
Mid-Level Data Fusion Techniques of LC-MS and HS-GC-MS for Distinguishing Green and Ripe Forsythiae Fructus


Abstract

1. Introduction
2. Results and Discussion
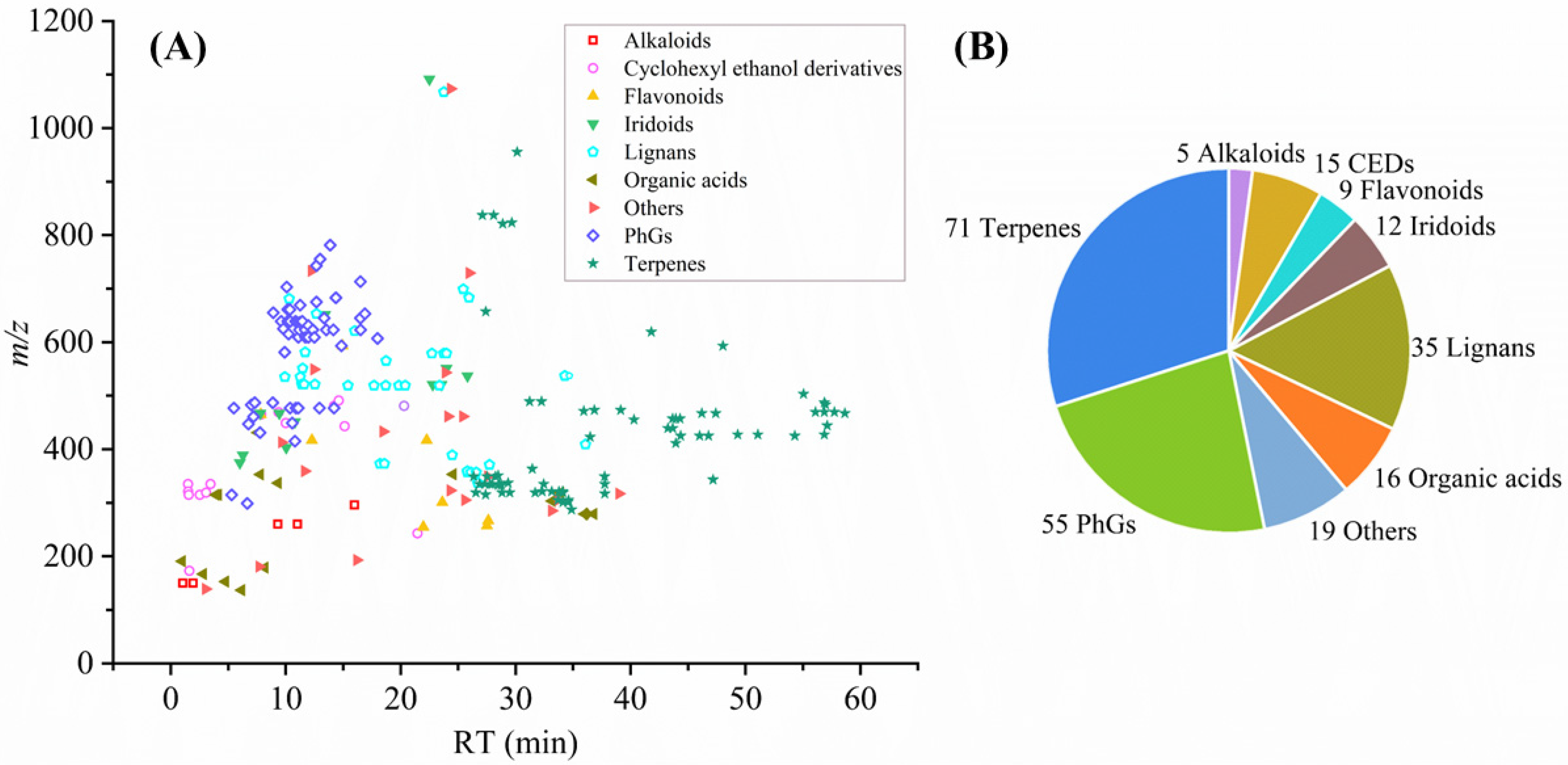
2.1. Characterization of Non-Volatile Metabolites in GFF and RFF
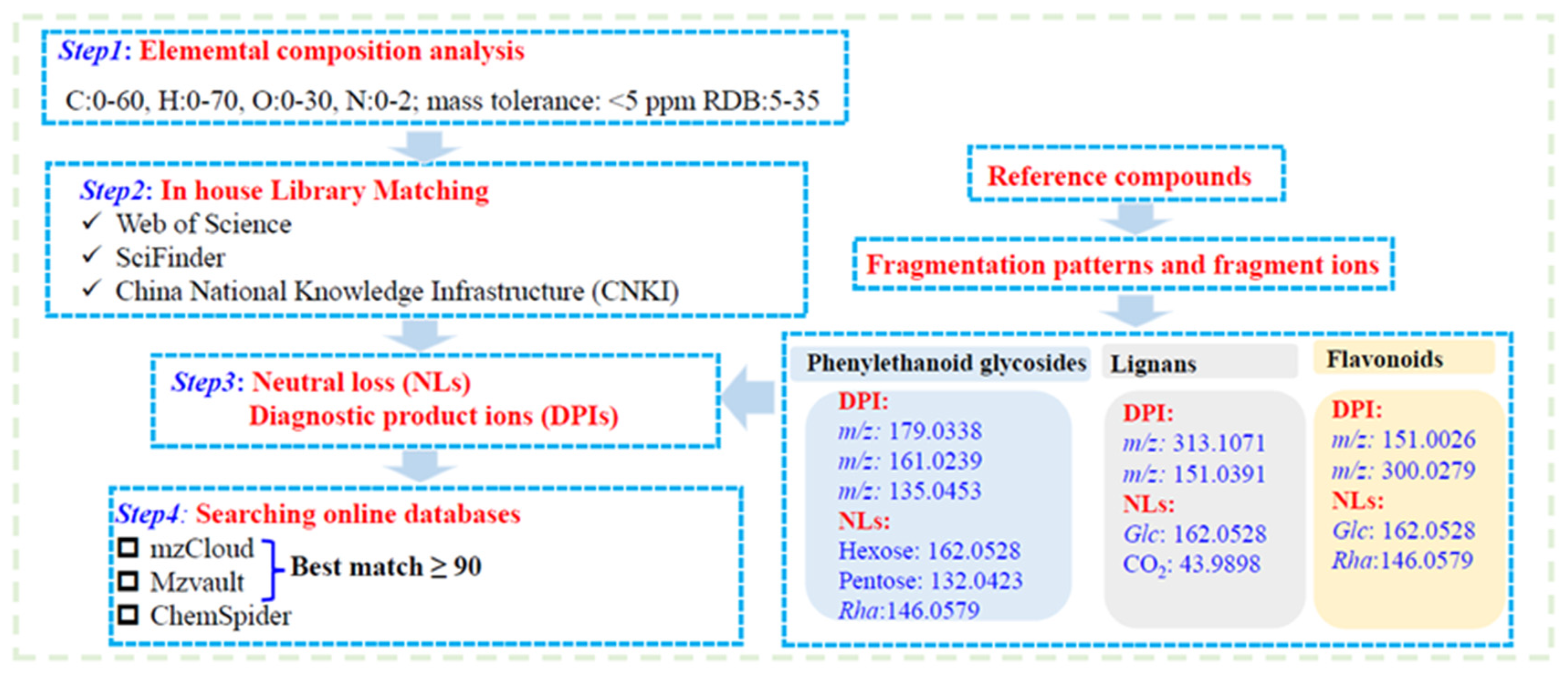
2.1.1. Identification of Phenylethanoid Glycosides
2.1.2. Identification of Lignans
2.1.3. Identification of Cyclohexyl Ethanol Derivatives
2.1.4. Identification of Terpenoid
2.1.5. Identification of Other Compounds
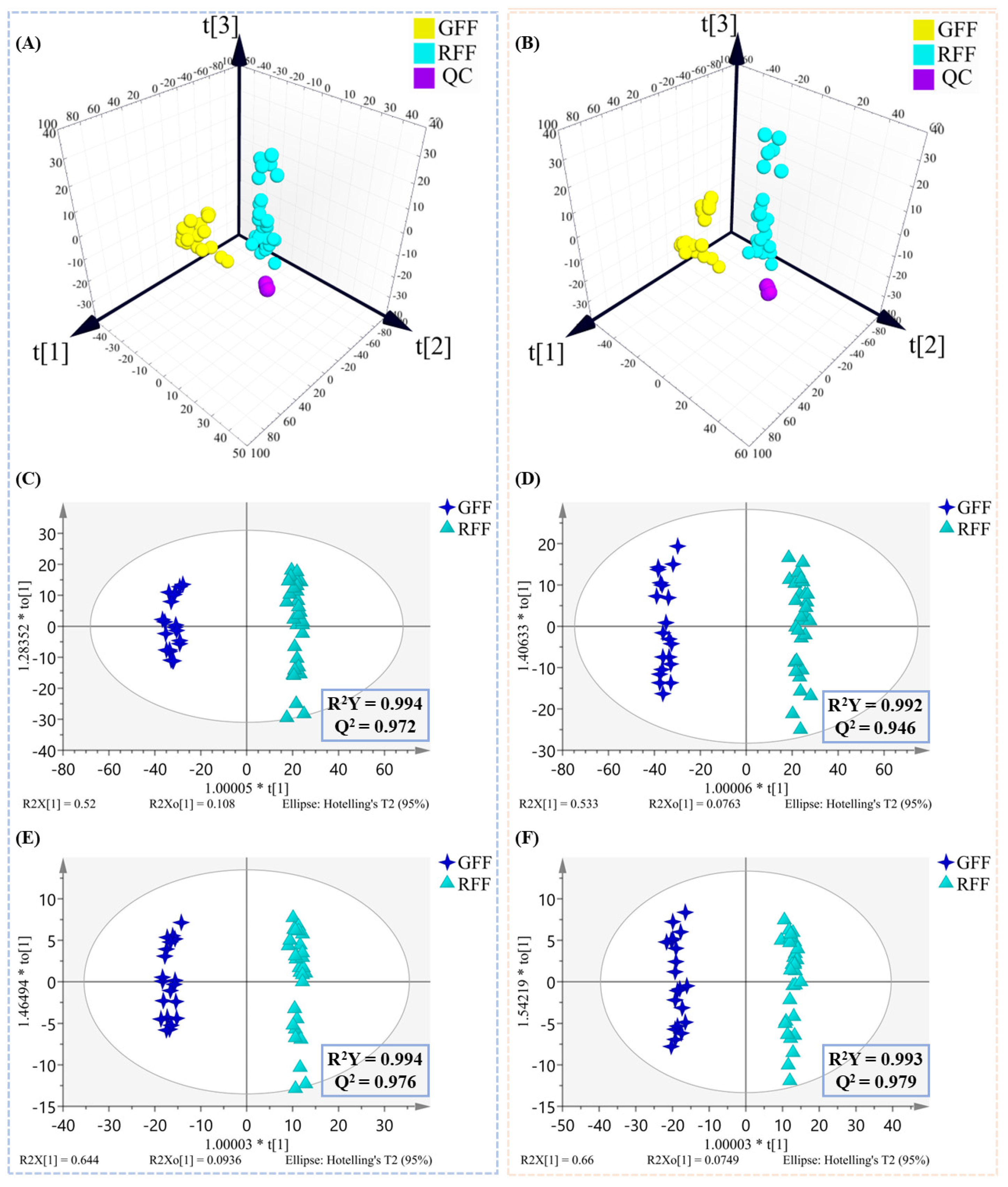
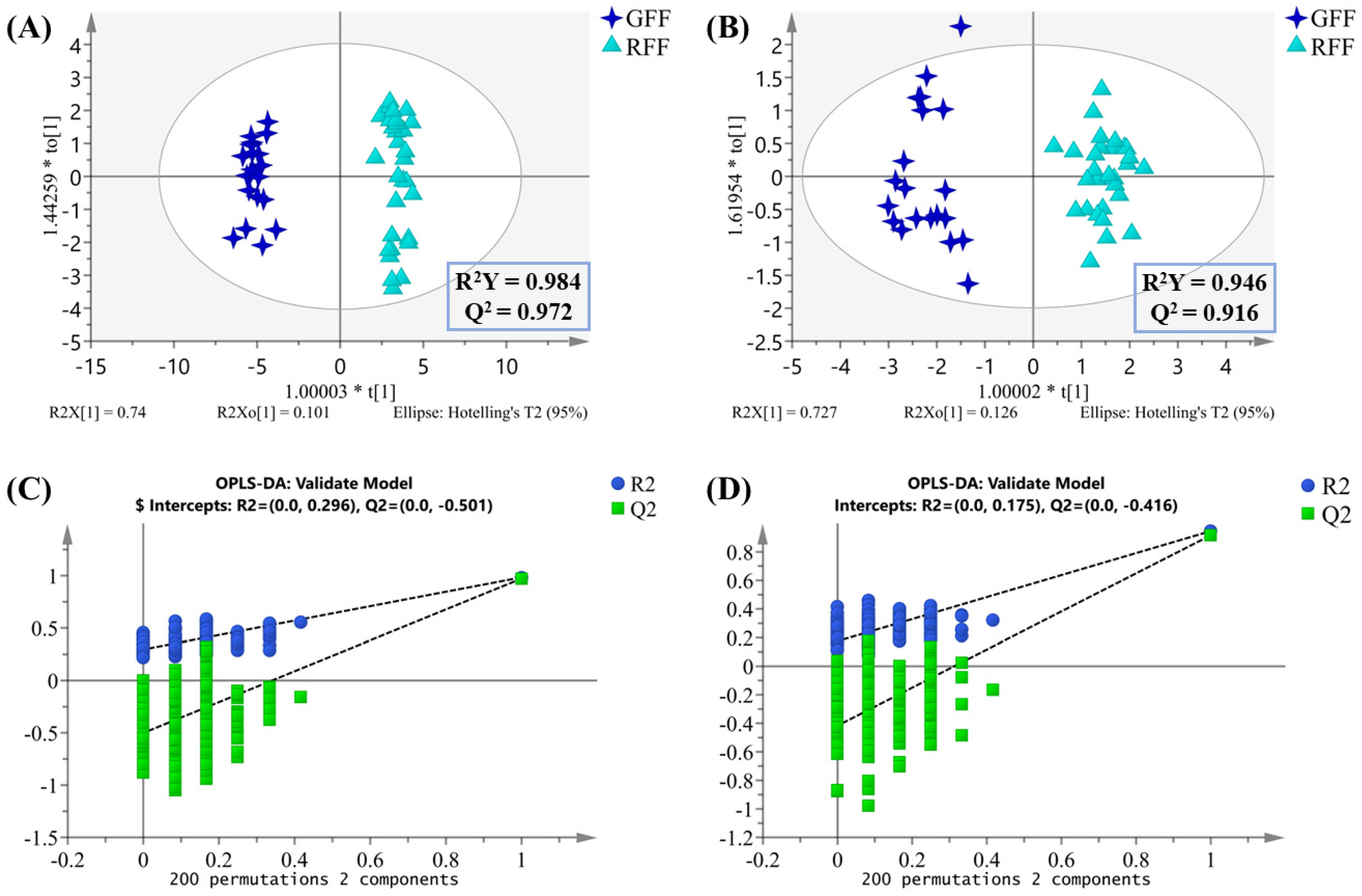
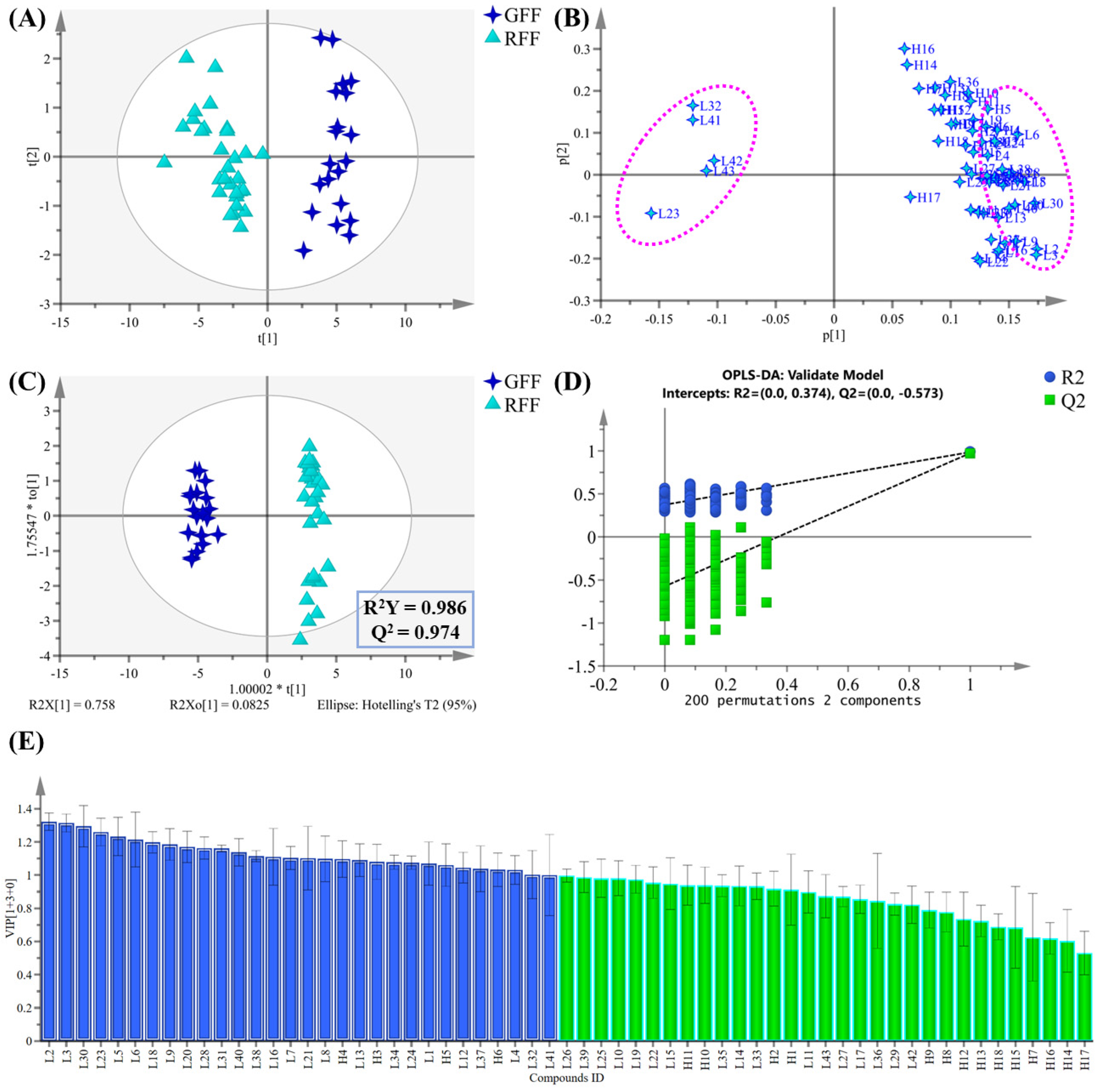
2.2. Multivariate Statistical Analysis of Non-Volatile Compounds
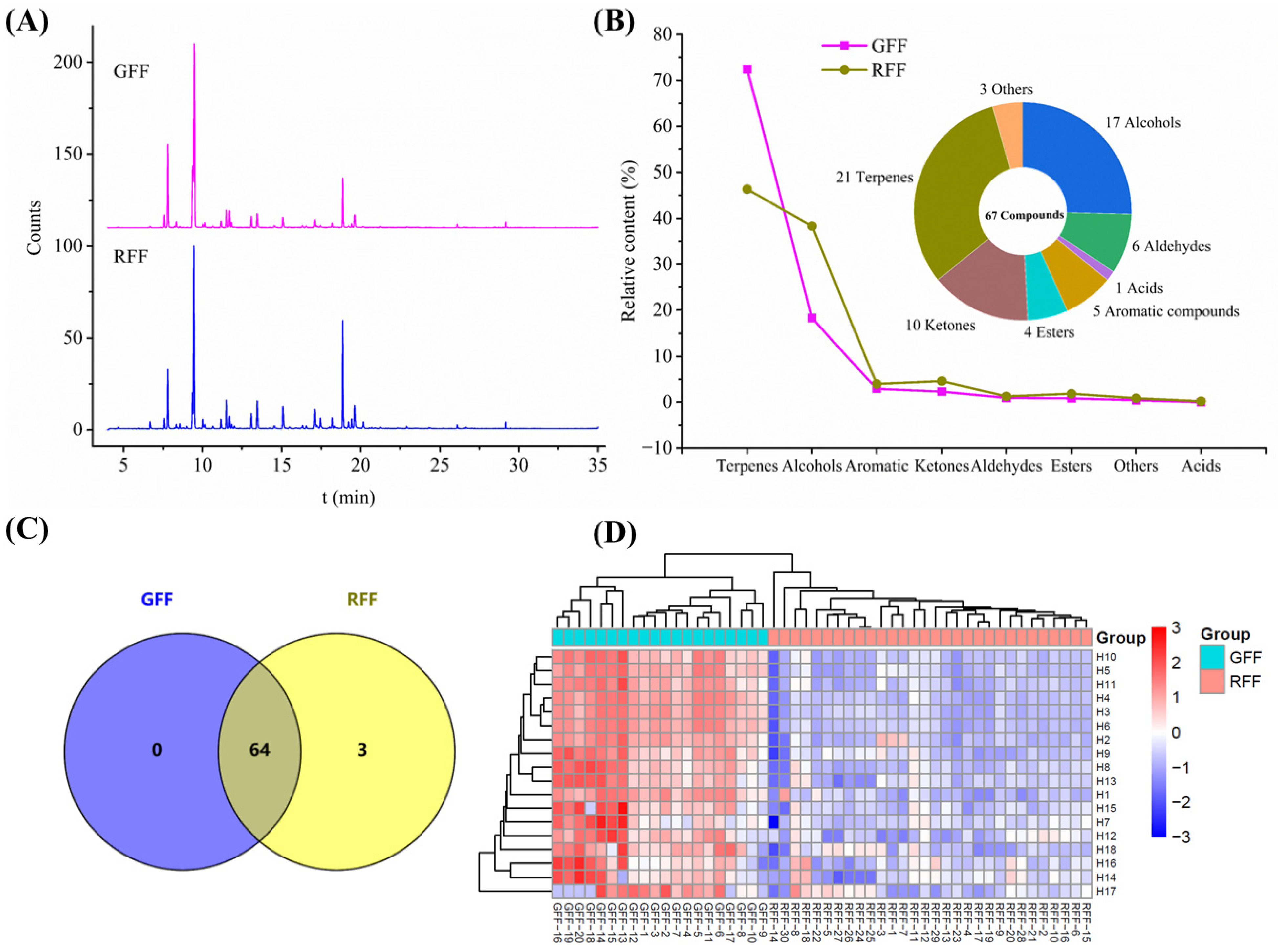
2.3. Characterization of the Volatile Metabolites in GFF and RFF
2.4. Multivariate Statistical Analysis of Volatile Compounds
2.5. Discriminant Ability Assessment of Differential Compounds
2.6. Mid-Level Data Fusion of LC-MS and HS-GC-MS
3. Materials and Methods
3.1. Chemicals and Materials
3.2. UPLC-Q/Orbitrap MS Analysis
3.2.1. Preparation of the Sample Solution
3.2.2. Data Acquisition
3.2.3. Data Pre-Processing
3.3. HS-GC-MS Analysis
3.3.1. Instruments and Conditions
3.3.2. Data Processing
3.4. Chemometric Analysis
3.5. Mid-Level Data Fusion Strategies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ji, Y.-L.; Feng, X.; Chang, Y.-Q.; Zheng, Y.-G.; Hou, F.-J.; Zhang, D.; Guo, L. Chemical Characterization of Different Parts of Forsythia suspensa and α-Glucosidase and Pancreatic Lipase Inhibitors Screening Based on UPLC-QTOF-MS/MS and Plant Metabolomics Analysis. Arab. J. Chem. 2024, 17, 105723. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, F.; Li, Z.; Qin, X.; Zhang, L. Comparison of Fruits of Forsythia suspensa at Two Different Maturation Stages by NMR-Based Metabolomics. Molecules 2015, 20, 10065–10081. [Google Scholar] [CrossRef]
- Dong, Z.; Lu, X.; Tong, X.; Dong, Y.; Tang, L.; Liu, M. Forsythiae Fructus: A Review on Its Phytochemistry, Quality Control, Pharmacology and Pharmacokinetics. Molecules 2017, 22, 1466. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.-Y.; Zhang, F.; Yang, Y.-N.; Feng, Z.-M.; Jiang, J.-S.; Zhang, P.-C. Neuroprotective and Anti-Inflammatory Phenylethanoidglycosides from the Fruits of Forsythia suspensa. Bioorg. Chem. 2021, 113, 105025. [Google Scholar] [CrossRef]
- Li, J.-J.; Chen, Z.-H.; Liu, C.-J.; Kang, Y.-S.; Tu, X.-P.; Liang, H.; Shi, W.; Zhang, F.-X. The Phytochemistry, Pharmacology, Pharmacokinetics, Quality Control, and Toxicity of Forsythiae Fructus: An Updated Systematic Review. Phytochemistry 2024, 222, 114096. [Google Scholar] [CrossRef]
- Pharmacopoeia Commission of the People’s Republic of China. Pharmacopoeia of People’s Republic of China; Chemical Industry Press: Beijing, China, 2020. [Google Scholar]
- Pan, M.; Su, Y.; Liu, X.; Xiang, S.; Ding, Y.; Li, Q. Identification of Forsythia suspensa (Thunb.) Vahl in Different Harvest Periods Using Intelligent Sensory Technologies, HPLC Characteristic Fingerprint Coupled with Chemometrics. Phytochem. Anal. 2022, 33, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Li, J.; Liu, W.; Jiao, X.; He, J.; Liu, J.; Ma, L.; Gao, X.; Chang, Y. Pharmacokinetic of 5 Components after Oral Administration of Fructus Forsythiae by HPLC-MS/MS and the Effects of Harvest Time and Administration Times. J. Chromatogr. B 2015, 993, 36–46. [Google Scholar] [CrossRef]
- Bao, J.; Ding, R.-B.; Liang, Y.; Liu, F.; Wang, K.; Jia, X.; Zhang, C.; Chen, M.; Li, P.; Su, H.; et al. Differences in Chemical Component and Anticancer Activity of Green and Ripe Forsythiae Fructus. Am. J. Chin. Med. 2017, 45, 1513–1536. [Google Scholar] [CrossRef]
