Abstract
Domain-specific O-fucosylation is an unusual type of glycosylation, where the fucose is directly attached to the serine or threonine residues in specific protein domains via an O-linkage. O-fucosylated proteins play critical roles in a wide variety of biological events and hold important therapeutic values, with the most studied being the Notch receptors and ADAMTS proteins. O-fucose glycans modulate the function of the proteins they modify and are closely associated with various diseases including cancer. In mammals, alongside the well-documented protein O-fucosyltransferase (POFUT) 1-mediated O-fucosylation of epidermal growth factor-like (EGF) repeats and POFUT2-mediated O-fucosylation of thrombospondin type 1 repeats (TSRs), a new type of O-fucosylation was recently identified on elastin microfibril interface (EMI) domains, mediated by POFUT3 and POFUT4 (formerly FUT10 and FUT11). In this review, we present an overview of our current knowledge of O-fucosylation, integrating the latest findings and with a particular focus on its biological functions and molecular mechanisms.
1. Introduction
Fucosylated glycan structures are broadly observed in a wide variety of organisms and play important roles in numerous physiological and pathological processes, including ABO blood group histocompatibility, immune modulation, protein folding, and cellular signal transduction. Two unique features distinguish fucose from other six-carbon sugars: an L-configuration and the absence of a hydroxyl group on the C-6 carbon. Fucose is typically found as terminal or core structures on glycans added by Golgi-localized fucosyltransferases (FUTs), but it can also be directly linked to proteins through an O-linkage to the hydroxyl groups of serines or threonines in proteins with specific domains. This domain-specific O-fucosylation is mediated by endoplasmic reticulum (ER)-localized protein O-fucosyltransferases (POFUTs). A growing body of research has demonstrated the biological importance of O-fucosylation. In this review, we will present an overview of our current knowledge of O-fucosylation, integrating the latest findings, with a particular focus on its biological functions and molecular mechanisms.
2. GDP-Fucose Synthesis and Transport
All FUTs utilize guanosine diphosphate (GDP)-fucose as a donor to transfer fucose to acceptor molecules. GDP-fucose is synthesized in the cytosol, then transported into the Golgi or ER, where it is used by Golgi-localized FUTs or ER-localized POFUTs, respectively. In mammals, GDP-fucose is synthesized via two pathways: the de novo pathway and the salvage pathway (Figure 1). The de novo pathway produces GDP-fucose from mannose or glucose, while the salvage pathway generates GDP-fucose from exogenous fucose or fucose salvaged from glycoconjugate degradation.
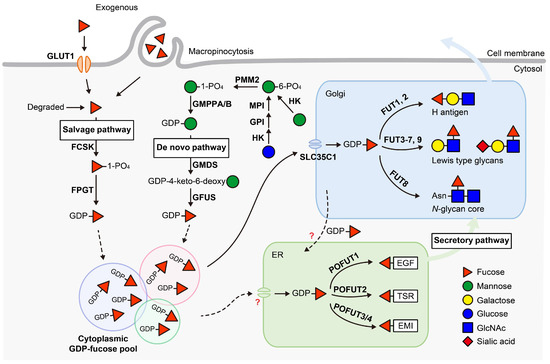
Figure 1.
GDP-fucose biosynthesis and transport. GDP-fucose is synthesized in the cytosol via two distinct pathways. The de novo pathway converts mannose or glucose into GDP-fucose, while the salvage pathway utilizes exogenous fucose or fucose salvaged from degraded glycoconjugates. The enzymes responsible for each step are listed and emphasized in bold. GDP-fucose derived from different origins is stored in distinct pools in cytosol and selectively transported into the Golgi by the GDP-fucose transporter SLC35C1 or into the ER by an as-yet-unknown transporter. The transport of GDP-fucose between the Golgi and ER GDP-fucose pools has been observed, most likely by retrograde transport. Once inside the Golgi or ER, GDP-fucose is transferred to specific substrates by 13 fucosyltransferases (FUTs) in mammals. In the Golgi, nine FUTs (FUT1-9) transfer GDP-fucose to glycan substrates. In the ER, four POFUTs (POFUT1-4) transfer GDP-fucose to proteins containing specific domain structures. These modified proteins are then either secreted or expressed on the cell surface. Dashed arrows indicate trafficking pathways with unknown mechanisms. “?” indicates unknown transporters. HK, hexokinase; GPI, glucose-6-phosphate isomerase; MPI, mannose-6-phosphate isomerase; PMM2, phosphomannomutase 2; GMPPA/B, GDP-mannose pyrophosphorylase A/B; GMDS, GDP-mannose 4,6-dehydratase; GFUS, GDP-L-fucose synthase; GLUT1, glucose transporter type 1; FCSK, L-fucose kinase; FPGT, fucose-1-phosphate guanylytransferase; EGF, epidermal growth factor-like repeat; TSR, thrombospondin type 1 repeat; EMI, elastin microfibril interface domain; FUT, fucosyltransferase; POFUT, protein O-fucosyltransferase.
2.1. De Novo Pathway
In the cytosol, glucose or mannose is synthesized into mannose-6-phosphate by a series of enzymes. Mannose-6-phosphate is then converted into mannose-1-phosphate by phosphomannomutase 2 (PMM2) and subsequently synthesized into GDP-mannose by GDP-mannose pyrophosphorylase A/B (GMPPA/B). In the de novo pathway, GDP-mannose that is derived from either mannose or glucose is converted into GDP-fucose. This transformation involves three reactions catalyzed by two enzymes and includes a keto-containing intermediate. GDP-mannose 4,6-dehydratase (GMDS) catalyzes the initial reaction, converting GDP-mannose into GDP-4-keto-6-deoxymannose [1]. This intermediate is epimerized to GDP-4-keto-6-deoxygalactose, then reduced to yield GDP-fucose, with both reactions catalyzed by GDP-L-fucose synthase (GFUS), a dual-functional enzyme acting as both an epimerase and a reductase [2].
2.2. Salvage Pathway
In the salvage pathway, the source of fucose is provided by exogenous fucose from the diet or fucose released from the lysosomal degradation of glycoconjugates [3,4]. Free fucose in the cytosol is firstly phosphorylated by L-fucose kinase (FCSK), forming fucose-1-phosphate, and is then converted to GDP-fucose by Fucose-1-phosphate guanylytransferase (FPGT) [5,6]. Exogenous fucose must be transported across cell membranes to present in the cytosol, the mechanism behind this transportation remained largely unknown for decades [7]. Recently, Ng et al., identified GLUT1 (encoded by SLC2A1) as a cell membrane fucose transporter using an siRNA screen of ~140 annotated transporter genes in HCT116 cells [8]. They also demonstrated that macropinocytosis plays a role in cellular fucose uptake.
2.3. GDP-Fucose Transport and Cellular GDP-Fucose Pool
GDP-fucose synthesized in the cytosol is then transported to the Golgi and ER. The major Golgi GDP-fucose transporter in mammals is SLC35C1 [9,10]. Lu et al. recently showed that SLC35C1 knockout in HEK293T cells led to a significant decrease, without complete loss, of O-fucosylation of epidermal growth factor-like (EGF) repeats and thrombospondin type 1 repeats (TSRs) in the ER, implicating crosstalk between the Golgi GDP-fucose pool and the ER GDP-fucose pool [11]. Additionally, SLC35C1 knockout in mice is perinatal lethal with skeletal defects typical of impaired Notch signaling. SLC35C2, a homologue of SLC35C1, was predicted to be a GDP-fucose transporter [12]. However, SLC35C2 knockout in HEK293T cells had no impact on Golgi fucosylation or ER O-fucosylation, and no developmental defects were observed in SLC35C2 null mice, indicating it is not a GDP-fucose transporter. Although an ER GDP-fucose transporter has been identified in Drosophila, its human ortholog, SLC35B4, is localized in the Golgi and has been demonstrated as a UDP-xylose/N-acetylglucosamine (GlcNAc) transporter [13]. The mammalian ER GDP-fucose transporter remains to be identified.
The salvage pathway is believed to contribute only a minor portion to the cellular GDP-fucose pool, based on studies in the 1970s that utilized exogenous 3H-fucose as a metabolic tracer [14,15]. However, oral fucose supplementation has proven to be an effective therapy for SLC35C1-CDG (LAD II) and GFUS-CDG patients with defects in GDP-fucose transportation or de novo synthesis (CDG: congenital disorders of glycosylation) [16]. This suggests that the salvage pathway has the capacity to enhance GDP-fucose production, thus compensating for deficiencies in the transport or de novo synthesis of GDP-fucose. Recent studies revealed that GDP-fucose originating from exogenous sources, salvaged from degradation, or de novo synthesized, is stored in separate and distinct pools rather than a single homogeneous pool [17,18]. By incubating cells with differentially labeled 13C-monosaccharides and measuring their contributions to glycans using gas chromatography-mass spectrometry (GC-MS), Sosicka et al. demonstrated that cells identify and selectively use different GDP-fucose pools depending on their heritage [17]. These pools communicate with each other to tightly maintain the total GDP-fucose pool at a constant level. Feedback inhibition of GMDS likely plays a role in this regulatory mechanism, as GDP-fucose is a potent competitive inhibitor for GMDS, allowing efficient fine-tuning of individual pool sizes [19,20,21].
3. Fucosyltransferases (FUTs)
FUTs catalyze the transfer of a fucose residue from the donor substrate GDP-fucose to acceptor molecules, including oligosaccharides, glycoproteins, glycolipids, and glycoRNAs [22,23,24,25]. To date, 13 FUTs have been identified in mammals based on conserved sequences that are involved in GDP-fucose binding (Figure 2). These FUTs are classified into α2-, α3/4-, α6-FUTs, and POFUTs, based on their distinct fucose linkages. While POFUTs are ER-localized proteins, all other FUTs are type II transmembrane proteins localized in the Golgi. Sequence alignment of the 13 human FUTs identified three distinct groups with high sequence homology: FUT1 and FUT2, FUT3–7 and FUT9 (with particularly high homology among FUT3, FUT5, and FUT6), and FUT10 and FUT11. In contrast, FUT8, POFUT1, and POFUT2 show limited homology with other FUTs (Supplemental Table S1).
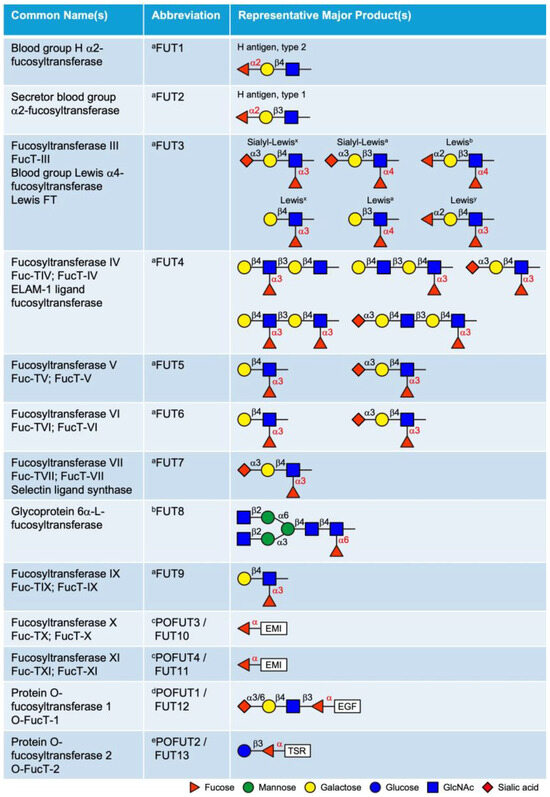
Figure 2.
List of 13 fucosyltransferases (FUTs) in mammals and their representative products. The linkage of the fucose added by each enzyme is labeled in red. a These enzymes add fucose to oligosaccharide chains on glycolipids, N-glycans, mucin O-glycans or glycoRNAs. b This enzyme adds fucose specifically to the innermost GlcNAc of the chitobiose unit in N-glycans. c This modification is observed only in EMI domains; see Figure 3C. d This modification is observed only in EGF repeats with the consensus sequence C2XXXX[S/T]C3; see Figure 3A. e This modification is observed only in TSRs with the consensus sequence C1−2XX[S/T]C2−3; see Figure 3B. EGF, epidermal growth factor-like repeat; TSR, thrombospondin type 1 repeat; EMI, elastin microfibril interface domain. Updated from Schneider et al., 2017 [24].
3.1. Golgi-Localized FUTs
The Golgi-localized FUTs include FUT1–9, all of which transfer fucose to glycan structures as terminal or core modifications. FUT1, FUT2, and Sec1 (non-functional protein encoded by a pseudogene) are α2-FUTs that belong to the CAZy GT11 family [26,27]. FUT1 and FUT2 are responsible for synthesizing the H antigens (O blood), which serve as fucosylated precursors for the A and B oligosaccharide structures in the ABH blood group system [28,29]. They exhibit distinct tissue expression patterns and acceptor specificities. FUT1 product (H antigen) is primarily expressed on erythrocyte membranes and vascular endothelium, with a preference for type 2 structures, whereas FUT2 (Se) is predominantly expressed in epithelial cells and exocrine secretions and is more active on type 1 structures. FUT3–7 and FUT9 are α3/4-FUTs that belong to the CAZy GT10 family, all of which have α3 activity, except FUT3 and FUT5, which also possess α4 activity [30,31,32,33,34,35]. These FUTs are involved in the synthesis of the complex series of Lewis antigen epitopes (i.e., Lewisx, Lewisy, Lewisa, Lewisb, Sialyl-Lewisx, Sialyl-Lewisa) with distinct acceptor specificities. The biological roles of the ABH and Lewis antigens have been previously reviewed in detail [36]. FUT8 is the only α6-FUT identified in mammals, belonging to the CAZy GT23 family [37,38]. FUT8 catalyzes N-glycan core fucosylation by transferring a fucose moiety to the innermost GlcNAc residue of the chitobiose unit. Core fucosylation plays critical roles in the regulation of antibody-dependent cellular cytotoxicity (ADCC) and serves as a biomarker for various types of cancer [39,40].
Additionally, based on sequence homology to known α3/4-FUTs, FUT10 and FUT11 were postulated to be α3-FUTs. However, as will be detailed below, substantial experimental evidence confirms that these enzymes function as POFUTs rather than α3-FUTs.
3.2. ER-Localized POFUTs
Unlike the Golgi-localized FUTs that transfer fucose to glycan structures, POFUTs add fucose directly to the hydroxyls of serines or threonines of proteins through an O-linkage. In mammals, four POFUTs have been identified: POFUT1 (CAZy GT65 family), POFUT2 (CAZy GT68 family), POFUT3 (formerly FUT10, CAZy GT10 family), and POFUT4 (formerly FUT11, CAZy GT10 family) [41,42,43,44,45]. POFUT1 contains a C-terminal KDEL-like sequence that retains it in the ER. While POFUT2 does not contain the ER retention sequence, it is proposed to be retained in the ER by association with other proteins that contain retention sequences. Similarly, POFUT3 and POFUT4 do not contain a KDEL-type ER retention sequence and likely bind to other ER proteins for retention in the ER. All POFUTs mediate domain-specific O-fucosylation and are highly specific for their respective substrates [45,46]. Unlike the Golgi-localized FUTs, all POFUTs require a folded domain structure for modification. The binding interface between each POFUT and its respective substrate displays a ‘hand-in-glove’ complementarity, with the substrate domain structure fitting into the enzyme’s groove, precisely positioning the hydroxyl group of serine or threonine for fucose transfer. This will be discussed in detail in Section 5.
3.2.1. POFUT1 (FUT12)
The unique fucose–protein linkage was initially identified in the urinary-type plasminogen activator (urokinase) using mass spectrometry [47,48]. This O-fucose monosaccharide was located on the single epidermal growth factor-like (EGF) domain of urokinase within a sequence C2XXGG[S/T]C3 (where C2 and C3 are the second and third cysteine in the EGF, S/T is the modified site). EGF domains are small protein domains with ~40 amino acids, featuring six highly conserved cysteines that form three disulfide bonds (Figure 3A). Soon after, more proteins with O-fucose monosaccharide or elongated tetrasaccharide were identified, all containing one or more EGF repeats, including t-PA [49], coagulation factor VII [50], factor XII [51], and factor IX [52]. These discoveries sparked an intensive investigation to identify the enzyme responsible for this modification. The Spellman group developed an enzymatic assay to test O-fucosyltransferase activity [53]. Interestingly, using extracts from Chinese hamster ovary (CHO) cells as the enzyme source, they observed activity when employing the complete EGF domain from human factor VII (bacterially expressed, unmodified) as the acceptor substrate, but not with synthetic peptides containing the consensus sequence. This indicates that the enzyme requires a properly folded EGF structure for its activity, which sets it apart from other glycan-modifying FUTs. Using this complete EGF domain from human factor VII (bacterially expressed) as bait, the enzyme (O-FucT-1) was successfully purified from CHO cells by the same group [54]. The sequence of O-FucT-1 was used in a screening of a human cDNA library and led to the identification of the human POFUT1 gene [55]. Homologues in mice (Pofut1), Drosophila (Ofut1), and C. elegans (pofut1) were also identified. The POFUT1 gene is highly conserved in mammals and shows nearly ubiquitous expression in all tissues examined, indicating the potential important biological roles of O-fucosylation.
Our knowledge on POFUT1-mediated EGF O-fucosylation has largely expanded as more protein targets have been uncovered and increased research efforts on exploring the biological functions. A refined consensus sequence C2XXXX[S/T]C3 was proposed to include a broader set of O-fucose sites [56,57,58]. Database searches with this consensus sequence in the context of an EGF repeat identified more than 100 putative protein targets for POFUT1 (splice variants are not included) (Table 1) [24,59]. However, the O-fucosylation status of many of these proteins is yet to be confirmed.
Heterozygous mutations of POFUT1 in human causes a rare skin condition known as Dowling–Degos Disease 2 (DDD2), characterized by skin pigmentation abnormalities [60]. Pofut1 knockout in mice is embryonic lethal with multiple severe developmental defects, including angiogenesis, hematopoiesis, neurogenesis and somitogenesis. These phenotypes are similar to those observed in Notch1 knockout mice embryos, indicating Notch as the primary biological target of POFUT1 in embryogenesis [61,62]. The Notch family proteins possess more predicted sites for POFUT1 modification than any other proteins and are the most extensively studied POFUT1 targets (Table 1) [63]. Notch is a key regulator in a variety of developmental processes [64]. O-fucosylation of Notch is essential for its proper functioning in processes like the Notch-dependent regulation of lymphopoiesis and myelopoiesis [65]. The extracellular domain of Notch is heavily decorated with O-fucose glycans (Figure 4A) [66,67]. A substantial number of O-fucose glycans are elongated to GlcNAcβ1-3Fucose disaccharide by the Fringe family of β3-N-acetylglucosaminyltransferases, and can be further elongated to a tetrasaccharide, Siaα2-6Galβ1-4GlcNAcβ1-3Fuc [56,67,68,69,70,71]. Interestingly, O-fucose glycans on distinct EGF repeats play varying roles in the regulation of Notch activity. O-fucose residues on EGF8 and EGF12 of NOTCH1 directly interact with ligands, Delta-like 1 and Jagged1, and their binding is further enhanced by Fringe modifications (Figure 5) [67,72,73,74]. However, Fringe modifications on EGF6 and EGF36 inhibit NOTCH1 activation by Jagged1 [67]. Through the regulation of POFUT1, Fringe, and several other protein O-glycosyltransferases associated with Notch, cells can dynamically fine-tune Notch activity to accommodate environmental changes. More details on the effects of O-fucose and other O-glycans on Notch function can be found in these recent reviews: [65,75,76,77,78].
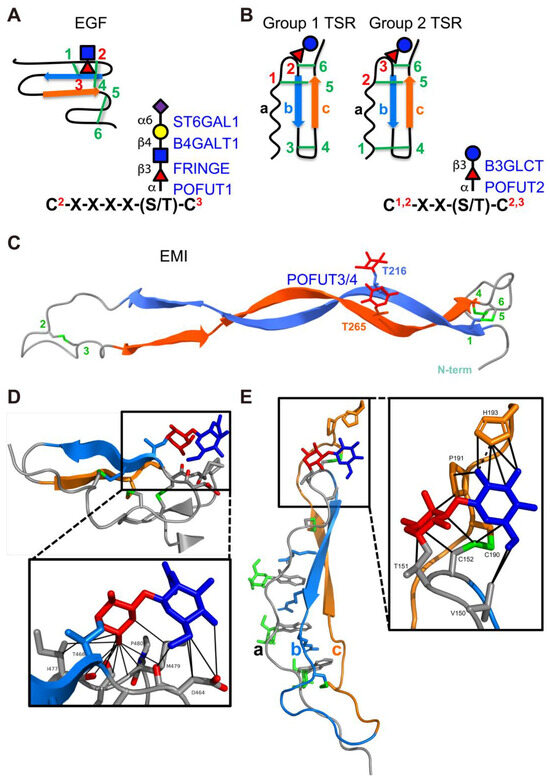
Figure 3.
EGF repeats, TSRs, and EMI domains are modified by O-fucose glycans. (A) (left) Disulfide bonding pattern of an EGF repeat. Cysteines are numbered, disulfide bonds are in green, β-strands are indicated as blue and orange arrows. O-fucose site is marked with fucose (red triangle), GlcNAc (blue square), Galactose (yellow circle), and Neu5Ac (purple diamond). (right) Consensus sequence for POFUT1 modification. C2 and C3 are the second and third conserved cysteine in the EGF repeat. X indicates any amino acid. Enzymes responsible for modification are in blue with linkages indicated. (B) (left) Two distinct disulfide bonding patterns for TSRs. Cysteines are numbered, disulfide bonds are in green, β-strands are indicated as blue and orange arrows. O-fucose site is marked with fucose (red triangle) and glucose (blue circle). (right) Consensus sequence for POFUT2 modification. The C’s can be C1 and C2 or C2 and C3, depending on whether the TSR is Group 1 or Group 2. Enzymes responsible for modification are in blue with linkages indicated. (C) AlphaFold2 predicted structure of the EMI domain from MMRN1 showing the disulfide bonding pattern. Cysteines are numbered, disulfide bonds are in green, β-strands are indicated as blue and orange arrows. The two O-fucose sites are indicated and marked with fucose residues in red. POFUT3 and POFUT4 are responsible for modification and indicated in blue. (D) Structure of NOTCH1 EGF12 modified with a GlcNAcβ1-3Fucose disaccharide (from PDB ID 4D0E). β-strands colored as in (A). Fucose in red, GlcNAc in blue, disulfide bonds in green, oxygen atoms highlighted in red. Box shows zoomed in region highlighting interactions of the disaccharide with underlying amino acids identified by MolProbity [79] (van der Waals, solid lines). Structures rendered in PyMOL (version 2.2.2). (E) Structure of Properdin TSR2 modified with Glucoseβ1-3Fucose disaccharide (from PDB ID 6RUS). The three strands (a, b, and c) of the TSRs are colored as in (B). Fucose in red, glucose in blue, mannose in green, disulfide bonds in green. Box shows zoomed in region highlighting interactions of the disaccharide with underlying amino acids identified by MolProbity [79] (H-bonds, dashed lines; van der Waals, solid lines). Structures rendered in PyMOL (version 3.0.4). Panels A, B and D are modified with permission from Ref. [44].
Figure 3.
EGF repeats, TSRs, and EMI domains are modified by O-fucose glycans. (A) (left) Disulfide bonding pattern of an EGF repeat. Cysteines are numbered, disulfide bonds are in green, β-strands are indicated as blue and orange arrows. O-fucose site is marked with fucose (red triangle), GlcNAc (blue square), Galactose (yellow circle), and Neu5Ac (purple diamond). (right) Consensus sequence for POFUT1 modification. C2 and C3 are the second and third conserved cysteine in the EGF repeat. X indicates any amino acid. Enzymes responsible for modification are in blue with linkages indicated. (B) (left) Two distinct disulfide bonding patterns for TSRs. Cysteines are numbered, disulfide bonds are in green, β-strands are indicated as blue and orange arrows. O-fucose site is marked with fucose (red triangle) and glucose (blue circle). (right) Consensus sequence for POFUT2 modification. The C’s can be C1 and C2 or C2 and C3, depending on whether the TSR is Group 1 or Group 2. Enzymes responsible for modification are in blue with linkages indicated. (C) AlphaFold2 predicted structure of the EMI domain from MMRN1 showing the disulfide bonding pattern. Cysteines are numbered, disulfide bonds are in green, β-strands are indicated as blue and orange arrows. The two O-fucose sites are indicated and marked with fucose residues in red. POFUT3 and POFUT4 are responsible for modification and indicated in blue. (D) Structure of NOTCH1 EGF12 modified with a GlcNAcβ1-3Fucose disaccharide (from PDB ID 4D0E). β-strands colored as in (A). Fucose in red, GlcNAc in blue, disulfide bonds in green, oxygen atoms highlighted in red. Box shows zoomed in region highlighting interactions of the disaccharide with underlying amino acids identified by MolProbity [79] (van der Waals, solid lines). Structures rendered in PyMOL (version 2.2.2). (E) Structure of Properdin TSR2 modified with Glucoseβ1-3Fucose disaccharide (from PDB ID 6RUS). The three strands (a, b, and c) of the TSRs are colored as in (B). Fucose in red, glucose in blue, mannose in green, disulfide bonds in green. Box shows zoomed in region highlighting interactions of the disaccharide with underlying amino acids identified by MolProbity [79] (H-bonds, dashed lines; van der Waals, solid lines). Structures rendered in PyMOL (version 3.0.4). Panels A, B and D are modified with permission from Ref. [44].
