
The Effect of Polyethylene Terephthalate Nanoplastics on Amyloid-β Peptide Fibrillation

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
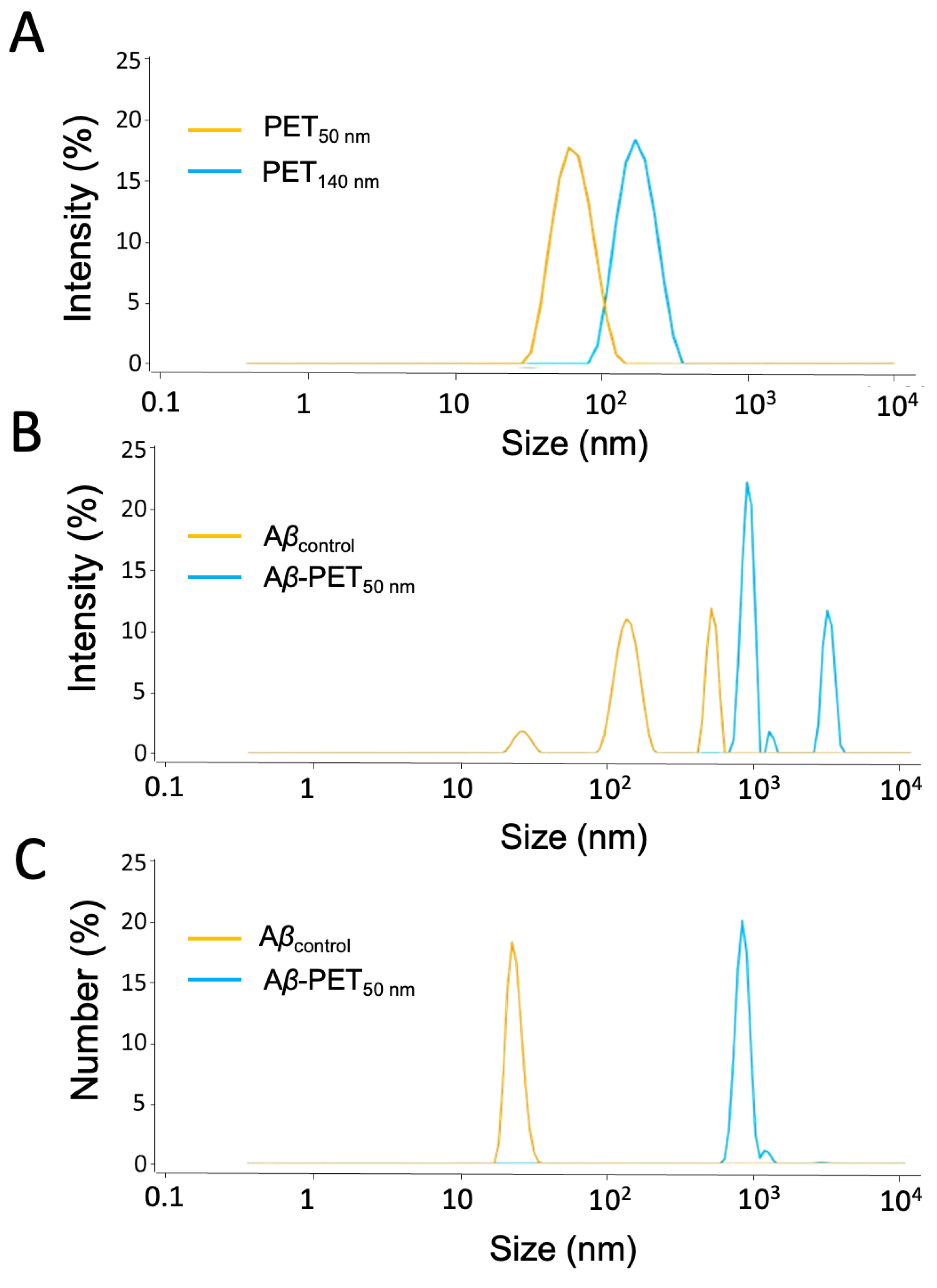
2.1. Characterization of PET NPs
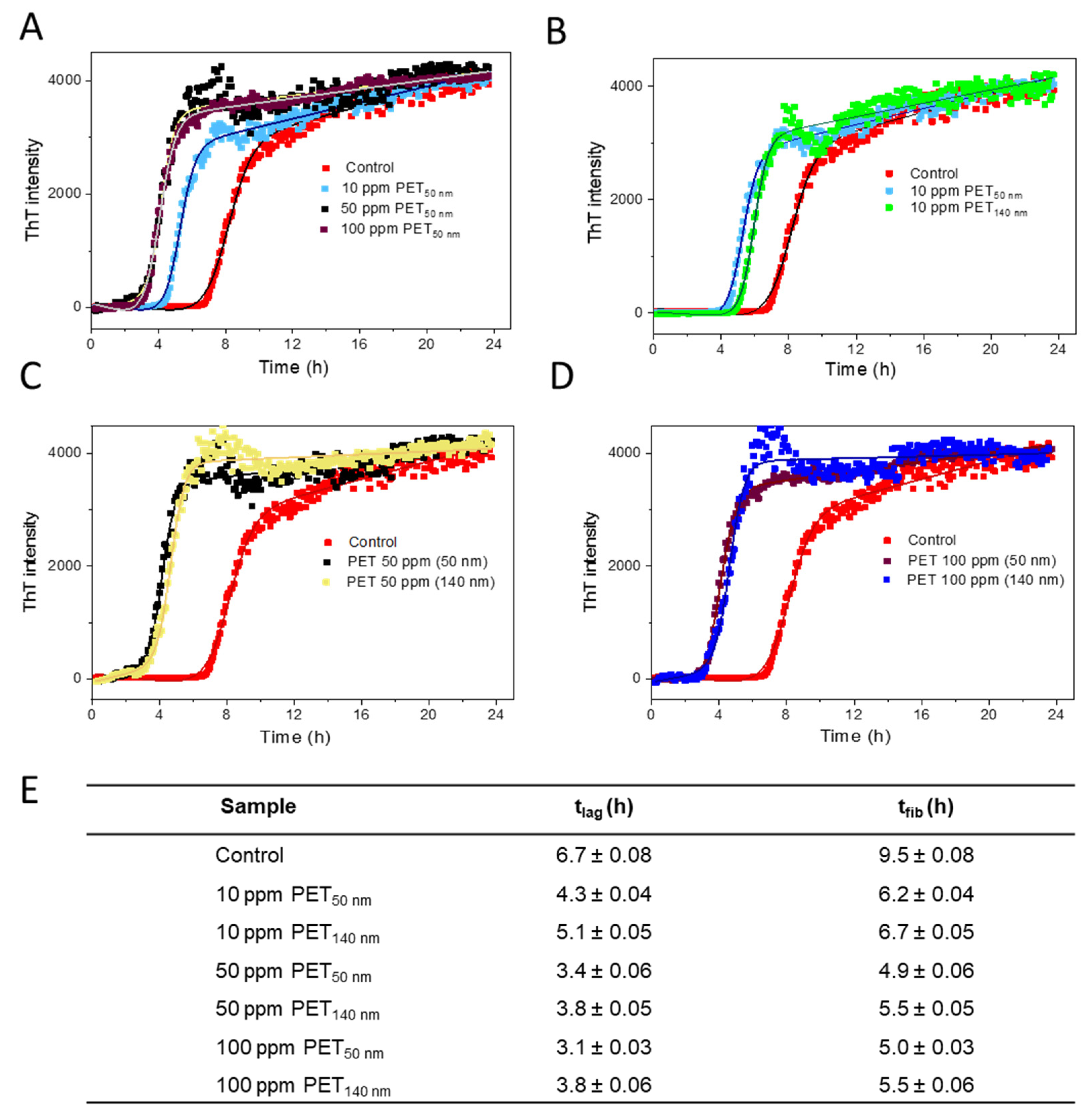
2.2. Aβ Fibrillation Kinetics
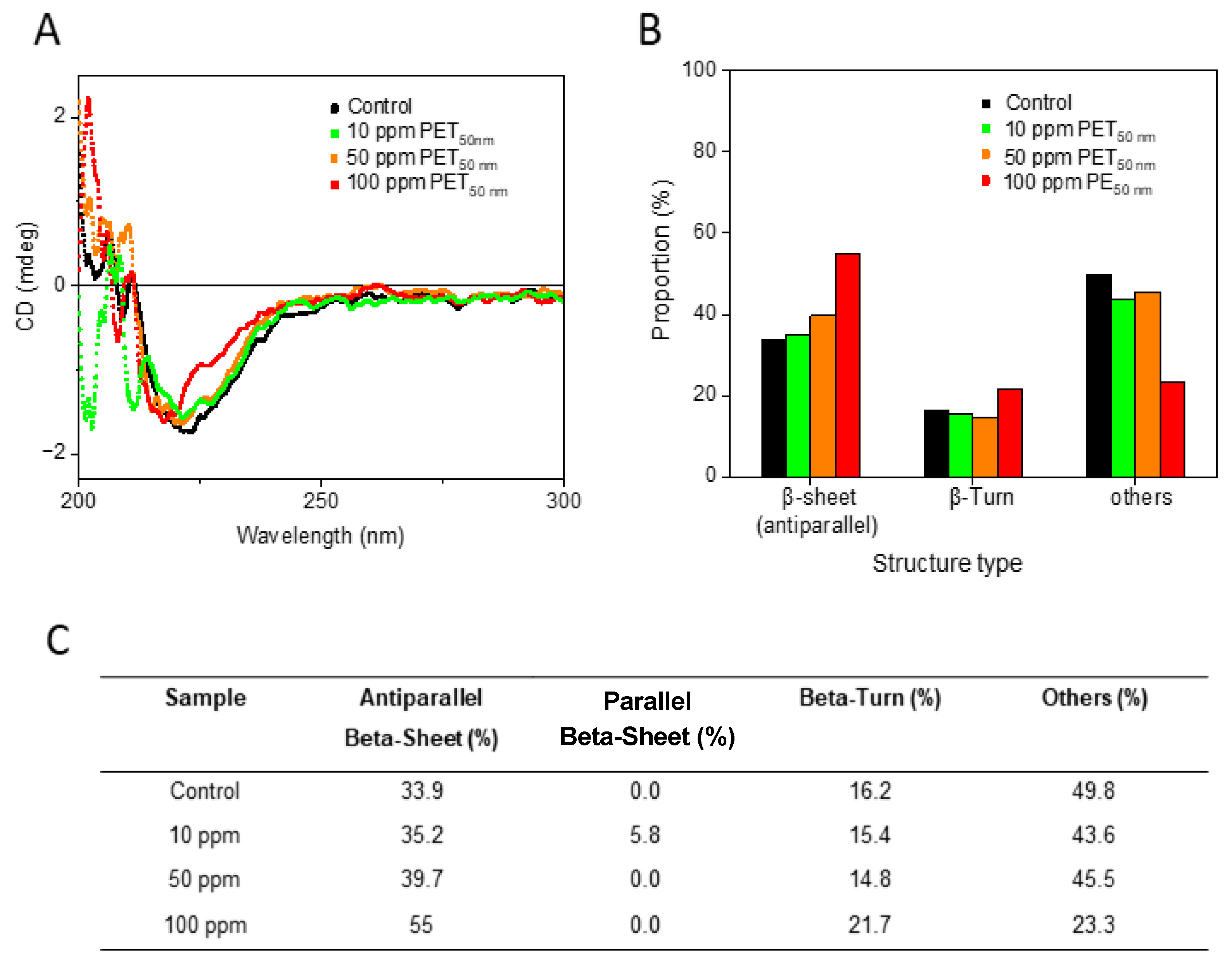
2.3. Secondary Structure Analysis
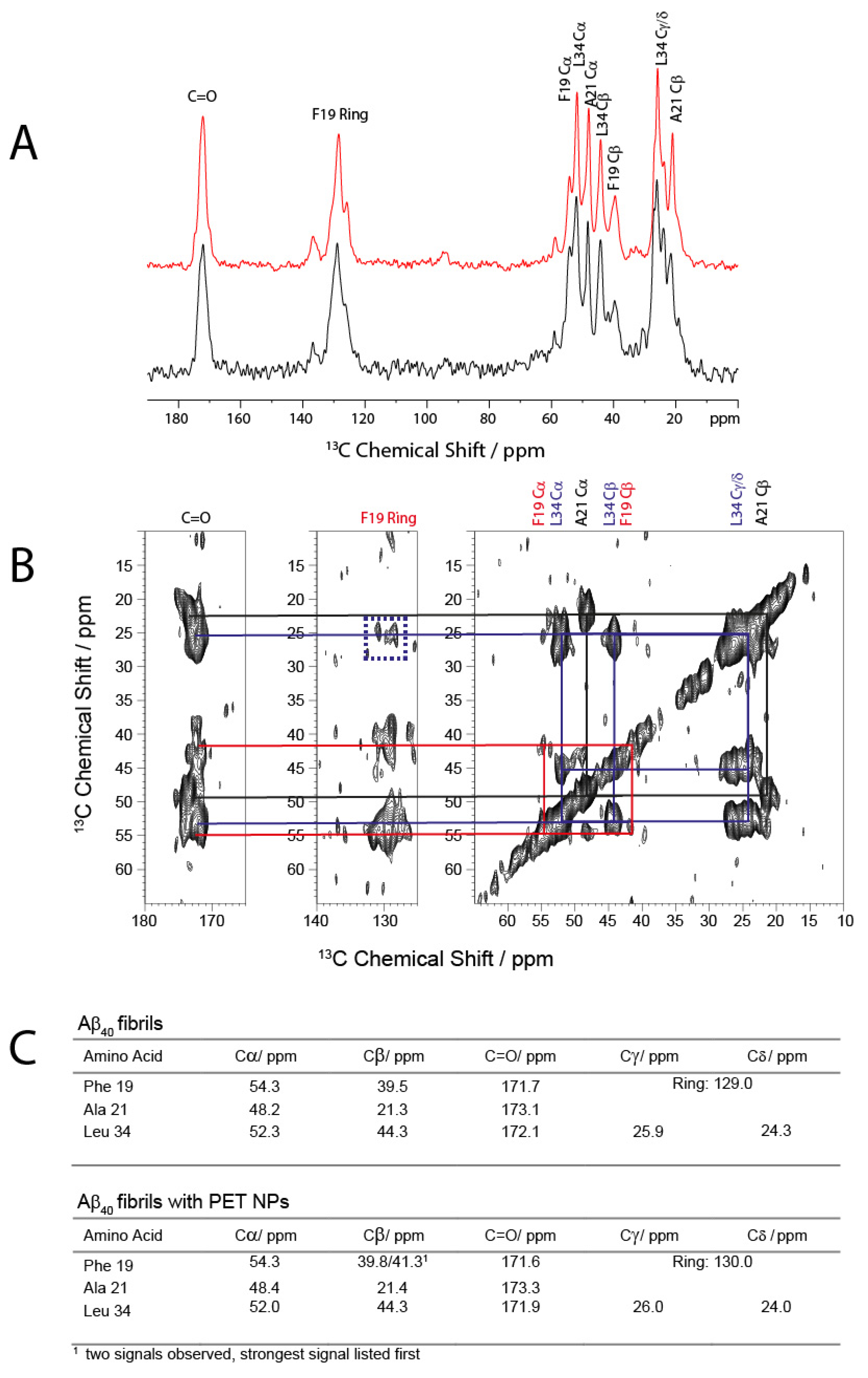
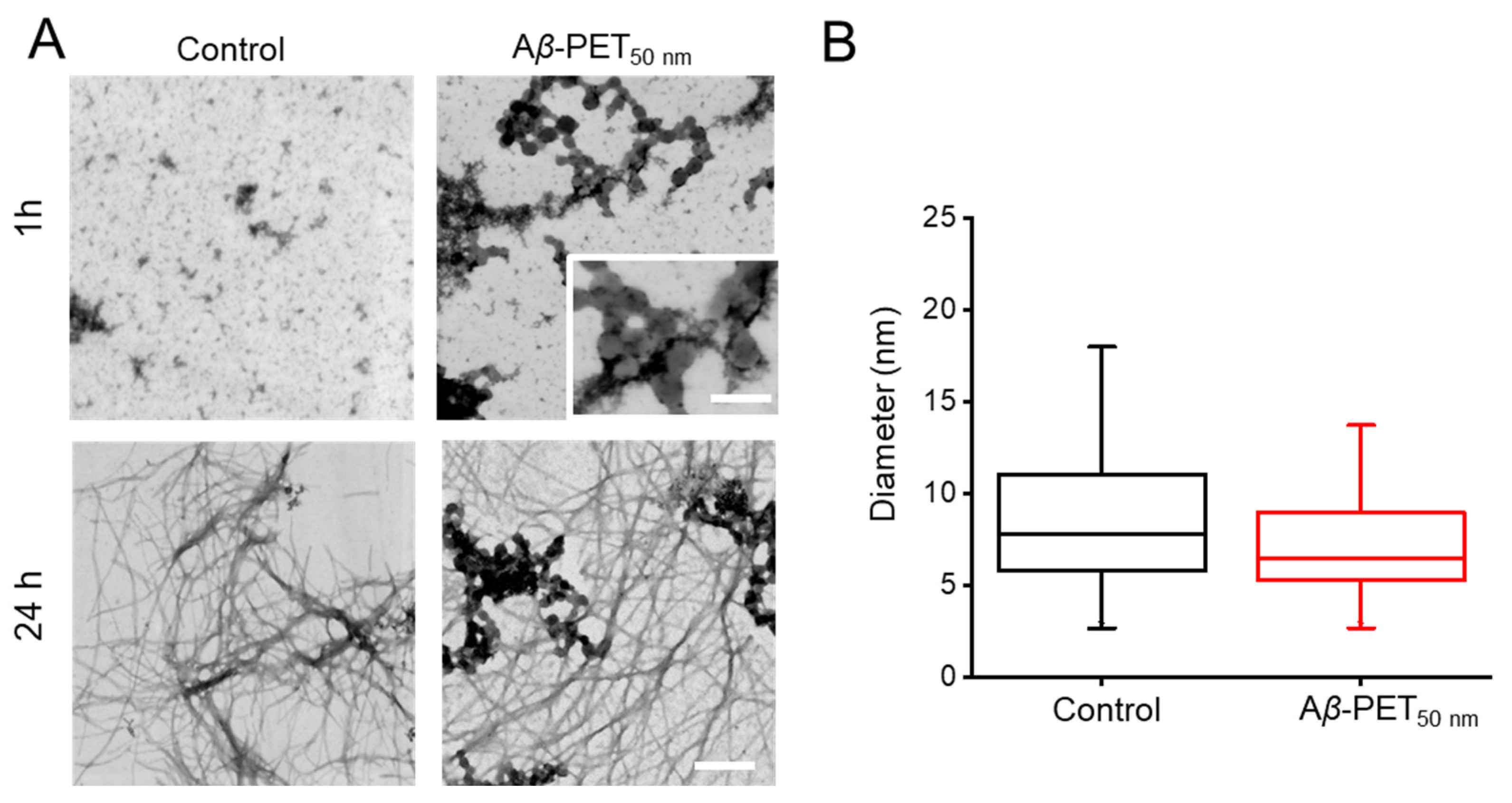
2.4. Morphology of Aβ Fibrils
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of PET NPs
3.3. Peptide Synthesis
3.4. Aβ40 Fibril Preparation
3.5. Dynamic Light Scattering (DLS)
3.6. Transmission Electron Microscopy
3.7. Circular Dichroism (CD) Spectroscopy
3.8. Solid-State NMR-Spectroscopy
3.9. Fluorescence Intensity Data Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- OECD. Policy Scenarios for Eliminating Plastic Pollution by 2040; OECD Publishing: Paris, France, 2024. [Google Scholar] [CrossRef]
- Jambeck, J.; Geyer, R.; Wilcox, C.; Siegler, T.; Perryman, M.; Andrady, A.; Narayan, R.; Law, K. Marine pollution. Plastic waste inputs from land into the ocean. Science 2015, 347, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Xu, X.; Guo, L.; Jin, R.; Lu, Y. Uptake and transport of micro/nanoplastics in terrestrial plants: Detection, mechanisms, and influencing factors. Sci. Total Environ. 2024, 907, 168155. [Google Scholar] [CrossRef] [PubMed]
- Wayman, C.; Niemann, H. The fate of plastic in the ocean environment–a minireview. Environ. Sci. Process. Impacts 2021, 23, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Dimassi, S.N.; Hahladakis, J.N.; Yahia, M.N.D.; Ahmad, M.I.; Sayadi, S.; Al-Ghouti, M.A. Degradation-fragmentation of marine plastic waste and their environmental implications: A critical review. Arab. J. Chem. 2022, 15, 104262. [Google Scholar] [CrossRef]
