
1′-Acetoxychavicol Acetate Selectively Downregulates Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) Expression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. ACA Inhibited TNF-α-Induced ICAM-1 Protein Expression in A549 Cells
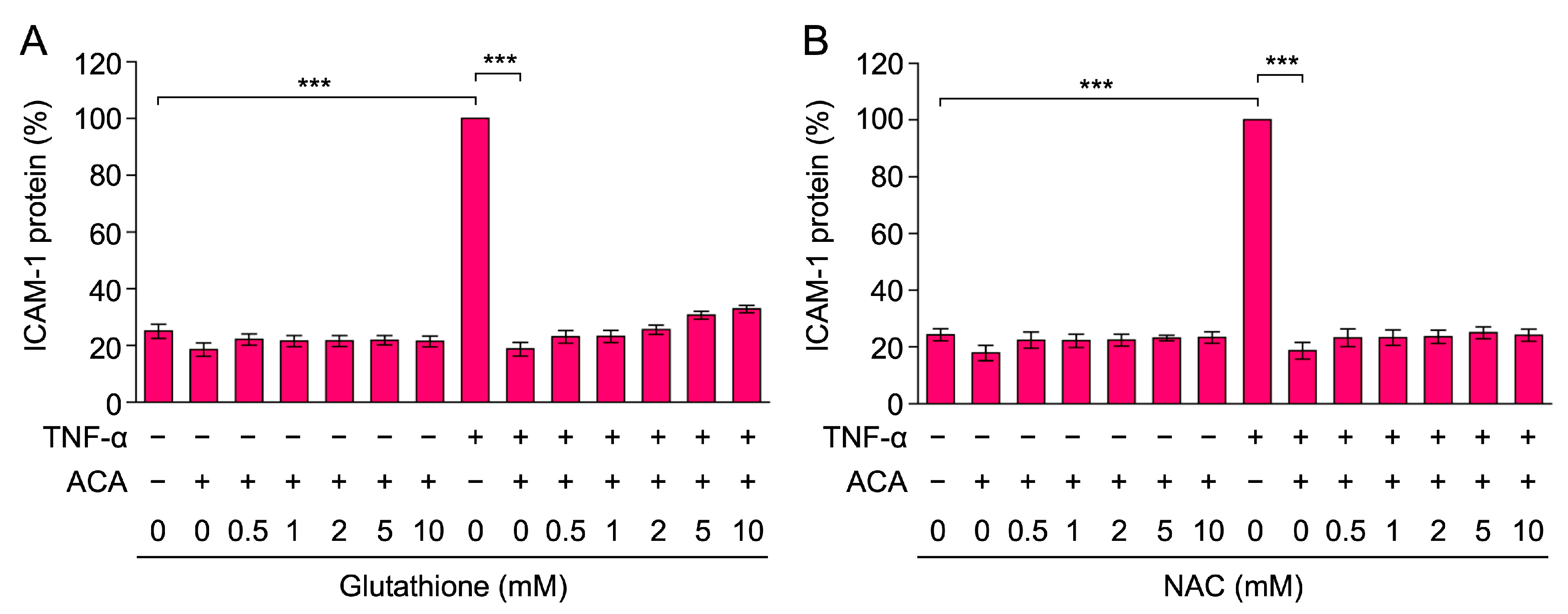
2.2. Glutathione and N-Acetyl-L-Cysteine (NAC) Did Not Reverse the Inhibitory Activity of ACA on TNF-α-Induced ICAM-1 Expression
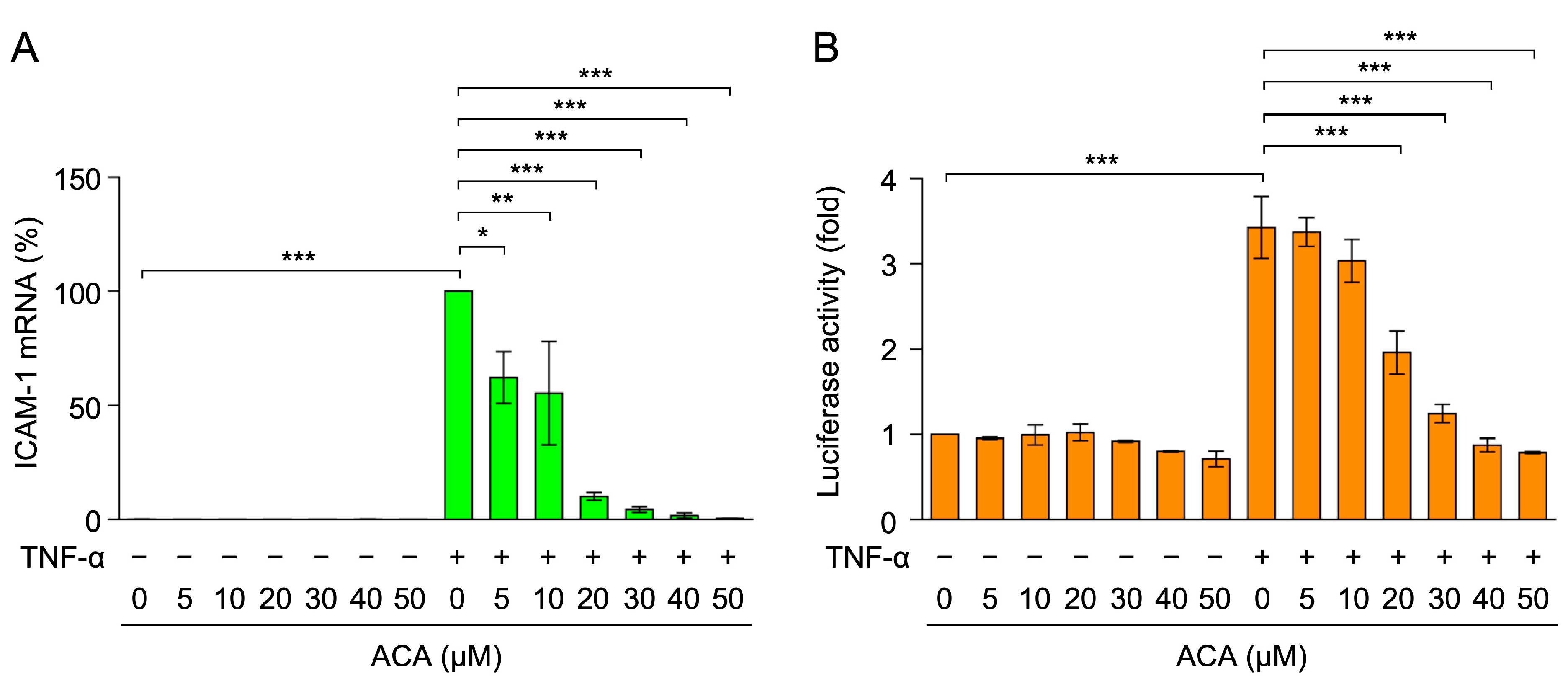
2.3. ACA Inhibited TNF-α-Induced ICAM-1 mRNA Expression in A549 Cells
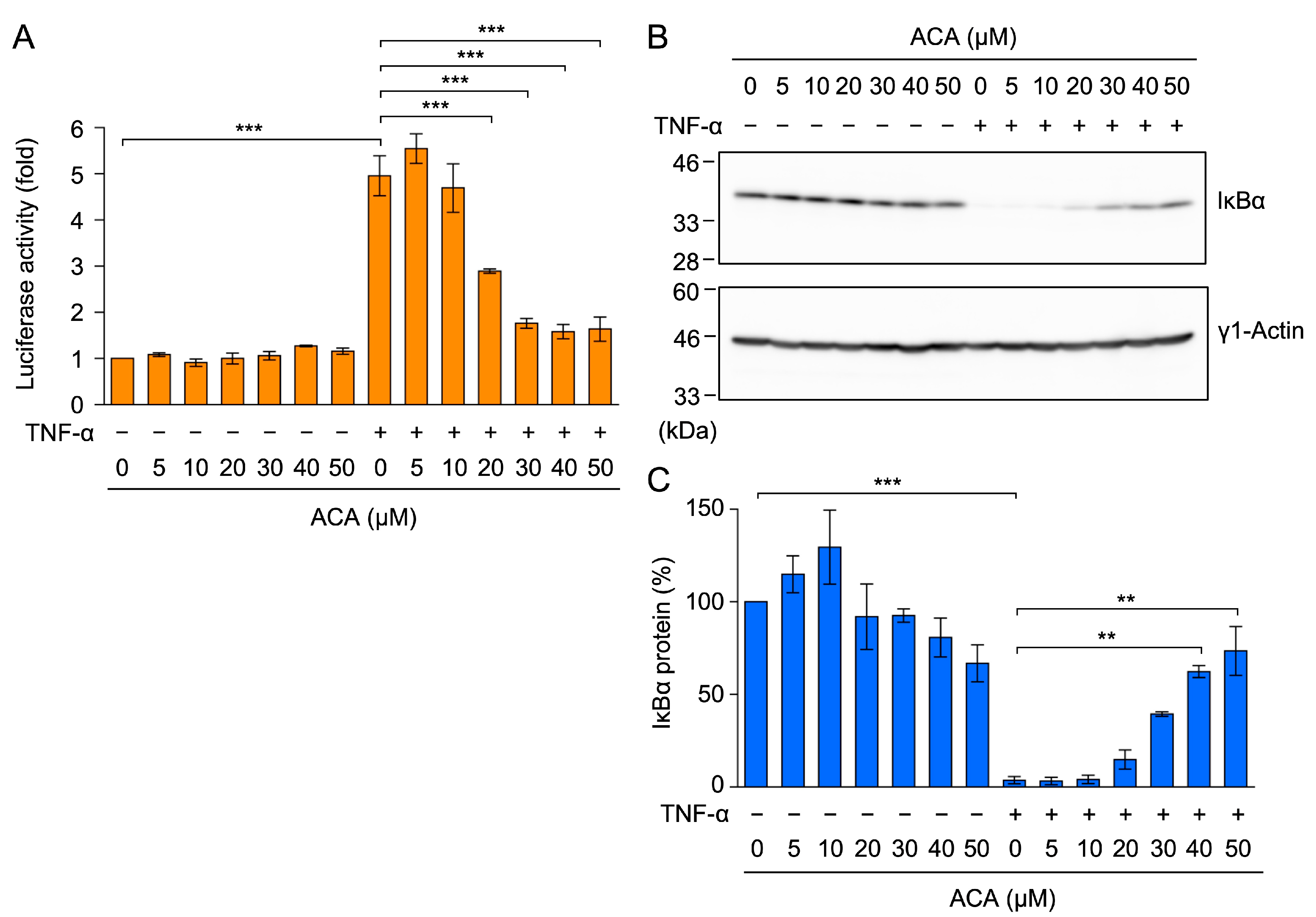
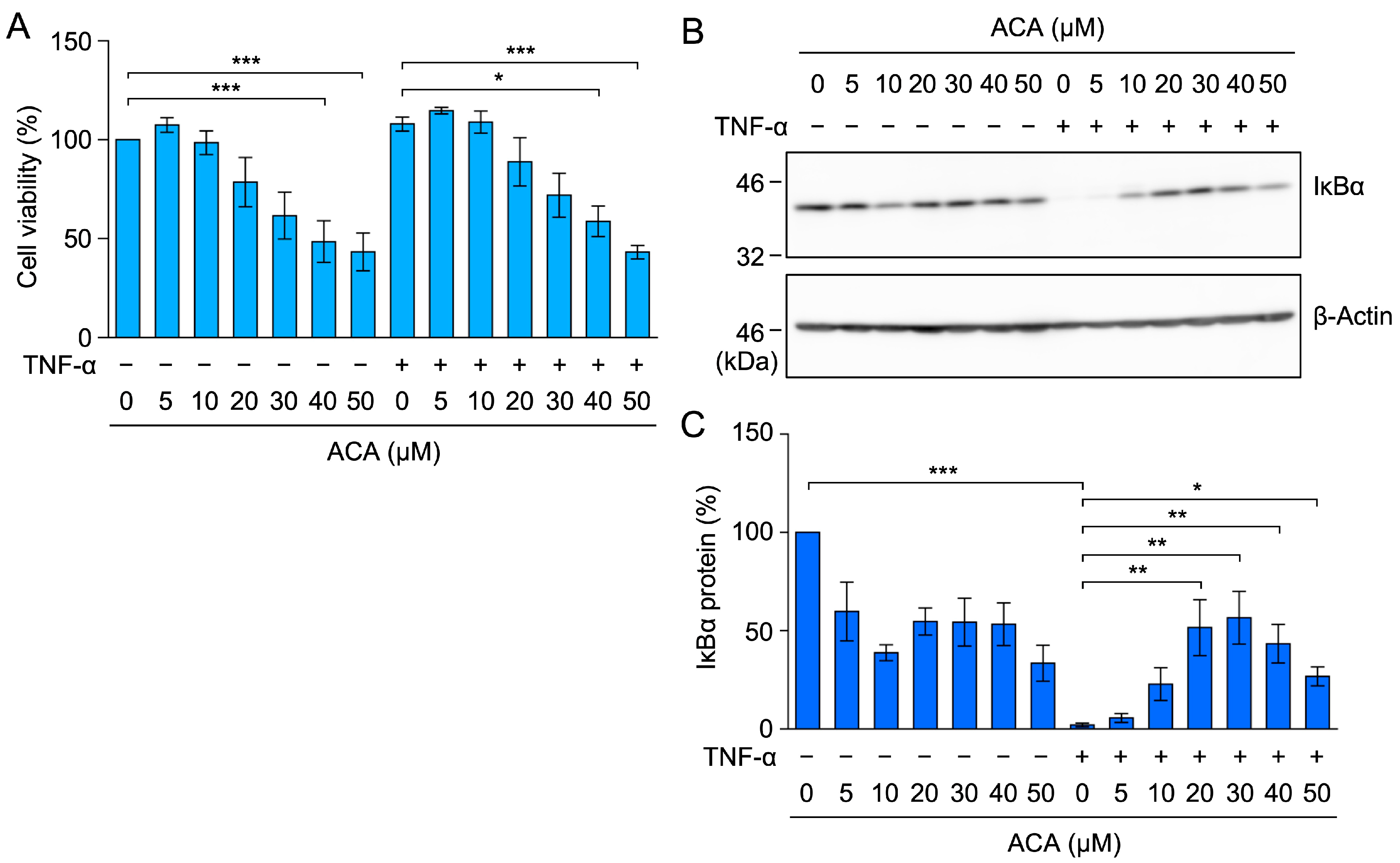
2.4. ACA Inhibited the TNF-α-Induced NF-κB Signaling Pathway in A549 Cells
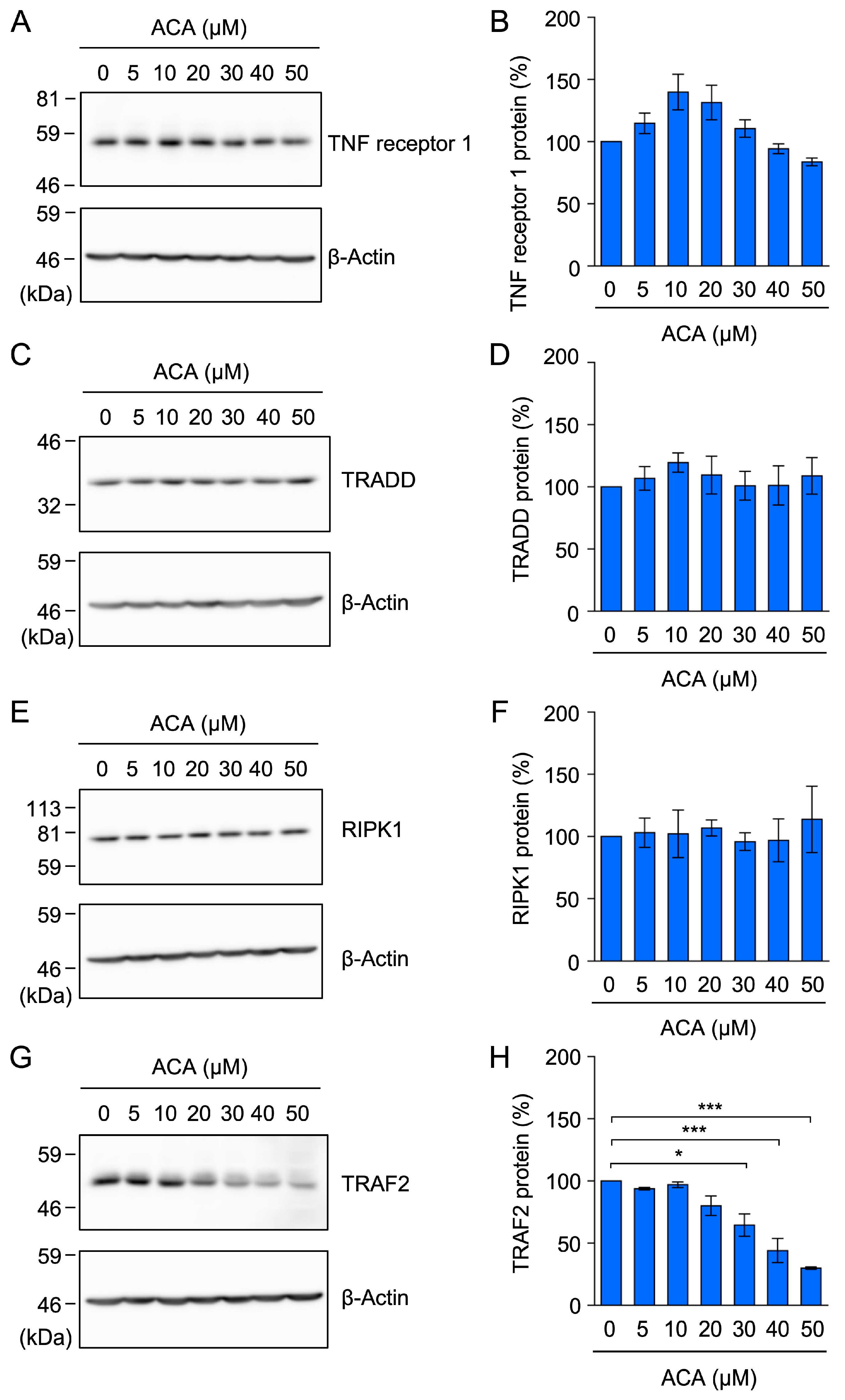
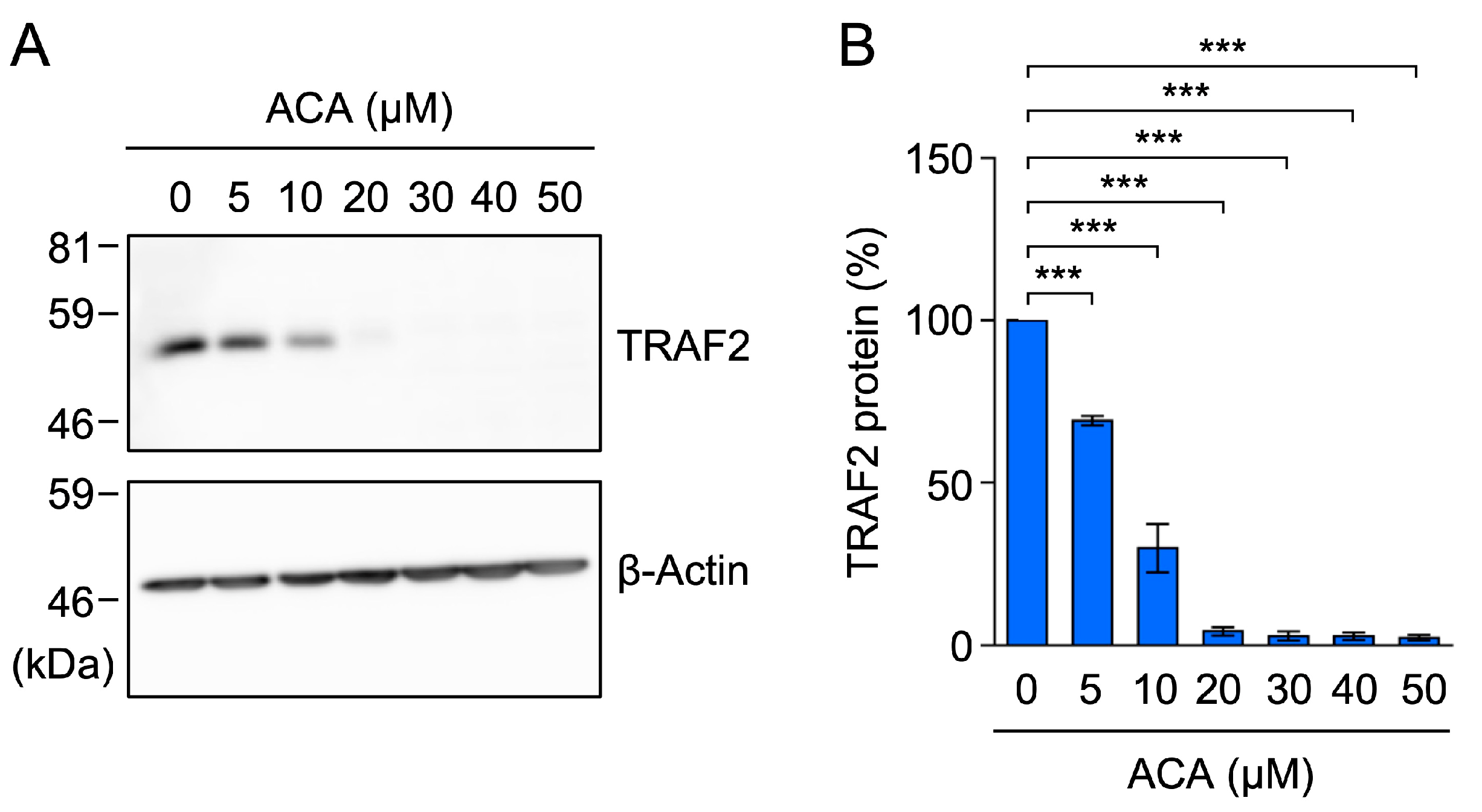
2.5. ACA Selectively Downregulated the Expression of TRAF2 in A549 Cells
2.6. ACA Decreased the Amount of the TRAF2 Protein in Whole-Cell Lysates of A549 Cells
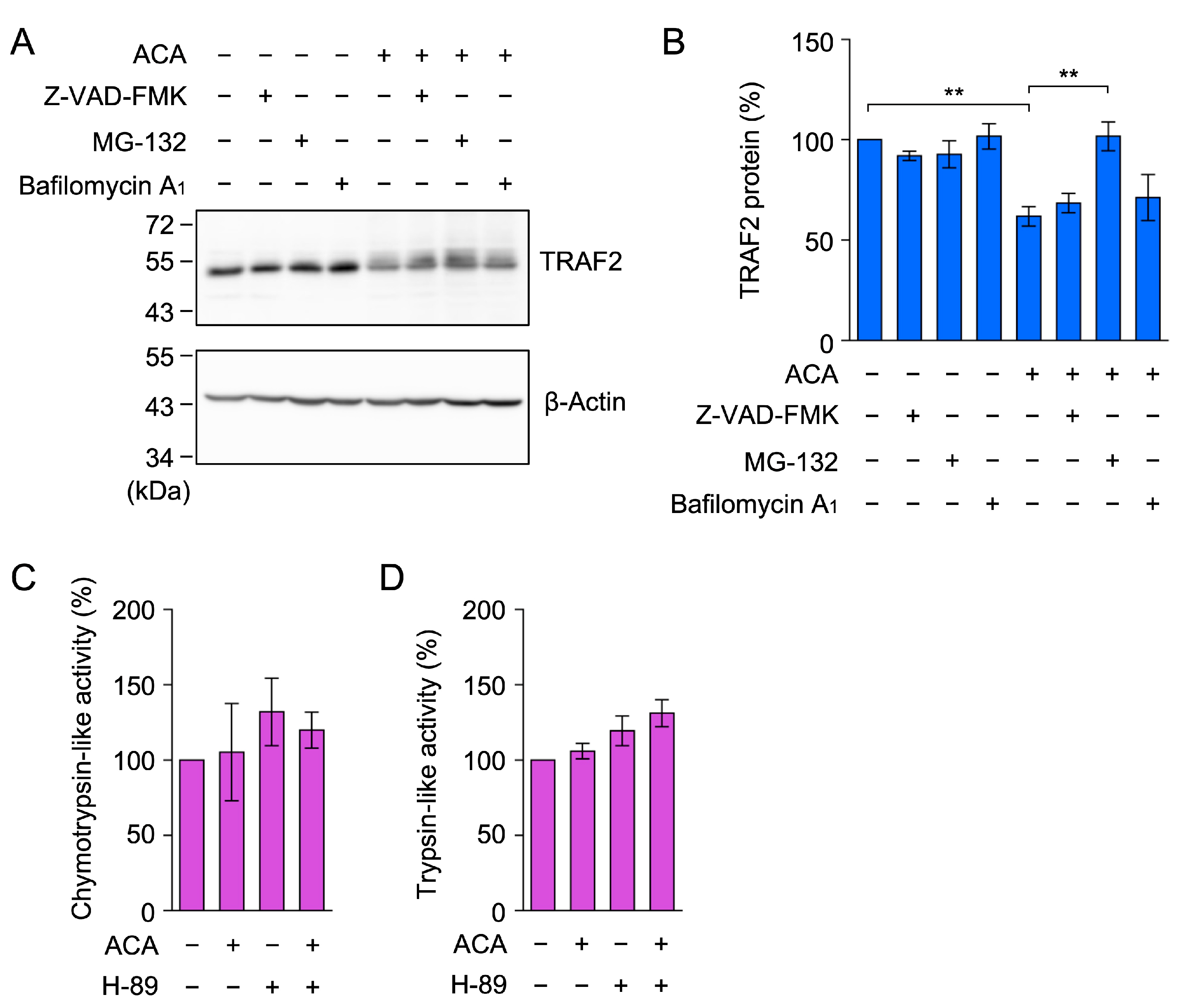
2.7. ACA Promoted the Proteasomal Degradation of TRAF2 in A549 Cells
2.8. ACA Inhibited the TNF-α-Induced NF-κB Signaling Pathway in HT-1080 Cells
2.9. ACA Downregulated TRAF2 Protein Expression in HT-1080 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Antibodies
4.4. Plasmids
4.5. Measurement of Cell Viability
4.6. Measurement of ICAM-1 Expression
4.7. Flow Cytometry
4.8. Measurement of mRNA Expression
4.9. Luciferase Reporter Assay
4.10. Preparation of Cell Lysates and Whole-Cell Lysates
4.11. Western Blotting
4.12. Proteasome Assay
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; He, C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018, 44, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, T.; Ley, K. Monocyte trafficking across the vessel wall. Cardiovasc. Res. 2015, 107, 321–330. [Google Scholar] [CrossRef]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef]
- Qiu, Z.; Wang, Y.; Zhang, Z.; Qin, R.; Peng, Y.; Tang, W.; Xi, Y.; Tian, G.; Zhang, Y. Roles of intercellular cell adhesion molecule-1 (ICAM-1) in colorectal cancer: Expression, functions, prognosis, tumorigenesis, polymorphisms and therapeutic implications. Front. Oncol. 2022, 12, 1052672. [Google Scholar] [CrossRef]
- Zhong, L.; Simard, M.J.; Huot, J. Endothelial microRNAs regulating the NF-κB pathway and cell adhesion molecules during inflammation. FASEB J. 2018, 32, 4070–4084. [Google Scholar] [CrossRef]
