [CrossRef]
- Sen, D.; Gilbert, W. Formation of Parallel Four-Stranded Complexes by Guanine-Rich Motifs in DNA and Its Implications for Meiosis. Nature 1988, 334, 364–366. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Klug, A. Telomeric DNA Dimerizes by Formation of Guanine Tetrads between Hairpin Loops. Nature 1989, 342, 825–829. [Google Scholar] [CrossRef]
- Williamson, J.R.; Raghuraman, M.K.; Cech, T.R. Monovalent Cation-Induced Structure of Telomeric DNA: The G-Quartet Model. Cell 1989, 59, 871–880. [Google Scholar] [CrossRef]
- Marzano, M.; D’Errico, S.; Greco, F.; Falanga, A.P.; Terracciano, M.; Di Prisco, D.; Piccialli, G.; Borbone, N.; Oliviero, G. Polymorphism of G-Quadruplexes Formed by Short Oligonucleotides Containing a 3′-3′ Inversion of Polarity: From G:C:G:C Tetrads to π–π Stacked G-Wires. Int. J. Biol. Macromol. 2023, 253, 127062. [Google Scholar] [CrossRef]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human Telomere, Oncogenic Promoter and 5′-UTR G-Quadruplexes: Diverse Higher Order DNA and RNA Targets for Cancer Therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef]
- Hershman, S.G.; Chen, Q.; Lee, J.Y.; Kozak, M.L.; Yue, P.; Wang, L.S.; Johnson, F.B. Genomic Distribution and Functional Analyses of Potential G-Quadruplex-Forming Sequences in Saccharomyces Cerevisiae. Nucleic Acids Res. 2008, 36, 144–156. [Google Scholar] [CrossRef]
- Murat, P.; Balasubramanian, S. Existence and Consequences of G-Quadruplex Structures in DNA. Curr. Opin. Genet. Dev. 2014, 25, 22–29. [Google Scholar] [CrossRef]
- Salazar, M.; Thompson, B.D.; Kerwin, S.M.; Hurley, L.H. Thermally Induced DNA·RNA Hybrid to G-Quadruplex Transitions: Possible Implications for Telomere Synthesis by Telomerase. Biochemistry 1996, 35, 16110–16115. [Google Scholar] [CrossRef] [PubMed]
- McLuckie, K.I.E.; Di Antonio, M.; Zecchini, H.; Xian, J.; Caldas, C.; Krippendorff, B.F.; Tannahill, D.; Lowe, C.; Balasubramanian, S. G-Quadruplex DNA as a Molecular Target for Induced Synthetic Lethality in Cancer Cells. J. Am. Chem. Soc. 2013, 135, 9640–9643. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, C.; Seimiya, H. G-Quadruplex in Cancer Biology and Drug Discovery. Biochem. Biophys. Res. Commun. 2020, 531, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Hänsel-Hertsch, R.; Simeone, A.; Shea, A.; Hui, W.W.I.; Zyner, K.G.; Marsico, G.; Rueda, O.M.; Bruna, A.; Martin, A.; Zhang, X.; et al. Landscape of G-Quadruplex DNA Structural Regions in Breast Cancer. Nat. Genet. 2020, 52, 878–883. [Google Scholar] [CrossRef]