- Chen, L.-H.; Zhang, Y.-B.; Yang, X.-W.; Xu, J.; Wang, Z.-J.; Sun, Y.-Z.; Xu, W.; Wang, Y.-P. Application of UPLC-Triple TOF-MS/MS Metabolomics Strategy to Reveal the Dynamic Changes of Triterpenoid Saponins during the Decocting Process of Asian Ginseng and American Ginseng. Food Chem. 2023, 424, 136425. [Google Scholar] [CrossRef]
- Yin, X.-L.; Peng, Z.-X.; Pan, Y.; Lv, Y.; Long, W.; Gu, H.-W.; Fu, H.; She, Y. UHPLC-QTOF-MS-Based Untargeted Metabolomic Authentication of Chinese Red Wines According to Their Grape Varieties. Food Res. Int. 2024, 178, 113923. [Google Scholar] [CrossRef]
- Jin, G.; Zhu, Y.; Cui, C.; Yang, C.; Hu, S.; Cai, H.; Ning, J.; Wei, C.; Li, A.; Hou, R. Tracing the Origin of Taiping Houkui Green Tea Using 1H NMR and HS-SPME-GC-MS Chemical Fingerprints, Data Fusion and Chemometrics. Food Chem. 2023, 425, 136538. [Google Scholar] [CrossRef]
- Yan, X.; Wang, Y.; Yu, X.; Xie, J.; Yu, Q.; Chen, Y. Exploring Authenticity and Potential Markers of Lushan Yunwu Green Tea by Fusing 1H NMR and UPLC-Q/TOF-MS Techniques. Food Control 2024, 163, 110492. [Google Scholar] [CrossRef]
- Rivera-Pérez, A.; Romero-González, R.; Garrido Frenich, A. Untargeted 1H NMR-Based Metabolomics and Multi-Technique Data Fusion: A Promising Combined Approach for Geographical and Processing Authentication of Thyme by Multivariate Statistical Analysis. Food Chem. 2023, 420, 136156. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Li, L.; Liu, Y.; Wang, Z.; Wei, B.; Zhang, Z. Untargeted Metabolomics Analysis Using UPLC-QTOF/MS and GC-MS to Unravel Changes in Antioxidant Activity and Compounds of Almonds before and after Roasting. Food Res. Int. 2024, 194, 114870. [Google Scholar] [CrossRef] [PubMed]
- Pantharos, P.; Sukcharoen, P.; Phadungrakwittaya, R.; Akarasereenont, P.; Booranasubkajorn, S.; Lumlerdkij, N. Utilization of UPLC-PDA and GC–MS/MS Coupled with Metabolomics Analysis to Identify Bioactive Metabolites in Medicinal Turmeric at Different Ages for the Quality Assurance. Phytomedicine 2022, 102, 154157. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.-X.; Tuliebieke, T.; Zhang, T.-T.; Lai, J.-X.; Rao, Y.; Zhang, L.-H.; Yang, W.-Z.; Jiang, M.-M.; Wang, Y.-F.; Tian, X.-X.; et al. A Strategy Integrating GC–MS, UPLC Profiling, and DNA Metabarcoding for Characterization and Discrimination of the Medicinal and Culinary Pieces from Four Curcuma Species. Microchem. J. 2024, 203, 110895. [Google Scholar] [CrossRef]
- Gan, Y. Using HS-GC-MS and Flash GC e-Nose in Combination with Chemometric Analysis and Machine Learning Algorithms to Identify the Varieties, Geographical Origins and Production Modes of Atractylodes lancea. Ind. Crops Prod. 2024, 209, 117955. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, S.-S.; Guo, S.; Hou, Y.; Bai, L.; Ho, C.-T.; Bai, N. A Comprehensive Analysis of the Chemical Composition of Fraxinus Mandshurica Roots Based on HPLC-DAD-MS, GC-MS and UPLC-ESI-Q-TOF-MS. J. Pharm. Biomed. Anal. 2023, 233, 115449. [Google Scholar] [CrossRef]
- Cuevas, F.J.; Pereira-Caro, G.; Moreno-Rojas, J.M.; Muñoz-Redondo, J.M.; Ruiz-Moreno, M.J. Assessment of Premium Organic Orange Juices Authenticity Using HPLC-HR-MS and HS-SPME-GC-MS Combining Data Fusion and Chemometrics. Food Control 2017, 82, 203–211. [Google Scholar] [CrossRef]
- Wu, X.-M.; Zuo, Z.-T.; Zhang, Q.-Z.; Wang, Y.-Z. Classification of Paris Species According to Botanical and Geographical Origins Based on Spectroscopic, Chromatographic, Conventional Chemometric Analysis and Data Fusion Strategy. Microchem. J. 2018, 143, 367–378. [Google Scholar] [CrossRef]