- Singh, M.; Thakur, M.; Mishra, M.; Yadav, M.; Vibhuti, R.; Memon, A.M.; Nagda, G.; Dwivedi, V.P.; Dakal, T.C.; Yadav, V. Gene regulation of intracellular adhesion molecule-1 (ICAM-1): A molecule with multiple functions. Immunol. Lett. 2021, 240, 123–136. [Google Scholar] [CrossRef]
- Wallach, D. The tumor necrosis factor family: Family conventions and private idiosyncrasies. Cold Spring Harb. Perspect. Biol. 2018, 10, a028431. [Google Scholar] [CrossRef]
- Ham, B.; Fernandez, M.C.; D’Costa, Z.; Brodt, P. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016, 11, 1–27. [Google Scholar] [PubMed]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the control of the life and death balance of macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Atretkhany, K.S.N.; Gogoleva, V.S.; Drutskaya, M.S.; Nedospasov, S.A. Distinct modes of TNF signaling through its two receptors in health and disease. J. Leukoc. Biol. 2020, 107, 893–905. [Google Scholar] [CrossRef]
- Scheidereit, C. IκB kinase complexes: Gateways to NF-κB activation and transcription. Oncogene 2006, 25, 6685–6705. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Bhoj, V.G.; Chen, Z.J. Ubiquitylation in innate and adaptive immunity. Nature 2009, 458, 430–437. [Google Scholar] [CrossRef]
- Sasaki, K.; Iwai, K. Roles of linear ubiquitinylation, a crucial regulator of NF-κB and cell death, in the immune system. Immunol. Rev. 2015, 266, 175–189. [Google Scholar] [CrossRef]
- Puar, Y.R.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Sethi, G.; Tergaonkar, V. Evidence for the involvement of the master transcription factor NF-κB in cancer initiation and progression. Biomedicines 2018, 6, 82. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Y.; Chen, X.; Ye, X.; Shen, X.; Lin, M.; Zeng, C.; Zhou, T.; Zhang, J. NF-κB in biology and targeted therapy: New insights and translational implications. Signal Transduct. Target Ther. 2024, 9, 53. [Google Scholar] [CrossRef]
- Chouni, A.; Paul, S. A review on phytochemical and pharmacological potential of Alpinia galanga. Pharmacogn. J. 2018, 10, 9–15. [Google Scholar] [CrossRef]