- Wang, E.; Thombre, R.; Shah, Y.; Latanich, R.; Wang, J. G-Quadruplexes as Pathogenic Drivers in Neurodegenerative Disorders. Nucleic Acids Res. 2021, 49, 4816–4830. [Google Scholar] [CrossRef]
- Cave, J.W.; Willis, D.E.; Cave, J.W.; Willis, D.E. G-Quadruplex Regulation of Neural Gene Expression. FEBS J. 2022, 289, 3284–3303. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Neidle, S. G-Quadruplex Nucleic Acids as Therapeutic Targets. Curr. Opin. Chem. Biol. 2009, 13, 345–353. [Google Scholar] [CrossRef]
- Marzano, M.; Prencipe, F.; Delre, P.; Mangiatordi, G.F.; Travagliante, G.; Ronga, L.; Piccialli, G.; Saviano, M.; D’Errico, S.; Tesauro, D.; et al. A CD Study of a Structure-Based Selection of N-Heterocyclic Bis-Carbene Gold(I) Complexes as Potential Ligands of the G-Quadruplex-Forming Human Telomeric HTel23 Sequence. Molecules 2024, 29, 5446. [Google Scholar] [CrossRef]
- Alessandrini, I.; Recagni, M.; Zaffaroni, N.; Folini, M. On the Road to Fight Cancer: The Potential of G-Quadruplex Ligands as Novel Therapeutic Agents. Int. J. Mol. Sci. 2021, 22, 5947. [Google Scholar] [CrossRef]
- Awadasseid, A.; Ma, X.; Wu, Y.; Zhang, W. G-Quadruplex Stabilization via Small-Molecules as a Potential Anti-Cancer Strategy. Biomed. Pharmacother. 2021, 139, 111550. [Google Scholar] [CrossRef]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-Quadruplexes: A Promising Target for Cancer Therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Hurley, L.H. G-Quadruplex DNA: A Potential Target for Anti-Cancer Drug Design. Trends Pharmacol. Sci. 2000, 21, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Vankayalapati, H.; Shin-Ya, K.; Wierzba, K.; Hurley, L.H. Telomestatin, a Potent Telomerase Inhibitor That Interacts Quite Specifically with the Human Telomeric Intramolecular G-Quadruplex. J. Am. Chem. Soc. 2002, 124, 2098–2099. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.F.; Zhao, P.; Xu, L.C.; Zheng, M.; Lu, J.Z.; Zhao, P.L.; Su, Q.L.; Chen, H.X.; Tang, D.T.; Chen, J.; et al. Synthesis, G-Quadruplexes DNA Binding, and Photocytotoxicity of Novel Cationic Expanded Porphyrins. Bioorg. Chem. 2015, 60, 110–117. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, M.C.; Zheng, M.; Jin, S.F.; Tang, D.T.; Chen, J.; Ma, Y.N.; Lin, J.Q.; Wang, X.H.; Liu, H.J. G-Quadruplex DNA Interactions, Docking and Cell Photocytotoxicity Research of Porphyrin Dyes. Dye. Pigm. 2016, 128, 41–48. [Google Scholar] [CrossRef]
- Sabharwal, N.C.; Mendoza, O.; Nicoludis, J.M.; Ruan, T.; Mergny, J.L.; Yatsunyk, L.A. Investigation of the Interactions between Pt(II) and Pd(II) Derivatives of 5,10,15,20-Tetrakis (N-Methyl-4-Pyridyl) Porphyrin and G-Quadruplex DNA. J. Biol. Inorg. Chem. 2016, 21, 227–239. [Google Scholar] [CrossRef]