- Ding, R.; Yu, L.; Wang, C.; Zhong, S.; Gu, R. Quality Assessment of Traditional Chinese Medicine Based on Data Fusion Combined with Machine Learning: A Review. Crit. Rev. Anal. Chem. 2024, 54, 2618–2635. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Birse, N.; Quinn, B.; Li, Y.; Jia, W.; McCarron, P.; Wu, D.; Da Silva, G.R.; Vanhaecke, L.; Van Ruth, S.; et al. Data Fusion and Multivariate Analysis for Food Authenticity Analysis. Nat. Commun. 2023, 14, 3309. [Google Scholar] [CrossRef]
- Rubini, M.; Sandak, J.; Robles, E.; Sandak, A.; Charrier, B. Mid-Level Data Fusion Techniques for Gum Rosin Discrimination According to Pinus Species, Tapping Methods, and Geographic Origins. Microchem. J. 2024, 112630. [Google Scholar] [CrossRef]
- Gao, X.; Dong, W.; Ying, Z.; Li, G.; Cheng, Q.; Zhao, Z.; Li, W. Rapid Discriminant Analysis for the Origin of Specialty Yam Based on Multispectral Data Fusion Strategies. Food Chem. 2024, 460, 140737. [Google Scholar] [CrossRef]
- Wang, F.N.; Ma, Z.Q.; Liu, Y.; Guo, Y.Z.; Gu, Z.W. New Phenylethanoid Glycosides from the Fruits of Forsythia suspense (Thunb.) Vahl. Molecules 2009, 14, 1324–1331. [Google Scholar] [CrossRef]
- Wu, L.; Georgiev, M.I.; Cao, H.; Nahar, L.; El-Seedi, H.R.; Sarker, S.D.; Xiao, J.; Lu, B. Therapeutic Potential of Phenylethanoid Glycosides: A Systematic Review. Med. Res. Rev. 2020, 40, 2605–2649. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, X.; Yin, X.; Wang, M.; Zhao, J.; Ren, Y. A Strategy Integrating Parent Ions List-Modified Mass Defect Filtering-Diagnostic Product Ions for Rapid Screening and Systematic Characterization of Flavonoids in Scutellaria Barbata Using Hybrid Quadrupole-Orbitrap High-Resolution Mass Spectrometry. J. Chromatogr. A 2022, 1674, 463149. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, A.-H.; Ye, M.; Yang, M.; Guo, D.-A. Characterization of Phenolic Compounds in the Fruits of Forsythia suspensa by High-Performance Liquid Chromatography Coupled with Electrospray Ionization Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 715–729. [Google Scholar] [CrossRef]
- Xia, Y.; Liang, J.; Guo, X.; Sun, H.; Kuang, H. UPLC-QTOF-MSE-based Diagnostic Product Ion Filtering to Unveil Unstable C6-C2 Glucoside Conjugates in Forsythia suspensa. J. Mass Spectrom. 2017, 52, 848–859. [Google Scholar] [CrossRef]
- Wei, Q.; Zhang, R.; Wang, Q.; Yan, X.J.; Yu, Q.W.; Yan, F.X.; Li, C.; Pei, Y.H. Iridoid, Phenylethanoid and Flavonoid Glycosides from Forsythia suspensa. Nat. Prod. Res. 2020, 34, 1320–1325. [Google Scholar] [CrossRef]
- Li, W.; Sun, L.; Zhao, L.; Yue, X.; Dai, S. New C9-Monoterpenoid Alkaloids Featuring a Rare Skeleton with Anti-Inflammatory and Antiviral Activities from Forsythia suspensa. Chem. Biodivers. 2022, 19, e202100668. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, W.; Dai, S.-J.; Liu, R.-X.; Xie, Z.-P.; Zhang, S.-M.; Yue, X.-D. Alkaloids Bearing Rare Skeletons from Forsythia suspensa with Anti-Inflammatory and Anti-Viral Activities In Vitro. Phytochemistry 2021, 186, 112739. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Mu, Q.; Li, W.; Yao, H.; Li, H.; Huang, H. Esculentic Acid, a Novel and Selective COX-2 Inhibitor with Anti-Inflammatory Effect In Vivo and In Vitro. Eur. J. Pharmacol. 2014, 740, 532–538. [Google Scholar] [CrossRef]
- Wang, S.; Shen, Y.; Qiu, R.; Chen, Z.; Chen, Z.; Chen, W. 18 β-Glycyrrhetinic Acid Exhibits Potent Antitumor Effects against Colorectal Cancer via Inhibition of Cell Proliferation and Migration. Int. J. Oncol. 2017, 51, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Montilla, M.P.; Agil, A.; Navarro, M.C.; Jiménez, M.I.; García-Granados, A.; Parra, A.; Cabo, M.M. Antioxidant Activity of Maslinic Acid, a Triterpene Derivative Obtained from Olea Europaea. Planta Med. 2003, 69, 472–474. [Google Scholar] [CrossRef]