- Kojima-Yuasa, A.; Matsui-Yuasa, I. Pharmacological effects of 1′-acetoxychavicol acetate, a major constituent in the rhizomes of Alpinia galanga and Alpinia conchigera. J. Med. Food 2020, 23, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Ohata, T.; Fukuda, K.; Murakami, A.; Ohigashi, H.; Sugimura, T.; Wakabayashi, K. Inhibition by 1′-acetoxychavicol acetate of lipopolysaccharide- and interferon-γ-induced nitric oxide production through suppression of inducible nitric oxide synthase gene expression in RAW264 cells. Carcinogenesis 1998, 19, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Matsumoto, K.; Koshimizu, K.; Ohigashi, H. Effects of selected food factors with chemopreventive properties on combined lipopolysaccharide- and interferon-γ-induced IκB degradation in RAW264.7 macrophages. Cancer Lett. 2003, 195, 17–25. [Google Scholar] [CrossRef]
- Ando, S.; Matsuda, H.; Morikawa, T.; Yoshikawa, M. 1′S-1′-Acetoxychavicol acetate as a new type inhibitor of interferon-β production in lipopolysaccharide-activated mouse peritoneal macrophages. Bioorg. Med. Chem. 2005, 13, 3289–3294. [Google Scholar] [CrossRef]
- Murakami, A.; Shigemori, T.; Ohigashi, H. Zingiberaceous and citrus constituents, 1′-acetoxychavicol acetate, zerumbone, auraptene, and nobiletin, suppress lipopolysaccharide-induced cyclooxygenase-2 expression in RAW264.7 murine macrophages through different modes of action. J. Nutr. 2005, 135, 2987S–2992S. [Google Scholar] [CrossRef]
- Ichikawa, H.; Murakami, A.; Aggarwal, B.B. 1′-Acetoxychavicol acetate inhibits RANKL-induced osteoclastic differentiation of RAW 264.7 monocytic cells by suppressing nuclear factor-κB activation. Mol. Cancer Res. 2006, 4, 275–281. [Google Scholar] [CrossRef]
- Ong, G.H.; Ori, D.; Kawasaki, T.; Kawai, T. Inhibition of lipopolysaccharide-induced inflammatory responses by 1′-acetoxychavicol acetate. Genes Cells 2022, 27, 482–492. [Google Scholar] [CrossRef]
- Ichikawa, H.; Takada, Y.; Murakami, A.; Aggarwal, B.B. Identification of a novel blocker of IκBα kinase that enhances cellular apoptosis and inhibits cellular invasion through suppression of NF-κB-regulated gene products. J. Immunol. 2005, 174, 7383–7392. [Google Scholar] [CrossRef]
- Ito, K.; Nakazato, T.; Xian, M.J.; Yamada, T.; Hozumi, N.; Murakami, A.; Ohigashi, H.; Ikeda, Y.; Kizaki, M. 1′-Acetoxychavicol acetate is a novel nuclear factor κB inhibitor with significant activity against multiple myeloma in vitro and in vivo. Cancer Res. 2005, 65, 4417–4424. [Google Scholar] [CrossRef]