- Sun, X.Y.; Zhao, P.; Jin, S.; Liu, M.C.; Wang, X.; Huang, Y.; Cheng, Z.; Yan, S.Q.; Li, Y.Y.; Chen, Y.Q.; et al. Shedding Lights on the Flexible-Armed Porphyrins: Human Telomeric G4 DNA Interaction and Cell Photocytotoxicity Research. J. Photochem. Photobiol. B Biol. 2017, 173, 606–617. [Google Scholar] [CrossRef]
- Joshi, S.; Singh, A.; Kukreti, S. Porphyrin Induced Structural Destabilization of a Parallel DNA G-Quadruplex in Human MRP1 Gene Promoter. J. Mol. Recognit. 2022, 35, e2950. [Google Scholar] [CrossRef]
- D’Urso, A.; Randazzo, R.; Rizzo, V.; Gangemi, C.M.A.; Romanucci, V.; Zarrelli, A.; Tomaselli, G.A.; Milardi, D.; Borbone, N.; Purrello, R.; et al. Stabilization vs. Destabilization of G-Quadruplex Superstructures: The Role of the Porphyrin Derivative Having Spermine Arms. Phys. Chem. Chem. Phys. 2017, 19, 17404–17410. [Google Scholar] [CrossRef]
- Falanga, A.P.; D’Urso, A.; Travagliante, G.; Gangemi, C.M.A.; Marzano, M.; D’Errico, S.; Terracciano, M.; Greco, F.; De Stefano, L.; Dardano, P.; et al. Higher-Order G-Quadruplex Structures and Porphyrin Ligands: Towards a Non-Ambiguous Relationship. Int. J. Biol. Macromol. 2024, 268, 131801. [Google Scholar] [CrossRef]
- Gehring, K.; Leroy, J.L.; Guéron, M. A Tetrameric DNA Structure with Protonated Cytosine-Cytosine Base Pairs. Nature 1993, 363, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.P.; Huppert, J.L.; Waller, Z.A.E. Identification of Multiple Genomic DNA Sequences Which Form I-Motif Structures at Neutral PH. Nucleic Acids Res. 2017, 45, 2951–2959. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Ding, Y.; Rogers, R.A.; Zhu, J.; Zhu, J.; Burton, A.D.; Carlisle, C.B.; Burrows, C.J. 4n-1 Is a “Sweet Spot” in DNA i-Motif Folding of 2′-Deoxycytidine Homopolymers. J. Am. Chem. Soc. 2017, 139, 4682–4689. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wei, C.; Jia, G.; Wang, X.; Feng, Z.; Li, C. Formation of I-Motif Structure at Neutral and Slightly Alkaline PH. Mol. Biosyst. 2010, 6, 580–586. [Google Scholar] [CrossRef]
- Abdelhamid, M.A.S.; Fábián, L.; Macdonald, C.J.; Cheesman, M.R.; Gates, A.J.; Waller, Z.A.E. Redox-Dependent Control of i-Motif DNA Structure Using Copper Cations. Nucleic Acids Res. 2018, 46, 5886–5893. [Google Scholar] [CrossRef]
- Kang, C.H.; Berger, I.; Lockshin, C.; Ratliff, R.; Moyzis, R.; Rich, A. Crystal Structure of Intercalated Four-Stranded d(C3T) at 1.4 A Resolution. Proc. Natl. Acad. Sci. USA 1994, 91, 11636–11640. [Google Scholar] [CrossRef]