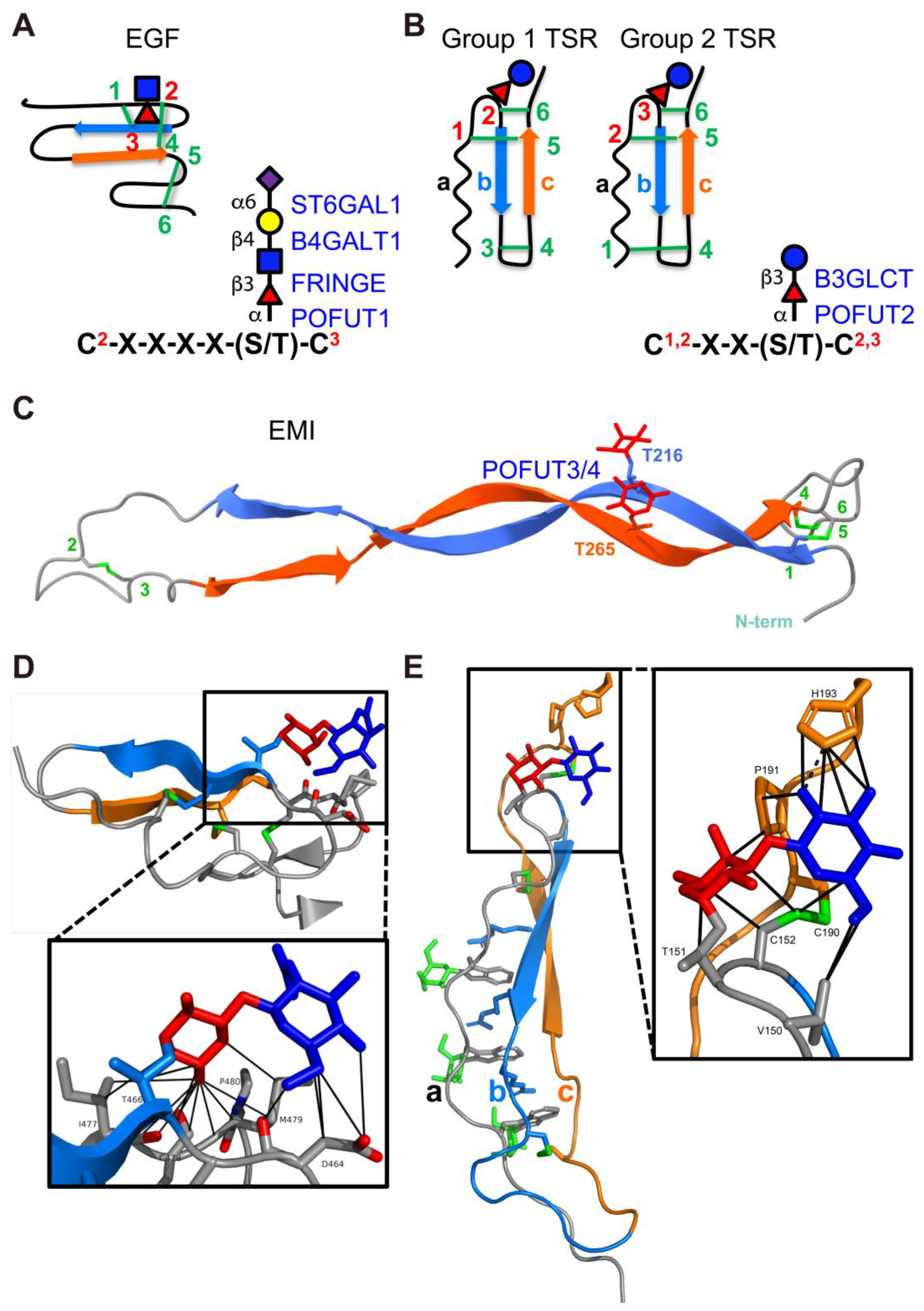

Table 1.
List of putative human protein substrates of POFUT1.
Table 1.
List of putative human protein substrates of POFUT1.
| UniProt ID | Gene Name | Protein Name | Consensus/Total | Subcellular Location | Known Human Pathology (If Any) |
|---|---|---|---|---|---|
| H0YMF1 | ACAN | Aggrecan core protein | 1/2 | ECM | - |
| Q14246 | ADGRE1 | Adhesion G protein-coupled receptor E1 | 4/6 | CM | - |
| Q9UHX3 | ADGRE2 | Adhesion G protein-coupled receptor E2 | 2/5 | CM | Vibratory urticaria (VBU) [80] |
| Q9BY15 | ADGRE3 | Adhesion G protein-coupled receptor E3 | 1/2 | ECM, CM | - |
| Q86SQ3 | ADGRE4P | Putative adhesion G protein-coupled receptor E4P | 1/2 | ECM, CM | - |
| P48960 | ADGRE5 | Adhesion G protein-coupled receptor E5 | 2/5 | ECM, CM | - |
| O00468 | AGRN | Agrin | 2/4 [81] | ECM | Myasthenic syndrome, congenital, 8 (CMS8) [82,83,84] |
| Q6UW56 | ATRAID | All-trans retinoic acid-induced differentiation factor | 1/1 | CM, N | - |
| Q96GW7 | BCAN | Brevican core protein | 1/1 | ECM, CM | - |
| Q9NPY3 | CD93 | Complement component C1q receptor | 1/5 | CM | - |
| Q9NYQ6 | CELSR1 | Cadherin EGF LAG seven-pass G-type receptor 1 | 2/8 | CM | Neural tube defects (NTD) [85]; Lymphatic malformation 9 (LMPHM9) [86,87] |
| Q9HCU4 | CELSR2 | Cadherin EGF LAG seven-pass G-type receptor 2 | 2/7 | CM | - |
| Q9NYQ7 | CELSR3 | Cadherin EGF LAG seven-pass G-type receptor 3 | 2/8 | CM | - |
| P0CG37 | CFC1 | Cryptic protein | 1/1 | ECM, CM | Heterotaxy, visceral, 2, autosomal (HTX2) [88,89] |
| P0CG36 | CFC1B | Cryptic family protein 1B | 1/1 | ECM | - |
| Q8WYK1 | CNTNAP5 | Contactin-associated protein-like 5 | 1/2 | CM | - |
| P82279 | CRB1 | Protein crumbs homolog 1 | 8/19 | ECM, CM | Retinitis pigmentosa 12 (RP12) [90,91]; Leber congenital amaurosis 8 (LCA8) [92]; Pigmented paravenous chorioretinal atrophy (PPCRA) [93] |
| Q5IJ48 | CRB2 | Protein crumbs homolog 2 | 9/15 | ECM, CM, CYT | Focal segmental glomerulosclerosis 9 (FSGS9) [94]; Retinitis pigmentosa (RP) [95]; Ventriculomegaly with cystic kidney disease (VMCKD) [96] |
| P13385 | CRIPTO | Protein Cripto | 1/1 [97,98] | ECM, CM | - |
| P51864 | CRIPTO3 | Putative protein CRIPTO3 | 1/1 | CM | - |
| O60494 | CUBN | Cubilin | 4/7 | CM | Imerslund-Grasbeck syndrome 1 (IGS1) [99,100,101]; Proteinuria, chronic benign (PROCHOB) [102] |
| P80370 | DLK1 | Protein delta homolog 1 | 3/6 [103] | CM, CYT | - |
| Q6UY11 | DLK2 | Protein delta homolog 2 | 1/6 | CM | - |
| O00548 | DLL1 | Delta-like protein 1 | 4/8 [56,58] | CM | Neurodevelopmental disorder with non-specific brain abnormalities and with or without seizures (NEDBAS) [104] |
| Q9NYJ7 | DLL3 | Delta-like protein 3 | 2/6 [105] | CM | Spondylocostal dysostosis 1, autosomal recessive (SCDO1) [106] |
| Q9NR61 | DLL4 | Delta-like protein 4 | 5/8 | CM | Adams-Oliver syndrome 6 (AOS6) [107] |
| Q8NFT8 | DNER | Delta and Notch-like epidermal growth factor-related receptor | 6/10 | CM | - |
| O43854 | EDIL3 | EGF-like repeat and discoidin I-like domain-containing protein 3 | 1/3 [108] | ECM | - |
| O95967 | EFEMP2 | EGF-containing fibulin-like extracellular matrix protein 2 | 1/6 | ECM | Cutis laxa, autosomal recessive, 1B (ARCL1B) [109] |
| P01133 | EGF | Pro-epidermal growth factor | 1/9 | CM | Hypomagnesemia 4 (HOMG4) [110] |
| Q9UHF1 | EGFL7 | Epidermal growth factor-like protein 7 | 1/2 | ECM | - |
| Q63HQ2 | EGFLAM | Pikachurin | 2/3 | ECM | - |
| Q5T1H1 | EYS | Protein eyes shut homolog | 11/27 | ECM, CYT | Retinitis pigmentosa 25 (RP25) [111,112] |
| P08709 | F7 | Coagulation factor VII | 1/2 [50] | ECM | Factor VII deficiency (FA7D) [113,114,115] |
| P00740 | F9 | Coagulation factor IX | 1/2 [52,116] | ECM | Hemophilia B (HEMB) [117,118]; Thrombophilia, X-linked, due to factor IX defect (THPH8) [119]; Warfarin sensitivity, X-linked (WARFS) [120] |
| P00748 | F12 | Coagulation factor XII | 1/2 [51] | ECM | Factor XII deficiency (FA12D) [121,122]; Angioedema, hereditary, 3 (HAE3) [123,124] |
| Q14517 | FAT1 | Protocadherin Fat 1 | 2/5 | CM, N | - |
| Q9NYQ8 | FAT2 | Protocadherin Fat 2 | 1/2 | CM | Spinocerebellar ataxia 45 (SCA45) [125] |
| Q8TDW7 | FAT3 | Protocadherin Fat 3 | 3/4 | CM | - |
| Q6V0I7 | FAT4 | Protocadherin Fat 4 | 5/6 | CM | Van Maldergem syndrome 2 (VMLDS2) [126]; Hennekam lymphangiectasia-lymphedema syndrome 2 (HKLLS2) [127] |
| P23142 | FBLN1 | Fibulin-1 | 3/9 | ECM | Complex type of synpolydactyly [128]; associated with human breast cancer [129] |
| P98095 | FBLN2 | Fibulin-2 | 2/10 | ECM | - |
| Q9UBX5 | FBLN5 | Fibulin-5 | 1/6 | ECM | Charcot-Marie-Tooth disease, demyelinating, 1H (CMT1H) [130]; Cutis laxa, autosomal dominant, 2 (ADCL2) [131]; Cutis laxa, autosomal recessive, 1A (ARCL1A) [132]; Macular degeneration, age-related, 3 (ARMD3) [133] |
| Q53RD9 | FBLN7 | Fibulin-7 | 2/3 | ECM | - |
| P35555 | FBN1 | Fibrillin-1 | 1/47 [134] | ECM | Marfan syndrome (MFS) [135,136]; Ectopia lentis 1, isolated, autosomal dominant (ECTOL1) [137]; Weill–Marchesani syndrome 2 (WMS2) [138]; Overlap connective tissue disease (OCTD) [139]; Stiff skin syndrome (SSKS) [140]; Geleophysic dysplasia 2 (GPHYSD2) [141]; Acromicric dysplasia (ACMICD) [141]; Marfanoid-progeroid-lipodystrophy syndrome (MFLS) [142] |
| P35556 | FBN2 | Fibrillin-2 | 2/47 | ECM | Contractural arachnodactyly, congenital (CCA) [143]; Macular degeneration, early-onset (EOMD) [144] |
| Q75N90 | FBN3 | Fibrillin-3 | 1/44 | ECM | - |
| Q14520 | HABP2 | Hyaluronan-binding protein 2 | 1/3 | ECM | Thyroid cancer, non-medullary, 5 (NMTC5) [145,146] |
| Q04756 | HGFAC | Hepatocyte growth factor activator | 2/2 | ECM | - |
| P98160 | HSPG2 | Basement membrane-specific heparan sulfate proteoglycan core protein | 3/4 | ECM | Schwartz-Jampel syndrome (SJS1) [147]; Dyssegmental dysplasia Silverman-Handmaker type (DDSH) [148] |
| P78504 | JAG1 | Jagged-1 | 11/16 [56,149] | CM | Alagille syndrome 1 (ALGS1) [150]; Tetralogy of Fallot (TOF) [151]; Deafness, congenital heart defects, and posterior embryotoxon (DCHE) [152]; Charcot-Marie-Tooth disease, axonal, 2HH (CMT2HH) [153] |
| Q9Y219 | JAG2 | Jagged-2 | 9/16 | CM | Muscular dystrophy, limb-girdle, autosomal recessive 27 (LGMDR27) [154] |
| Q07954 | LRP1 | Prolow-density lipoprotein receptor-related protein 1 | 5/22 | CM, CYT | Keratosis pilaris atrophicans (KPA) [155]; Developmental dysplasia of the hip 3 (DDH3) [156] |
| Q9NZR2 | LRP1B | Low-density lipoprotein receptor-related protein 1B | 4/22 | CM | - |
| Q14767 | LTBP2 | Latent-transforming growth factor beta-binding protein 2 | 1/20 | ECM | Glaucoma 3, primary congenital, D (GLC3D) [157]; Microspherophakia and/or megalocornea, with ectopia lentis and with or without secondary glaucoma (MSPKA) [158]; Weill–Marchesani syndrome 3 (WMS3) [159] |
| Q5VYJ5 | MALRD1 | MAM and LDL-receptor class A domain-containing protein 1 | 1/1 | CYT | - |
| O75095 | MEGF6 | Multiple epidermal growth factor-like domains protein 6 | 5/27 | ECM | - |
| Q7Z7M0 | MEGF8 | Multiple epidermal growth factor-like domains protein 8 | 2/5 | CM | Carpenter syndrome 2 (CRPT2) [160] |
| Q96KG7 | MEGF10 | Multiple epidermal growth factor-like domains protein 10 | 5/15 | CM | Congenital myopathy 10A, severe variant (CMYP10A) [161]; Congenital myopathy 10B, mild variant (CMYP10B) [162] |
| A6BM72 | MEGF11 | Multiple epidermal growth factor-like domains protein 11 | 8/14 | CM | - |
| Q13201 | MMRN1 | Multimerin-1 [45,163] | 1/1 | ECM | Factor V Quebec [164] |
| Q9UK23 | NAGPA | N-acetylglucosamine-1-phosphodiester alpha-N-acetylglucosaminidase | 1/2 | GA | Stuttering [165] |
| A0A087WY62 | NBPF26 | NBPF member 26 | 5/6 | CYT | - |
| O14594 | NCAN | Neurocan core protein | 2/2 | ECM | - |
| Q92832 | NELL1 | Protein kinase C-binding protein NELL1 | 1/5 | ECM, CYT | - |
| Q14112 | NID2 | Nidogen-2 | 1/5 | ECM | - |
| P46531 | NOTCH1 | Neurogenic locus notch homolog protein 1 | 21/36 [67,70] | CM | Aortic valve disease 1 (AOVD1) [166]; Adams-Oliver syndrome 5 (AOS5) [167] |
| Q04721 | NOTCH2 | Neurogenic locus notch homolog protein 2 | 20/36 [66,168] | CM | Alagille syndrome 2 (ALGS2) [169]; Hajdu-Cheney syndrome (HJCYS) [170] |
| Q7Z3S9 | NOTCH2NLA | Notch homolog 2 N-terminal-like protein A | 5/6 | ECM, CYT | Microcephaly, macrocephaly [171] |
| P0DPK3 | NOTCH2NLB | Notch homolog 2 N-terminal-like protein B | 4/6 | ECM | Microcephaly, macrocephaly [171] |
| P0DPK4 | NOTCH2NLC | Notch homolog 2 N-terminal-like protein C | 5/6 | ECM | Neuronal intranuclear inclusion disease (NIID) [172,173,174]; Oculopharyngodistal myopathy 3 (OPDM3) [175] |
| Q9UM47 | NOTCH3 | Neurogenic locus notch homolog protein 3 | 14/34 [176] | CM | Cerebral arteriopathy, autosomal dominant, with subcortical infarcts and leukoencephalopathy, 1 (CADASIL1) [177,178]; Myofibromatosis, infantile 2 (IMF2) [179]; Lateral meningocele syndrome (LMNS) [180] |
| Q99466 | NOTCH4 | Neurogenic locus notch homolog protein 4 | 18/29 | CM | - |
| Q96CW9 | NTNG2 | Netrin-G2 | 1/1 | CM | Neurodevelopmental disorder with behavioral abnormalities, absent speech, and hypotonia (NEDBASH) [181,182] |
| Q6UXH9 | PAMR1 | Inactive serine protease PAMR1 | 1/1 [183] | ECM | - |
| Q5VY43 | PEAR1 | Platelet endothelial aggregation receptor 1 | 3/9 | CM | Cardiovascular disease [184] |
| P00750 | PLAT | Tissue-type plasminogen activator | 1/1 [49] | ECM | Increased activity results in excessive bleeding; Defective release results in thrombosis or embolism [185] |
| P00749 | PLAU | Urokinase-type plasminogen activator | 1/1 [47,48] | ECM | Quebec platelet disorder (QPD) [186] |
| P04070 | PROC | Vitamin K-dependent protein C | 1/2 | ECM | Thrombophilia due to protein C deficiency, autosomal dominant (THPH3) [187]; Thrombophilia due to protein C deficiency, autosomal recessive (THPH4) [188] |
| P22891 | PROZ | Vitamin K-dependent protein Z | 1/2 | ECM | - |
| P78509 | RELN | Reelin | 2/8 | ECM | Lissencephaly 2 (LIS2) [189]; Epilepsy, familial temporal lobe, 7 (ETL7) [190] |
| Q96GP6 | SCARF2 | Scavenger receptor class F member 2 | 1/7 | CM | Van den Ende-Gupta syndrome (VDEGS) [191] |
| O75093 | SLIT1 | Slit homolog 1 protein | 2/9 | ECM | - |
| O94813 | SLIT2 | Slit homolog 2 protein | 3/7 | ECM | - |
| O75094 | SLIT3 | Slit homolog 3 protein | 3/9 | ECM | - |
| Q8TER0 | SNED1 | Sushi, nidogen and EGF-like domain-containing protein 1 | 10/15 | ECM | - |
| Q9NY15 | STAB1 | Stabilin-1 | 3/16 | CM | Hyperferritinemia (HRFT) [192] |
| Q8WWQ8 | STAB2 | Stabilin-2 | 6/17 | CM, CYT | - |
| Q6UWL2 | SUSD1 | Sushi domain-containing protein 1 | 2/3 | CM | - |
| Q4LDE5 | SVEP1 | Sushi, von Willebrand factor type A, EGF and pentraxin domain-containing protein 1 | 4/9 | ECM, CYT | Coronary artery disease [193]; artherosclerosis [194] |
| Q9UKZ4 | TENM1 | Teneurin-1 | 1/8 | CM | - |
| Q9NT68 | TENM2 | Teneurin-2 | 2/8 | CM | - |
| Q9P273 | TENM3 | Teneurin-3 | 1/7 | CM | Microphthalmia/Coloboma 9 (MCOPCB9) [195]; Microphthalmia, syndromic, 15 (MCOPS15) [196] |
| Q6N022 | TENM4 | Teneurin-4 | 2/8 | CM | Tremor, hereditary essential 5 (ETM5) [197] |
| P49746 | THBS3 | Thrombospondin-3 | 1/3 | ECM | - |
| P35590 | TIE1 | Tyrosine-protein kinase receptor Tie-1 | 1/3 | CM | Lymphatic malformation 11 (LMPHM11) [198] |
| P07911 | UMOD | Uromodulin | 3/4 | ECM, CM | Tubulointerstitial kidney disease, autosomal dominant, 1 (ADTKD1) [199,200] |
| Q5DID0 | UMODL1 | Uromodulin-like 1 | 1/3 | CM, CYT | - |
| Q6EMK4 | VASN | Vasorin | 1/1 | ECM, CM | - |
| P13611 | VCAN | Versican core protein | 2/2 | ECM | Wagner vitreoretinopathy (WGVRP) [201,202] |
| Q5GFL6 | VWA2 | von Willebrand factor A domain-containing protein 2 | 2/2 [203] | ECM | - |
| Q8N2E2 | VWDE | von Willebrand factor D and EGF domain-containing protein | 3/7 | ECM | - |
| Q9Y5W5 | WIF1 | Wnt inhibitory factor 1 | 2/5 | ECM | - |
| Q0PNF2 | - | FEX1 | 3/20 | - | - |
This list of putative POFUT1 substrates is updated from Luther et al., 2021 [59]. Proteins with experimentally confirmed O-fucosylation are highlighted in bold, with references provided. The number of EGF repeats containing POFUT1 consensus sequences C2XXXX[S/T]C3 out of the total EGF repeat number is listed. Any known human pathologies associated with the listed proteins (reported in UniProt) are indicated with the corresponding references. Major subcellular locations for the listed proteins (reported in UniProt) are indicated. ECM, extracellular matrix; CM, cell membrane; CYT, cytoplasm; N, nucleus; GA, Golgi apparatus.
3.2.2. POFUT2 (FUT13)
The discovery of O-fucose on thrombospondin type 1 repeats (TSRs) occurred later than the identifying of O-fucose on EGF domains. Similar to EGFs, TSRs are small protein domains of 50–70 amino acids that contain six cysteines forming three disulfide bonds, albeit with distinct bonding patterns (Figure 3B). The O-fucose on TSRs was initially identified as glucose–fucose disaccharides on thrombospondin-1 by Hofsteenge et al. while mapping sites for C-mannosylation [204]. An initial consensus sequence was proposed as C1SX[S/T]C2G, which has been further refined to C1XX[S/T]C2 (group 1 TSRs) and C2XX[S/T]C3 (group 2 TSRs) through the comparison of additional TSR O-fucose sites identified later and site-mutagenesis studies of TSR structures [205,206,207]. Note that group 1 TSRs and group 2 TSRs have different disulfide bonding patterns (group 1: C1-C5, C2-C6, C3-C4; group 2: C1-C4, C2-C5, C3-C6), but both can be O-fucosylated [208,209]. Tryptophan frequently appears upstream of the consensus sequence. While it does not appear to be necessary for O-fucosylation, tryptophan residues intercalate with the conserved arginine residues in TSRs, forming a characteristic tryptophan-arginine ladder that forms the core of the TSR fold (Figure 3E) [210]. Tryptophan also serves as a potential site for C-mannosylation (consensus sequence WXXW/C) that provides further support for the folding of TSR structures [210,211]. An enzyme distinct from POFUT1 was proposed for adding O-fucose to TSRs, since purified, recombinant POFUT1 was unable to catalysis this reaction [46]. Recombinant Drosophila Ofut2, a homologue of Drosophila Ofut1, was shown to have the capability to O-fucosylate TSRs [42]. Homologues of Ofut2 were also identified in the human (POFUT2), mouse (Pofut2), and C. elegans (pofut2) genomes.
Like POFUT1, POFUT2 is ER localized and requires a folded TSR structure for modification [46]. The fact that both POFUT1 and POFUT2 can discern folded substrate structures has led to the hypothesis that they participate in a non-canonical ER quality control process for protein folding, which will be discussed in more detail later. Pofut2 knockout in mice is embryonic lethal [212]. These embryos die in mid-gastrulation with extensive mesoderm differentiation, suggesting that POFUT2 is a key modulator of gastrulation. Database searches with POFUT2 O-fucose consensus sequence in the context of a TSR identified 49 putative protein targets (splice variants are not included) (Table 2) [24,59]. Notably, most of these targets are components of the extracellular matrix, with over half belonging to the A disintegrin and metalloproteinase with thrombospondin type 1 motifs (ADAMTS) family. Adamts9 knockout in mice leads to early embryonic lethality with phenotypes mirroring those observed in Pofut2 knockout embryos [213]. This strongly suggests that the gastrulation defects observed in Pofut2 knockout embryos are primarily due to the loss of O-fucosylation of ADAMTS9, emphasizing that O-fucosylation is essential for the biological function of POFUT2 protein targets. The O-fucose on TSR can be elongated with a glucose by β3-glucosyltransferase (B3GLCT). While no human mutations in POFUT2 have been identified, mutations in B3GLCT lead to a human developmental disorder named Peters-plus syndrome (PTRPLS), which will be discussed in more detail later [214].
Table 2.
List of putative human protein substrates of POFUT2.
Table 2.
List of putative human protein substrates of POFUT2.
| UniProt ID | Gene Name | Protein Name | Consensus/Total | Subcellular Location | Known Human Pathology (If Any) |
|---|---|---|---|---|---|
| Q9UHI8 | ADAMTS1 | A disintegrin and metalloproteinase with thrombospondin motifs 1 | 3/3 | ECM | - |
| O95450 | ADAMTS2 | A disintegrin and metalloproteinase with thrombospondin motifs 2 | 3/4 | ECM | Ehlers-Danlos syndrome, dermatosparaxis type (EDSDERMS) [215] |
| O15072 | ADAMTS3 | A disintegrin and metalloproteinase with thrombospondin motifs 3 | 3/4 | ECM | Hennekam lymphangiectasia-lymphedema syndrome 3 (HKLLS3) [216] |
| O75173 | ADAMTS4 | A disintegrin and metalloproteinase with thrombospondin motifs 4 | 1/1 | ECM | - |
| Q9UNA0 | ADAMTS5 | A disintegrin and metalloproteinase with thrombospondin motifs 5 | 2/2 [217] | ECM | - |
| Q9UKP5 | ADAMTS6 | A disintegrin and metalloproteinase with thrombospondin motifs 6 | 3/5 [218] | ECM | - |
| Q9UKP4 | ADAMTS7 | A disintegrin and metalloproteinase with thrombospondin motifs 7 | 6/8 | ECM | - |
| Q9UP79 | ADAMTS8 | A disintegrin and metalloproteinase with thrombospondin motifs 8 | 2/2 | ECM | - |
| Q9P2N4 | ADAMTS9 | A disintegrin and metalloproteinase with thrombospondin motifs 9 | 12/15 [219] | ECM | - |
| Q9H324 | ADAMTS10 | A disintegrin and metalloproteinase with thrombospondin motifs 10 | 3/5 [219] | ECM | Weill–Marchesani syndrome 1 (WMS1) [220,221] |
| P58397 | ADAMTS12 | A disintegrin and metalloproteinase with thrombospondin motifs 12 | 7/8 | ECM | - |
| Q76LX8 | ADAMTS13 | A disintegrin and metalloproteinase with thrombospondin motifs 13 | 7/8 [206] | ECM | Thrombotic thrombocytopenic purpura, hereditary (TTP) [222] |
| Q8WXS8 | ADAMTS14 | A disintegrin and metalloproteinase with thrombospondin motifs 14 | 3/4 | ECM | - |
| Q8TE58 | ADAMTS15 | A disintegrin and metalloproteinase with thrombospondin motifs 15 | 3/3 | ECM | Arthrogryposis, distal, 12 (DA12) [223] |
| Q8TE57 | ADAMTS16 | A disintegrin and metalloproteinase with thrombospondin motifs 16 | 6/6 | ECM | - |
| Q8TE56 | ADAMTS17 | A disintegrin and metalloproteinase with thrombospondin motifs 17 | 4/5 [224] | ECM | Weill–Marchesani syndrome 4 (WMS4) [225] |
| Q8TE60 | ADAMTS18 | A disintegrin and metalloproteinase with thrombospondin motifs 18 | 4/5 | ECM | Microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) [226] |
| Q8TE59 | ADAMTS19 | A disintegrin and metalloproteinase with thrombospondin motifs 19 | 4/5 | ECM | Cardiac valvular dysplasia 2 (CVDP2) [227,228] |
| P59510 | ADAMTS20 | A disintegrin and metalloproteinase with thrombospondin motifs 20 | 12/15 [229] | ECM | - |
| Q8N6G6 | ADAMTSL1 | ADAMTS-like protein 1 | 9/9 [207] | ECM | - |
| Q86TH1 | ADAMTSL2 | ADAMTS-like protein 2 | 6/7 [230] | ECM | Geleophysic dysplasia 1 (GPHYSD1) [231,232] |
| P82987 | ADAMTSL3 | ADAMTS-like protein 3 | 9/10 | ECM | - |
| Q6UY14 | ADAMTSL4 | ADAMTS-like protein 4 | 2/6 | ECM | Ectopia lentis 2, isolated, autosomal recessive (ECTOL2) [233]; Ectopia lentis et pupillae (ECTOLP) [234] |
| Q6ZMM2 | ADAMTSL5 | ADAMTS-like protein 5 | 1/1 | ECM | - |
| O14514 | ADGRB1 | Adhesion G protein-coupled receptor B1 | 4/5 [235] | CM | - |
| O60241 | ADGRB2 | Adhesion G protein-coupled receptor B2 | 4/4 | CM | - |
| O60242 | ADGRB3 | Adhesion G protein-coupled receptor B3 | 4/4 | CM | - |
| P13671 | C6 | Complement component C6 | 1/3 | ECM | Complement component 6 deficiency (C6D) [236] |
| O00622 | CCN1 | CCN family member 1 | 1/1 [237] | ECM | - |
| P29279 | CCN2 | CCN family member 2 | 1/1 [219] | ECM | - |
| P48745 | CCN3 | CCN family member 3 | 1/1 | ECM, CYT | - |
| O95388 | CCN4 | CCN family member 4 | 1/1 | ECM | - |
| O76076 | CCN5 | CCN family member 5 | 1/1 | ECM | - |
| O95389 | CCN6 | Cellular communication network factor 6 | 1/1 | ECM | Progressive pseudorheumatoid dysplasia (PPRD) [238] |
| P27918 | CFP | Properdin | 4/7 [239] | ECM | Properdin deficiency (PFD) [240] |
| Q8IUL8 | CILP2 | Cartilage intermediate layer protein 2 | 1/1 | ECM | - |
| Q96RW7 | HMCN1 | Hemicentin-1 | 6/6 | ECM, CYT | Macular degeneration, age-related, 1 (ARMD1) [241] |
| B1AKI9 | ISM1 | Isthmin-1 | 1/1 | ECM | - |
| O95428 | PAPLN | Papilin | 4/5 | ECM | - |
| Q13591 | SEMA5A | Semaphorin-5A | 2/7 | CM | - |
| Q9P283 | SEMA5B | Semaphorin-5B | 2/5 | CM | - |
| Q9HCB6 | SPON1 | Spondin-1 | 5/6 [242] | ECM | - |
| A2VEC9 | SSPOP | SCO-spondin | 16/25 [243] | ECM | - |
| P07996 | THBS1 | Thrombospondin-1 | 3/3 [204] | ECM | - |
| P35442 | THBS2 | Thrombospondin-2 | 3/3 [209] | ECM | Intervertebral disc disease (IDD) [244] |
| Q9NS62 | THSD1 | Thrombospondin type-1 domain-containing protein 1 | 1/1 | ECM | Lymphatic malformation 13 (LMPHM13) [245,246]; Aneurysm, intracranial berry, 12 (ANIB12) [247,248] |
| Q6ZMP0 | THSD4 | Thrombospondin type-1 domain-containing protein 4 | 3/6 | ECM | Aortic aneurysm, familial thoracic 12 (AAT12) [249] |
| Q9UPZ6 | THSD7A | Thrombospondin type-1 domain-containing protein 7A | 7/19 | ECM, CM | Pathogenic autoantigen in membranous nephropathy (MN) [250,251] |
| Q9C0I4 | THSD7B | Thrombospondin type-1 domain-containing protein 7B | 6/18 | CM | - |
This list of putative POFUT2 substrates is updated from Luther et al., 2021 [59]. Proteins with experimentally confirmed O-fucosylation are highlighted in bold, with references provided. The number of TSRs containing POFUT2 consensus sequences C1−2XX[S/T]C2−3 out of the total TSR number is listed. Any known human pathologies associated with the listed proteins (reported in UniProt) are indicated with the corresponding references. Major subcellular locations for the listed proteins (reported in UniProt) are indicated. ECM, extracellular matrix; CM, cell membrane; CYT, cytoplasm.