- Misawa, T.; Dodo, K.; Ishikawa, M.; Hashimoto, Y.; Sagawa, M.; Kizaki, M.; Aoyama, H. Structure-activity relationships of benzhydrol derivatives based on 1′-acetoxychavicol acetate (ACA) and their inhibitory activities on multiple myeloma cell growth via inactivation of the NF-κB pathway. Bioorg. Med. Chem. 2015, 23, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- Batra, V.; Syed, Z.; Gill, J.N.; Coburn, M.A.; Adegboyega, P.; DiGiovanni, J.; Mathis, J.M.; Shi, R.; Clifford, J.L.; Kleiner-Hancock, H.E. Effects of the tropical ginger compound, 1′-acetoxychavicol acetate, against tumor promotion in K5.Stat3C transgenic mice. J. Exp. Clin. Cancer Res. 2012, 31, 57. [Google Scholar] [CrossRef] [PubMed]
- In, L.L.; Arshad, N.M.; Ibrahim, H.; Azmi, M.N.; Awang, K.; Nagoor, N.H. 1′-Acetoxychavicol acetate inhibits growth of human oral carcinoma xenograft in mice and potentiates cisplatin effect via proinflammatory microenvironment alterations. BMC Complement. Altern. Med. 2012, 12, 179. [Google Scholar] [CrossRef]
- Kataoka, T. Chemical biology of inflammatory cytokine signaling. J. Antibiot. 2009, 62, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, J.; Kennedy, D.O.; Kojima, A.; Hasuma, T.; Yano, Y.; Otani, S.; Murakami, A.; Koshimizu, K.; Ohigashi, H.; Matsui-Yuasa, I. Involvement of protein tyrosine phosphorylation and reduction of cellular sulfhydryl groups in cell death induced by 1′-acetoxychavicol acetate in Ehrlich ascites tumor cells. Chem. Biol. Interact. 2002, 139, 215–230. [Google Scholar] [CrossRef]
- Campbell, C.T.; Prince, M.; Landry, G.M.; Kha, V.; Kleiner, H.E. Pro-apoptotic effects of 1′-acetoxychavicol acetate in human breast carcinoma cells. Toxicol. Lett. 2007, 173, 151–160. [Google Scholar] [CrossRef]
- Unahara, Y.; Kojima-Yuasa, A.; Higashida, M.; Kennedy, D.O.; Murakami, A.; Ohigashi, H.; Matsui-Yuasa, I. Cellular thiol status-dependent inhibition of tumor cell growth via modulation of p27(kip1) translocation and retinoblastoma protein phosphorylation by 1′-acetoxychavicol acetate. Amino Acids 2007, 33, 469–476. [Google Scholar] [CrossRef]
- Higashida, M.; Xu, S.; Kojima-Yuasa, A.; Kennedy, D.O.; Murakami, A.; Ohigashi, H.; Matsui-Yuasa, I. 1′-Acetoxychavicol acetate-induced cytotoxicity is accompanied by a rapid and drastic modulation of glutathione metabolism. Amino Acids 2009, 36, 107–113. [Google Scholar] [CrossRef]
- Xu, S.; Kojima-Yuasa, A.; Azuma, H.; Kennedy, D.O.; Konishi, Y.; Matsui-Yuasa, I. Comparison of glutathione reductase activity and the intracellular glutathione reducing effects of 13 derivatives of 1′-acetoxychavicol acetate in Ehrlich ascites tumor cells. Chem. Biol. Interact. 2010, 185, 235–240. [Google Scholar] [CrossRef]