- Zeraati, M.; Langley, D.B.; Schofield, P.; Moye, A.L.; Rouet, R.; Hughes, W.E.; Bryan, T.M.; Dinger, M.E.; Christ, D. I-Motif DNA Structures Are Formed in the Nuclei of Human Cells. Nat. Chem. 2018, 10, 631–637. [Google Scholar] [CrossRef]
- Sutherland, C.; Cui, Y.; Mao, H.; Hurley, L.H. A Mechanosensor Mechanism Controls the G-Quadruplex/i-Motif Molecular Switch in the MYC Promoter NHE III1. J. Am. Chem. Soc. 2016, 138, 14138–14151. [Google Scholar] [CrossRef]
- Luo, X.; Zhang, J.; Gao, Y.; Pan, W.; Yang, Y.; Li, X.; Chen, L.; Wang, C.; Wang, Y. Emerging Roles of I-Motif in Gene Expression and Disease Treatment. Front. Pharmacol. 2023, 14, 1136251. [Google Scholar] [CrossRef]
- Niu, K.; Zhang, X.; Deng, H.; Wu, F.; Ren, Y.; Xiang, H.; Zheng, S.; Liu, L.; Huang, L.; Zeng, B.; et al. BmILF and I-Motif Structure Are Involved in Transcriptional Regulation of BmPOUM2 in Bombyx Mori. Nucleic Acids Res. 2018, 46, 1710. [Google Scholar] [CrossRef]
- Kang, H.J.; Kendrick, S.; Hecht, S.M.; Hurley, L.H. The Transcriptional Complex between the BCL2 I-Motif and HnRNP LL Is a Molecular Switch for Control of Gene Expression That Can Be Modulated by Small Molecules. J. Am. Chem. Soc. 2014, 136, 4172–4185. [Google Scholar] [CrossRef] [PubMed]
- Benabou, S.; Aviñó, A.; Eritja, R.; González, C.; Gargallo, R. Fundamental Aspects of the Nucleic Acid I-Motif Structures. RSC Adv. 2014, 4, 26956–26980. [Google Scholar] [CrossRef]
- Rodriguez, J.; Domínguez, A.; Aviñó, A.; Borgonovo, G.; Eritja, R.; Mazzini, S.; Gargallo, R. Exploring the Stabilizing Effect on the I-Motif of Neighboring Structural Motifs and Drugs. Int. J. Biol. Macromol. 2023, 242, 124794. [Google Scholar] [CrossRef]
- Fedoroff, O.Y.; Rangan, A.; Chemeris, V.V.; Hurley, L.H. Cationic Porphyrins Promote the Formation of I-Motif DNA and Bind Peripherally by a Nonintercalative Mechanism. Biochemistry 2000, 39, 15083–15090. [Google Scholar] [CrossRef]
- Fernández, S.; Eritja, R.; Aviñó, A.; Jaumot, J.; Gargallo, R. Influence of PH, Temperature and the Cationic Porphyrin TMPyP4 on the Stability of the i-Motif Formed by the 5′-(C3TA2)4-3′ Sequence of the Human Telomere. Int. J. Biol. Macromol. 2011, 49, 729–736. [Google Scholar] [CrossRef]
- Qin, T.; Liu, K.; Song, D.; Yang, C.; Su, H. Porphyrin Bound to I-Motifs: Intercalation versus External Groove Binding. Chem. Asian J. 2017, 12, 1578–1586. [Google Scholar] [CrossRef]