- Takada, H.; Karapanagioti, H. Hazardous Chemicals Associated with Plastics in the Marine Environment; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Zhang, K.; Hamidian, A.H.; Tubić, A.; Zhang, Y.; Fang, J.K.H.; Wu, C.; Lam, P.K.S. Understanding plastic degradation and microplastic formation in the environment: A review. Environ. Pollut. 2021, 274, 116554. [Google Scholar] [CrossRef]
- Elahi, A.; Bukhari, D.A.; Shamim, S.; Rehman, A. Plastics degradation by microbes: A sustainable approach. J. King Saud. Univ. Sci. 2021, 33, 101538. [Google Scholar] [CrossRef]
- Liu, L.; Xu, M.; Ye, Y.; Zhang, B. On the degradation of (micro)plastics: Degradation methods, influencing factors, environmental impacts. Sci. Total Environ. 2022, 806 Pt 3, 151312. [Google Scholar] [CrossRef]
- Hua, X.; Wang, D. Cellular Uptake, Transport, and Organelle Response After Exposure to Microplastics and Nanoplastics: Current Knowledge and Perspectives for Environmental and Health Risks. Rev. Environ. Contam. Toxicol. 2022, 260, 12. [Google Scholar] [CrossRef]
- Yee, M.S.-L.; Hii, L.-W.; Looi, C.K.; Lim, W.-M.; Wong, S.-F.; Kok, Y.-Y.; Tan, B.-K.; Wong, C.-Y.; Leong, C.-O. Impact of Microplastics and Nanoplastics on Human Health. Nanomaterials 2021, 11, 496. [Google Scholar] [CrossRef]
- Giri, S.; Lamichhane, G.; Khadka, D.; Devkota, H.P. Microplastics contamination in food products: Occurrence, analytical techniques and potential impacts on human health. Curr. Res. Biotechnol. 2024, 7, 100190. [Google Scholar] [CrossRef]