- Ogura, H.; Tsukumo, Y.; Sugimoto, H.; Igarashi, M.; Nagai, K.; Kataoka, T. Ectodomain shedding of TNF receptor 1 induced by protein synthesis inhibitors regulates TNF-α-mediated activation of NF-κB and caspase-8. Exp. Cell Res. 2008, 314, 1406–1414. [Google Scholar] [CrossRef]
- Yaku, K.; Matsui-Yuasa, I.; Kojima-Yuasa, A. 1′-Acetoxychavicol acetate increases proteasome activity by activating cAMP-PKA signaling. Planta Med. 2018, 84, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Tanigaki, R.; Kimura, K.; Kataoka, T. Kujigamberol interferes with pro-inflammatory cytokine-induced expression of and N-glycan modifications to cell adhesion molecules at different stages in human umbilical vein endothelial cells. Int. Immunopharmacol. 2018, 62, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Kitamura, M. Bidirectional regulation of NF-κB by reactive oxygen species: A role of unfolded protein response. Free Radical Biol. Med. 2013, 65, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef]
- Pande, V.; Sousa, S.F.; Ramos, M.J. Direct covalent modification as a strategy to inhibit nuclear factor-kappa B. Curr. Med. Chem. 2009, 16, 4261–4273. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, R.; Li, W.; Ma, X.C.; Qiu, F.; Sun, C.P. IκB kinase β (IKKβ): Structure, transduction mechanism, biological function, and discovery of its inhibitors. Int. J. Biol. Sci. 2023, 19, 4181–4203. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Cohen, P. The TLR and IL-1 signaling network at a glance. J. Cell Sci. 2014, 127, 2383–2390. [Google Scholar]
- Min, W.; Bradley, J.R.; Galbraith, J.J.; Jones, S.J.; Ledgerwood, E.C.; Pober, J.S. The N-terminal domains target TNF receptor-associated factor-2 to the nucleus and display transcriptional regulatory activity. J. Immunol. 1998, 161, 319–324. [Google Scholar] [CrossRef]
- Thakar, N.Y.; Ovchinnikov, D.A.; Hastie, M.L.; Kobe, B.; Gorman, J.J.; Wolvetang, E.J. TRAF2 recruitment via T61 in CD30 drives NFκB activation and enhances hESC survival and proliferation. Mol. Biol. Cell 2015, 26, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- El Hokayem, J.; Brittain IV, G.C.; Nawaz, Z.; Bethea, J.R. Tumor necrosis factor receptor associated factors (TRAFs) 2 and 3 form a transcriptional complex with phosho-RNA polymerase II and p65 in CD40 ligand activated Neuro2a cells. Mol. Neurobiol. 2017, 54, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Back, S.H.; Kim, V.; Ryu, I.; Jang, S.K. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol. Cell. Biol. 2005, 25, 2450–2462. [Google Scholar] [CrossRef]