- Pan, F.; Zhang, Y.; Man, V.H.; Roland, C.; Sagui, C. E-Motif Formed by Extrahelical Cytosine Bases in DNA Homoduplexes of Trinucleotide and Hexanucleotide Repeats. Nucleic Acids Res. 2018, 46, 942–955. [Google Scholar] [CrossRef]
- Mirkin, S.M. Expandable DNA Repeats and Human Disease. Nature 2007, 447, 932–940. [Google Scholar] [CrossRef]
- Verma, A.K.; Khan, E.; Bhagwat, S.R.; Kumar, A. Exploring the Potential of Small Molecule-Based Therapeutic Approaches for Targeting Trinucleotide Repeat Disorders. Mol. Neurobiol. 2019, 57, 566–584. [Google Scholar] [CrossRef]
- Chang, C.; Jhan, C.-R.; Hou, M.-H. The Interaction of DNA-Binding Ligands with Trinucleotide-Repeat DNA: Implications for Therapy and Diagnosis of Neurological Disorders. Curr. Top. Med. Chem. 2015, 15, 1398–1408. [Google Scholar] [CrossRef]
- Chien, C.M.; Wu, P.C.; Satange, R.; Chang, C.C.; Lai, Z.L.; Hagler, L.D.; Zimmerman, S.C.; Hou, M.H. Structural Basis for Targeting T:T Mismatch with Triaminotriazine-Acridine Conjugate Induces a U-Shaped Head-to-Head Four-Way Junction in CTG Repeat DNA. J. Am. Chem. Soc. 2020, 142, 11165–11172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gao, H.; Yan, C.; Yang, T.; Zheng, X.; Xu, Q.; Wang, D.; Zhou, X.S.; Shao, Y. Selectively Recognizing Extrahelical Conformations of DNA Trinucleotide Repeats by a Hydroxylated Porphyrin Ligand. Anal. Chim. Acta 2022, 1190, 339265. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, I.; Bustamante, C. How RNA Folds. J. Mol. Biol. 1999, 293, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Fedoroff, O.Y.; Miller, J.M.; Ribeiro, N.S.; Reid, B.R. The DNA Strand in DNA·RNA Hybrid Duplexes Is Neither B-Form nor A-Form in Solution. Biochemistry 1993, 32, 4207–4215. [Google Scholar] [CrossRef]
- Blythe, A.J.; Fox, A.H.; Bond, C.S. The Ins and Outs of LncRNA Structure: How, Why and What Comes Next? Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 46–58. [Google Scholar] [CrossRef]
- Quigley, G.J.; Rich, A. Structural Domains of Transfer RNA Molecules. Science 1976, 194, 796–806. [Google Scholar] [CrossRef]
- Walter, A.E.; Turner, D.H.; Kim, J.; Lyttle, M.H.; Müller, P.; Mathews, D.H.; Zuker, M. Coaxial Stacking of Helixes Enhances Binding of Oligoribonucleotides and Improves Predictions of RNA Folding. Proc. Natl. Acad. Sci. USA 1994, 91, 9218–9222. [Google Scholar] [CrossRef]
- Lafontaine, D.A.; Norman, D.G.; Lilley, D.M.J. Structure, Folding and Activity of the VS Ribozyme: Importance of the 2-3-6 Helical Junction. EMBO J. 2001, 20, 1415–1424. [Google Scholar] [CrossRef]
- Laing, C.; Schlick, T. Analysis of Four-Way Junctions in RNA Structures. J. Mol. Biol. 2009, 390, 547–559. [Google Scholar] [CrossRef]
