
Synergistic Enhancement of Capacitive Performance in Porous Carbon by Phenolic Resin and Boric Acid

 , ,
, ,
Abstract

1. Introduction
2. Results and Discussion
2.1. Impact of PF Modification
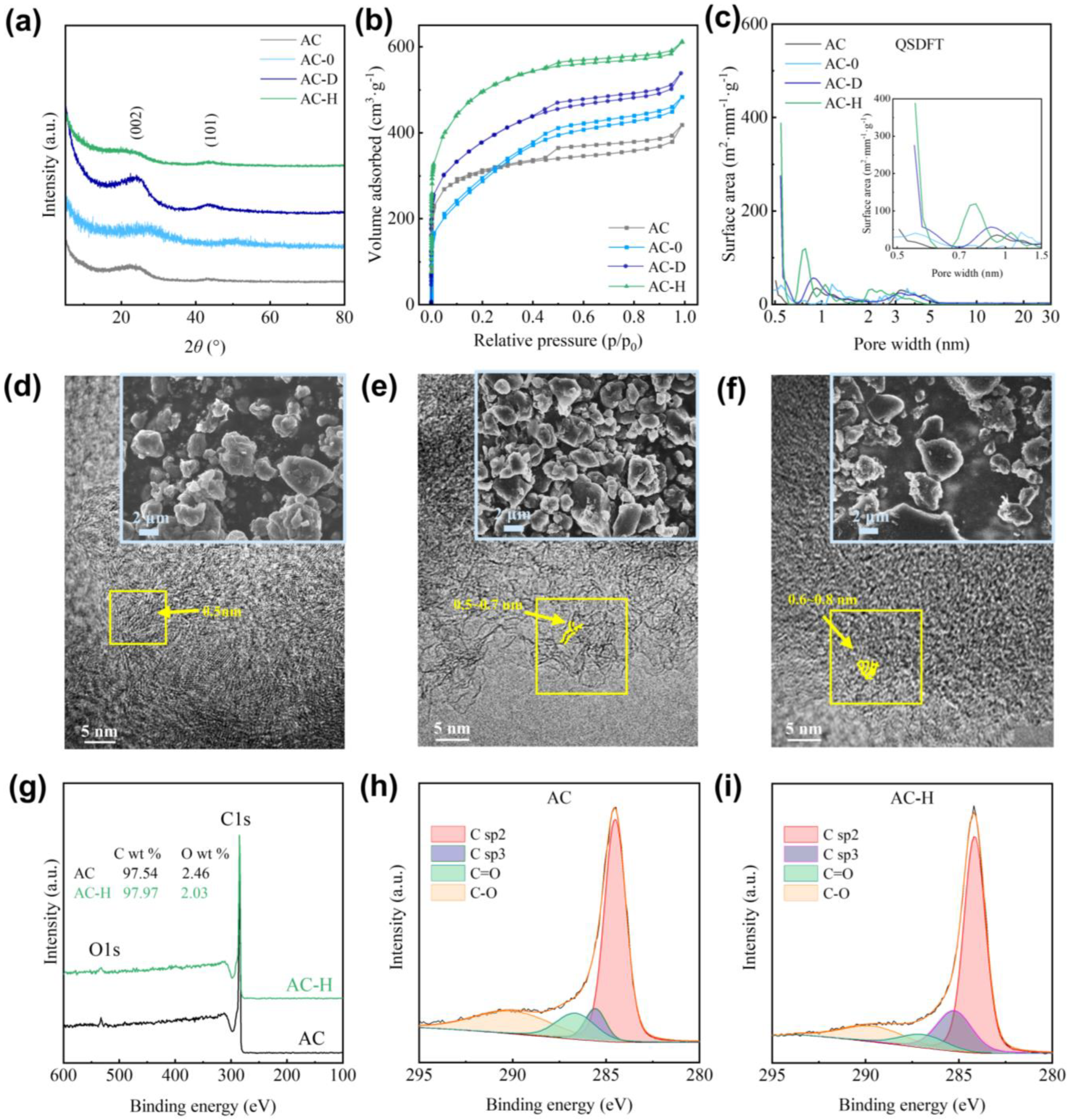
2.1.1. Structural Analysis of PF Modification