- Pironti, C.; Ricciardi, M.; Motta, O.; Miele, Y.; Proto, A.; Montano, L. Microplastics in the Environment: Intake through the Food Web, Human Exposure and Toxicological Effects. Toxics 2021, 9, 224. [Google Scholar] [CrossRef]
- Wright, S.L.; Kelly, F.J. Plastic and Human Health: A Micro Issue? Environ. Sci. Technol. 2017, 51, 6634–6647. [Google Scholar] [CrossRef]
- Hersh, A.M.; Alomari, S.; Tyler, B.M. Crossing the Blood-Brain Barrier: Advances in Nanoparticle Technology for Drug Delivery in Neuro-Oncology. Int. J. Mol. Sci. 2022, 23, 4153. [Google Scholar] [CrossRef]
- Shan, S.; Zhang, Y.; Zhao, H.; Zeng, T.; Zhao, X. Polystyrene nanoplastics penetrate across the blood-brain barrier and induce activation of microglia in the brain of mice. Chemosphere 2022, 298, 134261. [Google Scholar] [CrossRef] [PubMed]
- Sökmen, T.; Sulukan, E.; Türkoğlu, M.; Baran, A.; Özkaraca, M.; Ceyhun, S.B. Polystyrene nanoplastics (20 nm) are able to bioaccumulate and cause oxidative DNA damages in the brain tissue of zebrafish embryo (Danio rerio). Neurotoxicology 2020, 77, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Prüst, M.; Meijer, J.; Westerink, R.H.S. The plastic brain: Neurotoxicity of micro- and nanoplastics. Part. Fibre Toxicol. 2020, 17, 24. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.B.; Stock, V.; Cara-Carmona, J.; Lisicki, E.; Shopova, S.; Fessard, V.; Braeuning, A.; Sieg, H.; Böhmert, L. Micro- and nanoplastics–current state of knowledge with the focus on oral uptake and toxicity. Nanoscale Adv. 2020, 2, 4350–4367. [Google Scholar] [CrossRef]