- Brunel, C.; Marquet, R.; Romby, P.; Ehresmann, C. RNA Loop–Loop Interactions as Dynamic Functional Motifs. Biochimie 2002, 84, 925–944. [Google Scholar] [CrossRef]
- Staple, D.W.; Butcher, S.E. Pseudoknots: RNA Structures with Diverse Functions. PLoS Biol. 2005, 3, e213. [Google Scholar] [CrossRef] [PubMed]
- Giedroc, D.P.; Cornish, P.V. Frameshifting RNA Pseudoknots: Structure and Mechanism. Virus Res. 2009, 139, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Theimer, C.A.; Blois, C.A.; Feigon, J. Structure of the Human Telomerase RNA Pseudoknot Reveals Conserved Tertiary Interactions Essential for Function. Mol. Cell 2005, 17, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Collie, G.W.; Haider, S.M.; Neidle, S.; Parkinson, G.N. A Crystallographic and Modelling Study of a Human Telomeric RNA (TERRA) Quadruplex. Nucleic Acids Res. 2010, 38, 5569–5580. [Google Scholar] [CrossRef]
- Redon, S.; Reichenbach, P.; Lingner, J. The Non-Coding RNA TERRA Is a Natural Ligand and Direct Inhibitor of Human Telomerase. Nucleic Acids Res. 2010, 38, 5797–5806. [Google Scholar] [CrossRef]
- Draper, D.E. A Guide to Ions and RNA Structure. RNA 2004, 10, 335–343. [Google Scholar] [CrossRef]
- Flores, J.K.; Ataide, S.F. Structural Changes of RNA in Complex with Proteins in the SRP. Front. Mol. Biosci. 2018, 5, 330379. [Google Scholar] [CrossRef]
- Warf, M.B.; Berglund, J.A. The Role of RNA Structure in Regulating Pre-MRNA Splicing. Trends Biochem. Sci. 2009, 35, 169. [Google Scholar] [CrossRef]
- Serganov, A.; Patel, D.J. Ribozymes, Riboswitches and beyond: Regulation of Gene Expression without Proteins. Nat. Rev. Genet. 2007, 8, 776–790. [Google Scholar] [CrossRef]
- Lin, J.C.; Thirumalai, D. Relative Stability of Helices Determines the Folding Landscape of Adenine Riboswitch Aptamers. J. Am. Chem. Soc. 2008, 130, 14080–14081. [Google Scholar] [CrossRef]
- Butcher, S.E.; Pyle, A.M. The Molecular Interactions That Stabilize RNA Tertiary Structure: RNA Motifs, Patterns, and Networks. Acc. Chem. Res. 2011, 44, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Kertesz, M.; Spitale, R.C.; Segal, E.; Chang, H.Y. Understanding the Transcriptome through RNA Structure. Nat. Rev. Genet. 2011, 12, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Childs-Disney, J.L.; Yang, X.; Gibaut, Q.M.R.; Tong, Y.; Batey, R.T.; Disney, M.D. Targeting RNA Structures with Small Molecules. Nat. Rev. Drug Discov. 2022, 21, 736–762. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, R.; Onizuka, K.; Komatsu, K.R.; Miyashita, E.; Murase, H.; Ojima, K.; Ishikawa, S.; Ozawa, M.; Saito, H.; Nagatsugi, F. Large-Scale Analysis of Small Molecule-RNA Interactions Using Multiplexed RNA Structure Libraries. Commun. Chem. 2024, 7, 98. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, S.-J. Advances in Machine-Learning Approaches to RNA-Targeted Drug Design. Artif. Intell. Chem. 2024, 2, 100053. [Google Scholar] [CrossRef]