- Jing, F.; Wang, L.; Yang, M.; Wu, C.; Li, J.; Shi, L.; Feng, S.; Li, F. Visualizing the Spatial Distribution of Functional Metabolites in Forsythia suspensa at Different Harvest Stages by MALDI Mass Spectrometry Imaging. Fitoterapia 2022, 162, 105285. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Fu, Y.-J.; Zu, Y.-G.; Luo, M.; Wang, W.; Zhang, L.; Li, J. Enzyme-Assisted Microwave Hydro-Distillation Essential Oil from Fructus Forsythia, Chemical Constituents, and Its Antimicrobial and Antioxidant Activities. Food Chem. 2012, 134, 235–243. [Google Scholar] [CrossRef]
- Jiao, J.; Gai, Q.-Y.; Fu, Y.-J.; Zu, Y.-G.; Luo, M.; Zhao, C.-J.; Li, C.-Y. Microwave-Assisted Ionic Liquids Treatment Followed by Hydro-Distillation for the Efficient Isolation of Essential Oil from Fructus Forsythiae Seed. Sep. Purif. Technol. 2013, 107, 228–237. [Google Scholar] [CrossRef]
- Jiao, J.; Ma, D.-H.; Gai, Q.-Y.; Wang, W.; Luo, M.; Fu, Y.-J.; Ma, W. Rapid Analysis of Fructus Forsythiae Essential Oil by Ionic Liquids-Assisted Microwave Distillation Coupled with Headspace Single-Drop Microextraction Followed by Gas Chromatography-Mass Spectrometry. Anal. Chim. Acta 2013, 804, 143–150. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, X.; Deng, Y.; Zhang, R.; Fu, K.; Huang, J.; Huang, Q.; Zeng, C.; Liu, D.; Wang, W. Variations in Volatile Components and Biological Activities of Essential Oils from Citrus Aurantium ‘Changshanhuyou’ at Different Growth and Ripening Stages. Food Res. Int. 2024, 197, 115303. [Google Scholar] [CrossRef]
- Ray, A.; Mohanty, S.; Jena, S.; Sahoo, A.; Acharya, L.; Panda, P.C.; Sial, P.; Duraisamy, P.; Nayak, S. Drying Methods Affects Physicochemical Characteristics, Essential Oil Yield and Volatile Composition of Turmeric (Curcuma Longa L.). J. Appl. Res. Med. Aromat. Plants 2022, 26, 100357. [Google Scholar] [CrossRef]
- Famiani, F.; Farinelli, D.; Urbani, S.; Al Hariri, R.; Paoletti, A.; Rosati, A.; Esposto, S.; Selvaggini, R.; Taticchi, A.; Servili, M. Harvesting System and Fruit Storage Affect Basic Quality Parameters and Phenolic and Volatile Compounds of Oils from Intensive and Super-Intensive Olive Orchards. Sci. Hortic. 2020, 263, 109045. [Google Scholar] [CrossRef]
- Ricci, A.; Levante, A.; Cirlini, M.; Calani, L.; Bernini, V.; Del Rio, D.; Galaverna, G.; Neviani, E.; Lazzi, C. The Influence of Viable Cells and Cell-Free Extracts of Lactobacillus casei on Volatile Compounds and Polyphenolic Profile of Elderberry Juice. Front. Microbiol. 2018, 9, 2784. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, Y.; Ren, Y.; Xu, Y.; An, H.; Zhao, Q.; Wang, Y.; Li, H. Widely Metabolomic Combined with Transcriptome Analysis to Build a Bioactive Compound Regulatory Network for the Fruit Growth Cycle in Pseudocydonia sinensis. Food Chem. 2024, 456, 139933. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jiang, J.; Liang, Y.; Li, S.; Xia, Y.; Zhang, L.; Wang, X. Expansion and Functional Divergence of Terpene Synthase Genes in Angiosperms: A Driving Force of Terpene Diversity. Hortic. Res. 2025, 12, uhae272. [Google Scholar] [CrossRef]
- Xie, J.; Cao, Q.; Wang, W.; Zhang, H.; Deng, B. Understanding Changes in Volatile Compounds and Fatty Acids of Jincheng Orange Peel Oil at Different Growth Stages Using GC–MS. J. Integr. Agric. 2023, 22, 2282–2294. [Google Scholar] [CrossRef]
- Pei, Y.; Xue, Q.; Shu, P.; Xu, W.; Du, X.; Wu, M.; Liu, K.; Pirrello, J.; Bouzayen, M.; Hong, Y.; et al. Bifunctional Transcription Factors SlERF.H5 and H7 Activate Cell Wall and Repress Gibberellin Biosynthesis Genes in Tomato via a Conserved Motif. Dev. Cell 2024, 59, 1345–1359.e6. [Google Scholar] [CrossRef]
- Rivera-Pérez, A.; Romero-González, R.; Garrido Frenich, A. Application of an Innovative Metabolomics Approach to Discriminate Geographical Origin and Processing of Black Pepper by Untargeted UHPLC-Q-Orbitrap-HRMS Analysis and Mid-Level Data Fusion. Food Res. Int. 2021, 150, 110722. [Google Scholar] [CrossRef]