3.2.3. POFUT3 (FUT10) and POFUT4 (FUT11)
In 2002, Roos et al. used a whole-genome approach to search for enzymes that involved fucosylated glycan metabolism in Drosophila genome [252]. A Drosophila fucosyltransferase, FucTB, was among their findings. Using the FucTB sequence to search the human genome, two human orthologs were identified. These human genes were named FUT10 and FUT11, as they were identified after FUT9. Due to the presence of the two conserved motifs shared with other human α3-FUTs (FUT3–7 and FUT9), it was proposed that they were responsible for GDP-fucose donor binding, and FUT10 and FUT11 were initially presumed to be α3-FUTs.
The Drosophila FucTB gene was cloned together with FucTA and FucTC as homologs in 2001, but only FucTA demonstrated clear activity as a core α3-FUT for N-glycans [253]. No activity was detected for FucTB in generating either core-fucosylated N-glycans or Lewis-type glycans [253,254]. Efforts to experimentally demonstrate fucosyltransferase activity using free glycan substrates for the mouse homologs, Fuc-TX and Fuc-TXI, and the honeybee FucTB homolog were also unsuccessful, although Fuc-TXI was found to possess GDP-fucose hydrolase activity [255,256,257]. Studies exploring the fucosyltransferase activity of human FUT10 and FUT11 have yielded varied results. Mollicone et al. reported core α3-FUT activity for both FUT10 and FUT11, showing they modify the innermost GlcNAc moiety of the chitobiose unit on N-glycans via an α3-linkage using synthetic glycan substrates [258]. In contrast, Kumar et al. reported that FUT10 is involved in an α3-FUT activity for the synthesis of Lewis X (LeX) epitopes in neuroblastoma cells [259]. It is important to note that neither of these studies used purified enzymes to detect enzymatic activity; instead, crude lysates from FUT10/11-transfected cells were used as the enzyme source. This raises the possibility that the activity observed in their studies could be due to indirect effects from protein substrates modified by FUT10/11 or from other endogenous FUTs in the cell lysate. The substrate specificity of FUT10 and FUT11 remained unclear for a decade until recently (Table 3).
Table 3.
List of putative human protein substrates of POFUT3 and POFUT4.
In 2024 a new type of domain-specific O-fucosylation was identified in a proteomic study on human platelet releasate [163]. The fucose was added at a high stoichiometry on the elastin microfibril interface (EMI) domain of Multimerin-1 (MMRN1), a key site for protein–protein interaction (Figure 4C). EMI is a cysteine-rich domain with three conserved disulfide bonds, like the EGF or TSR domains but larger (Figure 3C). Neither POFUT1 nor POFUT2 was found to be responsible for this modification, suggesting the existence of a previously unidentified POFUT specific for the EMI O-fucosylation [45]. AlphaFold2 was employed to screen potential binding interactions between human FUTs and the EMI domain [45]. FUT10 and FUT11 demonstrated significant EMI binding that was not observed with other FUTs. The AlphaFold2 predictions were further validated through co-immunoprecipitation assays, confirming the interaction between FUT10/11 and EMI domains in a cellular context. In vitro enzymatic assays with purified, recombinant FUT10 and FUT11 revealed robust POFUT activity with EMI substrates. Furthermore, knocking out either FUT10 or FUT11 in HEK293T cells significantly reduced EMI O-fucosylation, while knocking out of both enzymes completely abolished the modification. These findings imply that FUT10 and FUT11 function as novel POFUTs for EMI domains, leading to their renaming as POFUT3 and POFUT4, respectively.
FUT10 and FUT11 have been annotated as α3-FUTs (see Q6P4F1 and Q495W5 in UniProt). They were classified to the CAZy GT10 family, along with the invertebrate and plant core α3-FUTs and all terminal α3/4-FUTs [27]. However, multiple clues suggest that they are in a distinct family that should be separated from other α3-FUTs. The α3/4-FUT family shares five conserved peptide motifs: motifs I–III contribute to acceptor substrate recognition, while motifs IV and V contribute to recognition and binding of the donor substrate GDP-fucose [258,267,268]. FUT10 and FUT11 show high sequence homology with other human GT10 family members (FUT3–7 and FUT9) in motifs IV and V, with strong conservation of residues essential for enzymatic activity and GDP-fucose binding [258]. This homology is also reflected in the 3D structural alignment of AlphaFold2 predicted structures of FUT10/11 with crystal structures of H. pylori FucT and human FUT9, where the C-terminal Rossmann-like domain that responsible for GDP-fucose binding shows a high degree of similarity [45]. In contrast to motifs IV and V, FUT10 and FUT11 show limited homology with other α3-FUTs in motifs I–III, both in primary peptide sequence and 3D structural alignment [45,252,256,258]. All α3/4-FUTs possess the conserved peptide sequence [F/I/V]HH[R/W][E/D] in motif II, a characteristic motif for lactosamine acceptor binding, as a single amino acid change (W to R) can convert the α4 activity of FUT3 to α3 activity [269]. Instead, FUT10 and FUT11 present a different conserved peptide motif FYGTDF at the equivalent position, indicating they do not use lactosamine as acceptors. Additionally, FUT10 and FUT11 contain a unique C-terminal extension predicted to generate additional contacts with EMI substrates for aiding in substrate selection [45]. These differences suggest that FUT10 and FUT11 have a distinct acceptor substrate compared to other α3/4-FUTs family members. Phylogenetic analysis indicates that FUT10 and FUT11 belong to an evolutionary distinct group compared to other α3/4-FUTs [252,258]. These enzymes are ancient, originating at about 830 MYA (Million Years Ago), and possess a split polyexonic genomic structure, which is clearly distinct from the monoexonic α3/4-FUTs that originated around 450 MYA. Note that POFUT1 and POFUT2 are also ancient enzymes (originated >1000 MYA) with a polyexonic structure [270].
Like POFUT1 and POFUT2, POFUT3 (FUT10) and POFUT4 (FUT11) are ER-localized enzymes [45]. They also require folded domain structures for efficient modification and participate in a non-canonical ER quality control pathway for protein secretion. In addition to these similarities, POFUT3/4-mediated EMI O-fucosylation displays several unique characteristics. First, unlike EGF repeats and TSRs which are often embedded within proteins as tandem repeats, the EMI domain is consistently singular and located at the N-terminus of proteins. Second, multiple fucose residues can be added to the EMI domain in a spatially adjacent manner. For instance, the EMI domain on MMRN1 can hold two fucose residues, while the EMI domain on MMRN2 can hold three [45]. These fucose residues on EMI domains are exclusively observed as monosaccharides, whereas those on EGF repeats and TSRs are frequently elongated. Screening more EMI-containing proteins for O-fucosylation would help decipher the code for fucose addition and refine the POFUT3/4 consensus sequence. There are 17 EMI-containing human proteins annotated in UniProt that are potential substrates for POFUT3/4 (Table 3). However, given the different subsets of EMI domain structures, it is possible only some are O-fucosylated [271,272,273].
No human genetic diseases have been identified with mutations in POFUT3 or POFUT4. However, aberrant expression of POFUT4 has been associated with several types of cancer [274,275,276,277,278,279,280]. In mice, POFUT3 and POFUT4 are expressed ubiquitously in embryos throughout development but show distinct tissue-specific expression patterns in adults [255,256,258]. Notably, they are strongly expressed in Purkinje cells in the brain, suggesting a potential role for these enzymes and their substrates in motor coordination [255]. POFUT3 is required for the maintenance of mouse embryonic stem (ES) cells and neural stem cells (NSCs) [259]. Overexpression of POFUT3 enhanced the self-renewal of NSCs, while knocking down POFUT3 induced the differentiation of NSCs and ES cells. Knocking down POFUT4 in zebrafish embryos resulted in malformations of the posterior trunk and tail, indicating its important roles in vertebrate development [256]. The POFUT3/4 protein substrates and potential mechanisms involved in these events remain unknown. Generating POFUT3 and/or POFUT4 knockout mice would provide deeper insights into their biological functions in embryonic development, neurological function, and stem cell maintenance.
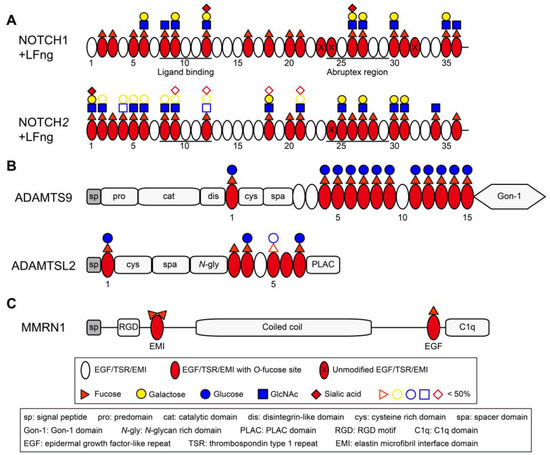
Figure 4.
Domain maps of representative EGF/TSR/EMI-containing proteins showing distribution of O-fucosylation. (A) Domain maps of mouse NOTCH1 EGF1–36 and NOTCH2 EGF1–36. The majority of EGF repeats are modified with O-fucose glycans with more than half elongated by Lfng (modified from Ref. [66] with permission). (B) Domain maps of human ADAMTS9 and mouse ADAMTSL2. The majority of TSRs in ADAMTS9 and ADAMTSL2 are modified with Glucoseβ1-3Fucose disaccharide by POFUT2 and B3GLCT (modified from Refs. [59,230] with permission) [213]. (C) Domain map of human MMRN1 showing the EMI domain at protein N-terminal and can be modified with two fucose residues.
Figure 4.
Domain maps of representative EGF/TSR/EMI-containing proteins showing distribution of O-fucosylation. (A) Domain maps of mouse NOTCH1 EGF1–36 and NOTCH2 EGF1–36. The majority of EGF repeats are modified with O-fucose glycans with more than half elongated by Lfng (modified from Ref. [66] with permission). (B) Domain maps of human ADAMTS9 and mouse ADAMTSL2. The majority of TSRs in ADAMTS9 and ADAMTSL2 are modified with Glucoseβ1-3Fucose disaccharide by POFUT2 and B3GLCT (modified from Refs. [59,230] with permission) [213]. (C) Domain map of human MMRN1 showing the EMI domain at protein N-terminal and can be modified with two fucose residues.
4. O-Fucosylation: Physiological and Pathological Significance
4.1. Regulation of Lymphoid and Myeloid Cell Development by Notch O-Fucosylation
As previously mentioned, Notch is a key regulator in various developmental processes, frequently influencing cell fate decisions at multiple stages as cells differentiate toward specific cell types. In hematopoiesis, proper Notch signaling is essential for the survival, proliferation, and cell fate determination at various stages of lineage commitment, including myeloid cell production in the bone marrow, T cells development in the thymus, and marginal zone (MZ) B cells development in the spleen. O-fucose glycans incorporated to Notch extracellular domain by POFUT1 and further elongated by Fringe modulate the strength of Notch signaling, thus playing critical roles in these Notch-regulated programs (Figure 4A).
Throughout adult life, blood cell production is maintained by a group of pluripotent hematopoietic stem cells (HSCs) located in the bone marrow. HSCs generate multipotent progenitor populations (MPPs) with increased lineage-specific potentials. Within this MPP pool, lymphoid-primed MPPs (LMPP or MPP4) subsequently give rise to common lymphoid progenitors (CLPs) and common myeloid progenitors (CMPs), which are committed to the lymphoid lineage (T, B, and natural killer cells) and the myeloid lineage (neutrophils, monocytes, erythrocytes, and more), respectively [281]. Notch signaling influences this decision by favoring a lymphoid over myeloid lineage outcome [282,283]. Conditional knockout Pofut1 in mice HSCs within the bone marrow leads to reduced T lymphopoiesis in the thymus and reduced marginal-zone B cells production in the spleen, whereas myeloid cells in bone marrow were increased [284]. Restoration of Notch1 signaling rescued these phenotypes. The Pofut1 knockout in HSCs completely abrogated the binding of cell surface Notch with Delta ligands and suppressed the activation of downstream Notch target genes. In another study, the selective deletion of the Notch ligand DLL4 in bone-producing osteocalcin (Ocn)-expressing cells, which highly express DLL4 in the bone marrow, results in a decrease in intrathymic T cell precursors and mature T cells production [285]. These findings suggest that Notch O-fucosylation is essential for maintaining hematopoietic lineage homeostasis by promoting lymphoid development and suppressing overt myelopoiesis. Interestingly, pan-Notch inhibition through the conditional deletion of RBP-Jκ led to more severe phenotypes than the deletion of Pofut1 in HSCs, suggesting other O-glycans on Notch may contribute to this Notch-regulated process [285]. The roles of other Notch O-glycans in hematopoiesis were recently reviewed in detail [65].
T cell maturation takes place in the thymus, where lymphocyte precursors released from the bone marrow migrate through blood vessels and colonize the thymus as thymus-seeding progenitors (TSPs). Upon entering the thymus, these cells initiate a differentiation process that includes crucial events such as T cell lineage commitment and the choice between αβ and γδ T-cell lineages, as they migrate from the corticomedullary junction towards the outer cortical zone. During this migration, the progenitor cells traverse a network of thymic stromal cells that express Notch ligands, initiating Notch signaling in the progenitor cells and guiding them through the differentiation programs. This requirement of Notch signaling in T cell development is well-documented. TSPs possess robust potential to generate non-T cell lineages, including B cells, myeloid cells, and dendritic cells (DC) [286]. NOTCH1-DLL4 signaling compels these cells to commit to a T cell fate while simultaneously suppressing their potential to adopt other lineages. Disrupting Notch signaling in HSCs through conditional deletion of Notch1 or RBPJκ blocks early-stage T cell development, causing T cell progenitors to differentiate into B cells and myeloid cells in the thymus [287,288,289,290]. By using fucose analogs (6-alkynyl fucose and 6-alkenyl fucose) that incorporate into Notch and specifically inhibit Delta-induced Notch signaling in an in vitro OP9 co-culture system, the differentiation of bone marrow stem cells into T cell progenitors was completely abolished, indicating the important roles of O-fucose in Notch-regulated T cell lineage commitment [291]. Interestingly, overexpression of Lunatic Fringe (Lfng) in the thymus, which enhances Notch binding with Delta ligands, unexpectedly inhibits Notch activation in TSPs [289]. This is because when Lfng is overexpressed in CD4/CD8 double-negative (DN) 3 and CD4/CD8 double-positive (DP) cells, it transforms them into ‘super-competitors’ for the limited intrathymic niches supplying Delta ligands [292]. This limits the access of early T-cell progenitors towards these ligands, which are needed for the required Notch signaling for their differentiation.
NOTCH2-DLL1 signaling is required for the development of marginal zone (MZ) B cells in the spleen [293,294]. Conditional deletion of Pofut1 in HSCs reduced marginal-zone B cells production in spleen [284]. Although neither Lfng nor Manic Fringe (Mfng) is indispensable for MZ B cell development, the loss of both Fringes significantly impairs the production of MZ B cells in spleen [295]. Therefore, Lfng and Mfng cooperatively enhance the NOTCH2-DLL1 signaling to promote the development of MZ B cells.
4.2. Cancer with Altered O-Fucosylation
Notch signaling plays diverse roles during development and tissue homeostasis. A growing number of cancers are linked to dysregulated Notch signaling, where Notch can act as either an oncogene or a tumor suppressor, depending on the cellular context [296]. As a crucial regulator of Notch signaling, altered O-fucosylation is commonly observed in cancer. Here, we highlight prototypical cancers where altered O-fucosylation plays a role in their pathological mechanisms. This alteration can involve changes in POFUT1 levels or activity, or mutations in Notch that impact its O-fucosylation.
POFUT1 is localized in the chromosomal region 20q11.21, which is frequently amplified in colorectal cancer (CRC) [297]. This copy number variations (CNVs) in CRC results in elevated POFUT1 expression in tumors compared to non-tumor adjacent tissues [298]. The high expression of POFUT1 in CRC from stage I is associated with the metastatic process. ShRNA-mediated knockdown of POFUT1 downregulates NOTCH1 signaling, leading to decreased cell proliferation, migration, and induced apoptosis in CRC cells in vitro, as well as suppression of CRC tumor growth and transplantation in vivo [299]. In less frequent CRC cases with no chromosomal amplification and POFUT1 overexpression, gain-of-function mutations in POFUT1 have been identified, potentially contributing to tumor progression [299]. POFUT1 is overexpressed in hepatocellular carcinoma (HCC), and its high expression correlates with poor prognosis in HCC patients [300,301]. Elevated POFUT1 levels accelerated the proliferation and migration of HCC cells and increased their binding to DLL1, consequently enhancing Notch signaling activity. Overexpression of POFUT1 is also observed in glioblastoma [302,303], gastric cancer [304], oral cancer [305], and breast cancer [306], correlating with increased Notch activity and aggressive phenotypes, such as increased cell proliferation, invasion, metastasis, or larger tumor size. Interestingly, in cancer types where Notch functions as a tumor suppressor, such as in muscle-invasive bladder cancer (MIBC), low levels of POFUT1 mRNA are associated with poor survival in MIBC patients after radical cystectomy [307,308]. These findings highlight the potential of POFUT1 as a diagnostic marker and therapeutic target for these cancers.
In anaplastic large cell lymphoma (ALCL), the Notch pathway ranks among the pathways that are most enriched in mutations through gene-set enrichment analysis. NOTCH1 p.T311P (eliminates O-fucose site on EGF8) and p.T349P (eliminates O-fucose site on EGF9) missense mutations were found in patients’ tumor samples by whole-exome sequencing [309]. Interestingly, overexpression of these mutations in HEK293T cells led to increased proliferation compared to cells with wild-type NOTCH1. While the detailed mechanism remains unclear, this suggests a correlation between alterations in specific O-fucose sites on NOTCH1 and cancer. Recently, by using glycoproteomic site mapping and cell-based Notch assays, nine cancer-associated Notch variants were investigated to determine their impact on Notch O-fucosylation, ligand binding and signaling activity [310]. Two variants led to a gain of function in NOTCH1 (G309R and N386T), six led to a loss of function (G310R, T311P, G347S, T349P, D464N, and A465T), and one had minimal effects (G230R). This suggests that point mutations in Notch can alter its O-fucosylation, consequently affecting Notch signaling activity and being linked to cancer-related processes.
Elevated expression of POFUT4 was observed in gastric cancer (GC) tissues compared to non-tumor adjacent tissues and is correlated with poor prognosis and survival in GC patients [274,276]. Knockdown of POFUT4 reduced the proliferation and migration of GC cells by suppressing the PI3K/AKT signaling pathway. Overexpression of POFUT4 and its correlation with poor prognosis were also reported in multiple other cancer types, including clear cell renal cell carcinoma (ccRCC) [279,280], colon adenocarcinoma (COAD) [278], hepatocellular carcinoma (HCC) [277], and pancreatic cancer (PC) [275]. Although the related protein substrates and mechanisms underlying POFUT4’s role in cancer development and progression remain unknown, these findings suggest that targeting POFUT4 holds therapeutic potential and may serve as a novel biomarker for cancer prognosis.
4.3. Peters-Plus Syndrome (PTRPLS)
Failure to add glucose to O-fucose on TSRs causes human Peters-plus syndrome (PTRPLS, OMIM #261540), an autosomal recessive congenital disorder of glycosylation (CDG) characterized by Peters’ anomaly of the eye (anterior eye chamber segment dysgenesis), craniofacial defects (widened forehead and cleft lip/palate), short stature, brachydactyly, and developmental delay [214]. PTRPLS is caused by loss-of-function mutations in β3-glucosyltransferase (B3GLCT), an enzyme responsible for extending O-fucose monosaccharide to the Glucoseβ1-3Fucose disaccharides on TSRs (Figure 3B) [311,312,313,314]. PTRPLS-like patients only share a subset of phenotypes observed in PTRPLS patients, and some of them also have missense mutations in B3GLCT [311]. In contrast to PTRPLS mutations that lead to a complete loss of enzymatic activity, PTRPLS-like mutations retained enzymatic activity with only minor destabilizing effects [315]. This partially explains the milder phenotypes observed in PTRPLS-like patients.
Over half of the putative POFUT2 targets are members of the ADAMTS/ADAMTS-like family, suggesting that these proteins could be the primary biological targets for B3GLCT (Table 2 and Figure 4B). These secreted ADAMTS proteases and non-catalytic members of the family (ADAMTS-like proteins) are important components of the extracellular matrix (ECM), and are implicated in diverse biological events, including embryogenesis and angiogenesis [316,317]. Numerous congenital disorders caused by mutations in ADAMTS/ADAMTS-like genes display overlapping phenotypes with PTRPLS patients. For instance, mutations in ADAMTSL2 cause human Geleophysic dysplasia 1 (GPHYSD1, OMIM #231050), where patients exhibit overlapped phenotypes such as short stature and short hands and feet [230,232]. Mutations in ADAMTS10 cause Weill–Marchesani syndrome 1 (WMS, OMIM #277600), with overlapped phenotypes including short stature, brachydactyly and eye anomalies [220]. These suggest that impaired O-fucosylation of the ADAMTS/ADAMTS-like family proteins is likely the primary biological mechanism responsible for the phenotypes observed in PTRPLS patients.
POFUT2 and B3GLCT are demonstrated to mediate a noncanonical ER quality-control process for the secretion of TSR-containing proteins (molecular details will be discussed later) [318]. POFUT2 is required for the secretion of almost all ADAMTS/ADAMTS-like proteins tested to date (ADAMTS6 [218], ADAMTS9 [219,229], ADAMTS10 [218], ADAMTS13 [318], ADAMTS17 [224], ADAMTS20 [229], ADAMTSL1 [318], ADAMTSL2 [318]), whereas the impact of B3GLCT on secretion varied among proteins. For instance, secretion of ADAMTS20, ADAMTSL1, and ADAMTSL2 is profoundly reduced with B3GLCT knockdown/out [229,318]. In contrast, ADAMTS9 and ADAMTS13 secretion is moderately reduced by 20%, and ADAMTS17 secretion is unaffected [219,224,229,318]. Considering the early embryo lethality observed in Pofut2-null mice and the less severe phenotypes in PTRPLS patients, it implies that the pathology of PTRPLS is due to defects in a specific subset of POFUT2 targets that are sensitive to the loss of extended glucose.
4.4. Spondylocostal Dysostosis 3 (SCDO3)
Failure to add GlcNAc to O-fucose on NOTCH1 EGF repeats causes Spondylocostal Dysostosis 3 (SCDO3, OMIM #609813), an autosomal recessive disorder in human that characterized by multiple defects in vertebral segmentation, including hemivertebrae, butterfly vertebrae, fusion or missing vertebrae, scoliosis, and rib anomalies [319]. SCDO3 is caused by loss-of-function mutations in Lunatic Fringe (Lfng), leading to mis-localization, reduced stability or impaired enzymatic activity of proteins [320]. More than twenty Lfng variants have been identified in SCDO3 patients, with the mutational spectrum evenly distributed across all exons [321]. Fringe modifications play a crucial role in Notch signaling by fine-tuning Notch-ligand interaction, enhancing signaling from Delta-like ligands and inhibiting signaling from Jagged ligands. Delta-like 1:NOTCH1 signaling is central to somite formation during embryonic development [322,323]. Lfng knockout in mice results in multiple somitogenesis defects similar to those reported in SCDO3 patients and causes perinatal lethality due to malformed rib cages [324,325]. In contrast, Rfng or Mfng knockouts in mice exhibit no obvious phenotypes, while triple knockouts of all three Fringes show the same phenotypes as Lfng knockout mice [326,327,328].