- Foster, N.; Singhal, A.K.; Smith, M.W.; Marcos, N.G.; Schray, K.J. Interactions of Porphyrins and Transfer RNA. Biochim. Biophys. Acta 1988, 950, 118–131. [Google Scholar] [CrossRef]
- Bustamante, C.; Gurrieri, S.; Pasternack, R.F.; Purrello, R.; Rizzarelli, E. Interaction of Water-Soluble Porphyrins with Single- and Double-Stranded Polyribonucleotides. Biopolymers 1994, 34, 1099–1104. [Google Scholar] [CrossRef]
- Uno, T.; Hamasaki, K.; Tanigawa, M.; Shimabayashi, S. Binding of Meso-Tetrakis(N-Methylpyridinium-4-Yl)Porphyrin to Double Helical RNA and DNA·RNA Hybrids. Inorg. Chem. 1997, 36, 1676–1683. [Google Scholar] [CrossRef]
- Ghazaryan, A.A.; Dalyan, Y.B.; Haroutiunian, S.G.; Tikhomirova, A.; Taulier, N.; Wells, J.W.; Chalikian, T.V. Thermodynamics of Interactions of Water-Soluble Porphyrins with RNA Duplexes. J. Am. Chem. Soc. 2006, 128, 1914–1921. [Google Scholar] [CrossRef]
- Briggs, B.N.; Gaier, A.J.; Fanwick, P.E.; Dogutan, D.K.; McMillin, D.R. Cationic Copper(II) Porphyrins Intercalate into Domains of Double-Stranded RNA. Biochemistry 2012, 51, 7496–7505. [Google Scholar] [CrossRef]
- Travagliante, G.; Gaeta, M.; Gangemi, C.M.A.; Alaimo, S.; Ferro, A.; Purrello, R.; D’Urso, A. Interactions between Achiral Porphyrins and a Mature MiRNA. Nanoscale 2024, 16, 5137–5148. [Google Scholar] [CrossRef] [PubMed]
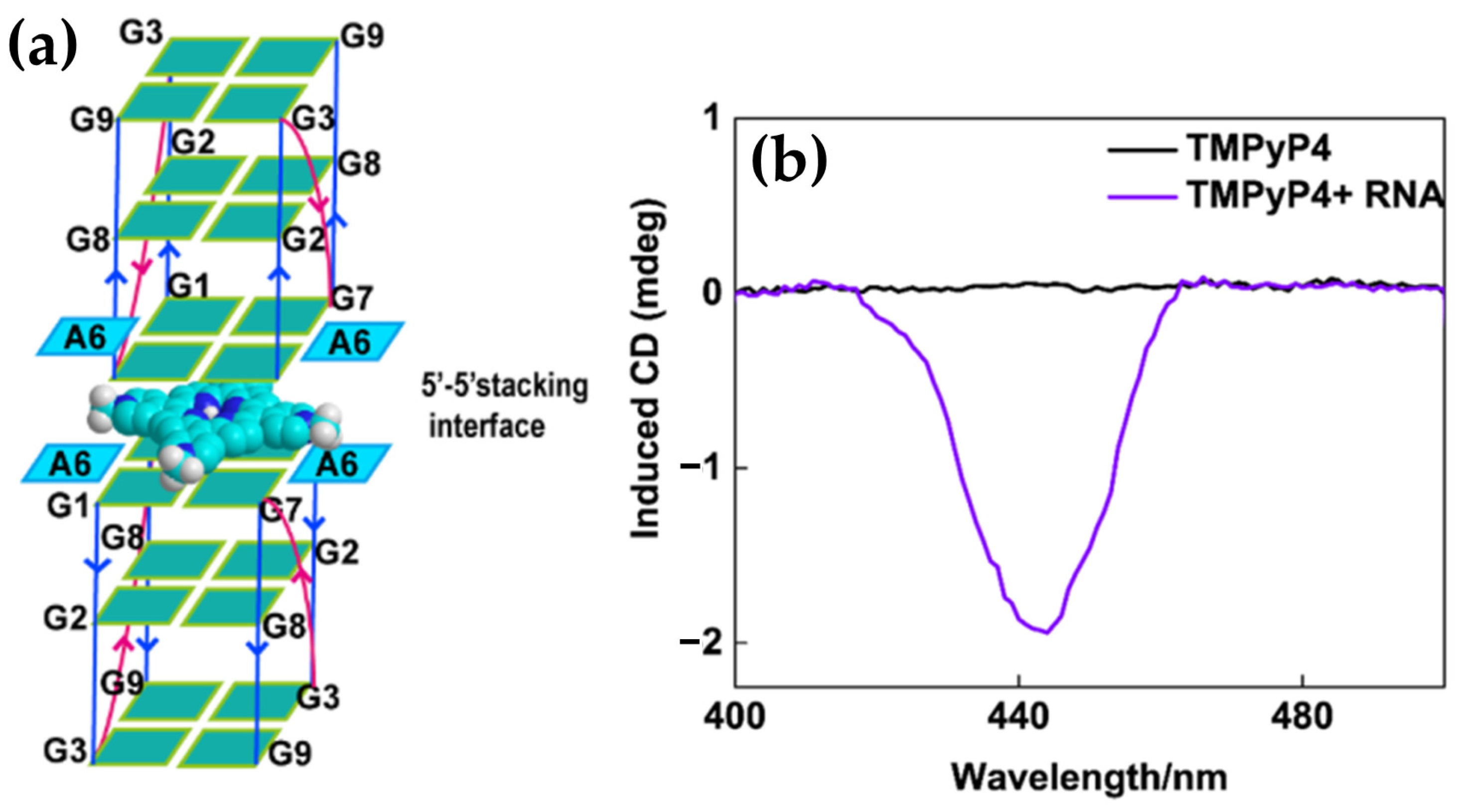
- Qi, Q.; Yang, C.; Xia, Y.; Guo, S.; Song, D.; Su, H. Preferential Binding of π-Ligand Porphyrin Targeting 5′-5′ Stacking Interface of Human Telomeric RNA G-Quadruplex Dimer. J. Phys. Chem. Lett. 2019, 10, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Zhang, Y.; Xia, Y.; Islam, B.; Liu, S.; Gervasio, F.L.; Mulholland, A.J.; Waller, Z.A.E.; Wei, D.; Haider, S. Mechanistic Insights into the Ligand-Induced Unfolding of an RNA G-Quadruplex. J. Am. Chem. Soc. 2022, 144, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Ellington, A.D. Diagnostic Applications of Nucleic Acid Circuits. Acc. Chem. Res. 2014, 47, 1825–1835. [Google Scholar] [CrossRef]
- Cohen, B.A.; Bergkvist, M. Targeted in Vitro Photodynamic Therapy via Aptamer-Labeled, Porphyrin-Loaded Virus Capsids. J. Photochem. Photobiol. B Biol. 2013, 121, 67–74. [Google Scholar] [CrossRef]