- Shan, M.-Q.; Qian, Y.; Yu, S.; Guo, S.-C.; Zhang, L.; Ding, A.-W.; Wu, Q.-N. Anti-Inflammatory Effect of Volatile Oil from Schizonepeta tenuifolia on Carrageenin-Induced Pleurisy in Rats and Its Application to Study of Appropriate Harvesting Time Coupled with Multi-Attribute Comprehensive Index Method. J. Ethnopharmacol. 2016, 194, 580–586. [Google Scholar] [CrossRef]
- Sieniawska, E.; Los, R.; Baj, T.; Malm, A.; Glowniak, K. Antimicrobial Efficacy of Mutellina purpurea Essential Oil and α-Pinene against Staphylococcus epidermidis Grown in Planktonic and Biofilm Cultures. Ind. Crops Prod. 2013, 51, 152–157. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound Name | VIP Score | RT (min) | Adduct | Formula | m/z | Error (ppm) | MS/MS |
|---|---|---|---|---|---|---|---|---|
| L1 | Quinic acid | 1.24 | 0.98 | [M−H]− | C7H12O6 | 191.0560 | −0.74 | 173.0463, 127.0405 |
| L2 | Hydroxytyrosol 1-O-glucoside | 1.52 | 1.56 | [M−H]− | C14H20O8 | 315.1085 | −0.41 | 153.0559, 135.0457, 89.0246 |
| L3 | Cornoside | 1.52 | 2.52 | [M−H]− | C14H20O8 | 315.1084 | −1.85 | 153.0571, 135.0455, 119.0350, 113.0244 |
| L4 | Rengynic acid-1-O-β-D-glucopyranoside | 1.24 | 3.46 | [M−H]− | C14H24O9 | 335.1346 | −0.34 | 179.0579, 161.0463, 131.0347, 119.0352 |
| L5 | Vanilloloside | 1.4 | 3.92 | [M−H]− | C14H20O8 | 315.1085 | −0.28 | 153.0554, 135.0459, 119.0353, 101.0250 |
| L6 | Forsythoside D | 1.43 | 5.49 | [M−H]− | C20H30O13 | 477.1605 | −1.71 | 449.4005, 315.1103, 179.0560, 161.0463, 135.0455 |
| L7 | Adoxosidic acid | 1.28 | 6.01 | [M−H]− | C16H24O10 | 375.1284 | −3.20 | 337.1743, 213.0771, 169.0872, 151.0768, 125.0611 |
| L8 | Forsythide | 1.23 | 6.28 | [M−H]− | C16H22O11 | 389.1077 | −3.05 | 345.1214, 183.0665, 165.0560, 121.0662, 119.0355 |
| L9 | Salidroside | 1.35 | 6.66 | [M−H]− | C14H20O7 | 299.1135 | −2.30 | 119.0350, 101.0248 |
| L10 | Rebouoside B | 1.11 | 6.76 | [M−H]− | C19H28O12 | 447.1499 | −2.07 | 315.1107, 191.0566, 149.0456, 135.0450, 131.0353 |
| L11 | Forsythoside E | 1.02 | 7.19 | [M−H]− | C20H30O12 | 461.1655 | −2.05 | 315.1081, 163.0613, 153.0561, 135.0454, 131.0353 |
| L12 | Darendoside A | 1.26 | 7.76 | [M−H]− | C19H28O11 | 431.1550 | −1.99 | 299.1140, 191.0568, 149.0460, 137.0603, 131.0353 |
| L13 | Forsythenside B | 1.24 | 7.86 | [M−H]− | C22H26O11 | 465.1396 | −1.15 | 161.0456, 153.0552, 149.0247, 135.0454 |
| L14 | 4-O-p-Coumaroylquinic acid | 1.1 | 9.33 | [M−H]− | C16H18O8 | 337.0929 | −0.05 | 191.0565, 173.0457, 163.0402, 145.0300 |
| L15 | Rengyoside D | 1.13 | 9.39 | [M−H]− | C22H30O11 | 469.1709 | −1.42 | 193.0505, 161.0617, 151.0403, 133.0296, 123.0459 |
| L16 | Hastatoside | 1.19 | 10.05 | [M−H]− | C17H24O11 | 403.1236 | −2.53 | 371.0999, 327.1099, 191.0359, 179.0715, 137.0610 |
| L17 | Calceolarioside A | 1.02 | 10.89 | [M−H]− | C23H26O11 | 477.1395 | −1.61 | 179.0354, 161.0250, 135.0456, 133.0298 |
| L18 | (+)-8-Hydroxypinoresinol 4-O-β-D-glucopyranoside | 1.2 | 11.27 | [M−H]− | C26H32O11 | 535.1816 | −0.66 | 373.1295, 343.1180, 313.1063, 179.0718, 163.0406, 151.0397, 109.0295 |
| L19 | Forsythiayanoside B | 1.44 | 11.47 | [M−H]− | C27H36O12 | 551.2130 | −0.64 | 359.1494, 191.0721, 163.0404 |
| L20 | Forsythialanside E | 1.19 | 11.69 | [M−H]− | C26H32O11 | 581.1874 | −0.31 | 373.1261, 343.1176, 163.0407 |
| L21 | Calceolarioside C | 1.28 | 11.97 | [M−H]− | C28H34O15 | 609.1819 | −1.01 | 447.1505, 315.1081, 179.0351, 161.0245, 153.0568 |
| L22 | Isoforsythiaside | 1.1 | 12.36 | [M−H]− | C29H36O15 | 623.1979 | −0.39 | 461.1692, 443.1567, 179.0353, 161.0246, 135.0454 |
| L23 | S-Suspensaside methyl ether | 1.54 | 12.67 | [M−H]− | C30H38O16 | 653.2085 | −0.33 | 621.1834, 179.0354 161.0246, 151.0404, 135.0454 |