- Kopatz, V.; Wen, K.; Kovács, T.; Keimowitz, A.S.; Pichler, V.; Widder, J.; Vethaak, A.D.; Hollóczki, O.; Kenner, L. Micro- and Nanoplastics Breach the Blood-Brain Barrier (BBB): Biomolecular Corona’s Role Revealed. Nanomaterials 2023, 13, 1404. [Google Scholar] [CrossRef]
- Gou, X.; Fu, Y.; Li, J.; Xiang, J.; Yang, M.; Zhang, Y. Impact of nanoplastics on Alzheimer ’s disease: Enhanced amyloid-β peptide aggregation and augmented neurotoxicity. J. Hazard. Mater. 2024, 465, 133518. [Google Scholar] [CrossRef]
- Dhaka, V.; Singh, S.; Anil, A.G.; Sunil Kumar Naik, T.S.; Garg, S.; Samuel, J.; Kumar, M.; Ramamurthy, P.C.; Singh, J. Occurrence, toxicity and remediation of polyethylene terephthalate plastics. A review. Environ. Chem. Lett. 2022, 20, 1777–1800. [Google Scholar] [CrossRef]
- Gwada, B.; Ogendi, G.; Makindi, S.; Trott, S. Composition of plastic waste discarded by households and its management approaches. Glob. J. Environ. Sci. Manag. 2019, 5, 83–94. [Google Scholar] [CrossRef]
- Bashirova, N.; Poppitz, D.; Klüver, N.; Scholz, S.; Matysik, J.; Alia, A. A mechanistic understanding of the effects of polyethylene terephthalate nanoplastics in the zebrafish (Danio rerio) embryo. Sci. Rep. 2023, 13, 1891. [Google Scholar] [CrossRef]
- Cipriani, G.; Dolciotti, C.; Picchi, L.; Bonuccelli, U. Alzheimer and his disease: A brief history. Neurol. Sci. 2011, 32, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Nakatuka, Y.; Yoshida, H.; Fukui, K.; Matuzawa, M. The effect of particle size distribution on effective zeta-potential by use of the sedimentation method. Adv. Powder Technol. 2015, 26, 650–656. [Google Scholar] [CrossRef]