- Lebedeva, N.S.; Gubarev, Y.A.; Koifman, M.O.; Koifman, O.I. The Application of Porphyrins and Their Analogues for Inactivation of Viruses. Molecules 2020, 25, 4368. [Google Scholar] [CrossRef]
- Ferino, A.; Nicoletto, G.; D’Este, F.; Zorzet, S.; Lago, S.; Richter, S.N.; Tikhomirov, A.; Shchekotikhin, A.; Xodo, L.E. Photodynamic Therapy for Ras-Driven Cancers: Targeting G-Quadruplex RNA Structures with Bifunctional Alkyl-Modified Porphyrins. J. Med. Chem. 2020, 63, 1245–1260. [Google Scholar] [CrossRef]
- Su, Y.B.; Zhao, X.; Chen, L.J.; Qian, H.L.; Yan, X.P. Fabrication of G-Quadruplex/Porphyrin Conjugated Gold/Persistent Luminescence Theranostic Nanoprobe for Imaging-Guided Photodynamic Therapy. Talanta 2021, 233, 122567. [Google Scholar] [CrossRef]
- Gibson, L.E.; Wright, D.W. Sensitive Method for Biomolecule Detection Utilizing Signal Amplification with Porphyrin Nanoparticles. Anal. Chem. 2016, 88, 5928–5933. [Google Scholar] [CrossRef]
- Liu, S.; Huo, Y.; Fan, L.; Ning, B.; Sun, T.; Gao, Z. Rapid and Ultrasensitive Detection of DNA and MicroRNA-21 Using a Zirconium Porphyrin Metal-Organic Framework-Based Switch Fluorescence Biosensor. Anal. Chim. Acta 2022, 1192, 339340. [Google Scholar] [CrossRef]
- Monteiro, A.R.; Ramos, C.I.V.; Fateixa, S.; Moura, N.M.M.; Neves, M.G.P.M.S.; Trindade, T. Hybrids Based on Graphene Oxide and Porphyrin as Tools for Detection and Stabilization of DNA G-Quadruplexes. ACS Omega 2018, 3, 11184–11191. [Google Scholar] [CrossRef]

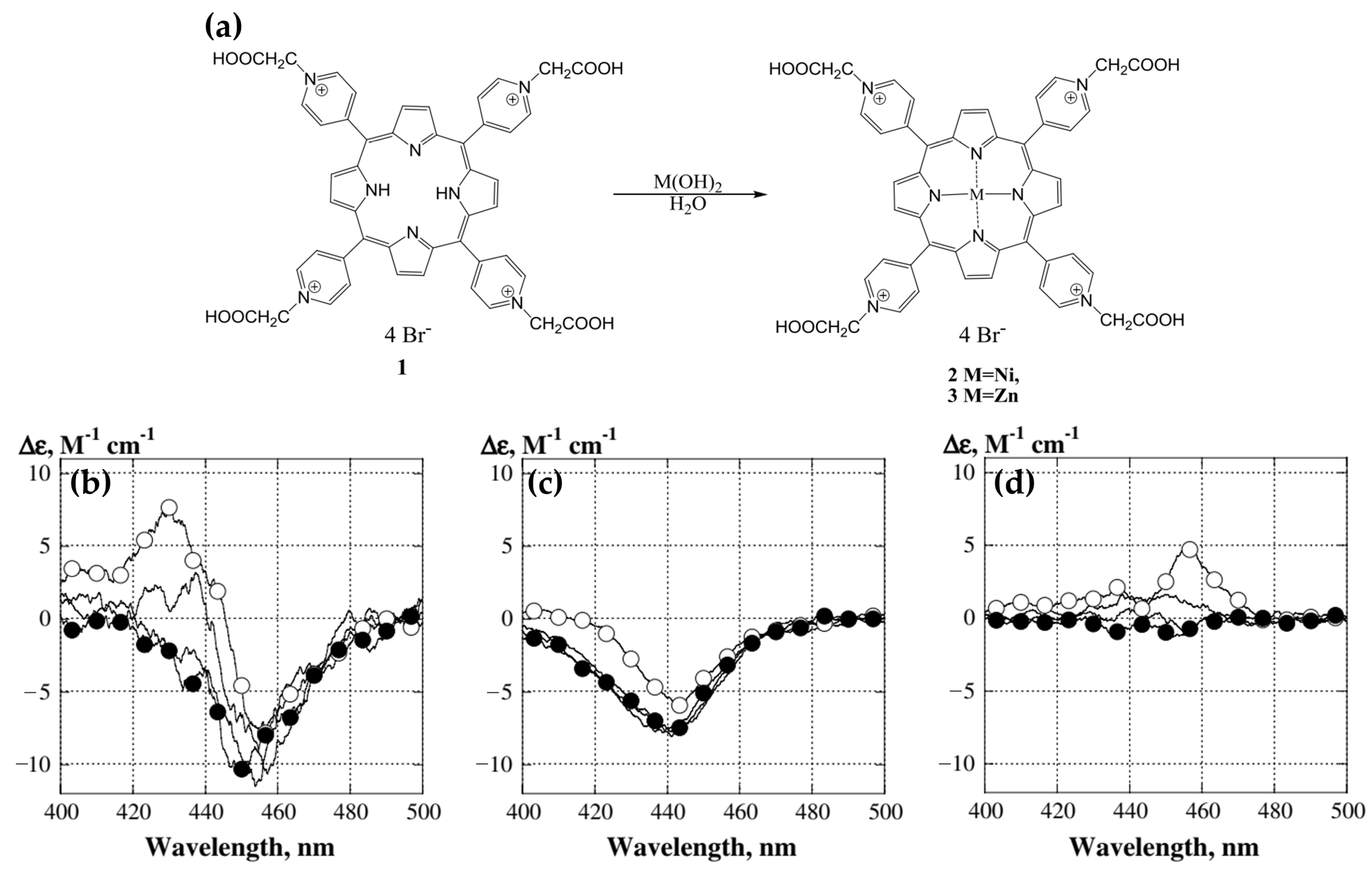
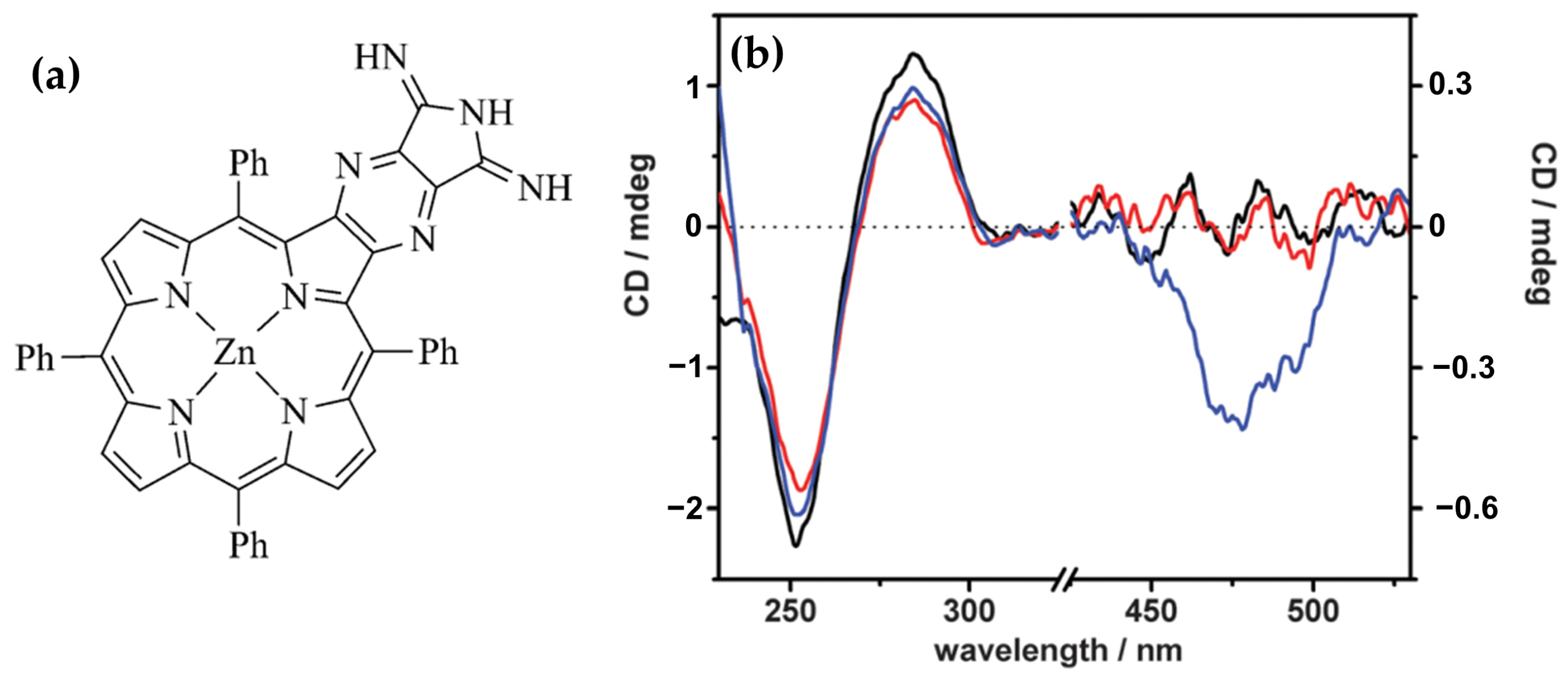
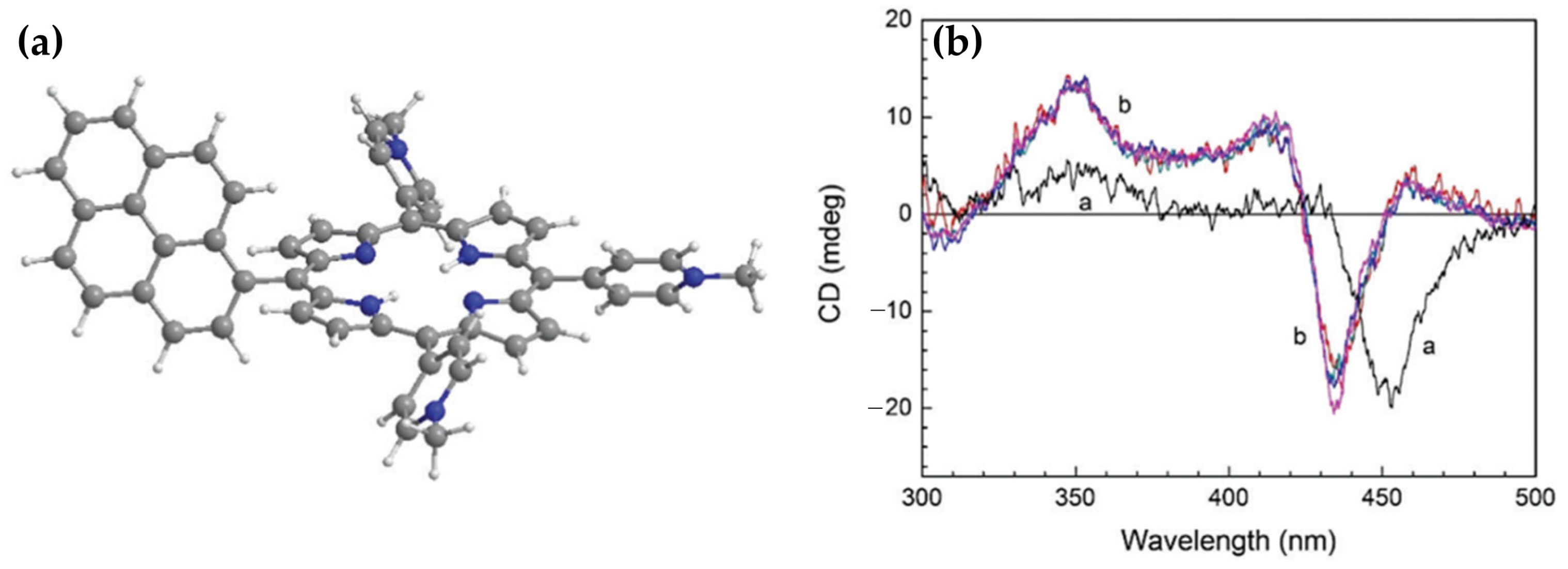

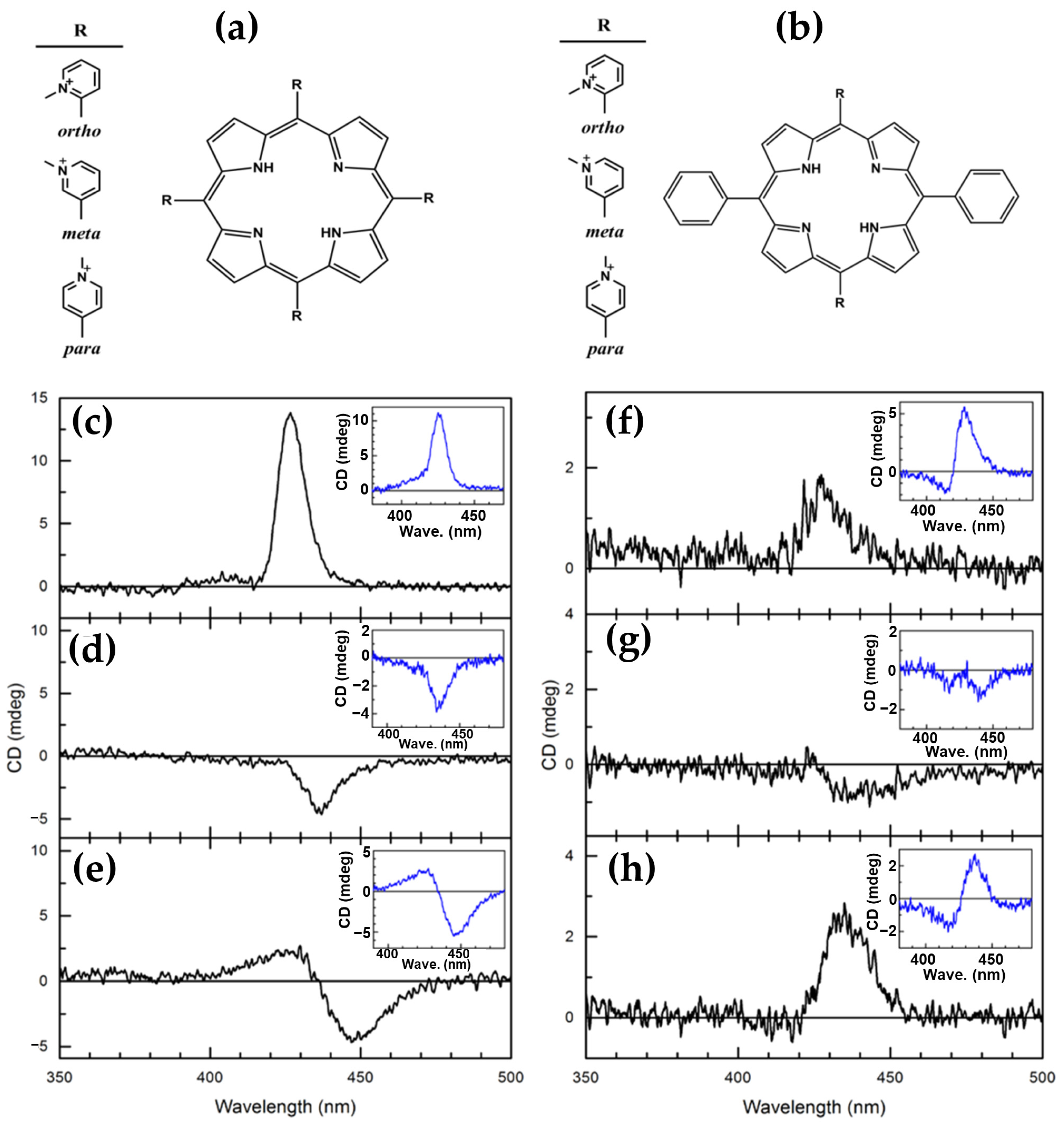






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Travagliante, G.; Gaeta, M.; Purrello, R.; D’Urso, A. Porphyrins as Chiroptical Conformational Probes for Biomolecules. Molecules 2025, 30, 1512. https://doi.org/10.3390/molecules30071512
Travagliante G, Gaeta M, Purrello R, D’Urso A. Porphyrins as Chiroptical Conformational Probes for Biomolecules. Molecules. 2025; 30(7):1512. https://doi.org/10.3390/molecules30071512
Chicago/Turabian StyleTravagliante, Gabriele, Massimiliano Gaeta, Roberto Purrello, and Alessandro D’Urso. 2025. "Porphyrins as Chiroptical Conformational Probes for Biomolecules" Molecules 30, no. 7: 1512. https://doi.org/10.3390/molecules30071512
APA StyleTravagliante, G., Gaeta, M., Purrello, R., & D’Urso, A. (2025). Porphyrins as Chiroptical Conformational Probes for Biomolecules. Molecules, 30(7), 1512. https://doi.org/10.3390/molecules30071512