- Kim, W.J.; Kim, J.H.; Jang, S.K. Anti-inflammatory lipid mediator 15d-PGJ2 inhibits translation through inactivation of eIF4A. EMBO J. 2007, 26, 5020–5032. [Google Scholar] [CrossRef] [PubMed]
- Habelhah, H.; Frew, I.J.; Laine, A.; Janes, P.W.; Relaix, F.; Sassoon, D.; Bowtell, D.D.L.; Ronai, Z. Stress-induced decrease in TRAF2 stability is mediated by Siah2. EMBO J. 2002, 21, 5756–5765. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Ashwell, J.D. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature 2002, 416, 345–349. [Google Scholar] [CrossRef]
- Dhingra, R.; Rabinovich-Nikitin, I.; Rothman, S.; Guberman, M.; Gang, H.; Margulets, V.; Jassal, D.S.; Alagarsamy, K.N.; Dhingra, S.; Ripoll, C.V.; et al. Proteasomal degradation of TRAF2 mediates mitochondrial dysfunction in doxorubicin-cardiomyopathy. Circulation 2022, 146, 934–954. [Google Scholar] [CrossRef]
- Vince, J.E.; Chau, D.; Callus, B.; Wong, W.W.L.; Hawkins, C.J.; Schneider, P.; McKinlay, M.; Benetatos, C.A.; Condon, S.M.; Chunduru, S.K.; et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFα. J. Cell Biol. 2008, 182, 171–184. [Google Scholar] [CrossRef]
- Li, L.; Soetandyo, N.; Wang, Q.; Ye, Y. The zinc finger protein A20 targets TRAF2 to the lysosomes for degradation. Biochim. Biophys. Acta 2009, 1793, 346–353. [Google Scholar] [CrossRef]
- Park, H.H. Structure of TRAF Family: Current understanding of receptor recognition. Front. Immunol. 2018, 9, 1999. [Google Scholar] [CrossRef]
- Arkee, T.; Bishop, G.A. TRAF family molecules in T cells: Multiple receptors and functions. J. Leukoc. Biol. 2020, 107, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.B.; Coon, T.A.; Glasser, J.R.; McVerry, B.J.; Zhao, J.; Zhao, Y.; Zou, C.; Ellis, B.; Sciurba, F.C.; Zhang, Y.; et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat. Immunol. 2013, 14, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Mallampalli, R.K.; Coon, T.A.; Glasser, J.R.; Wang, C.; Dunn, S.R.; Weathington, N.M.; Zhao, J.; Zou, C.; Zhao, Y.; Chen, B.B. Targeting F box protein Fbxo3 to control cytokine-driven inflammation. J. Immunol. 