- Swartzwelter, B.J.; Mayall, C.; Alijagic, A.; Barbero, F.; Ferrari, E.; Hernadi, S.; Michelini, S.; Navarro Pacheco, N.I.; Prinelli, A.; Swart, E.; et al. Cross-Species Comparisons of Nanoparticle Interactions with Innate Immune Systems: A Methodological Review. Nanomaterials 2021, 11, 1528. [Google Scholar] [CrossRef]
- Wang, J.; Cong, J.; Wu, J.; Chen, Y.; Fan, H.; Wang, X.; Duan, Z.; Wang, L. Nanoplastic-protein corona interactions and their biological effects: A review of recent advances and trends. Trends Anal. Chem. 2023, 166, 117206. [Google Scholar] [CrossRef]
- John, T.; Adler, J.; Elsner, C.; Petzold, J.; Krueger, M.; Martin, L.L.; Huster, D.; Risselada, H.J.; Abel, B. Mechanistic insights into the size-dependent effects of nanoparticles on inhibiting and accelerating amyloid fibril formation. J. Colloid. Interface Sci. 2022, 622, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Pino, P.D.; Pelaz, B.; Zhang, Q.; Maffre, P.; Nienhaus, G.U.; Parak, W.J. Protein corona formation around nanoparticles–from the past to the future. Mater. Horiz. 2014, 1, 301–313. [Google Scholar] [CrossRef]
- Hollóczki, O.; Gehrke, S. Nanoplastics can change the secondary structure of proteins. Sci. Rep. 2019, 9, 16013. [Google Scholar] [CrossRef]
- Ke, P.C.; Lin, S.; Parak, W.J.; Davis, T.P.; Caruso, F. A Decade of the Protein Corona. ACS Nano 2017, 11, 11773–11776. [Google Scholar] [CrossRef]
- Kopac, T. Protein corona, understanding the nanoparticle–protein interactions and future perspectives: A critical review. Int. J. Biol. Macromol. 2021, 169, 290–301. [Google Scholar] [CrossRef]
- Vianello, F.; Cecconello, A.; Magro, M. Toward the Specificity of Bare Nanomaterial Surfaces for Protein Corona Formation. Int. J. Mol. Sci. 2021, 22, 7625. [Google Scholar] [CrossRef] [PubMed]