| L24 | Plantainoside A | 1.3 | 12.93 | [M−H]− | C23H26O11 | 477.1395 | −1.44 | 315.1104, 179.0346, 161.0247, 135.0455, 133.0298 |
| L25 | Adoxosidic acid-6′-oleuroperic ester | 1.17 | 13.41 | [M−H]− | C30H36O16 | 651.1928 | −0.39 | 427.1253, 179.0351, 163.0611, 161.0246, 135.0453 |
| L26 | Forsythoside A | 1.19 | 13.54 | [M−H]− | C29H36O15 | 623.1978 | −0.50 | 461.1658, 315.1178, 179.0352, 161.0245, 135.0453 |
| L27 | Acteoside | 1.05 | 14.15 | [M−H]− | C29H36O15 | 623.1978 | −0.50 | 461.1663, 315.1077, 179.0348, 161.0245, 153.0556 |
| L28 | Calceolarioside B | 1.35 | 14.21 | [M−H]− | C23H26O11 | 477.1395 | −1.47 | 315.1085, 179.0352, 161.0246, 135.0452, 133.0297 |
| L29 | Kaempferol 3-O-rutinoside | 1.02 | 14.95 | [M−H]− | C27H30O15 | 593.1511 | −0.12 | 447.1546, 285.0403, 163.0035, 151.0037, 135.0085 |
| L30 | Forsythenside L | 1.57 | 15.12 | [M−H]− | C20H28O11 | 443.1551 | −1.86 | 427.8166, 149.0611 |
| L31 | (+)-Pinoresinol 4′-O-β-D-glucopyranoside | 1.39 | 15.42 | [M−H]− | C26H32O11 | 519.1867 | −0.98 | 357.1334, 342.1089, 311.1313, 163.0404, 161.0261, 151.0402, 136.0166 |
| L32 | Forsyshiyanine B | 1.09 | 15.98 | [M+H]+ | C18H17NO3 | 296.1281 | −0.10 | 132.0807, 117.0573 |
| L33 | Isoacteoside | 1.09 | 16.51 | [M−H]− | C29H36O15 | 623.1978 | −0.48 | 461.1663, 179.0351, 161.0245, 153.0553, 135.0453 |
| L34 | Fraxiresinol-4′-O-β-D-glucopyranoside | 1.26 | 18.73 | [M−H]− | C27H34O13 | 565.1925 | −0.37 | 357.1336, 327.1258, 151.0403, 137.0243, 135.0451 |
| L35 | Matairesinoside | 1.06 | 20.40 | [M−H]− | C26H32O11 | 519.1867 | −0.91 | 357.1335, 342.1098, 137.0614, 122.0375 |
| L36 | Forsydoitriside A | 1.08 | 22.50 | [M−H]− | C48H68O28 | 1091.3822 | −0.24 | 929.3305, 883.8681, 733.2557, 715.2502, 571.2033, 553.1916, 445.1343, 375.1285, 357.1173, 151.0765 |
| L37 | Suspenoidside A | 1.19 | 22.79 | [M−H]− | C25H30O12 | 521.1660 | −0.82 | 315.1242, 163.0400, 149.0121, 145.0293, 119.0502 |
| L38 | Acanthoside B | 1.33 | 23.76 | [M−H]− | C28H36O13 | 579.2081 | −0.41 | 371.1505, 356.1262, 121.0295 |
| L39 | Suspenoidside E | 1.24 | 23.99 | [M−H]− | C26H32O13 | 551.1767 | −0.62 | 345.1339, 327.1238, 193.0508, 179.0366, 149.0614 |
| L40 | Suspenoidsides D | 1.35 | 25.81 | [M−H]− | C25H30O13 | 537.1610 | −0.68 | 243.0665, 137.0250, 135.0089, 109.0298, 91.0191 |
| L41 | Esculentic acid | 1.19 | 32.25 | [M+H]+ | C30H48O5 | 489.3574 | −0.17 | 435.3245, 407.3309, 219.1744, 201.1638, 191.1795 |
| L42 | 18-β-Glycyrrhetinic acid | 1.04 | 35.94 | [M+H]+ | C30H46O4 | 471.3467 | −0.33 | 425.3417, 407.3317, 317.2097, 191.1799, 189.1639 |
| L43 | Maslinic acid | 1.09 | 39.15 | [M+H]+ | C30H48O4 | 473.3626 | 0.10 | 437.3422, 427.3572, 357.2803, 315.2331, 189.1631 |
| No. | RT a (min) | Compounds Name | Formula | CAS Number | RIcal b | RIref c | Class | Relative Content d (%) | |
|---|---|---|---|---|---|---|---|---|---|
| (Mean ± SD) | |||||||||
| GFF | RFF | ||||||||
| 1 | 4.66 | Furfural | C5H4O2 | 98-01-1 | 835 | 833 | Aldehydes | 0.25 ± 0.09 | 0.25 ± 0.11 |
| 2 | 5.68 | 1-Hexanol | C6H14O | 111-27-3 | 872 | 868 | Alcohols | - | 0.07 ± 0.05 |
| 3 | 6.66 | 3-Methylcyclopentyl acetate | C8H14O2 | 24070-70-0 | 901 | 905 | Esters | 0.27 ± 0.05 | 0.74 ± 0.17 |
| 4 | 7.35 | Cyclene | C10H16 | 508-32-7 | 922 | 925 | Terpenes | 0.05 ± 0.01 | 0.04 ± 0.02 |
| 5 | 7.56 | α-thujene | C10H16 | 2867-05-2 | 929 | 929 | Terpenes | 1.92 ± 0.22 | 1.63 ± 0.74 |
| 6 | 7.79 | α-Pinene | C10H16 | 80-56-8 | 935 | 937 | Terpenes | 13.09 ± 1.12 | 6.83 ± 1.21 |
| 7 | 8.34 | Camphene | C10H16 | 79-92-5 | 950 | 952 | Terpenes | 0.92 ± 0.11 | 0.46 ± 0.07 |
| 8 | 8.42 | 4-Methylenecyclohexanone | C7H10O | 29648-66-6 | 952 | \ | Ketones | - | 0.11 ± 0.04 |
| 9 | 8.57 | 2,4-Thujadiene | C10H14 | 36262-09-6 | 956 | 956 | Others | 0.24 ± 0.06 | 0.45 ± 0.16 |