2013, 191, 5247–5255. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, D.; Zhao, J.; Weathington, N.M.; Shang, D.; Zhao, Y. The deubiquitinating enzyme USP48 stabilizes TRAF2 and reduces E-cadherin-mediated adherens junctions. FASEB J. 2018, 32, 230–242. [Google Scholar] [CrossRef]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef]
- Sun, L.L.; Wang, J.; Zhao, Z.J.; Liu, N.; Wang, A.L.; Ren, H.Y.; Yang, F.; Diao, K.X.; Fu, W.N.; Wan, E.H.; et al. Suppressive role of miR-502-5p in breast cancer via downregulation of TRAF2. Oncol. Rep. 2014, 31, 2085–2092. [Google Scholar] [CrossRef]
- Peramuhendige, P.; Marino, S.; Bishop, R.T.; de Ridder, D.; Khogeer, A.; Baldini, I.; Capulli, M.; Rucci, N.; Idris, A.I. TRAF2 in osteotropic breast cancer cells enhances skeletal tumour growth and promotes osteolysis. Sci. Rep. 2018, 8, 39. [Google Scholar] [CrossRef]
- Yan, R.; Zhu, H.; Huang, P.; Yang, M.; Shen, M.; Pan, Y.; Zhang, C.; Zhou, X.; Li, H.; Ke, X.; et al. Liquidambaric acid inhibits Wnt/β-catenin signaling and colon cancer via targeting TNF receptor-associated factor 2. Cell Rep. 2022, 38, 110319. [Google Scholar] [CrossRef]
- Vredevoogd, D.W.; Kuilman, T.; Ligtenberg, M.A.; Boshuizen, J.; Stecker, K.E.; de Bruijn, B.; Krijgsman, O.; Huang, X.; Kenski, J.C.N.; Lacroix, R.; et al. Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. Cell 2019, 178, 585–599. [Google Scholar] [CrossRef]
- Vo, N.T.; Kusagawa, E.; Nakano, K.; Moriwaki, C.; Miyake, Y.; Haruyama, S.; Fukuhara, S.; Nguyen, N.T.; Dang, P.H.; Nguyen, M.T.T.; et al. Biological evaluation of alkyl triphenylphosphonium ostruthin derivatives as potential anti-inflammatory agents targeting the nuclear factor κB signaling pathway in human lung adenocarcinoma A549 cells. BioChem 2021, 1, 107–121. [Google Scholar] [CrossRef]
- Dohrman, A.; Kataoka, T.; Cuenin, S.; Russell, J.Q.; Tschopp, J.; Budd, R.C. Cellular FLIP (long form) regulates CD8+ T cell activation through caspase-8-dependent NF-κB activation. J. Immunol. 2005, 174, 5270–5278. [Google Scholar] [CrossRef] [PubMed]
- Vu, Q.V.; Baba, K.; Sakaki, S.; Kawaguchi, K.; Hirano, H.; Osada, H.; Kataoka, T. Alantolactone derivatives inhibit the tumor necrosis factor α-induced nuclear factor κB pathway by a different mechanism from alantolactone. Eur. J. Pharmacol. 2024, 969, 176458. [Google Scholar] [CrossRef] [PubMed]