- Walkey, C.D.; Chan, W.C.W. Understanding and controlling the interaction of nanomaterials with proteins in a physiological environment. Chem. Soc. Rev. 2012, 41, 2780–2799. [Google Scholar] [CrossRef] [PubMed]
- Linse, S. Mechanism of amyloid protein aggregation and the role of inhibitors. Pure Appl. Chem. 2019, 91, 211–229. [Google Scholar] [CrossRef]
- Tycko, R. Molecular structure of amyloid fibrils: Insights from solid-state NMR. Q. Rev. Biophys. 2006, 39, 1–55. [Google Scholar] [CrossRef]
- Chatani, E.; Yamamoto, N. Recent progress on understanding the mechanisms of amyloid nucleation. Biophys. Rev. 2018, 10, 527–534. [Google Scholar] [CrossRef]
- Lee, C.C.; Nayak, A.; Sethuraman, A.; Belfort, G.; McRae, G.J. A three-stage kinetic model of amyloid fibrillation. Biophys. J. 2007, 92, 3448–3458. [Google Scholar] [CrossRef]
- Zaman, M.; Ahmad, E.; Qadeer, A.; Rabbani, G.; Khan, R.H. Nanoparticles in relation to peptide and protein aggregation. Int. J. Nanomed. 2014, 9, 899–912. [Google Scholar] [CrossRef]
- Linse, S.; Cabaleiro-Lago, C.; Xue, W.-F.; Lynch, I.; Lindman, S.; Thulin, E.; Radford, S.E.; Dawson, K.A. Nucleation of protein fibrillation by nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 8691–8696. [Google Scholar] [CrossRef]
- Windheim, J.; Colombo, L.; Battajni, N.C.; Russo, L.; Cagnotto, A.; Diomede, L.; Bigini, P.; Vismara, E.; Fiumara, F.; Gabbrielli, S.; et al. Micro- and Nanoplastics’ Effects on Protein Folding and Amyloidosis. Int. J. Mol. Sci. 2022, 23, 10329. [Google Scholar] [CrossRef]