| 10 | 8.74 | (2E)-2-Heptenal | C7H12O | 18829-55-5 | 960 | 958 | Aldehydes | 0.04 ± 0.01 | 0.09 ± 0.04 |
| 11 | 8.83 | Benzaldehyde | C7H6O | 100-52-7 | 962 | 962 | Aromatic | 0.06 ± 0.01 | 0.12 ± 0.02 |
| 12 | 9.37 | Sabinene | C10H16 | 3387-41-5 | 975 | 974 | Terpenes | 8.98 ± 1.04 | 4.88 ± 1.18 |
| 13 | 9.46 | β-Pinene | C10H16 | 127-91-3 | 977 | 979 | Terpenes | 39.96 ± 1.65 | 23.52 ± 2.64 |
| 14 | 9.84 | Hexanoic acid | C6H12O2 | 142-62-1 | 985 | 990 | Acids | - | 0.2 ± 0.07 |
| 15 | 10.02 | 6-Methylhept-5-en-2-one | C8H14O | 110-93-0 | 989 | 986 | Ketones | 0.6 ± 0.11 | 1.07 ± 0.15 |
| 16 | 10.15 | β-Myrcene | C10H16 | 123-35-3 | 992 | 991 | Terpenes | 0.72 ± 0.18 | 0.69 ± 0.08 |
| 17 | 10.65 | α-Phellandrene | C10H16 | 99-83-2 | 1003 | 1005 | Terpenes | 0.3 ± 0.04 | 0.4 ± 0.05 |
| 18 | 11.18 | α-Terpinene | C10H16 | 99-86-5 | 1016 | 1017 | Terpenes | 1.15 ± 0.14 | 1.52 ± 0.37 |
| 19 | 11.53 | o-Cymene | C10H14 | 527-84-4 | 1024 | 1022 | Aromatic | 2.59 ± 0.31 | 3.23 ± 0.57 |
| 20 | 11.70 | (R)-Isocarvestrene | C10H16 | 1461-27-4 | 1028 | 1027 | Terpenes | 2.22 ± 0.32 | 1.83 ± 0.2 |
| 21 | 11.83 | Eucalyptol | C10H18O | 470-82-6 | 1031 | 1032 | Alcohols | 0.88 ± 0.08 | 0.72 ± 0.09 |
| 22 | 12.00 | Benzyl alcohol | C7H8O | 100-51-6 | 1035 | 1036 | Aromatic | 0.17 ± 0.02 | 0.39 ± 0.1 |
| 23 | 12.39 | Phenylacetaldehyde | C8H8O | 122-78-1 | 1044 | 1045 | Aldehydes | 0.07 ± 0.01 | 0.09 ± 0.03 |
| 24 | 12.66 | trans-Thujenol | C10H16O | 97631-68-0 | 1049 | 1039 | Alcohols | 0.05 ± 0.01 | 0.05 ± 0.01 |
| 25 | 13.08 | γ-Terpinene | C10H16 | 99-85-4 | 1058 | 1060 | Terpenes | 1.84 ± 0.24 | 2.38 ± 0.53 |
| 26 | 13.46 | (Z)-sabinene hydrate | C10H18O | 15537-55-0 | 1066 | 1070 | Alcohols | 2.33 ± 0.15 | 5.53 ± 1.68 |
| 27 | 13.99 | Benzyl formate | C8H8O2 | 104-57-4 | 1076 | 1080 | Esters | 0.04 ± 0.02 | 0.08 ± 0.02 |
| 28 | 14.54 | Terpinolene | C10H16 | 586-62-9 | 1086 | 1088 | Terpenes | 0.42 ± 0.04 | 0.59 ± 0.14 |
| 29 | 15.06 | (E)-sabinene hydrate | C10H18O | 17699-16-0 | 1170 | 1070 | Alcohols | 1.89 ± 0.18 | 5.16 ± 1.7 |
| 30 | 15.29 | Linalool | C10H18O | 78-70-6 | 1100 | 1099 | Alcohols | 0.09 ± 0.01 | 0.16 ± 0.04 |
| 31 | 15.46 | 3,4-Dimethylstyrene | C10H12 | 27831-13-6 | 1104 | 1100 | Others | 0.16 ± 0.03 | 0.39 ± 0.1 |
| 32 | 15.87 | Fenchol | C10H16O | 1632-73-1 | 1114 | 1113 | Alcohols | 0.05 ± 0.04 | 0.18 ± 0.05 |
| 33 | 15.93 | Phenylethyl Alcohol | C8H10O | 60-12-8 | 1115 | 1116 | Aromatic | 0.1 ± 0.03 | 0.2 ± 0.04 |
| 34 | 16.06 | Thujone | C10H16O | 546-80-5 | 1118 | 1103 | Ketones | 0.07 ± 0.01 | 0.1 ± 0.02 |
| 35 | 16.30 | 4-Terpinenyl acetate | C12H20O2 | 4821-04-9 | 1124 | 1301 | Esters | 0.35 ± 0.06 | 0.68 ± 0.11 |
| 36 | 16.55 | α-Campholenal | C10H16O | 4501-58-0 | 1130 | 1125 | Aldehydes | 0.31 ± 0.06 | 0.43 ± 0.15 |
| 37 | 17.08 | Laevo-pinocarveol | C10H16O | 547-61-5 | 1142 | 1139 | Alcohols | 1.49 ± 0.36 | 2.91 ± 0.72 |
| 38 | 17.20 | (E)-Para-2-menthen-1-ol | C10H18O | 29803-81-4 | 1144 | 1140 | Alcohols | 0.35 ± 0.07 | 0.84 ± 0.09 |
| 39 | 17.43 | Camphene hydrate | C10H18O | 465-31-6 | 1151 | 1148 | Alcohols | 0.5 ± 0.12 | 1.48 ± 0.45 |
| 40 | 17.97 | Sabinone | C10H14O | 67690-48-6 | 1160 | 1163 | Ketones | 0.08 ± 0.02 | 0.11 ± 0.03 |
| 41 | 18.03 | Sabina ketone | C9H14O | 513-20-2 | 1163 | 1156 | Ketones | 0.1 ± 0.03 | 0.27 ± 0.07 |
| 42 | 18.20 | Pinocarvone | C10H14O | 30460-92-5 | 1166 | 1164 | Ketones | 0.79 ± 0.18 | 1.25 ± 0.45 |
| 43 | 18.35 | endo-Borneol | C10H18O | 507-70-0 | 1169 | 1167 | Alcohols | 0.16 ± 0.03 | 0.43 ± 0.1 |
| 44 | 18.53 | cis-Sabinol | C10H16O | 3310-02-9 | 1172 | 1175 | Alcohols | 0.1 ± 0.03 | 0.16 ± 0.03 |
| 45 | 18.70 | Pinocamphone | C10H16O | 15358-88-0 | 1176 | 1173 | Ketones | 0.05 ± 0.03 | 0.11 ± 0.04 |