4.5. Other Biological Processes Where O-Fucosylation May Play a Role
In addition to the biological events discussed above, O-fucose glycans have been identified in numerous other biologically important proteins, where they play crucial roles in modulating protein function. POFUT1-mediated O-fucosylation of Agrin, a key regulator of postsynaptic differentiation at the neuromuscular junction (NMJ), has been demonstrated to determine its acetylcholine receptor clustering activity [81]. The O-fucose moiety on TSR3 of BAI1 (ADGRB1), added by POFUT2, directly interacts with the RTN4 receptor, enabling high-affinity interactions that regulate neuronal development (Figure 5C) [235]. Mutating the POFUT3/4 O-fucose sites within the EMI domain of MMRN1, an essential platelet component supporting platelet adhesion and thrombus formation, significantly reduces its secretion. While mutating the POFUT1 O-fucose site within the EGF domain leads to a complete loss of MMRN1 secretion, implying an as-yet-unidentified role for O-fucosylation in platelet functions [163]. We envision that a deeper profiling of the O-fucose proteome and investigation into the mechanisms by which O-fucose affects protein functionality will uncover more biological significance of O-fucosylation in the future.
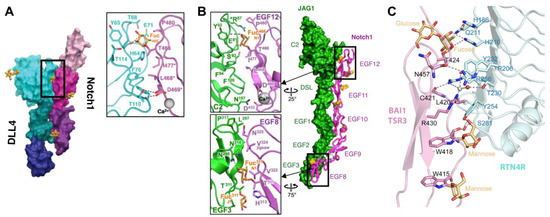
Figure 5.
O-fucose glycans form direct contacts with binding partners. (A) Co-crystal structure of NOTCH1 EGF11–13 (magenta/purple) and DLL4 N-EGF1 (blue/green) (modified with permission from Ref. [72]). Zoomed panel shows direct interaction between fucose residue on NOTCH1 EGF12 with residues in DLL4. (B) Co-crystal structure of NOTCH1 EGF8–12 (magenta) and JAG1 N-EGF3 (green) (modified with permission from Ref. [73]). Zoomed panels show direct interactions between fucose residues on EGF8 and EGF12 with residues in JAG1. (C) Co-crystal structure of RTN4R (cyan) and BAI1 TSR3 (pink) (modified with permission from Ref. [235]). O-fucose forms direct interactions with residues in RTN4R.
Figure 5.
O-fucose glycans form direct contacts with binding partners. (A) Co-crystal structure of NOTCH1 EGF11–13 (magenta/purple) and DLL4 N-EGF1 (blue/green) (modified with permission from Ref. [72]). Zoomed panel shows direct interaction between fucose residue on NOTCH1 EGF12 with residues in DLL4. (B) Co-crystal structure of NOTCH1 EGF8–12 (magenta) and JAG1 N-EGF3 (green) (modified with permission from Ref. [73]). Zoomed panels show direct interactions between fucose residues on EGF8 and EGF12 with residues in JAG1. (C) Co-crystal structure of RTN4R (cyan) and BAI1 TSR3 (pink) (modified with permission from Ref. [235]). O-fucose forms direct interactions with residues in RTN4R.
5. Molecular Mechanisms of How O-Fucose Regulates Protein Functions
5.1. O-Fucosylation Forms Direct Contacts with Binding Partners
Out of the 36 EGF repeats in the extracellular domain of mouse NOTCH1, 20 contain the O-fucose consensus sequence, and 17 have been confirmed to be modified at high stoichiometries (Figure 4A) [67]. Similar results were observed in Drosophila NOTCH and mouse NOTCH2 [66,329]. These heavily decorated O-fucose glycans are required for the proper function of Notch. Through the examination of individual O-fucose effects on NOTCH1 activity via site-directed mutagenesis, it was found that O-fucose on EGF8 and EGF12 had the most significant impact on the binding of NOTCH1 with DLL1 and JAG1 ligands [67]. Similar results were obtained with NOTCH2 [66]. In vitro assays using purified human NOTCH1 EGF11–13 also demonstrated that the addition of O-fucose enhanced its binding to DLL1 and JAG1 ligands [74].
Evidence confirming the direct interaction of O-fucose on NOTCH1 EGF8 and EGF12 with ligands is provided by two co-crystal structures: one between a portion of NOTCH1 ligand-binding domain (EGF11–13) and a portion of DLL4 [72], and the other between the NOTCH1 ligand-binding domain (EGF8–12) and a portion of JAG1 (Figure 5A,B) [73]. Both co-crystal structures showed direct contacts between O-fucose on EGF12 with the backbone and side-chain residues of both ligands. The NOTCH1-JAG1 structure also showed a direct contact of O-fucose on EGF8 with JAG1. Unlike many other glycan modifications, O-fucose residues display thermal mobility comparable to amino acids, acting as ‘surrogate amino acids’ to form specific and essential contacts with residues on binding partners, thereby facilitating ligand binding. This direct intermolecular interaction generated by O-fucose glycans in protein–protein interactions was also observed in the co-crystal structure of BAI1 with RTN4 receptor (Figure 5C) [235]. The Glucoseβ1-3Fucose disaccharide of the BAI1 TSR3 domain directly interacts with the RTN4 receptor, enabling high-affinity binding and regulating neuronal development.
5.2. O-Fucosylation Generates Intramolecular Interactions That Stabilize Protein Domains and Facilitate Secretion
The appropriate pairing of cysteines to form correct disulfide bonds is a crucial step during protein folding in the ER, and this presents a particular challenge for proteins that contain multiple, tandem cysteine-rich domains such as EGFs and TSRs. It provides a strategic rationale for cells to evolve dedicated quality-control mechanisms to meet the specialized requirements for the folding of these domains. As mentioned above, POFUT1–4 are localized in the ER and only modify properly folded domain structures. These led to the hypothesis that the ER-localized POFUTs participate in a non-canonical ER quality control system for the folding and secretion of proteins containing cysteine-rich domains.
In co-crystal structures of mouse POFUT1 with EGF domains from Notch, the binding interface between POFUT1 and EGF showed a high degree of complementarity, with approximately one-third of the EGF’s surface area being deeply embedded within the groove of POFUT1 (Figure 6A) [330]. The EGF region that contains O-fucose consensus motif C2XXXX[S/T]C3 is buried at the bottom and the hydroxyl group of serine or threonine to be modified is precisely oriented to the position necessary for the fucose transfer. Similarly, in co-crystal structures of Caenorhabditis elegans POFUT2 (CePOFUT2) with human TSR1, nearly half of the TSR1 surface is embedded within the groove of CePOFUT2 (Figure 6A) [205]. Likewise, the AlphaFold2-multimer predicted structures of POFUT3/4 and the MMRN1 EMI domain revealed a highly confident interface complementarity, with fucose sites positioned in close proximity to the putative GDP-binding pocket (Figure 6B) [45]. These ‘hand-in-glove’ complementary interactions between enzymes and their respective protein domain structures elegantly explain why the enzyme necessitates a folded structure and a specific consensus sequence for modification. This suggests that the POFUTs are folding sensors for their respective domains.
Glycan modifications are usually too flexible to be observed in crystal structures. However, O-fucose glycans are remarkably visible. Their thermal stability (based on B factors) is comparable to that of the underlying amino acids due to numerous intramolecular interactions [72,74]. In the crystal structures of human NOTCH1 EGF11–13 modified with GlcNAcβ1-3Fucose disaccharide, the glycan residues lay down on the surface of EGF12, forming multiple intramolecular contacts with neighboring amino acid residues (Figure 3D) [74]. Through a combination of MD simulations, X-ray crystallography, and NMR, the O-fucose glycans on TSR3 of THBS1 were shown to make intramolecular interactions with adjacent amino acids resulting in shielding of the C2-C6 disulfide bond to protect it from reduction [331]. Such interactions were also identified by MolProbity in several other TSR-containing proteins, such as ADAMTS13 and Properdin (Figure 3E) [44,79]. For the EMI domain, although no crystal structures are available, the AlphaFold2-predicted structure of the MMRN1 EMI domain shows that the two fucose sites are spatially adjacent but reside on two separate, antiparallel β-strands (Figure 3C) [45]. It is possible that the fucose residues generate intramolecular interactions with spatially adjacent amino acid residues on the other β-strand, bringing the two β-strands closer together, thereby restricting their flexibility and compacting the EMI structure.
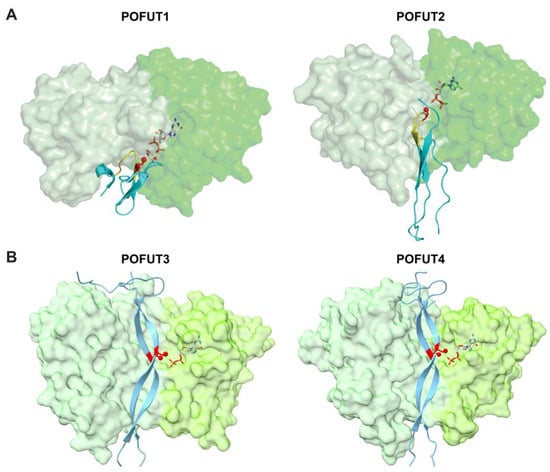
Figure 6.
The cleft in each POFUT shows a high degree of complementarity with its respective substrate domains. (A) Crystal structures of POFUT1 with EGF domain (left panel, overlay of PDB IDs 5KXH and 5KY3) and POFUT2 with TSR (right panel, PDB ID 5FOE). (B) AlphaFold2-multimer predicted structure of human POFUT3 (FUT10) (left panel, residues 81–479) and human POFUT4 (FUT11) (right panel, residues 73–492) with human MMRN1 EMI domain. POFUT1, POFUT2, POFUT3 and POFUT4 enzymes are displayed in surface representations with A domains in light green and B domains in darker green. Folded domains (EGF repeats for POFUT1; TSR for POFUT2; EMI for POFUT3 and POFUT4) are in cyan cartoon with respective consensus sequences of POFUT1 and POFUT2 in yellow. Acceptor amino acid is shown in red sticks, with the acceptor oxygen as a red sphere. Donor nucleotides, white sticks. Panel A is modified with permission from Ref. [332].
Figure 6.
The cleft in each POFUT shows a high degree of complementarity with its respective substrate domains. (A) Crystal structures of POFUT1 with EGF domain (left panel, overlay of PDB IDs 5KXH and 5KY3) and POFUT2 with TSR (right panel, PDB ID 5FOE). (B) AlphaFold2-multimer predicted structure of human POFUT3 (FUT10) (left panel, residues 81–479) and human POFUT4 (FUT11) (right panel, residues 73–492) with human MMRN1 EMI domain. POFUT1, POFUT2, POFUT3 and POFUT4 enzymes are displayed in surface representations with A domains in light green and B domains in darker green. Folded domains (EGF repeats for POFUT1; TSR for POFUT2; EMI for POFUT3 and POFUT4) are in cyan cartoon with respective consensus sequences of POFUT1 and POFUT2 in yellow. Acceptor amino acid is shown in red sticks, with the acceptor oxygen as a red sphere. Donor nucleotides, white sticks. Panel A is modified with permission from Ref. [332].
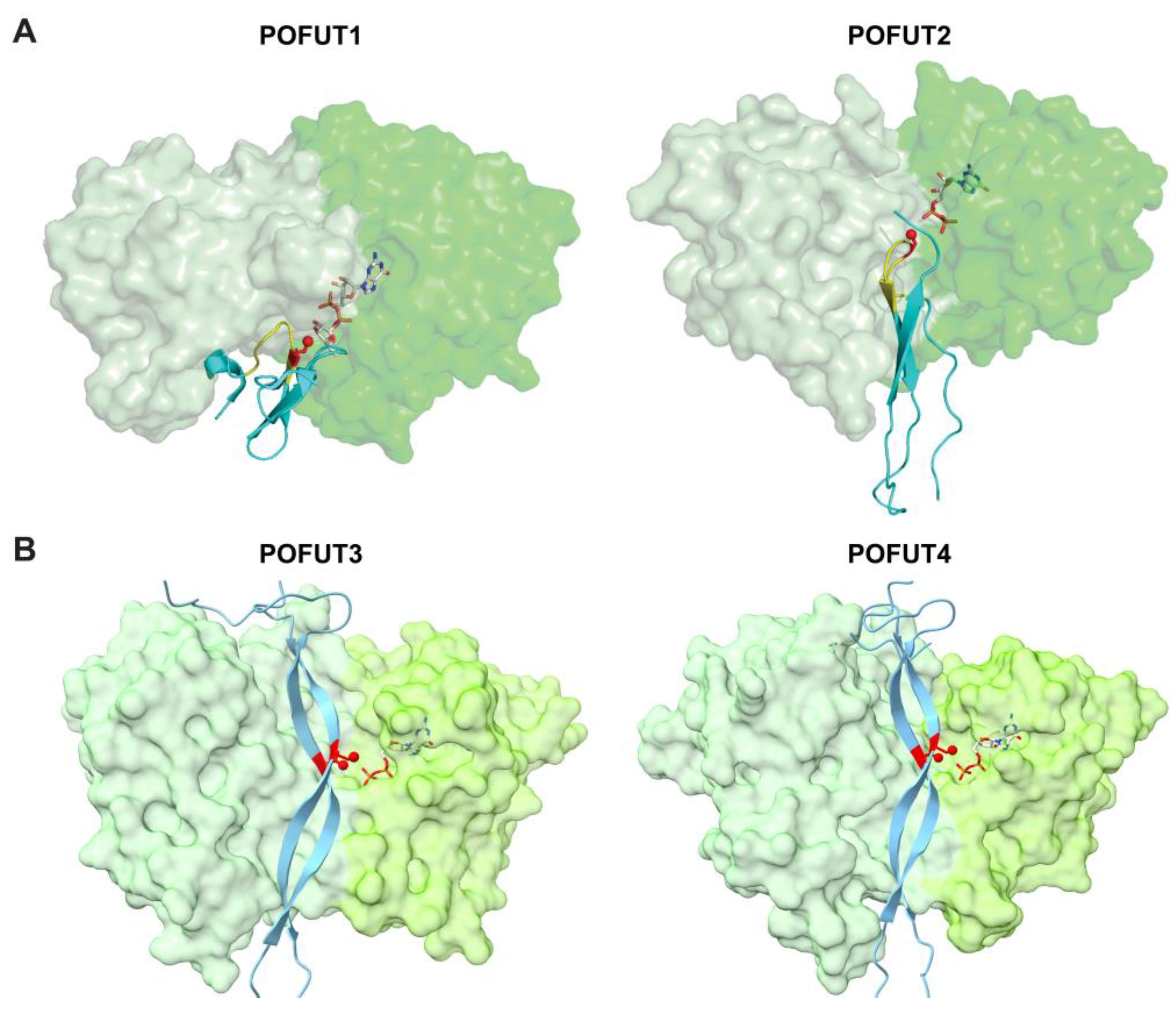
The addition of an O-fucose to EGF or TSR domain stabilizes the folded structures and protects them from unexpected unfolding due to environmental factors. O-fucosylated EGF repeats and TSRs showed a significantly reduced unfolding rate in reductive unfolding assays compared to their unmodified counterparts [318,333]. Additionally, the refolding of unmodified TSRs was accelerated in the presence of POFUT2 and GDP-fucose in vitro, whereas the addition of POFUT2 alone did not have the same effect [318]. This implies that the transferred O-fucose, rather than POFUT2, contributes to the acceleration of the folding rate. This stabilizing effect generated by the O-fucose glycans drives the domain into an energy well, preventing the domains from re-entry into the folding cycle, and consequently facilitating protein folding and secretion.
In line with this, several studies have shown that disruptions in O-fucosylation lead to defects in protein secretion. As mentioned above, knockdown or knockout of POFUT2 in cells eliminates the secretion of almost all ADAMTS/ADAMTS-like proteins tested to date in HEK293T cells. Mutating O-fucose sites within the EMI domain or knockout of both POFUT3 and POFUT4 resulted in a substantial reduction in the secretion of EMI-containing proteins [45,163]. Knockdown of Drosophila Ofut1 led to reduced cell surface expression of NOTCH and its accumulation in the ER [334]. Interestingly, this secretion defect can be partially rescued by an enzymatically inactive form of POFUT1 (R240A mutant), suggesting that Ofut1 might possess chaperone activity independent of its enzymatic function. However, mice carrying POFUT1 with point mutations at the equivalent position exhibited severe defects in somite formation, resembling those observed in Pofut1 null mice [335]. Additionally, the POFUT1 R240A mutant showed significantly reduced effectiveness in rescuing Notch signaling in POFUT1 knockout U2OS cells when compared to the wildtype POFUT1 [336]. The chaperone activity of POFUT1 may depend on the specific context within different organisms and can yield distinct biological functions. POFUT1 knockout in different cellular contexts led to varying outcomes in reducing cell surface Notch levels. For instance, POFUT1 knockout in HEK293T cells resulted in ~60% reduced surface expression of NOTCH1, whereas Pofut1 knockout in mouse embryonic stem cells displayed a similar level of cell surface NOTCH compared to the wild type [333,337]. The variation observed may stem from the distinct regulation of other chaperones that compensate for the folding and secretion defects in Notch within different cellular contexts.
6. Inhibitors/Modulators of O-Fucosylation
To date, no specific inhibitors for POFUT1, POFUT2, POFUT3, or POFUT4 have been identified. As discussed above, POFUTs play crucial roles in diseases, including various types of cancer. Having specific inhibitors or modulators for these enzymes would hold significant therapeutic potential and serve as valuable research tools. A virtual compound library screening with available POFUT structures could serve as a starting point for identifying potential inhibitors.
While not directly targeting POFUTs, specific fucose analogs like 5-thio-fucose (5T-Fuc) [338], 2-deoxy-2-fluoro-fucose (2F-Fuc) [339], 6-deoxy-6-fluoro-fucose (6F-Fuc) [340], 6,6,6-trifluoro-fucose (Fucostatin) [341], 6-alkynyl-fucose (6-AF) [342], 7-alkynyl-fucose (7-AF) [343], and β-carbafucose [344] are pan-inhibitors of cellular fucosylation. These compounds either antagonizing enzymes of the de novo GDP-fucose biosynthetic pathway or destabilizing the oxocarbenium ion-like transition states during the transfer reaction. Furthermore, 6-AF can enter the salvage pathway and be converted into GDP-6-AF, which is well-tolerated by POFUT1 and POFUT2 for incorporation into their respective protein targets [291,345,346]. The incorporated 6-AF can alter the functions of proteins, as exemplified in the case of Notch, where the incorporation of 6-AF selectively inhibits DLL ligands-mediated Notch signaling but not JAG ligands [291]. 6-AF inhibits most other FUTs, including POFUT3 and 4 [45,346]. Therefore, fucose analogs could serve as valuable tools for O-fucosylation research and hold therapeutic potential for their capacity to modulate the functions of proteins.
7. Conclusions
Given the biological importance and therapeutic potential of O-fucosylation, identifying proteins that are modified with O-fucose and understanding its role in modulating protein function would be of great value. Notable progress has been made in the last decade, including site-mapping of more EGF/TSR-containing proteins, successful crystallization of key POFUT target proteins (such as NOTCH1 and BAI1) and several POFUTs, deeper insights into the molecular mechanisms by which O-fucose glycans regulate protein function, and the discovery of a novel domain-specific O-fucosylation mediated by two uncharacterized POFUTs. Despite these remarkable progresses, much work remains to be done. First, while extensive research has focused on prominent O-fucosylated proteins such as Notch and the ADAMTS family, investigation into other proteins is needed. The O-fucosylation status of many putative POFUT substrates remains unconfirmed, yet they may have important biological functions. A comprehensive profiling of the O-fucose proteome is necessary. Second, the discovery of EMI O-fucosylation by POFUT3/4 has opened a new avenue for research, with much remaining to be explored in this emerging field. Generating POFUT3 and/or POFUT4 knockout mice and tissue-specific conditional knockout mice would be valuable for studying POFUT3/4-related diseases. Third, we are just beginning to understand the molecular mechanisms of how O-fucose modulates protein functions. While the role of O-fucose on NOTCH1 EGF8 and EGF12 in ligand binding is well-studied, fucose residues on other EGF repeats are also necessary for proper Notch function and may have distinct regulatory mechanisms. Additionally, little is known about how the O-fucose on TSRs and EMIs affects protein intermolecular interactions, while TSRs and EMIs are frequently localized within the regions involved in protein–protein interactions. Lastly, specific POFUT inhibitors are still lacking but would greatly benefit both research and therapeutic drug development. We envision that efforts to answer these questions will deepen our understanding of O-fucosylation and further advance its application in disease treatment.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules30071470/s1, Table S1: Multiple sequence alignments of FUTs.