- Cabaleiro-Lago, C.; Quinlan-Pluck, F.; Lynch, I.; Dawson, K.A.; Linse, S. Dual effect of amino modified polystyrene nanoparticles on amyloid β protein fibrillation. ACS Chem. Neurosci. 2010, 1, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-f.; Xu, T.-h.; Yan, Y.; Zhou, Y.-r.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D. Chemical shifts as a tool for structure determination. Methods Enzym. 1994, 239, 363–392. [Google Scholar] [CrossRef]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A structural model for Alzheimer’s beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Petkova, A.T.; Tycko, R. Polymorphic fibril formation by residues 10-40 of the Alzheimer’s beta-amyloid peptide. Biophys. J. 2006, 90, 4618–4629. [Google Scholar] [CrossRef]
- Bertini, I.; Gonnelli, L.; Luchinat, C.; Mao, J.; Nesi, A. A new structural model of Aβ40 fibrils. J. Am. Chem. Soc. 2011, 133, 16013–16022. [Google Scholar] [CrossRef]
- Scheidt, H.A.; Morgado, I.; Rothemund, S.; Huster, D. Dynamics of amyloid β fibrils revealed by solid-state NMR. J. Biol. Chem. 2012, 287, 2017–2021. [Google Scholar] [CrossRef] [PubMed]
- Adler, J.; Scheidt, H.A.; Krüger, M.; Thomas, L.; Huster, D. Local interactions influence the fibrillation kinetics, structure and dynamics of Aβ(1–40) but leave the general fibril structure unchanged. Phys. Chem. Chem. Phys. 2014, 16, 7461–7471. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, B.; Huster, D. How Single site mutations can help understanding structure formation of amyloid β1-40. Macromol. Biosci. 2023, 23, e2200489. [Google Scholar] [CrossRef]
- Welzel, K.; Müller, R.-J.; Deckwer, W.-D. Enzymatischer Abbau von Polyester-Nanopartikeln. Chem. Ing. Tech. 2002, 74, 1496–1500. [Google Scholar] [CrossRef]