- Vu, Q.V.; Vu, N.T.; Baba, K.; Sasaki, S.; Tamura, R.; Morimoto, K.; Hirano, H.; Osada, H.; Kataoka, T. Porphyrin derivatives inhibit tumor necrosis factor α-induced gene expression and reduce the expression and increase the cross-linked forms of cellular components of the nuclear factor κB signaling pathway. Eur. J. Pharmacol. 2024, 977, 176747. [Google Scholar] [CrossRef] [PubMed]
- Vo, N.T.; Sasaki, S.; Miyake, Y.; Nguyen, N.T.; Dang, P.H.; Nguyen, M.T.T.; Kataoka, T. α-Conidendrin inhibits the expression of intercellular adhesion molecule-1 induced by tumor necrosis factor-α in human lung adenocarcinoma A549 cells. Eur. J. Pharmacol. 2021, 890, 173651. [Google Scholar] [CrossRef]
- Wan, M.; Liu, J.; Ouyang, X. Nucleotide-binding oligomerization domain 1 regulates Porphyromonas gingivalis-induced vascular cell adhesion molecule 1 and intercellular adhesion molecule 1 expression in endothelial cells through NF-κB pathway. J. Periodontal. Res. 2015, 50, 189–196. [Google Scholar] [CrossRef]
- Zhang, Y.; Lian, F.; Zhu, Y.; Xia, M.; Wang, Q.; Ling, W.; Wang, X.D. Cyanidin-3-O-β-glucoside inhibits LPS-induced expression of inflammatory mediators through decreasing IκBα phosphorylation in THP-1 cells. Inflamm. Res. 2010, 59, 723–730. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moriwaki, C.; Takahashi, S.; Thi Vu, N.; Miyake, Y.; Kataoka, T. 1′-Acetoxychavicol Acetate Selectively Downregulates Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) Expression. Molecules 2025, 30, 1243. https://doi.org/10.3390/molecules30061243
Moriwaki C, Takahashi S, Thi Vu N, Miyake Y, Kataoka T. 1′-Acetoxychavicol Acetate Selectively Downregulates Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) Expression. Molecules. 2025; 30(6):1243. https://doi.org/10.3390/molecules30061243
Chicago/Turabian StyleMoriwaki, Chihiro, Shingo Takahashi, Nhat Thi Vu, Yasunobu Miyake, and Takao Kataoka. 2025. "1′-Acetoxychavicol Acetate Selectively Downregulates Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) Expression" Molecules 30, no. 6: 1243. https://doi.org/10.3390/molecules30061243
APA StyleMoriwaki, C., Takahashi, S., Thi Vu, N., Miyake, Y., & Kataoka, T. (2025). 1′-Acetoxychavicol Acetate Selectively Downregulates Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) Expression. Molecules, 30(6), 1243. https://doi.org/10.3390/molecules30061243