Author Contributions
Conceptualization, H.H. and R.S.H.; formal analysis, B.M.E.; writing—original draft preparation, H.H.; writing—review and editing, H.H., R.S.H., and M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a grant from the National Institute of General Medical Sciences (R35GM148433) awarded to R.S.H. and a grant from the National Health and Medical Research Council (GNT2030089) awarded to M.L.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank all the current and past members of the Haltiwanger laboratory for technical advice and critical comments on this paper. We thank the assistance provided by ChatGPT (version 3.5) for language and writing refinement during the preparation of this paper.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ginsburg, V. Formation of guanosine diphosphate L-fucose from guanosine diphosphate D-mannose. J. Biol. Chem. 1960, 235, 2196–2201. [Google Scholar] [PubMed]
- Tonetti, M.; Sturla, L.; Bisso, A.; Benatti, U.; De Flora, A. Synthesis of GDP-L-fucose by the human FX protein. J. Biol. Chem. 1996, 271, 27274–27279. [Google Scholar] [PubMed]
- Coffey, J.W.; Miller, O.N.; Sellinger, O.Z. The metabolism of L-fucose in the rat. J. Biol. Chem. 1964, 239, 4011–4017. [Google Scholar] [PubMed]
- Kaufman, R.L.; Ginsburg, V. The metabolism of L-fucose by HeLa cells. Exp. Cell Res. 1968, 50, 127–132. [Google Scholar]
- Ishihara, H.; Heath, E.C. The metabolism of L-fucose. IV. The biosynthesis of guanosine diphosphate L-fucose in porcine liver. J. Biol. Chem. 1968, 243, 1110–1115. [Google Scholar]
- Ishihara, H.; Massaro, D.J.; Heath, E.C. The metabolism of L-fucose. 3. The enzymatic synthesis of beta-L-fucose 1-phosphate. J. Biol. Chem. 1968, 243, 1103–1109. [Google Scholar]
- Wiese, T.J.; Dunlap, J.A.; Yorek, M.A. L-fucose is accumulated via a specific transport system in eukaryotic cells. J. Biol. Chem. 1994, 269, 22705–22711. [Google Scholar]
- Ng, B.G.; Sosicka, P.; Xia, Z.; Freeze, H.H. GLUT1 is a highly efficient L-fucose transporter. J. Biol. Chem. 2023, 299, 102738. [Google Scholar]
- Lühn, K.; Laskowska, A.; Pielage, J.; Klämbt, C.; Ipe, U.; Vestweber, D.; Wild, M.K. Identification and molecular cloning of a functional GDP-fucose transporter in Drosophila melanogaster. Exp. Cell Res. 2004, 301, 242–250. [Google Scholar]
- Lühn, K.; Wild, M.K.; Eckhardt, M.; Gerardy-Schahn, R.; Vestweber, D. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat. Genet. 2001, 28, 69–72. [Google Scholar]
- Lu, L.; Varshney, S.; Yuan, Y.; Wei, H.X.; Tanwar, A.; Sundaram, S.; Nauman, M.; Haltiwanger, R.S.; Stanley, P. In vivo evidence for GDP-fucose transport in the absence of transporter SLC35C1 and putative transporter SLC35C2. J. Biol. Chem. 2023, 299, 105406. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hou, X.; Shi, S.; Körner, C.; Stanley, P. SLC35C2 promotes Notch1 fucosylation and is required for optimal Notch signaling in mammalian cells. J. Biol. Chem. 2010, 285, 36245–36254. [Google Scholar] [CrossRef] [PubMed]
- Ashikov, A.; Routier, F.; Fuhlrott, J.; Helmus, Y.; Wild, M.; Gerardy-Schahn, R.; Bakker, H. The human solute carrier gene SLC35B4 encodes a bifunctional nucleotide sugar transporter with specificity for UDP-xylose and UDP-N-acetylglucosamine. J. Biol. Chem. 2005, 280, 27230–27235. [Google Scholar] [CrossRef] [PubMed]
- Yurchenco, P.D.; Atkinson, P.H. Fucosyl-glycoprotein and precursor polls in HeLa cells. Biochemistry 1975, 14, 3107–3114. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; Atkinson, P.H. Equilibration of fucosyl glycoprotein pools in HeLa cells. Biochemistry 1977, 16, 944–953. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Hullen, A.; Koller, A.; Kotzot, D.; Grote, V.; Rapp, E.; Hofbauer, P.; Brugger, K.; Thiel, C.; Mayr, J.A.; et al. A spoonful of L-fucose-an efficient therapy for GFUS-CDG, a new glycosylation disorder. EMBO Mol. Med. 2021, 13, e14332. [Google Scholar] [CrossRef]
- Sosicka, P.; Ng, B.G.; Pepi, L.E.; Shajahan, A.; Wong, M.; Scott, D.A.; Matsumoto, K.; Xia, Z.J.; Lebrilla, C.B.; Haltiwanger, R.S.; et al. Origin of cytoplasmic GDP-fucose determines its contribution to glycosylation reactions. J. Cell Biol. 2022, 221, e202205038. [Google Scholar] [CrossRef]
- Skurska, E.; Szulc, B.; Maszczak-Seneczko, D.; Wiktor, M.; Wiertelak, W.; Makowiecka, A.; Olczak, M. Incorporation of fucose into glycans independent of the GDP-fucose transporter SLC35C1 preferentially utilizes salvaged over de novo GDP-fucose. J. Biol. Chem. 2022, 298, 102206. [Google Scholar] [CrossRef]
- Bisso, A.; Sturla, L.; Zanardi, D.; De Flora, A.; Tonetti, M. Structural and enzymatic characterization of human recombinant GDP-D-mannose-4,6-dehydratase. FEBS Lett. 1999, 456, 370–374. [Google Scholar] [CrossRef]
- Sullivan, F.X.; Kumar, R.; Kriz, R.; Stahl, M.; Xu, G.Y.; Rouse, J.; Chang, X.J.; Boodhoo, A.; Potvin, B.; Cumming, D.A. Molecular cloning of human GDP-mannose 4,6-dehydratase and reconstitution of GDP-fucose biosynthesis in vitro. J. Biol. Chem. 1998, 273, 8193–8202. [Google Scholar] [CrossRef]
- Richards, W.L.; Kilker, R.D.; Serif, G.S. Metabolite control of L-fucose utilization. J. Biol. Chem. 1978, 253, 8359–8361. [Google Scholar] [PubMed]
- Becker, D.J.; Lowe, J.B. Fucose: Biosynthesis and biological function in mammals. Glycobiology 2003, 13, 41R–53R. [Google Scholar] [PubMed]
- Ma, B.; Simala-Grant, J.L.; Taylor, D.E. Fucosylation in prokaryotes and eukaryotes. Glycobiology 2006, 16, 158R–184R. [Google Scholar]
- Schneider, M.; Al-Shareffi, E.; Haltiwanger, R.S. Biological functions of fucose in mammals. Glycobiology 2017, 27, 601–618. [Google Scholar] [PubMed]
- Flynn, R.A.; Pedram, K.; Malaker, S.A.; Batista, P.J.; Smith, B.A.H.; Johnson, A.G.; George, B.M.; Majzoub, K.; Villalta, P.W.; Carette, J.E.; et al. Small RNAs are modified with N-glycans and displayed on the surface of living cells. Cell 2021, 184, 3109–3124.e22. [Google Scholar] [CrossRef]
- Oriol, R.; Mollicone, R. Fucosyltransferases 1, 2. GDP-Fucose Galactoside α2-Fucosyltransferases. FUT1 or H Blood Group, FUT2 or ABH Secretor Status and Sec1 (FUT1, FUT2, Sec1). In Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 515–530. [Google Scholar]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Lowe, J.B. The blood group-specific human glycosyltransferases. Bailliere’s Clin. Haematol. 1993, 6, 465–492. [Google Scholar]
- Scharberg, E.A.; Olsen, C.; Bugert, P. The H blood group system. Immunohematology 2016, 32, 112–118. [Google Scholar]
- Kannagi, R. Fucosyltransferase 5. GDP-Fucose Lactosamine α3/4-Fucosyltransferase (FUT5). In Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 549–558. [Google Scholar]
- Kannagi, R. Fucosyltransferase 6. GDP-Fucose Lactosamine α3-Fucosyltransferase (FUT6). In Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 559–571. [Google Scholar]
- Kudo, T.; Narimatsu, H. Fucosyltransferase 3. GDP-Fucose Lactosamine α1,3/4-Fucosyltransferase. Lea and Leb Histo-Blood Groups (FUT3, Lewis Enzyme). In Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 531–539. [Google Scholar]
- Kudo, T.; Narimatsu, H. Fucosyltransferase 4. GDP-Fucose Lactosamine α1,3-Fucosyltransferase. Myeloid Specific (FUT4). In Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 541–547. [Google Scholar]
- Kudo, T.; Narimatsu, H. Fucosyltransferase 7. GDP-Fucose Lactosamine α1,3-Fucosyltransferase. Sialyl-Lex Specific (FUT7). In Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 573–580. [Google Scholar]
- Kudo, T.; Narimatsu, H. Fucosyltransferase 9. GDP-Fucose Lactosamine α1,3-Fucosyltransferase. Lex Specific (FUT9). In Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 597–603. [Google Scholar]
- Stowell, C.P.; Stowell, S.R. Biologic roles of the ABH and Lewis histo-blood group antigens Part I: Infection and immunity. Vox Sang. 2019, 114, 426–442. [Google Scholar] [CrossRef]
- Ihara, H.; Tsukamoto, H.; Gu, J.; Miyoshi, E.; Taniguchi, N.; Ikeda, Y. Fucosyltransferase 8. GDP-Fucose N-Glycan Core α6-Fucosyltransferase (FUT8). In Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 581–596. [Google Scholar]
- Boruah, B.M.; Kadirvelraj, R.; Liu, L.; Ramiah, A.; Li, C.; Zong, G.; Bosman, G.P.; Yang, J.Y.; Wang, L.X.; Boons, G.J.; et al. Characterizing human α-1,6-fucosyltransferase (FUT8) substrate specificity and structural similarities with related fucosyltransferases. J. Biol. Chem. 2020, 295, 17027–17045. [Google Scholar] [CrossRef]
- Bastian, K.; Scott, E.; Elliott, D.J.; Munkley, J. FUT8 Alpha-(1,6)-Fucosyltransferase in Cancer. Int. J. Mol. Sci. 2021, 22, 455. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, C.; Grau, S.; Jäger, C.; Sondermann, P.; Brünker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [PubMed]
- Luo, Y.; Haltiwanger, R.S. O-fucosylation of notch occurs in the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 11289–11294. [Google Scholar] [PubMed]
- Luo, Y.; Koles, K.; Vorndam, W.; Haltiwanger, R.S.; Panin, V.M. Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. J. Biol. Chem. 2006, 281, 9393–9399. [Google Scholar]
- Kakuda, S.; Haltiwanger, R.S. Fucosyltransferases 12, 13: Protein O-Fucosyltransferases 1 and 2 (POFUT1, POFUT2). In Handbook of Glycosyltransferases and Related Genes; Springer: Tokyo, Japan, 2014; pp. 623–633. [Google Scholar]
- Holdener, B.C.; Haltiwanger, R.S. Protein O-fucosylation: Structure and function. Curr. Opin. Struct. Biol. 2019, 56, 78–86. [Google Scholar]
- Hao, H.; Yuan, Y.; Ito, A.; Eberand, B.M.; Tjondro, H.; Cielesh, M.; Norris, N.; Moreno, C.L.; Maxwell, J.W.C.; Neely, G.G.; et al. FUT10 and FUT11 are protein O-fucosyltransferases that modify protein EMI domains. Nat. Chem. Biol. 2025. [Google Scholar] [CrossRef]
- Luo, Y.; Nita-Lazar, A.; Haltiwanger, R.S. Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J. Biol. Chem. 2006, 281, 9385–9392. [Google Scholar]
- Kentzer, E.J.; Buko, A.; Menon, G.; Sarin, V.K. Carbohydrate composition and presence of a fucose-protein linkage in recombinant human pro-urokinase. Biochem. Biophys. Res. Commun. 1990, 171, 401–406. [Google Scholar]
- Buko, A.M.; Kentzer, E.J.; Petros, A.; Menon, G.; Zuiderweg, E.R.; Sarin, V.K. Characterization of a posttranslational fucosylation in the growth factor domain of urinary plasminogen activator. Proc. Natl. Acad. Sci. USA 1991, 88, 3992–3996. [Google Scholar]
- Harris, R.J.; Leonard, C.K.; Guzzetta, A.W.; Spellman, M.W. Tissue plasminogen activator has an O-linked fucose attached to threonine-61 in the epidermal growth factor domain. Biochemistry 1991, 30, 2311–2314. [Google Scholar]
- Bjoern, S.; Foster, D.C.; Thim, L.; Wiberg, F.C.; Christensen, M.; Komiyama, Y.; Pedersen, A.H.; Kisiel, W. Human plasma and recombinant factor VII. Characterization of O-glycosylations at serine residues 52 and 60 and effects of site-directed mutagenesis of serine 52 to alanine. J. Biol. Chem. 1991, 266, 11051–11057. [Google Scholar] [PubMed]
- Harris, R.J.; Ling, V.T.; Spellman, M.W. O-linked fucose is present in the first epidermal growth factor domain of factor XII but not protein C. J. Biol. Chem. 1992, 267, 5102–5107. [Google Scholar] [PubMed]
- Harris, R.J.; van Halbeek, H.; Glushka, J.; Basa, L.J.; Ling, V.T.; Smith, K.J.; Spellman, M.W. Identification and structural analysis of the tetrasaccharide NeuAc alpha(2-->6)Gal beta(1-->4)GlcNAc beta(1-->3)Fuc alpha 1-->O-linked to serine 61 of human factor IX. Biochemistry 1993, 32, 6539–6547. [Google Scholar] [PubMed]
- Wang, Y.; Lee, G.F.; Kelley, R.F.; Spellman, M.W. Identification of a GDP-L-fucose:polypeptide fucosyltransferase and enzymatic addition of O-linked fucose to EGF domains. Glycobiology 1996, 6, 837–842. [Google Scholar]
- Wang, Y.; Spellman, M.W. Purification and characterization of a GDP-fucose:polypeptide fucosyltransferase from Chinese hamster ovary cells. J. Biol. Chem. 1998, 273, 8112–8118. [Google Scholar]
- Wang, Y.; Shao, L.; Shi, S.; Harris, R.J.; Spellman, M.W.; Stanley, P.; Haltiwanger, R.S. Modification of epidermal growth factor-like repeats with O-fucose. Molecular cloning and expression of a novel GDP-fucose protein O-fucosyltransferase. J. Biol. Chem. 2001, 276, 40338–40345. [Google Scholar]
- Panin, V.M.; Shao, L.; Lei, L.; Moloney, D.J.; Irvine, K.D.; Haltiwanger, R.S. Notch ligands are substrates for protein O-fucosyltransferase-1 and Fringe. J. Biol. Chem. 2002, 277, 29945–29952. [Google Scholar]
- Shao, L.; Moloney, D.J.; Haltiwanger, R. Fringe modifies O-fucose on mouse Notch1 at epidermal growth factor-like repeats within the ligand-binding site and the Abruptex region. J. Biol. Chem. 2003, 278, 7775–7782. [Google Scholar]
- Müller, J.; Rana, N.A.; Serth, K.; Kakuda, S.; Haltiwanger, R.S.; Gossler, A. O-fucosylation of the notch ligand mDLL1 by POFUT1 is dispensable for ligand function. PLoS ONE 2014, 9, e88571. [Google Scholar]
- Luther, K.B.; Haltiwanger, R.S. 1.07—O-Fucosylation of Proteins. In Comprehensive Glycoscience, 2nd ed.; Barchi, J.J., Ed.; Elsevier: Oxford, UK, 2021; pp. 182–203. [Google Scholar]
- Li, M.; Cheng, R.; Liang, J.; Yan, H.; Zhang, H.; Yang, L.; Li, C.; Jiao, Q.; Lu, Z.; He, J.; et al. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling-Degos disease. Am. J. Hum. Genet. 2013, 92, 895–903. [Google Scholar]
- Shi, S.; Stanley, P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 5234–5239. [Google Scholar] [PubMed]
- Okamura, Y.; Saga, Y. Pofut1 is required for the proper localization of the Notch receptor during mouse development. Mech. Dev. 2008, 125, 663–673. [Google Scholar] [PubMed]
- Varshney, S.; Stanley, P. Multiple roles for O-glycans in Notch signalling. FEBS Lett. 2018, 592, 3819–3834. [Google Scholar] [PubMed]
- Artavanis-Tsakonas, S.; Muskavitch, M.A. Notch: The past, the present, and the future. Curr. Top. Dev. Biol. 2010, 92, 1–29. [Google Scholar]
- Stanley, P.; Tanwar, A. Regulation of myeloid and lymphoid cell development by O-glycans on Notch. Front. Mol. Biosci. 2022, 9, 979724. [Google Scholar]
- Kakuda, S.; LoPilato, R.K.; Ito, A.; Haltiwanger, R.S. Canonical Notch ligands and Fringes have distinct effects on NOTCH1 and NOTCH2. J. Biol. Chem. 2020, 295, 14710–14722. [Google Scholar]
- Kakuda, S.; Haltiwanger, R.S. Deciphering the Fringe-Mediated Notch Code: Identification of Activating and Inhibiting Sites Allowing Discrimination between Ligands. Dev. Cell 2017, 40, 193–201. [Google Scholar]
- Moloney, D.J.; Panin, V.M.; Johnston, S.H.; Chen, J.; Shao, L.; Wilson, R.; Wang, Y.; Stanley, P.; Irvine, K.D.; Haltiwanger, R.S.; et al. Fringe is a glycosyltransferase that modifies Notch. Nature 2000, 406, 369–375. [Google Scholar]
- Brückner, K.; Perez, L.; Clausen, H.; Cohen, S. Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature 2000, 406, 411–415. [Google Scholar]
- Moloney, D.J.; Shair, L.H.; Lu, F.M.; Xia, J.; Locke, R.; Matta, K.L.; Haltiwanger, R.S. Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. J. Biol. Chem. 2000, 275, 9604–9611. [Google Scholar]
- Chen, J.; Moloney, D.J.; Stanley, P. Fringe modulation of Jagged1-induced Notch signaling requires the action of beta 4galactosyltransferase-1. Proc. Natl. Acad. Sci. USA 2001, 98, 13716–13721. [Google Scholar] [PubMed]
- Luca, V.C.; Jude, K.M.; Pierce, N.W.; Nachury, M.V.; Fischer, S.; Garcia, K.C. Structural biology. Structural basis for Notch1 engagement of Delta-like 4. Science 2015, 347, 847–853. [Google Scholar] [PubMed]
- Luca, V.C.; Kim, B.C.; Ge, C.; Kakuda, S.; Wu, D.; Roein-Peikar, M.; Haltiwanger, R.S.; Zhu, C.; Ha, T.; Garcia, K.C. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 2017, 355, 1320–1324. [Google Scholar] [PubMed]
- Taylor, P.; Takeuchi, H.; Sheppard, D.; Chillakuri, C.; Lea, S.M.; Haltiwanger, R.S.; Handford, P.A. Fringe-mediated extension of O-linked fucose in the ligand-binding region of Notch1 increases binding to mammalian Notch ligands. Proc. Natl. Acad. Sci. USA 2014, 111, 7290–7295. [Google Scholar]
- Wang, W.; Okajima, T.; Takeuchi, H. Significant Roles of Notch O-Glycosylation in Cancer. Molecules 2022, 27, 1783. [Google Scholar] [CrossRef]
- Saiki, W.; Ma, C.; Okajima, T.; Takeuchi, H. Current Views on the Roles of O-Glycosylation in Controlling Notch-Ligand Interactions. Biomolecules 2021, 11, 309. [Google Scholar] [CrossRef]
- Pandey, A.; Niknejad, N.; Jafar-Nejad, H. Multifaceted regulation of Notch signaling by glycosylation. Glycobiology 2021, 31, 8–28. [Google Scholar]
- Matsumoto, K.; Luther, K.B.; Haltiwanger, R.S. Diseases related to Notch glycosylation. Mol. Aspects Med. 2021, 79, 100938. [Google Scholar]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar]
- Boyden, S.E.; Desai, A.; Cruse, G.; Young, M.L.; Bolan, H.C.; Scott, L.M.; Eisch, A.R.; Long, R.D.; Lee, C.C.; Satorius, C.L.; et al. Vibratory Urticaria Associated with a Missense Variant in ADGRE2. N. Engl. J. Med. 2016, 374, 656–663. [Google Scholar]
- Kim, M.L.; Chandrasekharan, K.; Glass, M.; Shi, S.; Stahl, M.C.; Kaspar, B.; Stanley, P.; Martin, P.T. O-fucosylation of muscle agrin determines its ability to cluster acetylcholine receptors. Mol. Cell. Neurosci. 2008, 39, 452–464. [Google Scholar]
- Huzé, C.; Bauché, S.; Richard, P.; Chevessier, F.; Goillot, E.; Gaudon, K.; Ben Ammar, A.; Chaboud, A.; Grosjean, I.; Lecuyer, H.A.; et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am. J. Hum. Genet. 2009, 85, 155–167. [Google Scholar] [PubMed]
- Maselli, R.A.; Fernandez, J.M.; Arredondo, J.; Navarro, C.; Ngo, M.; Beeson, D.; Cagney, O.; Williams, D.C.; Wollmann, R.L.; Yarov-Yarovoy, V.; et al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum. Genet. 2012, 131, 1123–1135. [Google Scholar] [PubMed]
- Nicole, S.; Chaouch, A.; Torbergsen, T.; Bauché, S.; de Bruyckere, E.; Fontenille, M.J.; Horn, M.A.; van Ghelue, M.; Løseth, S.; Issop, Y.; et al. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain 2014, 137 Pt 9, 2429–2443. [Google Scholar] [PubMed]
- Robinson, A.; Escuin, S.; Doudney, K.; Vekemans, M.; Stevenson, R.E.; Greene, N.D.; Copp, A.J.; Stanier, P. Mutations in the planar cell polarity genes CELSR1 and SCRIB are associated with the severe neural tube defect craniorachischisis. Hum. Mutat. 2012, 33, 440–447. [Google Scholar]
- Gonzalez-Garay, M.L.; Aldrich, M.B.; Rasmussen, J.C.; Guilliod, R.; Lapinski, P.E.; King, P.D.; Sevick-Muraca, E.M. A novel mutation in CELSR1 is associated with hereditary lymphedema. Vasc. Cell 2016, 8, 1. [Google Scholar]
- Maltese, P.E.; Michelini, S.; Ricci, M.; Maitz, S.; Fiorentino, A.; Serrani, R.; Lazzerotti, A.; Bruson, A.; Paolacci, S.; Benedetti, S.; et al. Increasing evidence of hereditary lymphedema caused by CELSR1 loss-of-function variants. Am. J. Med. Genet. A 2019, 179, 1718–1724. [Google Scholar]
- Bamford, R.N.; Roessler, E.; Burdine, R.D.; Saplakoğlu, U.; dela Cruz, J.; Splitt, M.; Goodship, J.A.; Towbin, J.; Bowers, P.; Ferrero, G.B.; et al. Loss-of-function mutations in the EGF-CFC gene CFC1 are associated with human left-right laterality defects. Nat. Genet. 2000, 26, 365–369. [Google Scholar]
- Goldmuntz, E.; Bamford, R.; Karkera, J.D.; dela Cruz, J.; Roessler, E.; Muenke, M. CFC1 mutations in patients with transposition of the great arteries and double-outlet right ventricle. Am. J. Hum. Genet. 2002, 70, 776–780. [Google Scholar]
- den Hollander, A.I.; Heckenlively, J.R.; van den Born, L.I.; de Kok, Y.J.; van der Velde-Visser, S.D.; Kellner, U.; Jurklies, B.; van Schooneveld, M.J.; Blankenagel, A.; Rohrschneider, K.; et al. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am. J. Hum. Genet. 2001, 69, 198–203. [Google Scholar]
- den Hollander, A.I.; ten Brink, J.B.; de Kok, Y.J.; van Soest, S.; van den Born, L.I.; van Driel, M.A.; van de Pol, D.J.; Payne, A.M.; Bhattacharya, S.S.; Kellner, U.; et al. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat. Genet. 1999, 23, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Lotery, A.J.; Jacobson, S.G.; Fishman, G.A.; Weleber, R.G.; Fulton, A.B.; Namperumalsamy, P.; Héon, E.; Levin, A.V.; Grover, S.; Rosenow, J.R.; et al. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch. Ophthalmol. 2001, 119, 415–420. [Google Scholar] [CrossRef] [PubMed]
- McKay, G.J.; Clarke, S.; Davis, J.A.; Simpson, D.A.; Silvestri, G. Pigmented paravenous chorioretinal atrophy is associated with a mutation within the crumbs homolog 1 (CRB1) gene. Investig. Ophthalmol. Vis. Sci. 2005, 46, 322–328. [Google Scholar] [CrossRef]
- Ebarasi, L.; Ashraf, S.; Bierzynska, A.; Gee, H.Y.; McCarthy, H.J.; Lovric, S.; Sadowski, C.E.; Pabst, W.; Vega-Warner, V.; Fang, H.; et al. Defects of CRB2 cause steroid-resistant nephrotic syndrome. Am. J. Hum. Genet. 2015, 96, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jiang, C.; Yang, D.; Sun, R.; Wang, M.; Sun, H.; Xu, M.; Zhou, L.; Chen, M.; Xie, P.; et al. CRB2 mutation causes autosomal recessive retinitis pigmentosa. Exp. Eye Res. 2019, 180, 164–173. [Google Scholar] [CrossRef]
- Slavotinek, A.; Kaylor, J.; Pierce, H.; Cahr, M.; DeWard, S.J.; Schneidman-Duhovny, D.; Alsadah, A.; Salem, F.; Schmajuk, G.; Mehta, L. CRB2 mutations produce a phenotype resembling congenital nephrosis, Finnish type, with cerebral ventriculomegaly and raised alpha-fetoprotein. Am. J. Hum. Genet. 2015, 96, 162–169. [Google Scholar] [CrossRef]
- Yan, Y.T.; Liu, J.J.; Luo, Y.; E, C.; Haltiwanger, R.S.; Abate-Shen, C.; Shen, M.M. Dual roles of Cripto as a ligand and coreceptor in the nodal signaling pathway. Mol. Cell. Biol. 2002, 22, 4439–4449. [Google Scholar] [CrossRef]