- Micsonai, A.; Bulyáki, É.; Kardos, J. BeStSel: From Secondary Structure Analysis to Protein Fold Prediction by Circular Dichroism Spectroscopy. In Structural Genomics: General Applications; Chen, Y.W., Yiu, C.-P.B., Eds.; Springer: Berlin/Heidelberg, Germany, 2021; pp. 175–189. [Google Scholar]
- Nielsen, L.; Khurana, R.; Coats, A.; Frokjaer, S.; Brange, J.; Vyas, S.; Uversky, V.N.; Fink, A.L. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 2001, 40, 6036–6046. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bashirova, N.; Schölzel, F.; Hornig, D.; Scheidt, H.A.; Krueger, M.; Salvan, G.; Huster, D.; Matysik, J.; Alia, A. The Effect of Polyethylene Terephthalate Nanoplastics on Amyloid-β Peptide Fibrillation. Molecules 2025, 30, 1432. https://doi.org/10.3390/molecules30071432
Bashirova N, Schölzel F, Hornig D, Scheidt HA, Krueger M, Salvan G, Huster D, Matysik J, Alia A. The Effect of Polyethylene Terephthalate Nanoplastics on Amyloid-β Peptide Fibrillation. Molecules. 2025; 30(7):1432. https://doi.org/10.3390/molecules30071432
Chicago/Turabian StyleBashirova, Narmin, Franziska Schölzel, Dominik Hornig, Holger A. Scheidt, Martin Krueger, Georgeta Salvan, Daniel Huster, Joerg Matysik, and A. Alia. 2025. "The Effect of Polyethylene Terephthalate Nanoplastics on Amyloid-β Peptide Fibrillation" Molecules 30, no. 7: 1432. https://doi.org/10.3390/molecules30071432
APA StyleBashirova, N., Schölzel, F., Hornig, D., Scheidt, H. A., Krueger, M., Salvan, G., Huster, D., Matysik, J., & Alia, A. (2025). The Effect of Polyethylene Terephthalate Nanoplastics on Amyloid-β Peptide Fibrillation. Molecules, 30(7), 1432. https://doi.org/10.3390/molecules30071432