| 46 | 18.86 | Terpinen-4-ol | C10H18O | 562-74-3 | 1180 | 1177 | Alcohols | 6.77 ± 1.13 | 15.31 ± 1.67 |
| 47 | 19.03 | Myrtanal | C10H16O | 4764-14-1 | 1183 | 1188 | Aldehydes | 0.11 ± 0.02 | 0.17 ± 0.04 |
| 48 | 19.25 | 4-Isopropyl-2-cyclohexen-1-one | C9H14O | 500-02-7 | 1187 | 1184 | Ketones | 0.36 ± 0.06 | 0.8 ± 0.21 |
| 49 | 19.44 | α-Terpineol | C10H18O | 98-55-5 | 1191 | 1189 | Alcohols | 0.51 ± 0.12 | 1.19 ± 0.25 |
| 50 | 19.65 | (±)-Myrtenol | C10H16O | 515-00-4 | 1195 | 1195 | Alcohols | 2.99 ± 0.76 | 3.96 ± 1.27 |
| 51 | 20.16 | (S)-Verbenone | C10H14O | 80-57-9 | 1208 | 1205 | Ketones | 0.24 ± 0.11 | 0.85 ± 0.26 |
| 52 | 20.55 | Carveol | C10H16O | 99-48-9 | 1220 | 1219 | Alcohols | 0.09 ± 0.02 | 0.13 ± 0.04 |
| 53 | 20.72 | 2-Hydroxycineole | C10H18O2 | 18679-48-6 | 1224 | 1228 | Alcohols | 0.06 ± 0.02 | 0.14 ± 0.05 |
| 54 | 21.26 | Cuminaldehyde | C10H12O | 122-03-2 | 1241 | 1239 | Aldehydes | 0.17 ± 0.04 | 0.23 ± 0.07 |
| 55 | 21.83 | Piperitone | C10H16O | 89-81-6 | 1257 | 1253 | Ketones | 0.03 ± 0.01 | 0.07 ± 0.03 |
| 56 | 22.51 | β-Cubebene | C15H24 | 13744-15-5 | 1392 | 1389 | Terpenes | 0.06 ± 0.01 | 0.09 ± 0.03 |
| 57 | 22.92 | Bornyl acetate | C12H20O2 | 5655-61-8 | 1287 | 1284 | Esters | 0.16 ± 0.03 | 0.34 ± 0.05 |
| 58 | 23.49 | 3-tert-Butylphenol | C10H14O | 585-34-2 | 1302 | 1296 | Aromatic | 0.03 ± 0.01 | 0.05 ± 0.02 |
| 59 | 25.23 | α-Cubebene | C15H24 | 17699-14-8 | 1355 | 1351 | Terpenes | 0.04 ± 0.01 | 0.09 ± 0.01 |
| 60 | 26.09 | Copaene | C15H24 | 3856-25-5 | 1380 | 1376 | Terpenes | 0.3 ± 0.08 | 0.48 ± 0.12 |
| 61 | 26.50 | (+)-epi-Bicyclosesquiphellandrene | C15H24 | 54274-73-6 | 1392 | \ | Terpenes | 0.05 ± 0.01 | 0.09 ± 0.06 |
| 62 | 27.24 | Santalene | C15H24 | 512-61-8 | 1424 | 1420 | Terpenes | 0.03 ± 0.01 | 0.05 ± 0.02 |
| 63 | 28.07 | γ-Muurolene | C15H24 | 30021-74-0 | 1466 | 1477 | Terpenes | 0.01 ± 0.01 | 0.03 ± 0.01 |
| 64 | 28.47 | Germacrene D | C15H24 | 23986-74-5 | 1486 | 1481 | Terpenes | 0.04 ± 0.02 | 0.05 ± 0.03 |
| 65 | 28.79 | (R)-β-himachalene | C15H24 | 1461-03-6 | 1502 | 1500 | Terpenes | 0.01 ± 0.01 | 0.03 ± 0.01 |
| 66 | 29.17 | (+)-δ-Cadinene | C15H24 | 483-76-1 | 1529 | 1524 | Terpenes | 0.34 ± 0.1 | 0.65 ± 0.26 |
| 67 | 29.47 | α-Calacorene | C15H20 | 21391-99-1 | 1550 | 1542 | Others | 0.02 ± 0.01 | 0.03 ± 0.01 |
| Terpenes | 72.46 | 46.35 | |||||||
| Alcohols | 18.30 | 38.33 | |||||||
| Aromatic | 2.94 | 3.99 | |||||||
| Ketones | 2.32 | 4.62 | |||||||
| Aldehydes | 0.94 | 1.26 | |||||||
| Esters | 0.82 | 1.85 | |||||||
| Others | 0.42 | 0.87 | |||||||
| Acids | 0.00 | 0.20 | |||||||
| Total identified compounds | 98.20 | 97.46 | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Q.; Yuan, H.; Liu, S.; Liang, L.; Luo, J.; Wang, M.; Li, B.; Wang, W. Mid-Level Data Fusion Techniques of LC-MS and HS-GC-MS for Distinguishing Green and Ripe Forsythiae Fructus. Molecules 2025, 30, 1404. https://doi.org/10.3390/molecules30071404
Xie Q, Yuan H, Liu S, Liang L, Luo J, Wang M, Li B, Wang W. Mid-Level Data Fusion Techniques of LC-MS and HS-GC-MS for Distinguishing Green and Ripe Forsythiae Fructus. Molecules. 2025; 30(7):1404. https://doi.org/10.3390/molecules30071404
Chicago/Turabian StyleXie, Qingling, Hanwen Yuan, Shiqi Liu, Ling Liang, Jiangyi Luo, Mengyun Wang, Bin Li, and Wei Wang. 2025. "Mid-Level Data Fusion Techniques of LC-MS and HS-GC-MS for Distinguishing Green and Ripe Forsythiae Fructus" Molecules 30, no. 7: 1404. https://doi.org/10.3390/molecules30071404
APA StyleXie, Q., Yuan, H., Liu, S., Liang, L., Luo, J., Wang, M., Li, B., & Wang, W. (2025). Mid-Level Data Fusion Techniques of LC-MS and HS-GC-MS for Distinguishing Green and Ripe Forsythiae Fructus. Molecules, 30(7), 1404. https://doi.org/10.3390/molecules30071404