- Schiffer, S.G.; Foley, S.; Kaffashan, A.; Hronowski, X.; Zichittella, A.E.; Yeo, C.Y.; Miatkowski, K.; Adkins, H.B.; Damon, B.; Whitman, M.; et al. Fucosylation of Cripto is required for its ability to facilitate nodal signaling. J. Biol. Chem. 2001, 276, 37769–37778. [Google Scholar] [CrossRef]
- Aminoff, M.; Carter, J.E.; Chadwick, R.B.; Johnson, C.; Gräsbeck, R.; Abdelaal, M.A.; Broch, H.; Jenner, L.B.; Verroust, P.J.; Moestrup, S.K.; et al. Mutations in CUBN, encoding the intrinsic factor-vitamin B12 receptor, cubilin, cause hereditary megaloblastic anaemia 1. Nat. Genet. 1999, 21, 309–313. [Google Scholar] [CrossRef]
- Kristiansen, M.; Aminoff, M.; Jacobsen, C.; de La Chapelle, A.; Krahe, R.; Verroust, P.J.; Moestrup, S.K. Cubilin P1297L mutation associated with hereditary megaloblastic anemia 1 causes impaired recognition of intrinsic factor-vitamin B(12) by cubilin. Blood 2000, 96, 405–409. [Google Scholar] [CrossRef]
- Storm, T.; Zeitz, C.; Cases, O.; Amsellem, S.; Verroust, P.J.; Madsen, M.; Benoist, J.F.; Passemard, S.; Lebon, S.; Jønsson, I.M.; et al. Detailed investigations of proximal tubular function in Imerslund-Gräsbeck syndrome. BMC Med. Genet. 2013, 14, 111. [Google Scholar]
- Bedin, M.; Boyer, O.; Servais, A.; Li, Y.; Villoing-Gaudé, L.; Tête, M.J.; Cambier, A.; Hogan, J.; Baudouin, V.; Krid, S.; et al. Human C-terminal CUBN variants associate with chronic proteinuria and normal renal function. J. Clin. Investig. 2020, 130, 335–344. [Google Scholar]
- Krogh, T.N.; Bachmann, E.; Teisner, B.; Skjødt, K.; Højrup, P. Glycosylation analysis and protein structure determination of murine fetal antigen 1 (mFA1)--the circulating gene product of the delta-like protein (dlk), preadipocyte factor 1 (Pref-1) and stromal-cell-derived protein 1 (SCP-1) cDNAs. Eur. J. Biochem. 1997, 244, 334–342. [Google Scholar] [PubMed]
- Fischer-Zirnsak, B.; Segebrecht, L.; Schubach, M.; Charles, P.; Alderman, E.; Brown, K.; Cadieux-Dion, M.; Cartwright, T.; Chen, Y.; Costin, C.; et al. Haploinsufficiency of the Notch Ligand DLL1 Causes Variable Neurodevelopmental Disorders. Am. J. Hum. Genet. 2019, 105, 631–639. [Google Scholar] [PubMed]
- Serth, K.; Schuster-Gossler, K.; Kremmer, E.; Hansen, B.; Marohn-Köhn, B.; Gossler, A. O-fucosylation of DLL3 is required for its function during somitogenesis. PLoS ONE 2015, 10, e0123776. [Google Scholar]
- Bulman, M.P.; Kusumi, K.; Frayling, T.M.; McKeown, C.; Garrett, C.; Lander, E.S.; Krumlauf, R.; Hattersley, A.T.; Ellard, S.; Turnpenny, P.D. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat. Genet. 2000, 24, 438–441. [Google Scholar]
- Meester, J.A.; Southgate, L.; Stittrich, A.B.; Venselaar, H.; Beekmans, S.J.; den Hollander, N.; Bijlsma, E.K.; Helderman-van den Enden, A.; Verheij, J.B.; Glusman, G.; et al. Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome. Am. J. Hum. Genet. 2015, 97, 475–482. [Google Scholar]
- Schürpf, T.; Chen, Q.; Liu, J.H.; Wang, R.; Springer, T.A.; Wang, J.H. The RGD finger of Del-1 is a unique structural feature critical for integrin binding. FASEB J. 2012, 26, 3412–3420. [Google Scholar]
- Hucthagowder, V.; Sausgruber, N.; Kim, K.H.; Angle, B.; Marmorstein, L.Y.; Urban, Z. Fibulin-4: A novel gene for an autosomal recessive cutis laxa syndrome. Am. J. Hum. Genet. 2006, 78, 1075–1080. [Google Scholar]
- Groenestege, W.M.; Thébault, S.; van der Wijst, J.; van den Berg, D.; Janssen, R.; Tejpar, S.; van den Heuvel, L.P.; van Cutsem, E.; Hoenderop, J.G.; Knoers, N.V.; et al. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J. Clin. Investig. 2007, 117, 2260–2267. [Google Scholar]
- Abd El-Aziz, M.M.; Barragan, I.; O’Driscoll, C.A.; Goodstadt, L.; Prigmore, E.; Borrego, S.; Mena, M.; Pieras, J.I.; El-Ashry, M.F.; Safieh, L.A.; et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat. Genet. 2008, 40, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Collin, R.W.; Littink, K.W.; Klevering, B.J.; van den Born, L.I.; Koenekoop, R.K.; Zonneveld, M.N.; Blokland, E.A.; Strom, T.M.; Hoyng, C.B.; den Hollander, A.I.; et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am. J. Hum. Genet. 2008, 83, 594–603. [Google Scholar] [PubMed]
- Hao, X.; Cheng, X.; Ye, J.; Wang, Y.; Yang, L.; Wang, M.; Jin, Y. Severe coagulation factor VII deficiency caused by a novel homozygous mutation (p. Trp284Gly) in loop 140s. Blood Coagul. Fibrinolysis 2016, 27, 461–463. [Google Scholar] [PubMed]
- O’Brien, D.P.; Gale, K.M.; Anderson, J.S.; McVey, J.H.; Miller, G.J.; Meade, T.W.; Tuddenham, E.G. Purification and characterization of factor VII 304-Gln: A variant molecule with reduced activity isolated from a clinically unaffected male. Blood 1991, 78, 132–140. [Google Scholar]
- Wulff, K.; Herrmann, F.H. Twenty two novel mutations of the factor VII gene in factor VII deficiency. Hum. Mutat. 2000, 15, 489–496. [Google Scholar]
- Nishimura, H.; Takao, T.; Hase, S.; Shimonishi, Y.; Iwanaga, S. Human factor IX has a tetrasaccharide O-glycosidically linked to serine 61 through the fucose residue. J. Biol. Chem. 1992, 267, 17520–17525. [Google Scholar]
- Bertina, R.M.; van der Linden, I.K.; Mannucci, P.M.; Reinalda-Poot, H.H.; Cupers, R.; Poort, S.R.; Reitsma, P.H. Mutations in hemophilia Bm occur at the Arg180-Val activation site or in the catalytic domain of factor IX. J. Biol. Chem. 1990, 265, 10876–10883. [Google Scholar]
- Ludwig, M.; Sabharwal, A.K.; Brackmann, H.H.; Olek, K.; Smith, K.J.; Birktoft, J.J.; Bajaj, S.P. Hemophilia B caused by five different nondeletion mutations in the protease domain of factor IX. Blood 1992, 79, 1225–1232. [Google Scholar]
- Simioni, P.; Tormene, D.; Tognin, G.; Gavasso, S.; Bulato, C.; Iacobelli, N.P.; Finn, J.D.; Spiezia, L.; Radu, C.; Arruda, V.R. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N. Engl. J. Med. 2009, 361, 1671–1675. [Google Scholar]
- Chu, K.; Wu, S.M.; Stanley, T.; Stafford, D.W.; High, K.A. A mutation in the propeptide of Factor IX leads to warfarin sensitivity by a novel mechanism. J. Clin. Investig. 1996, 98, 1619–1625. [Google Scholar]
- Bernardi, F.; Marchetti, G.; Patracchini, P.; del Senno, L.; Tripodi, M.; Fantoni, A.; Bartolai, S.; Vannini, F.; Felloni, L.; Rossi, L.; et al. Factor XII gene alteration in Hageman trait detected by TaqI restriction enzyme. Blood 1987, 69, 1421–1424. [Google Scholar] [PubMed]
- Schloesser, M.; Hofferbert, S.; Bartz, U.; Lutze, G.; Lämmle, B.; Engel, W. The novel acceptor splice site mutation 11396(G-->A) in the factor XII gene causes a truncated transcript in cross-reacting material negative patients. Hum. Mol. Genet. 1995, 4, 1235–1237. [Google Scholar] [PubMed]
- Cichon, S.; Martin, L.; Hennies, H.C.; Müller, F.; Van Driessche, K.; Karpushova, A.; Stevens, W.; Colombo, R.; Renné, T.; Drouet, C.; et al. Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am. J. Hum. Genet. 2006, 79, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Dewald, G.; Bork, K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem. Biophys. Res. Commun. 2006, 343, 1286–1289. [Google Scholar]
- Nibbeling, E.A.R.; Duarri, A.; Verschuuren-Bemelmans, C.C.; Fokkens, M.R.; Karjalainen, J.M.; Smeets, C.; de Boer-Bergsma, J.J.; van der Vries, G.; Dooijes, D.; Bampi, G.B.; et al. Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain 2017, 140, 2860–2878. [Google Scholar] [CrossRef]
- Cappello, S.; Gray, M.J.; Badouel, C.; Lange, S.; Einsiedler, M.; Srour, M.; Chitayat, D.; Hamdan, F.F.; Jenkins, Z.A.; Morgan, T.; et al. Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat. Genet. 2013, 45, 1300–1308. [Google Scholar] [CrossRef]
- Alders, M.; Al-Gazali, L.; Cordeiro, I.; Dallapiccola, B.; Garavelli, L.; Tuysuz, B.; Salehi, F.; Haagmans, M.A.; Mook, O.R.; Majoie, C.B.; et al. Hennekam syndrome can be caused by FAT4 mutations and be allelic to Van Maldergem syndrome. Hum. Genet. 2014, 133, 1161–1167. [Google Scholar]
- Debeer, P.; Schoenmakers, E.F.; Twal, W.O.; Argraves, W.S.; De Smet, L.; Fryns, J.P.; Van De Ven, W.J. The fibulin-1 gene (FBLN1) is disrupted in a t(12;22) associated with a complex type of synpolydactyly. J. Med. Genet. 2002, 39, 98–104. [Google Scholar] [CrossRef]
- Greene, L.M.; Twal, W.O.; Duffy, M.J.; McDermott, E.W.; Hill, A.D.; O’Higgins, N.J.; McCann, A.H.; Dervan, P.A.; Argraves, W.S.; Gallagher, W.M. Elevated expression and altered processing of fibulin-1 protein in human breast cancer. Br. J. Cancer 2003, 88, 871–878. [Google Scholar]
- Auer-Grumbach, M.; Weger, M.; Fink-Puches, R.; Papić, L.; Fröhlich, E.; Auer-Grumbach, P.; El Shabrawi-Caelen, L.; Schabhüttl, M.; Windpassinger, C.; Senderek, J.; et al. Fibulin-5 mutations link inherited neuropathies, age-related macular degeneration and hyperelastic skin. Brain 2011, 134 Pt 6, 1839–1852. [Google Scholar]
- Markova, D.; Zou, Y.; Ringpfeil, F.; Sasaki, T.; Kostka, G.; Timpl, R.; Uitto, J.; Chu, M.L. Genetic heterogeneity of cutis laxa: A heterozygous tandem duplication within the fibulin-5 (FBLN5) gene. Am. J. Hum. Genet. 2003, 72, 998–1004. [Google Scholar] [PubMed]
- Loeys, B.; Van Maldergem, L.; Mortier, G.; Coucke, P.; Gerniers, S.; Naeyaert, J.M.; De Paepe, A. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum. Mol. Genet. 2002, 11, 2113–2118. [Google Scholar] [PubMed]
- Stone, E.M.; Braun, T.A.; Russell, S.R.; Kuehn, M.H.; Lotery, A.J.; Moore, P.A.; Eastman, C.G.; Casavant, T.L.; Sheffield, V.C. Missense variations in the fibulin 5 gene and age-related macular degeneration. N. Engl. J. Med. 2004, 351, 346–353. [Google Scholar]
- Neupane, S.; Williamson, D.B.; Roth, R.A.; Halabi, C.M.; Haltiwanger, R.S.; Holdener, B.C. Poglut2/3 double knockout in mice results in neonatal lethality with reduced levels of fibrillin in lung tissues. J. Biol. Chem. 2024, 300, 107445. [Google Scholar]
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [PubMed]
- Dietz, H.C.; Saraiva, J.M.; Pyeritz, R.E.; Cutting, G.R.; Francomano, C.A. Clustering of fibrillin (FBN1) missense mutations in Marfan syndrome patients at cysteine residues in EGF-like domains. Hum. Mutat. 1992, 1, 366–374. [Google Scholar]
- Lönnqvist, L.; Child, A.; Kainulainen, K.; Davidson, R.; Puhakka, L.; Peltonen, L. A novel mutation of the fibrillin gene causing ectopia lentis. Genomics 1994, 19, 573–576. [Google Scholar]
- Faivre, L.; Gorlin, R.J.; Wirtz, M.K.; Godfrey, M.; Dagoneau, N.; Samples, J.R.; Le Merrer, M.; Collod-Beroud, G.; Boileau, C.; Munnich, A.; et al. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J. Med. Genet. 2003, 40, 34–36. [Google Scholar]
- Glesby, M.J.; Pyeritz, R.E. Association of mitral valve prolapse and systemic abnormalities of connective tissue. A phenotypic continuum. JAMA 1989, 262, 523–528. [Google Scholar]
- Loeys, B.L.; Gerber, E.E.; Riegert-Johnson, D.; Iqbal, S.; Whiteman, P.; McConnell, V.; Chillakuri, C.R.; Macaya, D.; Coucke, P.J.; De Paepe, A.; et al. Mutations in fibrillin-1 cause congenital scleroderma: Stiff skin syndrome. Sci. Transl. Med. 2010, 2, 23ra20. [Google Scholar]
- Le Goff, C.; Mahaut, C.; Wang, L.W.; Allali, S.; Abhyankar, A.; Jensen, S.; Zylberberg, L.; Collod-Beroud, G.; Bonnet, D.; Alanay, Y.; et al. Mutations in the TGFβ binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am. J. Hum. Genet. 2011, 89, 7–14. [Google Scholar] [PubMed]
- Graul-Neumann, L.M.; Kienitz, T.; Robinson, P.N.; Baasanjav, S.; Karow, B.; Gillessen-Kaesbach, G.; Fahsold, R.; Schmidt, H.; Hoffmann, K.; Passarge, E. Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3’ terminus of the FBN1-gene. Am. J. Med. Genet. A 2010, 152, 2749–2755. [Google Scholar]
- Putnam, E.A.; Zhang, H.; Ramirez, F.; Milewicz, D.M. Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat. Genet. 1995, 11, 456–458. [Google Scholar] [PubMed]
- Ratnapriya, R.; Zhan, X.; Fariss, R.N.; Branham, K.E.; Zipprer, D.; Chakarova, C.F.; Sergeev, Y.V.; Campos, M.M.; Othman, M.; Friedman, J.S.; et al. Rare and common variants in extracellular matrix gene Fibrillin 2 (FBN2) are associated with macular degeneration. Hum. Mol. Genet. 2014, 23, 5827–5837. [Google Scholar]
- Gara, S.K.; Jia, L.; Merino, M.J.; Agarwal, S.K.; Zhang, L.; Cam, M.; Patel, D.; Kebebew, E. Germline HABP2 Mutation Causing Familial Nonmedullary Thyroid Cancer. N. Engl. J. Med. 2015, 373, 448–455. [Google Scholar]
- Willeit, J.; Kiechl, S.; Weimer, T.; Mair, A.; Santer, P.; Wiedermann, C.J.; Roemisch, J. Marburg I polymorphism of factor VII--activating protease: A prominent risk predictor of carotid stenosis. Circulation 2003, 107, 667–670. [Google Scholar]
- Nicole, S.; Davoine, C.S.; Topaloglu, H.; Cattolico, L.; Barral, D.; Beighton, P.; Hamida, C.B.; Hammouda, H.; Cruaud, C.; White, P.S.; et al. Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia). Nat. Genet. 2000, 26, 480–483. [Google Scholar]
- Arikawa-Hirasawa, E.; Wilcox, W.R.; Le, A.H.; Silverman, N.; Govindraj, P.; Hassell, J.R.; Yamada, Y. Dyssegmental dysplasia, Silverman-Handmaker type, is caused by functional null mutations of the perlecan gene. Nat. Genet. 2001, 27, 431–434. [Google Scholar]
- Thakurdas, S.M.; Lopez, M.F.; Kakuda, S.; Fernandez-Valdivia, R.; Zarrin-Khameh, N.; Haltiwanger, R.S.; Jafar-Nejad, H. Jagged1 heterozygosity in mice results in a congenital cholangiopathy which is reversed by concomitant deletion of one copy of Poglut1 (Rumi). Hepatology 2016, 63, 550–565. [Google Scholar]
- Li, L.; Krantz, I.D.; Deng, Y.; Genin, A.; Banta, A.B.; Collins, C.C.; Qi, M.; Trask, B.J.; Kuo, W.L.; Cochran, J.; et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet. 1997, 16, 243–251. [Google Scholar]
- Eldadah, Z.A.; Hamosh, A.; Biery, N.J.; Montgomery, R.A.; Duke, M.; Elkins, R.; Dietz, H.C. Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Hum. Mol. Genet. 2001, 10, 163–169. [Google Scholar] [PubMed]
- Le Caignec, C.; Lefevre, M.; Schott, J.J.; Chaventre, A.; Gayet, M.; Calais, C.; Moisan, J.P. Familial deafness, congenital heart defects, and posterior embryotoxon caused by cysteine substitution in the first epidermal-growth-factor-like domain of jagged 1. Am. J. Hum. Genet. 2002, 71, 180–186. [Google Scholar] [PubMed]
- Sullivan, J.M.; Motley, W.W.; Johnson, J.O.; Aisenberg, W.H.; Marshall, K.L.; Barwick, K.E.; Kong, L.; Huh, J.S.; Saavedra-Rivera, P.C.; McEntagart, M.M.; et al. Dominant mutations of the Notch ligand Jagged1 cause peripheral neuropathy. J. Clin. Investig. 2020, 130, 1506–1512. [Google Scholar] [PubMed]
- Coppens, S.; Barnard, A.M.; Puusepp, S.; Pajusalu, S.; Õunap, K.; Vargas-Franco, D.; Bruels, C.C.; Donkervoort, S.; Pais, L.; Chao, K.R.; et al. A form of muscular dystrophy associated with pathogenic variants in JAG2. Am. J. Hum. Genet. 2021, 108, 840–856. [Google Scholar]
- Klar, J.; Schuster, J.; Khan, T.N.; Jameel, M.; Mäbert, K.; Forsberg, L.; Baig, S.A.; Baig, S.M.; Dahl, N. Whole exome sequencing identifies LRP1 as a pathogenic gene in autosomal recessive keratosis pilaris atrophicans. J. Med. Genet. 2015, 52, 599–606. [Google Scholar]
- Yan, W.; Zheng, L.; Xu, X.; Hao, Z.; Zhang, Y.; Lu, J.; Sun, Z.; Dai, J.; Shi, D.; Guo, B.; et al. Heterozygous LRP1 deficiency causes developmental dysplasia of the hip by impairing triradiate chondrocytes differentiation due to inhibition of autophagy. Proc. Natl. Acad. Sci. USA 2022, 119, e2203557119. [Google Scholar]
- Ali, M.; McKibbin, M.; Booth, A.; Parry, D.A.; Jain, P.; Riazuddin, S.A.; Hejtmancik, J.F.; Khan, S.N.; Firasat, S.; Shires, M.; et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am. J. Hum. Genet. 2009, 84, 664–671. [Google Scholar]
- Kumar, A.; Duvvari, M.R.; Prabhakaran, V.C.; Shetty, J.S.; Murthy, G.J.; Blanton, S.H. A homozygous mutation in LTBP2 causes isolated microspherophakia. Hum. Genet. 2010, 128, 365–371. [Google Scholar]
- Haji-Seyed-Javadi, R.; Jelodari-Mamaghani, S.; Paylakhi, S.H.; Yazdani, S.; Nilforushan, N.; Fan, J.B.; Klotzle, B.; Mahmoudi, M.J.; Ebrahimian, M.J.; Chelich, N.; et al. LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum. Mutat. 2012, 33, 1182–1187. [Google Scholar]
- Twigg, S.R.; Lloyd, D.; Jenkins, D.; Elçioglu, N.E.; Cooper, C.D.; Al-Sannaa, N.; Annagür, A.; Gillessen-Kaesbach, G.; Hüning, I.; Knight, S.J.; et al. Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am. J. Hum. Genet. 2012, 91, 897–905. [Google Scholar]
- Logan, C.V.; Lucke, B.; Pottinger, C.; Abdelhamed, Z.A.; Parry, D.A.; Szymanska, K.; Diggle, C.P.; van Riesen, A.; Morgan, J.E.; Markham, G.; et al. Mutations in MEGF10, a regulator of satellite cell myogenesis, cause early onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD). Nat. Genet. 2011, 43, 1189–1192. [Google Scholar] [PubMed]
- Boyden, S.E.; Mahoney, L.J.; Kawahara, G.; Myers, J.A.; Mitsuhashi, S.; Estrella, E.A.; Duncan, A.R.; Dey, F.; DeChene, E.T.; Blasko-Goehringer, J.M.; et al. Mutations in the satellite cell gene MEGF10 cause a recessive congenital myopathy with minicores. Neurogenetics 2012, 13, 115–124. [Google Scholar] [PubMed]
- Houlahan, C.B.; Kong, Y.; Johnston, B.; Cielesh, M.; Chau, T.H.; Fenwick, J.; Coleman, P.R.; Hao, H.; Haltiwanger, R.S.; Thaysen-Andersen, M.; et al. Analysis of the Healthy Platelet Proteome Identifies a New Form of Domain-Specific O-Fucosylation. Mol. Cell. Proteom. 2024, 23, 100717. [Google Scholar]
- Hayward, C.P.; Rivard, G.E.; Kane, W.H.; Drouin, J.; Zheng, S.; Moore, J.C.; Kelton, J.G. An autosomal dominant, qualitative platelet disorder associated with multimerin deficiency, abnormalities in platelet factor V, thrombospondin, von Willebrand factor, and fibrinogen and an epinephrine aggregation defect. Blood 1996, 87, 4967–4978. [Google Scholar]
- Kang, C.; Riazuddin, S.; Mundorff, J.; Krasnewich, D.; Friedman, P.; Mullikin, J.C.; Drayna, D. Mutations in the lysosomal enzyme-targeting pathway and persistent stuttering. N. Engl. J. Med. 2010, 362, 677–685. [Google Scholar]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar]
- Stittrich, A.B.; Lehman, A.; Bodian, D.L.; Ashworth, J.; Zong, Z.; Li, H.; Lam, P.; Khromykh, A.; Iyer, R.K.; Vockley, J.G.; et al. Mutations in NOTCH1 cause Adams-Oliver syndrome. Am. J. Hum. Genet. 2014, 95, 275–284. [Google Scholar]
- Shimizu, K.; Chiba, S.; Saito, T.; Kumano, K.; Takahashi, T.; Hirai, H. Manic fringe and lunatic fringe modify different sites of the Notch2 extracellular region, resulting in different signaling modulation. J. Biol. Chem. 2001, 276, 25753–25758. [Google Scholar]
- McDaniell, R.; Warthen, D.M.; Sanchez-Lara, P.A.; Pai, A.; Krantz, I.D.; Piccoli, D.A.; Spinner, N.B. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am. J. Hum. Genet. 2006, 79, 169–173. [Google Scholar]
- Simpson, M.A.; Irving, M.D.; Asilmaz, E.; Gray, M.J.; Dafou, D.; Elmslie, F.V.; Mansour, S.; Holder, S.E.; Brain, C.E.; Burton, B.K.; et al. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011, 43, 303–305. [Google Scholar]
- Fiddes, I.T.; Lodewijk, G.A.; Mooring, M.; Bosworth, C.M.; Ewing, A.D.; Mantalas, G.L.; Novak, A.M.; van den Bout, A.; Bishara, A.; Rosenkrantz, J.L.; et al. Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell 2018, 173, 1356–1369.e22. [Google Scholar] [PubMed]
- Tian, Y.; Wang, J.L.; Huang, W.; Zeng, S.; Jiao, B.; Liu, Z.; Chen, Z.; Li, Y.; Wang, Y.; Min, H.X.; et al. Expansion of Human-Specific GGC Repeat in Neuronal Intranuclear Inclusion Disease-Related Disorders. Am. J. Hum. Genet. 2019, 105, 166–176. [Google Scholar] [PubMed]
- Ishiura, H.; Shibata, S.; Yoshimura, J.; Suzuki, Y.; Qu, W.; Doi, K.; Almansour, M.A.; Kikuchi, J.K.; Taira, M.; Mitsui, J.; et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 2019, 51, 1222–1232. [Google Scholar] [PubMed]
- Sone, J.; Mitsuhashi, S.; Fujita, A.; Mizuguchi, T.; Hamanaka, K.; Mori, K.; Koike, H.; Hashiguchi, A.; Takashima, H.; Sugiyama, H.; et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 2019, 51, 1215–1221. [Google Scholar]
- Ogasawara, M.; Iida, A.; Kumutpongpanich, T.; Ozaki, A.; Oya, Y.; Konishi, H.; Nakamura, A.; Abe, R.; Takai, H.; Hanajima, R.; et al. CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. Acta Neuropathol. Commun. 2020, 8, 204. [Google Scholar]
- Arboleda-Velasquez, J.F.; Rampal, R.; Fung, E.; Darland, D.C.; Liu, M.; Martinez, M.C.; Donahue, C.P.; Navarro-Gonzalez, M.F.; Libby, P.; D’Amore, P.A.; et al. CADASIL mutations impair Notch3 glycosylation by Fringe. Hum. Mol. Genet. 2005, 14, 1631–1639. [Google Scholar]
- Dichgans, M.; Filippi, M.; Brüning, R.; Iannucci, G.; Berchtenbreiter, C.; Minicucci, L.; Uttner, I.; Crispin, A.; Ludwig, H.; Gasser, T.; et al. Quantitative MRI in CADASIL: Correlation with disability and cognitive performance. Neurology 1999, 52, 1361–1367. [Google Scholar]
- Joutel, A.; Vahedi, K.; Corpechot, C.; Troesch, A.; Chabriat, H.; Vayssière, C.; Cruaud, C.; Maciazek, J.; Weissenbach, J.; Bousser, M.G.; et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 1997, 350, 1511–1515. [Google Scholar]
- Martignetti, J.A.; Tian, L.; Li, D.; Ramirez, M.C.; Camacho-Vanegas, O.; Camacho, S.C.; Guo, Y.; Zand, D.J.; Bernstein, A.M.; Masur, S.K.; et al. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am. J. Hum. Genet. 2013, 92, 1001–1007. [Google Scholar]
- Gripp, K.W.; Robbins, K.M.; Sobreira, N.L.; Witmer, P.D.; Bird, L.M.; Avela, K.; Makitie, O.; Alves, D.; Hogue, J.S.; Zackai, E.H.; et al. Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome. Am. J. Med. Genet. A 2015, 167, 271–281. [Google Scholar]
- Abu-Libdeh, B.; Ashhab, M.; Shahrour, M.; Daana, M.; Dudin, A.; Elpeleg, O.; Edvardson, S.; Harel, T. Homozygous frameshift variant in NTNG2, encoding a synaptic cell adhesion molecule, in individuals with developmental delay, hypotonia, and autistic features. Neurogenetics 2019, 20, 209–213. [Google Scholar]
- Dias, C.M.; Punetha, J.; Zheng, C.; Mazaheri, N.; Rad, A.; Efthymiou, S.; Petersen, A.; Dehghani, M.; Pehlivan, D.; Partlow, J.N.; et al. Homozygous Missense Variants in NTNG2, Encoding a Presynaptic Netrin-G2 Adhesion Protein, Lead to a Distinct Neurodevelopmental Disorder. Am. J. Hum. Genet. 2019, 105, 1048–1056. [Google Scholar] [PubMed]
- Pennarubia, F.; Germot, A.; Pinault, E.; Maftah, A.; Legardinier, S. The single EGF-like domain of mouse PAMR1 is modified by O-Glucose, O-Fucose and O-GlcNAc. Glycobiology 2021, 31, 55–68. [Google Scholar] [PubMed]
- Elenbaas, J.S.; Pudupakkam, U.; Ashworth, K.J.; Kang, C.J.; Patel, V.; Santana, K.; Jung, I.-H.; Lee, P.C.; Burks, K.H.; Amrute, J.M.; et al. SVEP1 is an endogenous ligand for the orphan receptor PEAR1. Nat. Commun. 2023, 14, 850. [Google Scholar] [PubMed]
- Byeon, I.J.; Llinás, M. Solution structure of the tissue-type plasminogen activator kringle 2 domain complexed to 6-aminohexanoic acid an antifibrinolytic drug. J. Mol. Biol. 1991, 222, 1035–1051. [Google Scholar]
- Paterson, A.D.; Rommens, J.M.; Bharaj, B.; Blavignac, J.; Wong, I.; Diamandis, M.; Waye, J.S.; Rivard, G.E.; Hayward, C.P. Persons with Quebec platelet disorder have a tandem duplication of PLAU, the urokinase plasminogen activator gene. Blood 2010, 115, 1264–1266. [Google Scholar]
- Romeo, G.; Hassan, H.J.; Staempfli, S.; Roncuzzi, L.; Cianetti, L.; Leonardi, A.; Vicente, V.; Mannucci, P.M.; Bertina, R.; Peschle, C.; et al. Hereditary thrombophilia: Identification of nonsense and missense mutations in the protein C gene. Proc. Natl. Acad. Sci. USA 1987, 84, 2829–2832. [Google Scholar]
- Grundy, C.B.; Chisholm, M.; Kakkar, V.V.; Cooper, D.N. A novel homozygous missense mutation in the protein C (PROC) gene causing recurrent venous thrombosis. Hum. Genet. 1992, 89, 683–684. [Google Scholar]
- Hong, S.E.; Shugart, Y.Y.; Huang, D.T.; Shahwan, S.A.; Grant, P.E.; Hourihane, J.O.; Martin, N.D.; Walsh, C.A. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 2000, 26, 93–96. [Google Scholar]
- Dazzo, E.; Fanciulli, M.; Serioli, E.; Minervini, G.; Pulitano, P.; Binelli, S.; Di Bonaventura, C.; Luisi, C.; Pasini, E.; Striano, S.; et al. Heterozygous reelin mutations cause autosomal-dominant lateral temporal epilepsy. Am. J. Hum. Genet. 2015, 96, 992–1000. [Google Scholar]
- Anastasio, N.; Ben-Omran, T.; Teebi, A.; Ha, K.C.; Lalonde, E.; Ali, R.; Almureikhi, M.; Der Kaloustian, V.M.; Liu, J.; Rosenblatt, D.S.; et al. Mutations in SCARF2 are responsible for Van Den Ende-Gupta syndrome. Am. J. Hum. Genet. 2010, 87, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Monfrini, E.; Pelucchi, S.; Hollmén, M.; Viitala, M.; Mariani, R.; Bertola, F.; Majore, S.; Di Fonzo, A.; Piperno, A. A form of inherited hyperferritinemia associated with bi-allelic pathogenic variants of STAB1. Am. J. Hum. Genet. 2023, 110, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Stitziel, N.O.; Stirrups, K.E.; Masca, N.G.; Erdmann, J.; Ferrario, P.G.; König, I.R.; Weeke, P.E.; Webb, T.R.; Auer, P.L.; Schick, U.M.; et al. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N. Engl. J. Med. 2016, 374, 1134–1144. [Google Scholar] [PubMed]
- Jung, I.H.; Elenbaas, J.S.; Alisio, A.; Santana, K.; Young, E.P.; Kang, C.J.; Kachroo, P.; Lavine, K.J.; Razani, B.; Mecham, R.P.; et al. SVEP1 is a human coronary artery disease locus that promotes atherosclerosis. Sci. Transl. Med. 2021, 13, eabe0357. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Mohammed, J.Y.; Al-Hazzaa, S.; Alkuraya, F.S. Homozygous null mutation in ODZ3 causes microphthalmia in humans. Genet. Med. 2012, 14, 900–904. [Google Scholar] [CrossRef]
- Chassaing, N.; Ragge, N.; Plaisancié, J.; Patat, O.; Geneviève, D.; Rivier, F.; Malrieu-Eliaou, C.; Hamel, C.; Kaplan, J.; Calvas, P. Confirmation of TENM3 involvement in autosomal recessive colobomatous microphthalmia. Am. J. Med. Genet. A 2016, 170, 1895–1898. [Google Scholar] [CrossRef]
- Hor, H.; Francescatto, L.; Bartesaghi, L.; Ortega-Cubero, S.; Kousi, M.; Lorenzo-Betancor, O.; Jiménez-Jiménez, F.J.; Gironell, A.; Clarimón, J.; Drechsel, O.; et al. Missense mutations in TENM4, a regulator of axon guidance and central myelination, cause essential tremor. Hum. Mol. Genet. 2015, 24, 5677–5686. [Google Scholar] [CrossRef]
- Michelini, S.; Ricci, M.; Veselenyiova, D.; Kenanoglu, S.; Kurti, D.; Baglivo, M.; Fiorentino, A.; Basha, S.H.; Priya, S.; Serrani, R.; et al. TIE1 as a Candidate Gene for Lymphatic Malformations with or without Lymphedema. Int. J. Mol. Sci. 2020, 21, 6780. [Google Scholar] [CrossRef]
- Hart, T.C.; Gorry, M.C.; Hart, P.S.; Woodard, A.S.; Shihabi, Z.; Sandhu, J.; Shirts, B.; Xu, L.; Zhu, H.; Barmada, M.M.; et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J. Med. Genet. 2002, 39, 882–892. [Google Scholar] [CrossRef]
- Rampoldi, L.; Caridi, G.; Santon, D.; Boaretto, F.; Bernascone, I.; Lamorte, G.; Tardanico, R.; Dagnino, M.; Colussi, G.; Scolari, F.; et al. Allelism of MCKD, FJHN and GCKD caused by impairment of uromodulin export dynamics. Hum. Mol. Genet. 2003, 12, 3369–3384. [Google Scholar] [CrossRef]
- Miyamoto, T.; Inoue, H.; Sakamoto, Y.; Kudo, E.; Naito, T.; Mikawa, T.; Mikawa, Y.; Isashiki, Y.; Osabe, D.; Shinohara, S.; et al. Identification of a novel splice site mutation of the CSPG2 gene in a Japanese family with Wagner syndrome. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2726–2735. [Google Scholar]
- Kloeckener-Gruissem, B.; Neidhardt, J.; Magyar, I.; Plauchu, H.; Zech, J.C.; Morlé, L.; Palmer-Smith, S.M.; Macdonald, M.J.; Nas, V.; Fry, A.E.; et al. Novel VCAN mutations and evidence for unbalanced alternative splicing in the pathogenesis of Wagner syndrome. Eur. J. Hum. Genet. 2013, 21, 352–356. [Google Scholar] [PubMed]
- Gebauer, J.M.; Müller, S.; Hanisch, F.G.; Paulsson, M.; Wagener, R. O-glucosylation and O-fucosylation occur together in close proximity on the first epidermal growth factor repeat of AMACO (VWA2 protein). J. Biol. Chem. 2008, 283, 17846–17854. [Google Scholar] [PubMed]
- Hofsteenge, J.; Huwiler, K.G.; Macek, B.; Hess, D.; Lawler, J.; Mosher, D.F.; Peter-Katalinic, J. C-mannosylation and O-fucosylation of the thrombospondin type 1 module. J. Biol. Chem. 2001, 276, 6485–6498. [Google Scholar]
- Valero-González, J.; Leonhard-Melief, C.; Lira-Navarrete, E.; Jiménez-Osés, G.; Hernández-Ruiz, C.; Pallarés, M.C.; Yruela, I.; Vasudevan, D.; Lostao, A.; Corzana, F.; et al. A proactive role of water molecules in acceptor recognition by protein O-fucosyltransferase 2. Nat. Chem. Biol. 2016, 12, 240–246. [Google Scholar] [CrossRef]
- Ricketts, L.M.; Dlugosz, M.; Luther, K.B.; Haltiwanger, R.S.; Majerus, E.M. O-fucosylation is required for ADAMTS13 secretion. J. Biol. Chem. 2007, 282, 17014–17023. [Google Scholar]
- Wang, L.W.; Dlugosz, M.; Somerville, R.P.; Raed, M.; Haltiwanger, R.S.; Apte, S.S. O-fucosylation of thrombospondin type 1 repeats in ADAMTS-like-1/punctin-1 regulates secretion: Implications for the ADAMTS superfamily. J. Biol. Chem. 2007, 282, 17024–17031. [Google Scholar] [CrossRef]
- Adams, J.C.; Tucker, R.P. The thrombospondin type 1 repeat (TSR) superfamily: Diverse proteins with related roles in neuronal development. Dev. Dyn. 2000, 218, 280–299. [Google Scholar] [CrossRef]
- Leonhard-Melief, C.; Haltiwanger, R.S. O-fucosylation of thrombospondin type 1 repeats. Methods Enzymol. 2010, 480, 401–416. [Google Scholar]
- Shcherbakova, A.; Preller, M.; Taft, M.H.; Pujols, J.; Ventura, S.; Tiemann, B.; Buettner, F.F.; Bakker, H. C-mannosylation supports folding and enhances stability of thrombospondin repeats. eLife 2019, 8, e52978. [Google Scholar] [CrossRef]
- Furmanek, A.; Hofsteenge, J. Protein C-mannosylation: Facts and questions. Acta Biochim. Pol. 2000, 47, 781–789. [Google Scholar] [PubMed]
- Du, J.; Takeuchi, H.; Leonhard-Melief, C.; Shroyer, K.R.; Dlugosz, M.; Haltiwanger, R.S.; Holdener, B.C. O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Dev. Biol. 2010, 346, 25–38. [Google Scholar] [PubMed]
- Benz, B.A.; Nandadasa, S.; Takeuchi, M.; Grady, R.C.; Takeuchi, H.; LoPilato, R.K.; Kakuda, S.; Somerville, R.P.T.; Apte, S.S.; Haltiwanger, R.S.; et al. Genetic and biochemical evidence that gastrulation defects in Pofut2 mutants result from defects in ADAMTS9 secretion. Dev. Biol. 2016, 416, 111–122. [Google Scholar] [PubMed]
- Lesnik Oberstein, S.A.; Kriek, M.; White, S.J.; Kalf, M.E.; Szuhai, K.; den Dunnen, J.T.; Breuning, M.H.; Hennekam, R.C. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am. J. Hum. Genet. 2006, 79, 562–566. [Google Scholar]
- Colige, A.; Sieron, A.L.; Li, S.W.; Schwarze, U.; Petty, E.; Wertelecki, W.; Wilcox, W.; Krakow, D.; Cohn, D.H.; Reardon, W.; et al. Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Am. J. Hum. Genet. 1999, 65, 308–317. [Google Scholar]
- Brouillard, P.; Dupont, L.; Helaers, R.; Coulie, R.; Tiller, G.E.; Peeden, J.; Colige, A.; Vikkula, M. Loss of ADAMTS3 activity causes Hennekam lymphangiectasia-lymphedema syndrome 3. Hum. Mol. Genet. 2017, 26, 4095–4104. [Google Scholar]
- Wang, L.W.; Leonhard-Melief, C.; Haltiwanger, R.S.; Apte, S.S. Post-translational modification of thrombospondin type-1 repeats in ADAMTS-like 1/punctin-1 by C-mannosylation of tryptophan. J. Biol. Chem. 2009, 284, 30004–30015. [Google Scholar]
- Neupane, S.; Berardinelli, S.J.; Cameron, D.C.; Grady, R.C.; Komatsu, D.E.; Percival, C.J.; Takeuchi, M.; Ito, A.; Liu, T.W.; Nairn, A.V.; et al. O-fucosylation of thrombospondin type 1 repeats is essential for ECM remodeling and signaling during bone development. Matrix Biol. 2022, 107, 77–96. [Google Scholar]
- Dubail, J.; Vasudevan, D.; Wang, L.W.; Earp, S.E.; Jenkins, M.W.; Haltiwanger, R.S.; Apte, S.S. Impaired ADAMTS9 secretion: A potential mechanism for eye defects in Peters Plus Syndrome. Sci. Rep. 2016, 6, 33974. [Google Scholar]
- Dagoneau, N.; Benoist-Lasselin, C.; Huber, C.; Faivre, L.; Mégarbané, A.; Alswaid, A.; Dollfus, H.; Alembik, Y.; Munnich, A.; Legeai-Mallet, L.; et al. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am. J. Hum. Genet. 2004, 75, 801–806. [Google Scholar]
- Kutz, W.E.; Wang, L.W.; Dagoneau, N.; Odrcic, K.J.; Cormier-Daire, V.; Traboulsi, E.I.; Apte, S.S. Functional analysis of an ADAMTS10 signal peptide mutation in Weill-Marchesani syndrome demonstrates a long-range effect on secretion of the full-length enzyme. Hum. Mutat. 2008, 29, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick, J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.; Gruppo, R.; et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001, 413, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Boschann, F.; Cogulu, M.; Pehlivan, D.; Balachandran, S.; Vallecillo-Garcia, P.; Grochowski, C.M.; Hansmeier, N.R.; Coban Akdemir, Z.H.; Prada-Medina, C.A.; Aykut, A.; et al. Biallelic variants in ADAMTS15 cause a novel form of distal arthrogryposis. Genet. Med. 2022, 24, 2187–2193. [Google Scholar] [CrossRef] [PubMed]
- Hubmacher, D.; Schneider, M.; Berardinelli, S.J.; Takeuchi, H.; Willard, B.; Reinhardt, D.P.; Haltiwanger, R.S.; Apte, S.S. Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease. Sci. Rep. 2017, 7, 41871. [Google Scholar] [CrossRef]
- Morales, J.; Al-Sharif, L.; Khalil, D.S.; Shinwari, J.M.; Bavi, P.; Al-Mahrouqi, R.A.; Al-Rajhi, A.; Alkuraya, F.S.; Meyer, B.F.; Al Tassan, N. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am. J. Hum. Genet. 2009, 85, 558–568. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Alshammari, M.J.; Khan, A.O.; Mohamed, J.Y.; Alhabib, F.A.; Alkuraya, F.S. The syndrome of microcornea, myopic chorioretinal atrophy, and telecanthus (MMCAT) is caused by mutations in ADAMTS18. Hum. Mutat. 2013, 34, 1195–1199. [Google Scholar] [CrossRef]
- Massadeh, S.; Alhashem, A.; van de Laar, I.; Alhabshan, F.; Ordonez, N.; Alawbathani, S.; Khan, S.; Kabbani, M.S.; Chaikhouni, F.; Sheereen, A.; et al. ADAMTS19-associated heart valve defects: Novel genetic variants consolidating a recognizable cardiac phenotype. Clin. Genet. 2020, 98, 56–63. [Google Scholar] [CrossRef]
- Wünnemann, F.; Ta-Shma, A.; Preuss, C.; Leclerc, S.; van Vliet, P.P.; Oneglia, A.; Thibeault, M.; Nordquist, E.; Lincoln, J.; Scharfenberg, F.; et al. Loss of ADAMTS19 causes progressive non-syndromic heart valve disease. Nat. Genet. 2020, 52, 40–47. [Google Scholar] [CrossRef]
- Holdener, B.C.; Percival, C.J.; Grady, R.C.; Cameron, D.C.; Berardinelli, S.J.; Zhang, A.; Neupane, S.; Takeuchi, M.; Jimenez-Vega, J.C.; Uddin, S.M.Z.; et al. ADAMTS9 and ADAMTS20 are differentially affected by loss of B3GLCT in mouse model of Peters plus syndrome. Hum. Mol. Genet. 2019, 28, 4053–4066. [Google Scholar] [CrossRef]
- Zhang, A.; Berardinelli, S.J.; Leonhard-Melief, C.; Vasudevan, D.; Liu, T.W.; Taibi, A.; Giannone, S.; Apte, S.S.; Holdener, B.C.; Haltiwanger, R.S. O-Fucosylation of ADAMTSL2 is required for secretion and is impacted by geleophysic dysplasia-causing mutations. J. Biol. Chem. 2020, 295, 15742–15753. [Google Scholar] [CrossRef]
- Allali, S.; Le Goff, C.; Pressac-Diebold, I.; Pfennig, G.; Mahaut, C.; Dagoneau, N.; Alanay, Y.; Brady, A.F.; Crow, Y.J.; Devriendt, K.; et al. Molecular screening of ADAMTSL2 gene in 33 patients reveals the genetic heterogeneity of geleophysic dysplasia. J. Med. Genet. 2011, 48, 417–421. [Google Scholar] [PubMed]
- Le Goff, C.; Morice-Picard, F.; Dagoneau, N.; Wang, L.W.; Perrot, C.; Crow, Y.J.; Bauer, F.; Flori, E.; Prost-Squarcioni, C.; Krakow, D.; et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat. Genet. 2008, 40, 1119–1123. [Google Scholar] [PubMed]
- Ahram, D.; Sato, T.S.; Kohilan, A.; Tayeh, M.; Chen, S.; Leal, S.; Al-Salem, M.; El-Shanti, H. A homozygous mutation in ADAMTSL4 causes autosomal-recessive isolated ectopia lentis. Am. J. Hum. Genet. 2009, 84, 274–278. [Google Scholar] [PubMed]
- Christensen, A.E.; Fiskerstrand, T.; Knappskog, P.M.; Boman, H.; Rødahl, E. A novel ADAMTSL4 mutation in autosomal recessive ectopia lentis et pupillae. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6369–6373. [Google Scholar]
- Wang, J.; Miao, Y.; Wicklein, R.; Sun, Z.; Wang, J.; Jude, K.M.; Fernandes, R.A.; Merrill, S.A.; Wernig, M.; Garcia, K.C.; et al. RTN4/NoGo-receptor binding to BAI adhesion-GPCRs regulates neuronal development. Cell 2021, 184, 5869–5885.e25. [Google Scholar]
- Ikinciogullari, A.; Tekin, M.; Dogu, F.; Reisli, I.; Tanir, G.; Yi, Z.; Garrison, N.; Brilliant, M.H.; Babacan, E. Meningococccal meningitis and complement component 6 deficiency associated with oculocutaneous albinism. Eur. J. Pediatr. 2005, 164, 177–179. [Google Scholar] [CrossRef]
- Niwa, Y.; Suzuki, T.; Dohmae, N.; Simizu, S. O-Fucosylation of CCN1 is required for its secretion. FEBS Lett. 2015, 589, 3287–3293. [Google Scholar]
- Hurvitz, J.R.; Suwairi, W.M.; Van Hul, W.; El-Shanti, H.; Superti-Furga, A.; Roudier, J.; Holderbaum, D.; Pauli, R.M.; Herd, J.K.; Van Hul, E.V.; et al. Mutations in the CCN gene family member WISP3 cause progressive pseudorheumatoid dysplasia. Nat. Genet. 1999, 23, 94–98. [Google Scholar] [CrossRef]
- Hess, D.; Keusch, J.J.; Oberstein, S.A.; Hennekam, R.C.; Hofsteenge, J. Peters Plus syndrome is a new congenital disorder of glycosylation and involves defective Omicron-glycosylation of thrombospondin type 1 repeats. J. Biol. Chem. 2008, 283, 7354–7360. [Google Scholar]
- Fredrikson, G.N.; Westberg, J.; Kuijper, E.J.; Tijssen, C.C.; Sjöholm, A.G.; Uhlén, M.; Truedsson, L. Molecular characterization of properdin deficiency type III: Dysfunction produced by a single point mutation in exon 9 of the structural gene causing a tyrosine to aspartic acid interchange. J. Immunol. 1996, 157, 3666–3671. [Google Scholar]
- Schultz, D.W.; Klein, M.L.; Humpert, A.J.; Luzier, C.W.; Persun, V.; Schain, M.; Mahan, A.; Runckel, C.; Cassera, M.; Vittal, V.; et al. Analysis of the ARMD1 locus: Evidence that a mutation in HEMICENTIN-1 is associated with age-related macular degeneration in a large family. Hum. Mol. Genet. 2003, 12, 3315–3323. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez de Peredo, A.; Klein, D.; Macek, B.; Hess, D.; Peter-Katalinic, J.; Hofsteenge, J. C-mannosylation and o-fucosylation of thrombospondin type 1 repeats. Mol. Cell. Proteom. 2002, 1, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Neupane, S.; Goto, J.; Berardinelli, S.J.; Ito, A.; Haltiwanger, R.S.; Holdener, B.C. Hydrocephalus in mouse B3glct mutants is likely caused by defects in multiple B3GLCT substrates in ependymal cells and subcommissural organ. Glycobiology 2021, 31, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Chiba, K.; Karasugi, T.; Nakajima, M.; Kawaguchi, Y.; Mikami, Y.; Furuichi, T.; Mio, F.; Miyake, A.; Miyamoto, T.; et al. A functional polymorphism in THBS2 that affects alternative splicing and MMP binding is associated with lumbar-disc herniation. Am. J. Hum. Genet. 2008, 82, 1122–1129. [Google Scholar] [CrossRef]
- Al Rawi, W.N.; Ibrahim, F.H.; El Nakeib, O.A.S.; Al Zidgali, F.M. Manifestations of thrombospondin type-1 domain-containing protein 1 gene mutation in an extremely premature infant with nonimmune hydrops fetalis. Am. J. Med. Genet. A 2021, 185, 1598–1601. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Tulbah, M.; Kurdi, W.; Nemer, M.; Alsahan, N.; Al Mardawi, E.; Khalifa, O.; Hashem, A.; Kurdi, A.; Babay, Z.; et al. Identification of embryonic lethal genes in humans by autozygosity mapping and exome sequencing in consanguineous families. Genome Biol. 2015, 16, 116. [Google Scholar] [CrossRef]
- Rui, Y.N.; Xu, Z.; Fang, X.; Menezes, M.R.; Balzeau, J.; Niu, A.; Hagan, J.P.; Kim, D.H. The Intracranial Aneurysm Gene THSD1 Connects Endosome Dynamics to Nascent Focal Adhesion Assembly. Cell Physiol. Biochem. 2017, 43, 2200–2211. [Google Scholar] [CrossRef]
- Santiago-Sim, T.; Fang, X.; Hennessy, M.L.; Nalbach, S.V.; DePalma, S.R.; Lee, M.S.; Greenway, S.C.; McDonough, B.; Hergenroeder, G.W.; Patek, K.J.; et al. THSD1 (Thrombospondin Type 1 Domain Containing Protein 1) Mutation in the Pathogenesis of Intracranial Aneurysm and Subarachnoid Hemorrhage. Stroke 2016, 47, 3005–3013. [Google Scholar] [CrossRef]
- Elbitar, S.; Renard, M.; Arnaud, P.; Hanna, N.; Jacob, M.P.; Guo, D.C.; Tsutsui, K.; Gross, M.S.; Kessler, K.; Tosolini, L.; et al. Pathogenic variants in THSD4, encoding the ADAMTS-like 6 protein, predispose to inherited thoracic aortic aneurysm. Genet. Med. 2021, 23, 111–122. [Google Scholar] [CrossRef]
- Seifert, L.; Hoxha, E.; Eichhoff, A.M.; Zahner, G.; Dehde, S.; Reinhard, L.; Koch-Nolte, F.; Stahl, R.A.K.; Tomas, N.M. The Most N-Terminal Region of THSD7A Is the Predominant Target for Autoimmunity in THSD7A-Associated Membranous Nephropathy. J. Am. Soc. Nephrol. 2018, 29, 1536–1548. [Google Scholar] [CrossRef]
- Tomas, N.M.; Beck, L.H., Jr.; Meyer-Schwesinger, C.; Seitz-Polski, B.; Ma, H.; Zahner, G.; Dolla, G.; Hoxha, E.; Helmchen, U.; Dabert-Gay, A.S.; et al. Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N. Engl. J. Med. 2014, 371, 2277–2287. [Google Scholar] [PubMed]
- Roos, C.; Kolmer, M.; Mattila, P.; Renkonen, R. Composition of Drosophila melanogaster proteome involved in fucosylated glycan metabolism. J. Biol. Chem. 2002, 277, 3168–3175. [Google Scholar] [PubMed]
- Fabini, G.; Freilinger, A.; Altmann, F.; Wilson, I.B. Identification of core alpha 1,3-fucosylated glycans and cloning of the requisite fucosyltransferase cDNA from Drosophila melanogaster: Potential basis of the neural anti-horseadish peroxidase epitope. J. Biol. Chem. 2001, 276, 28058–28067. [Google Scholar] [PubMed]
- Rendic, D.; Linder, A.; Paschinger, K.; Borth, N.; Wilson, I.B.; Fabini, G. Modulation of neural carbohydrate epitope expression in Drosophila melanogaster cells. J. Biol. Chem. 2006, 281, 3343–3353. [Google Scholar]
- Baboval, T.; Smith, F.I. Comparison of human and mouse Fuc-TX and Fuc-TXI genes, and expression studies in the mouse. Mamm. Genome 2002, 13, 538–541. [Google Scholar]
- Patnaik, S. Characterization of Fut10 and Fut11, Putative Alpha-1–3/4 Fucosyltransferase Genes Important for Vertebrate Development. Nat. Preced. 2007. [Google Scholar] [CrossRef]
- Rendić, D.; Klaudiny, J.; Stemmer, U.; Schmidt, J.; Paschinger, K.; Wilson, I.B. Towards abolition of immunogenic structures in insect cells: Characterization of a honey-bee (Apis mellifera) multi-gene family reveals both an allergy-related core alpha1,3-fucosyltransferase and the first insect Lewis-histo-blood-group-related antigen-synthesizing enzyme. Biochem. J. 2007, 402, 105–115. [Google Scholar]
- Mollicone, R.; Moore, S.E.; Bovin, N.; Garcia-Rosasco, M.; Candelier, J.J.; Martinez-Duncker, I.; Oriol, R. Activity, splice variants, conserved peptide motifs, and phylogeny of two new alpha1,3-fucosyltransferase families (FUT10 and FUT11). J. Biol. Chem. 2009, 284, 4723–4738. [Google Scholar]
- Kumar, A.; Torii, T.; Ishino, Y.; Muraoka, D.; Yoshimura, T.; Togayachi, A.; Narimatsu, H.; Ikenaka, K.; Hitoshi, S. The Lewis X-related α1,3-fucosyltransferase, Fut10, is required for the maintenance of stem cell populations. J. Biol. Chem. 2013, 288, 28859–28868. [Google Scholar] [CrossRef]
- Capuano, A.; Bucciotti, F.; Farwell, K.D.; Tippin Davis, B.; Mroske, C.; Hulick, P.J.; Weissman, S.M.; Gao, Q.; Spessotto, P.; Colombatti, A.; et al. Diagnostic Exome Sequencing Identifies a Novel Gene, EMILIN1, Associated with Autosomal-Dominant Hereditary Connective Tissue Disease. Hum. Mutat. 2016, 37, 84–97. [Google Scholar]
- Iacomino, M.; Doliana, R.; Marchese, M.; Capuano, A.; Striano, P.; Spessotto, P.; Bosisio, G.; Iodice, R.; Manganelli, F.; Lanteri, P.; et al. Distal motor neuropathy associated with novel EMILIN1 mutation. Neurobiol. Dis. 2020, 137, 104757. [Google Scholar]
- Boutboul, S.; Black, G.C.; Moore, J.E.; Sinton, J.; Menasche, M.; Munier, F.L.; Laroche, L.; Abitbol, M.; Schorderet, D.F. A subset of patients with epithelial basement membrane corneal dystrophy have mutations in TGFBI/BIGH3. Hum. Mutat. 2006, 27, 553–557. [Google Scholar] [PubMed]
- Chakravarthi, S.V.; Kannabiran, C.; Sridhar, M.S.; Vemuganti, G.K. TGFBI gene mutations causing lattice and granular corneal dystrophies in Indian patients. Investig. Ophthalmol. Vis. Sci. 2005, 46, 121–125. [Google Scholar]
- Fujiki, K.; Hotta, Y.; Nakayasu, K.; Yokoyama, T.; Takano, T.; Yamaguchi, T.; Kanai, A. A new L527R mutation of the betaIGH3 gene in patients with lattice corneal dystrophy with deep stromal opacities. Hum. Genet. 1998, 103, 286–289. [Google Scholar]
- Rozzo, C.; Fossarello, M.; Galleri, G.; Sole, G.; Serru, A.; Orzalesi, N.; Serra, A.; Pirastu, M. A common beta ig-h3 gene mutation (delta f540) in a large cohort of Sardinian Reis Bücklers corneal dystrophy patients. Mutations in brief no. 180. Online. Hum. Mutat. 1998, 12, 215–216. [Google Scholar]
- Yamamoto, S.; Okada, M.; Tsujikawa, M.; Shimomura, Y.; Nishida, K.; Inoue, Y.; Watanabe, H.; Maeda, N.; Kurahashi, H.; Kinoshita, S.; et al. A kerato-epithelin (betaig-h3) mutation in lattice corneal dystrophy type IIIA. Am. J. Hum. Genet. 1998, 62, 719–722. [Google Scholar]
- Dupuy, F.; Germot, A.; Julien, R.; Maftah, A. Structure/function study of Lewis alpha3- and alpha3/4-fucosyltransferases: The alpha1,4 fucosylation requires an aromatic residue in the acceptor-binding domain. Glycobiology 2004, 14, 347–356. [Google Scholar]
- Dupuy, F.; Germot, A.; Marenda, M.; Oriol, R.; Blancher, A.; Julien, R.; Maftah, A. Alpha1,4-fucosyltransferase activity: A significant function in the primate lineage has appeared twice independently. Mol. Biol. Evol. 2002, 19, 815–824. [Google Scholar]
- Dupuy, F.; Petit, J.M.; Mollicone, R.; Oriol, R.; Julien, R.; Maftah, A. A single amino acid in the hypervariable stem domain of vertebrate alpha1,3/1,4-fucosyltransferases determines the type 1/type 2 transfer. Characterization of acceptor substrate specificity of the lewis enzyme by site-directed mutagenesis. J. Biol. Chem. 1999, 274, 12257–12262. [Google Scholar]
- Loriol, C.; Dupuy, F.; Rampal, R.; Dlugosz, M.A.; Haltiwanger, R.S.; Maftah, A.; Germot, A. Molecular evolution of protein O-fucosyltransferase genes and splice variants. Glycobiology 2006, 16, 736–747. [Google Scholar]
- Callebaut, I.; Mignotte, V.; Souchet, M.; Mornon, J.P. EMI domains are widespread and reveal the probable orthologs of the Caenorhabditis elegans CED-1 protein. Biochem. Biophys. Res. Commun. 2003, 300, 619–623. [Google Scholar] [PubMed]
- Doliana, R.; Bot, S.; Bonaldo, P.; Colombatti, A. EMI, a novel cysteine-rich domain of EMILINs and other extracellular proteins, interacts with the gC1q domains and participates in multimerization. FEBS Lett. 2000, 484, 164–168. [Google Scholar] [PubMed]
- Colombatti, A.; Spessotto, P.; Doliana, R.; Mongiat, M.; Bressan, G.M.; Esposito, G. The EMILIN/Multimerin family. Front. Immunol. 2011, 2, 93. [Google Scholar]
- Cao, W.; Zeng, Z.; Lan, J.; Yang, Y.; Lu, M.; Lei, S. Knockdown of FUT11 inhibits the progression of gastric cancer via the PI3K/AKT pathway. Heliyon 2023, 9, e17600. [Google Scholar]
- Cao, W.; Zeng, Z.; Pan, R.; Wu, H.; Zhang, X.; Chen, H.; Nie, Y.; Yu, Z.; Lei, S. Hypoxia-Related Gene FUT11 Promotes Pancreatic Cancer Progression by Maintaining the Stability of PDK1. Front. Oncol. 2021, 11, 675991. [Google Scholar]
- Huang, Y.; Yang, X.; Wei, M.; Yang, X.; Yuan, Z.; Huang, J.; Wei, J.; Tian, L. FUT11 expression in gastric cancer: Its prognostic significance and role in immune regulation. Discov. Oncol. 2024, 15, 250. [Google Scholar]
- Ruan, W.; Yang, Y.; Yu, Q.; Huang, T.; Wang, Y.; Hua, L.; Zeng, Z.; Pan, R. FUT11 is a target gene of HIF1α that promotes the progression of hepatocellular carcinoma. Cell Biol. Int. 2021, 45, 2275–2286. [Google Scholar]
- Wang, P.; Liu, X.; Yu, J.; Meng, Z.; Lv, Z.; Shang, C.; Geng, Q.; Wang, D.; Xue, D.; Li, L. Fucosyltransferases Regulated by Fusobacterium Nucleatum and Act as Novel Biomarkers in Colon Adenocarcinoma. J. Inflamm. Res. 2023, 16, 747–768. [Google Scholar]
- Zhang, P.; Tang, W.; Jiang, Y.; Lyu, F.; Liu, Z.; Xiao, Y.; Wang, D. Dry and wet experiments reveal the significant role of FUT11 in clear cell renal cell carcinoma. Int. Immunopharmacol. 2022, 113 Pt B, 109447. [Google Scholar]
- Zodro, E.; Jaroszewski, M.; Ida, A.; Wrzesiński, T.; Kwias, Z.; Bluyssen, H.; Wesoly, J. FUT11 as a potential biomarker of clear cell renal cell carcinoma progression based on meta-analysis of gene expression data. Tumour Biol. 2014, 35, 2607–2617. [Google Scholar]
- Iwasaki, H.; Akashi, K. Hematopoietic developmental pathways: On cellular basis. Oncogene 2007, 26, 6687–6696. [Google Scholar] [PubMed]
- Stier, S.; Cheng, T.; Dombkowski, D.; Carlesso, N.; Scadden, D.T. Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood 2002, 99, 2369–2378. [Google Scholar] [PubMed]
- Ohishi, K.; Varnum-Finney, B.; Bernstein, I.D. Delta-1 enhances marrow and thymus repopulating ability of human CD34(+)CD38(-) cord blood cells. J. Clin. Investig. 2002, 110, 1165–1174. [Google Scholar]
- Yao, D.; Huang, Y.; Huang, X.; Wang, W.; Yan, Q.; Wei, L.; Xin, W.; Gerson, S.; Stanley, P.; Lowe, J.B.; et al. Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood 2011, 117, 5652–5662. [Google Scholar] [PubMed]
- Yu, V.W.; Saez, B.; Cook, C.; Lotinun, S.; Pardo-Saganta, A.; Wang, Y.H.; Lymperi, S.; Ferraro, F.; Raaijmakers, M.H.; Wu, J.Y.; et al. Specific bone cells produce DLL4 to generate thymus-seeding progenitors from bone marrow. J. Exp. Med. 2015, 212, 759–774. [Google Scholar]
- Bhandoola, A.; Sambandam, A. From stem cell to T cell: One route or many? Nat. Rev. Immunol. 2006, 6, 117–126. [Google Scholar]
- Maillard, I.; Fang, T.; Pear, W.S. Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annu. Rev. Immunol. 2005, 23, 945–974. [Google Scholar]
- Tanigaki, K.; Honjo, T. Regulation of lymphocyte development by Notch signaling. Nat. Immunol. 2007, 8, 451–456. [Google Scholar]
- Koch, U.; Lacombe, T.A.; Holland, D.; Bowman, J.L.; Cohen, B.L.; Egan, S.E.; Guidos, C.J. Subversion of the T/B lineage decision in the thymus by lunatic fringe-mediated inhibition of Notch-1. Immunity 2001, 15, 225–236. [Google Scholar]
- Feyerabend, T.B.; Terszowski, G.; Tietz, A.; Blum, C.; Luche, H.; Gossler, A.; Gale, N.W.; Radtke, F.; Fehling, H.J.; Rodewald, H.R. Deletion of Notch1 converts pro-T cells to dendritic cells and promotes thymic B cells by cell-extrinsic and cell-intrinsic mechanisms. Immunity 2009, 30, 67–79. [Google Scholar]
- Schneider, M.; Kumar, V.; Nordstrøm, L.U.; Feng, L.; Takeuchi, H.; Hao, H.; Luca, V.C.; Garcia, K.C.; Stanley, P.; Wu, P.; et al. Inhibition of Delta-induced Notch signaling using fucose analogs. Nat. Chem. Biol. 2018, 14, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Visan, I.; Tan, J.B.; Yuan, J.S.; Harper, J.A.; Koch, U.; Guidos, C.J. Regulation of T lymphopoiesis by Notch1 and Lunatic fringe-mediated competition for intrathymic niches. Nat. Immunol. 2006, 7, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Yahata, T.; Negishi, N.; Nakano, Y.; Habu, S.; Hozumi, K.; Ando, K. Expression of Delta-like 1 in the splenic non-hematopoietic cells is essential for marginal zone B cell development. Immunol. Lett. 2008, 121, 33–37. [Google Scholar] [PubMed]
- Saito, T.; Chiba, S.; Ichikawa, M.; Kunisato, A.; Asai, T.; Shimizu, K.; Yamaguchi, T.; Yamamoto, G.; Seo, S.; Kumano, K.; et al. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity 2003, 18, 675–685. [Google Scholar] [CrossRef]
- Tan, J.B.; Xu, K.; Cretegny, K.; Visan, I.; Yuan, J.S.; Egan, S.E.; Guidos, C.J. Lunatic and manic fringe cooperatively enhance marginal zone B cell precursor competition for delta-like 1 in splenic endothelial niches. Immunity 2009, 30, 254–263. [Google Scholar]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef]
- Postma, C.; Hermsen, M.A.; Coffa, J.; Baak, J.P.; Mueller, J.D.; Mueller, E.; Bethke, B.; Schouten, J.P.; Stolte, M.; Meijer, G.A. Chromosomal instability in flat adenomas and carcinomas of the colon. J. Pathol. 2005, 205, 514–521. [Google Scholar]
- Chabanais, J.; Labrousse, F.; Chaunavel, A.; Germot, A.; Maftah, A. POFUT1 as a Promising Novel Biomarker of Colorectal Cancer. Cancers 2018, 10, 411. [Google Scholar] [CrossRef]
- Deschuyter, M.; Pennarubia, F.; Pinault, E.; Legardinier, S.; Maftah, A. Functional Characterization of POFUT1 Variants Associated with Colorectal Cancer. Cancers 2020, 12, 1430. [Google Scholar] [CrossRef]
- Ma, L.; Dong, P.; Liu, L.; Gao, Q.; Duan, M.; Zhang, S.; Chen, S.; Xue, R.; Wang, X. Overexpression of protein O-fucosyltransferase 1 accelerates hepatocellular carcinoma progression via the Notch signaling pathway. Biochem. Biophys. Res. Commun. 2016, 473, 503–510. [Google Scholar] [CrossRef]
- Sawey, E.T.; Chanrion, M.; Cai, C.; Wu, G.; Zhang, J.; Zender, L.; Zhao, A.; Busuttil, R.W.; Yee, H.; Stein, L.; et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell 2011, 19, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, J.; Ma, X.; Wang, M.; Zhou, L. POFUT1 acts as a tumor promoter in glioblastoma by enhancing the activation of Notch signaling. J. Bioenerg. Biomembr. 2021, 53, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Kroes, R.A.; Dawson, G.; Moskal, J.R. Focused microarray analysis of glyco-gene expression in human glioblastomas. J. Neurochem. 2007, 103 (Suppl. 1), 14–24. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Wang, Z.; Huang, B.; Zhang, J.; Ge, Y.; Fan, Q.; Wang, Z. Bioinformatics insight into glycosyltransferase gene expression in gastric cancer: POFUT1 is a potential biomarker. Biochem. Biophys. Res. Commun. 2017, 483, 171–177. [Google Scholar] [CrossRef]
- Yokota, S.; Ogawara, K.; Kimura, R.; Shimizu, F.; Baba, T.; Minakawa, Y.; Higo, M.; Kasamatsu, A.; Endo-Sakamoto, Y.; Shiiba, M.; et al. Protein O-fucosyltransferase 1: A potential diagnostic marker and therapeutic target for human oral cancer. Int. J. Oncol. 2013, 43, 1864–1870. [Google Scholar] [CrossRef]
- Milde-Langosch, K.; Karn, T.; Schmidt, M.; zu Eulenburg, C.; Oliveira-Ferrer, L.; Wirtz, R.M.; Schumacher, U.; Witzel, I.; Schutze, D.; Muller, V. Prognostic relevance of glycosylation-associated genes in breast cancer. Breast Cancer Res. Treat. 2014, 145, 295–305. [Google Scholar] [CrossRef]
- Wahby, S.; Jarczyk, J.; Fierek, A.; Heinkele, J.; Weis, C.A.; Eckstein, M.; Martini, T.; Porubsky, S.; Hafner, M.; Erben, P. POFUT1 mRNA expression as an independent prognostic parameter in muscle-invasive bladder cancer. Transl. Oncol. 2021, 14, 100900. [Google Scholar] [CrossRef]
- Rampias, T.; Vgenopoulou, P.; Avgeris, M.; Polyzos, A.; Stravodimos, K.; Valavanis, C.; Scorilas, A.; Klinakis, A. A new tumor suppressor role for the Notch pathway in bladder cancer. Nat. Med. 2014, 20, 1199–1205. [Google Scholar] [CrossRef]
- Larose, H.; Prokoph, N.; Matthews, J.D.; Schlederer, M.; Högler, S.; Alsulami, A.F.; Ducray, S.P.; Nuglozeh, E.; Fazaludeen, F.M.S.; Elmouna, A.; et al. Whole Exome Sequencing reveals NOTCH1 mutations in anaplastic large cell lymphoma and points to Notch both as a key pathway and a potential therapeutic target. Haematologica 2021, 106, 1693–1704. [Google Scholar] [CrossRef]
- Pennarubia, F.; Ito, A.; Takeuchi, M.; Haltiwanger, R.S. Cancer-associated Notch receptor variants lead to O-fucosylation defects that deregulate Notch signaling. J. Biol. Chem. 2022, 298, 102616. [Google Scholar] [CrossRef]
- Weh, E.; Reis, L.M.; Tyler, R.C.; Bick, D.; Rhead, W.J.; Wallace, S.; McGregor, T.L.; Dills, S.K.; Chao, M.C.; Murray, J.C.; et al. Novel B3GALTL mutations in classic Peters plus syndrome and lack of mutations in a large cohort of patients with similar phenotypes. Clin. Genet. 2014, 86, 142–148. [Google Scholar] [PubMed]
- Aliferis, K.; Marsal, C.; Pelletier, V.; Doray, B.; Weiss, M.M.; Tops, C.M.; Speeg-Schatz, C.; Lesnik, S.A.; Dollfus, H. A novel nonsense B3GALTL mutation confirms Peters plus syndrome in a patient with multiple malformations and Peters anomaly. Ophthalmic Genet. 2010, 31, 205–208. [Google Scholar] [PubMed]
- Kozma, K.; Keusch, J.J.; Hegemann, B.; Luther, K.B.; Klein, D.; Hess, D.; Haltiwanger, R.S.; Hofsteenge, J. Identification and characterization of abeta1,3-glucosyltransferase that synthesizes the Glc-beta1,3-Fuc disaccharide on thrombospondin type 1 repeats. J. Biol. Chem. 2006, 281, 36742–36751. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Sato, M.; Kiyohara, K.; Sogabe, M.; Shikanai, T.; Kikuchi, N.; Togayachi, A.; Ishida, H.; Ito, H.; Kameyama, A.; et al. Molecular cloning and characterization of a novel human beta1,3-glucosyltransferase, which is localized at the endoplasmic reticulum and glucosylates O-linked fucosylglycan on thrombospondin type 1 repeat domain. Glycobiology 2006, 16, 1194–1206. [Google Scholar] [CrossRef]
- Zhang, A.; Venkat, A.; Taujale, R.; Mull, J.L.; Ito, A.; Kannan, N.; Haltiwanger, R.S. Peters plus syndrome mutations affect the function and stability of human beta1,3-glucosyltransferase. J. Biol. Chem. 2021, 297, 100843. [Google Scholar]
- Mead, T.J.; Apte, S.S. ADAMTS proteins in human disorders. Matrix Biol. 2018, 71–72, 225–239. [Google Scholar]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar]
- Vasudevan, D.; Takeuchi, H.; Johar, S.S.; Majerus, E.; Haltiwanger, R.S. Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr. Biol. 2015, 25, 286–295. [Google Scholar]
- Sparrow, D.B.; Chapman, G.; Wouters, M.A.; Whittock, N.V.; Ellard, S.; Fatkin, D.; Turnpenny, P.D.; Kusumi, K.; Sillence, D.; Dunwoodie, S.L. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am. J. Hum. Genet. 2006, 78, 28–37. [Google Scholar] [CrossRef]
- Wengryn, P.; Fenrich, F.; Silveira, K.D.C.; Oborn, C.; Mizumoto, S.; Beke, A.; Soltys, C.L.; Yamada, S.; Kannu, P. Integrative analysis of Lunatic Fringe variants associated with spondylocostal dysostosis type-III. FASEB J. 2024, 38, e23753. [Google Scholar] [CrossRef]
- Wang, L.; Mizumoto, S.; Zhang, R.; Zhang, Y.; Liu, Y.; Cheng, W.; Li, X.; Dan, M.; Zhang, C.; Gao, X.; et al. Identification of a novel LFNG variant in a Chinese fetus with spondylocostal dysostosis and a systematic review. J. Hum. Genet. 2024, 69, 321–327. [Google Scholar] [PubMed]
- Nóbrega, A.; Maia-Fernandes, A.C.; Andrade, R.P. Altered Cogs of the Clock: Insights into the Embryonic Etiology of Spondylocostal Dysostosis. J. Dev. Biol. 2021, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Wahi, K.; Bochter, M.S.; Cole, S.E. The many roles of Notch signaling during vertebrate somitogenesis. Semin. Cell Dev. Biol. 2016, 49, 68–75. [Google Scholar] [PubMed]
- Evrard, Y.A.; Lun, Y.; Aulehla, A.; Gan, L.; Johnson, R.L. lunatic fringe is an essential mediator of somite segmentation and patterning. Nature 1998, 394, 377–381. [Google Scholar]
- Zhang, N.; Gridley, T. Defects in somite formation in lunatic fringe-deficient mice. Nature 1998, 394, 374–377. [Google Scholar]
- Moran, J.L.; Levorse, J.M.; Vogt, T.F. Limbs move beyond the radical fringe. Nature 1999, 399, 742–743. [Google Scholar]
- Zhang, N.; Norton, C.R.; Gridley, T. Segmentation defects of Notch pathway mutants and absence of a synergistic phenotype in lunatic fringe/radical fringe double mutant mice. Genesis 2002, 33, 21–28. [Google Scholar]
- Moran, J.L.; Shifley, E.T.; Levorse, J.M.; Mani, S.; Ostmann, K.; Perez-Balaguer, A.; Walker, D.M.; Vogt, T.F.; Cole, S.E. Manic fringe is not required for embryonic development, and fringe family members do not exhibit redundant functions in the axial skeleton, limb, or hindbrain. Dev. Dyn. 2009, 238, 1803–1812. [Google Scholar]
- Harvey, B.M.; Rana, N.A.; Moss, H.; Leonardi, J.; Jafar-Nejad, H.; Haltiwanger, R.S. Mapping Sites of O-Glycosylation and Fringe Elongation on Drosophila Notch. J. Biol. Chem. 2016, 291, 16348–16360. [Google Scholar]
- Li, Z.; Han, K.; Pak, J.E.; Satkunarajah, M.; Zhou, D.; Rini, J.M. Recognition of EGF-like domains by the Notch-modifying O-fucosyltransferase POFUT1. Nat. Chem. Biol. 2017, 13, 757–763. [Google Scholar]
- Berardinelli, S.J.; Eletsky, A.; Valero-Gonzalez, J.; Ito, A.; Manjunath, R.; Hurtado-Guerrero, R.; Prestegard, J.H.; Woods, R.J.; Haltiwanger, R.S. O-fucosylation stabilizes the TSR3 motif in thrombospondin-1 by interacting with nearby amino acids and protecting a disulfide bond. J. Biol. Chem. 2022, 298, 102047. [Google Scholar] [CrossRef]
- Moremen, K.W.; Haltiwanger, R.S. Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat. Chem. Biol. 2019, 15, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Yu, H.; Hao, H.; Takeuchi, M.; Ito, A.; Li, H.; Haltiwanger, R.S. O-Glycosylation modulates the stability of epidermal growth factor-like repeats and thereby regulates Notch trafficking. J. Biol. Chem. 2017, 292, 15964–15973. [Google Scholar] [CrossRef] [PubMed]
- Okajima, T.; Xu, A.; Lei, L.; Irvine, K.D. Chaperone activity of protein O-fucosyltransferase 1 promotes notch receptor folding. Science 2005, 307, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Ajima, R.; Suzuki, E.; Saga, Y. Pofut1 point-mutations that disrupt O-fucosyltransferase activity destabilize the protein and abolish Notch1 signaling during mouse somitogenesis. PLoS ONE 2017, 12, e0187248. [Google Scholar] [CrossRef]
- McMillan, B.J.; Zimmerman, B.; Egan, E.D.; Lofgren, M.; Xu, X.; Hesser, A.; Blacklow, S.C. Structure of human POFUT1, its requirement in ligand-independent oncogenic Notch signaling, and functional effects of Dowling-Degos mutations. Glycobiology 2017, 27, 777–786. [Google Scholar] [CrossRef]
- Stahl, M.; Uemura, K.; Ge, C.; Shi, S.; Tashima, Y.; Stanley, P. Roles of Pofut1 and O-fucose in mammalian Notch signaling. J. Biol. Chem. 2008, 283, 13638–13651. [Google Scholar] [CrossRef]
- Zandberg, W.F.; Kumarasamy, J.; Pinto, B.M.; Vocadlo, D.J. Metabolic inhibition of sialyl-Lewis X biosynthesis by 5-thiofucose remodels the cell surface and impairs selectin-mediated cell adhesion. J. Biol. Chem. 2012, 287, 40021–40030. [Google Scholar] [CrossRef]
- Okeley, N.M.; Alley, S.C.; Anderson, M.E.; Boursalian, T.E.; Burke, P.J.; Emmerton, K.M.; Jeffrey, S.C.; Klussman, K.; Law, C.L.; Sussman, D.; et al. Development of orally active inhibitors of protein and cellular fucosylation. Proc. Natl. Acad. Sci. USA 2013, 110, 5404–5409. [Google Scholar] [CrossRef]
- Rillahan, C.D.; Antonopoulos, A.; Lefort, C.T.; Sonon, R.; Azadi, P.; Ley, K.; Dell, A.; Haslam, S.M.; Paulson, J.C. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol. 2012, 8, 661–668. [Google Scholar] [CrossRef]
- Allen, J.G.; Mujacic, M.; Frohn, M.J.; Pickrell, A.J.; Kodama, P.; Bagal, D.; San Miguel, T.; Sickmier, E.A.; Osgood, S.; Swietlow, A.; et al. Facile Modulation of Antibody Fucosylation with Small Molecule Fucostatin Inhibitors and Cocrystal Structure with GDP-Mannose 4,6-Dehydratase. ACS Chem. Biol. 2016, 11, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Kizuka, Y.; Nakano, M.; Yamaguchi, Y.; Nakajima, K.; Oka, R.; Sato, K.; Ren, C.T.; Hsu, T.L.; Wong, C.H.; Taniguchi, N. An Alkynyl-Fucose Halts Hepatoma Cell Migration and Invasion by Inhibiting GDP-Fucose-Synthesizing Enzyme FX, TSTA3. Cell Chem. Biol. 2017, 24, 1467–1478.e5. [Google Scholar] [CrossRef] [PubMed]
- Kizuka, Y.; Funayama, S.; Shogomori, H.; Nakano, M.; Nakajima, K.; Oka, R.; Kitazume, S.; Yamaguchi, Y.; Sano, M.; Korekane, H.; et al. High-Sensitivity and Low-Toxicity Fucose Probe for Glycan Imaging and Biomarker Discovery. Cell Chem. Biol. 2016, 23, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Gilormini, P.A.; Thota, V.N.; Fers-Lidou, A.; Ashmus, R.A.; Nodwell, M.; Brockerman, J.; Kuo, C.W.; Wang, Y.; Gray, T.E.; Nitin; et al. A metabolic inhibitor blocks cellular fucosylation and enables production of afucosylated antibodies. Proc. Natl. Acad. Sci. USA 2024, 121, e2314026121. [Google Scholar] [CrossRef]
- Al-Shareffi, E.; Chaubard, J.L.; Leonhard-Melief, C.; Wang, S.K.; Wong, C.H.; Haltiwanger, R.S. 6-alkynyl fucose is a bioorthogonal analog for O-fucosylation of epidermal growth factor-like repeats and thrombospondin type-1 repeats by protein O-fucosyltransferases 1 and 2. Glycobiology 2013, 23, 188–198. [Google Scholar] [CrossRef]
- Ma, C.; Takeuchi, H.; Hao, H.; Yonekawa, C.; Nakajima, K.; Nagae, M.; Okajima, T.; Haltiwanger, R.S.; Kizuka, Y. Differential Labeling of Glycoproteins with Alkynyl Fucose Analogs. Int. J. Mol. Sci. 2020, 21, 6007. [Google Scholar] [CrossRef]
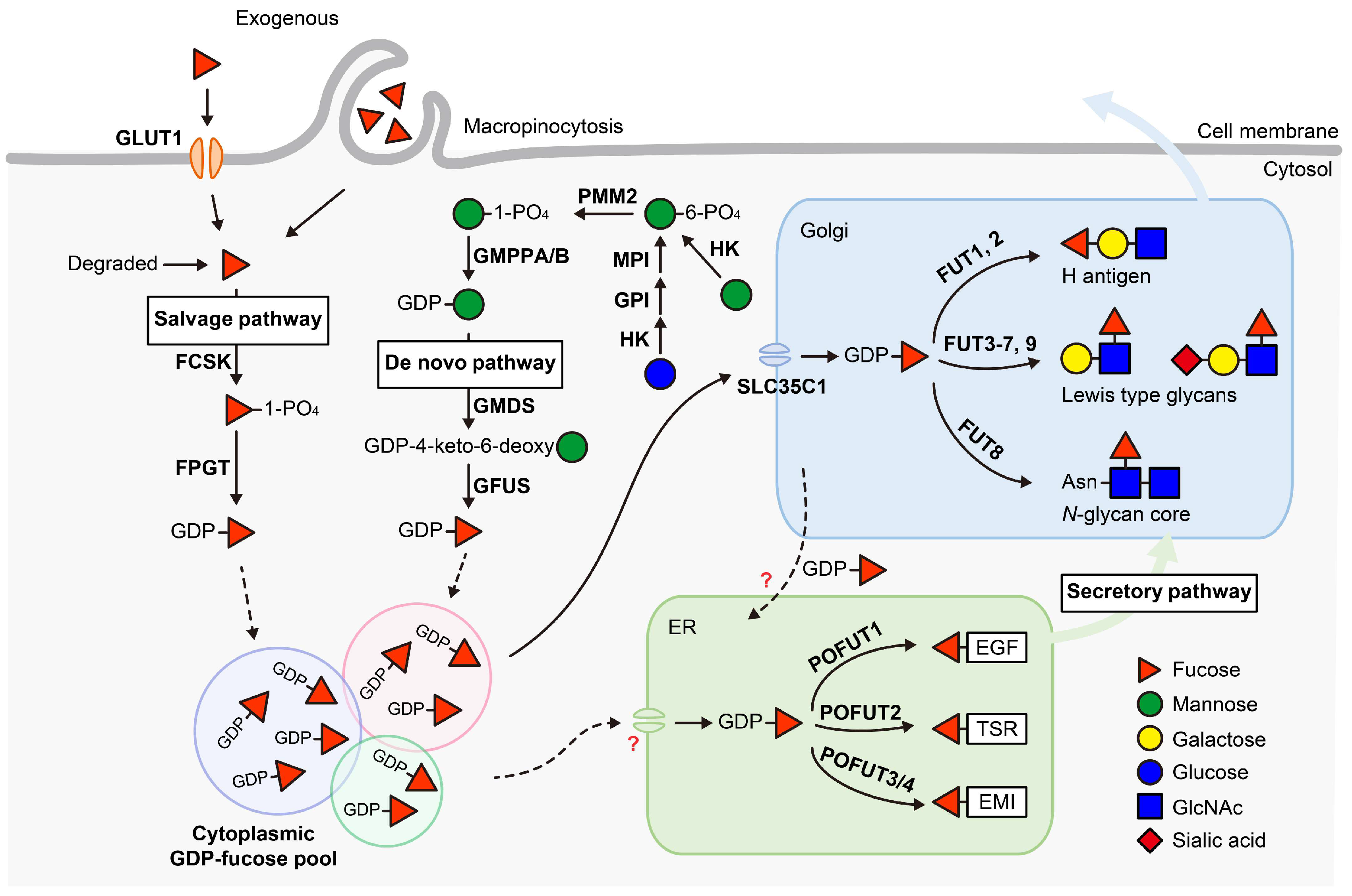
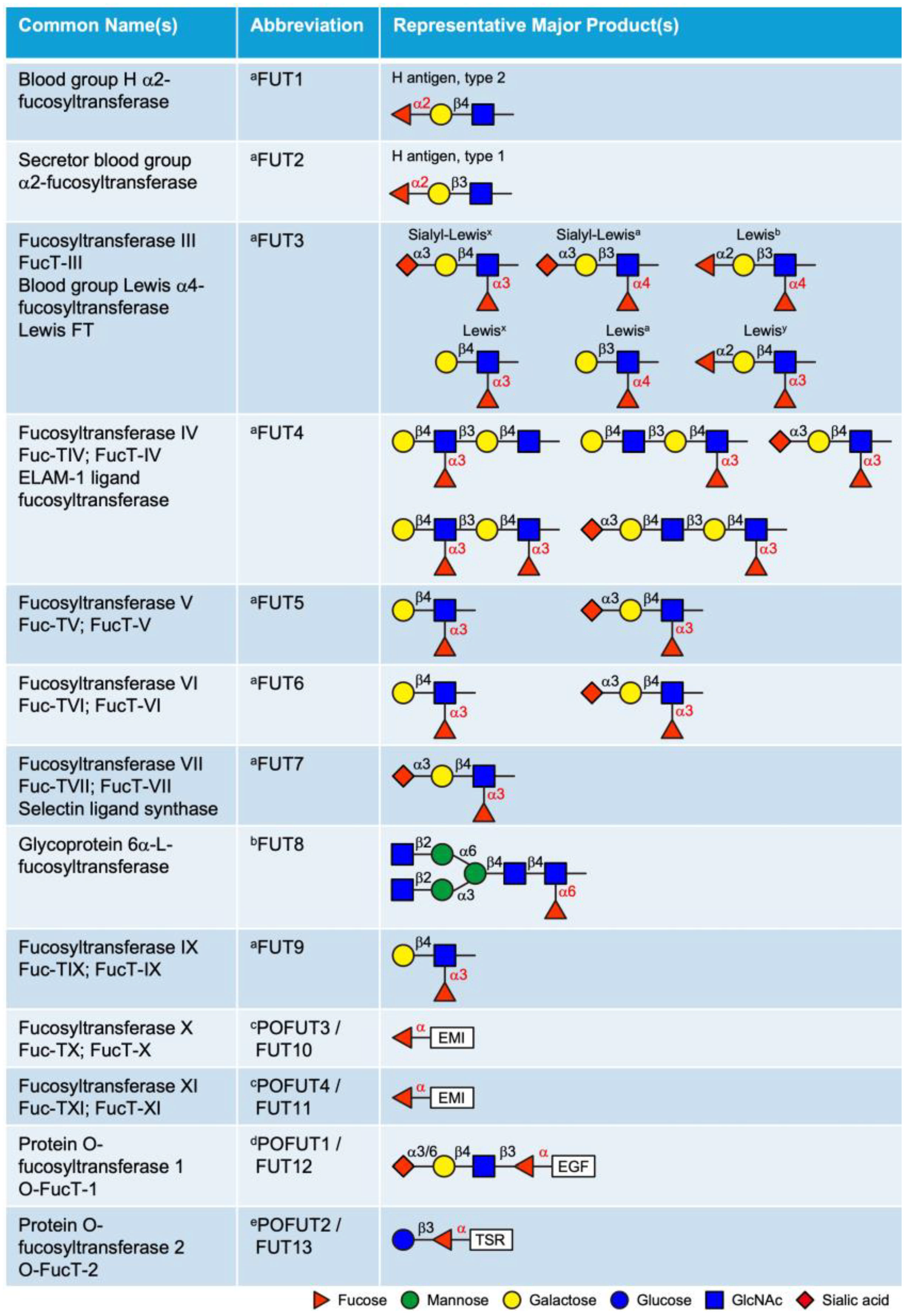
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).