2.1.2. Microstructural Morphology Analysis of PF Modification
2.1.3. Surface Properties Analysis of PF Modification
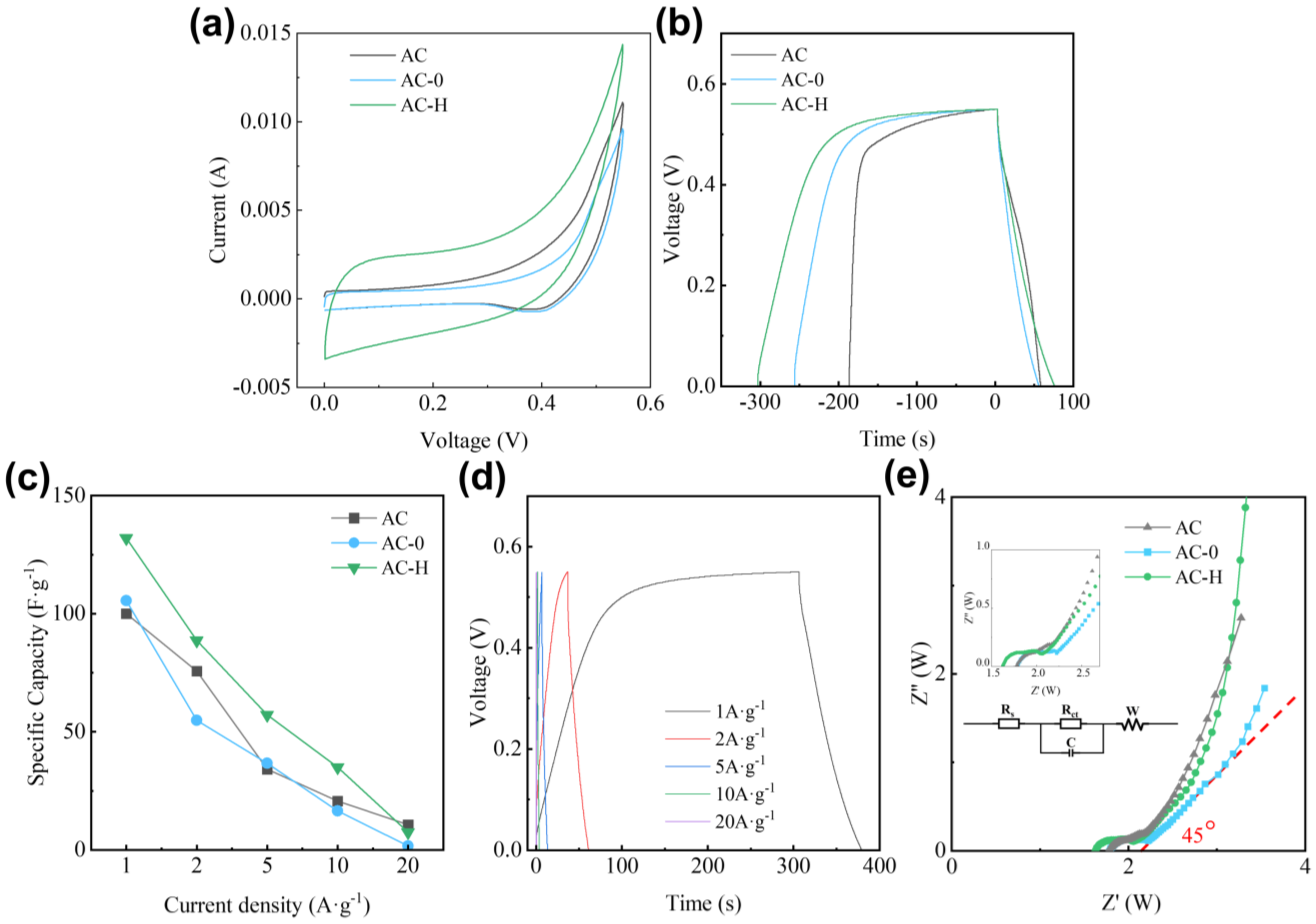
2.1.4. Electrochemical Performance Analysis of PF Modification
2.2. Impact of BA and PF Co-Modification
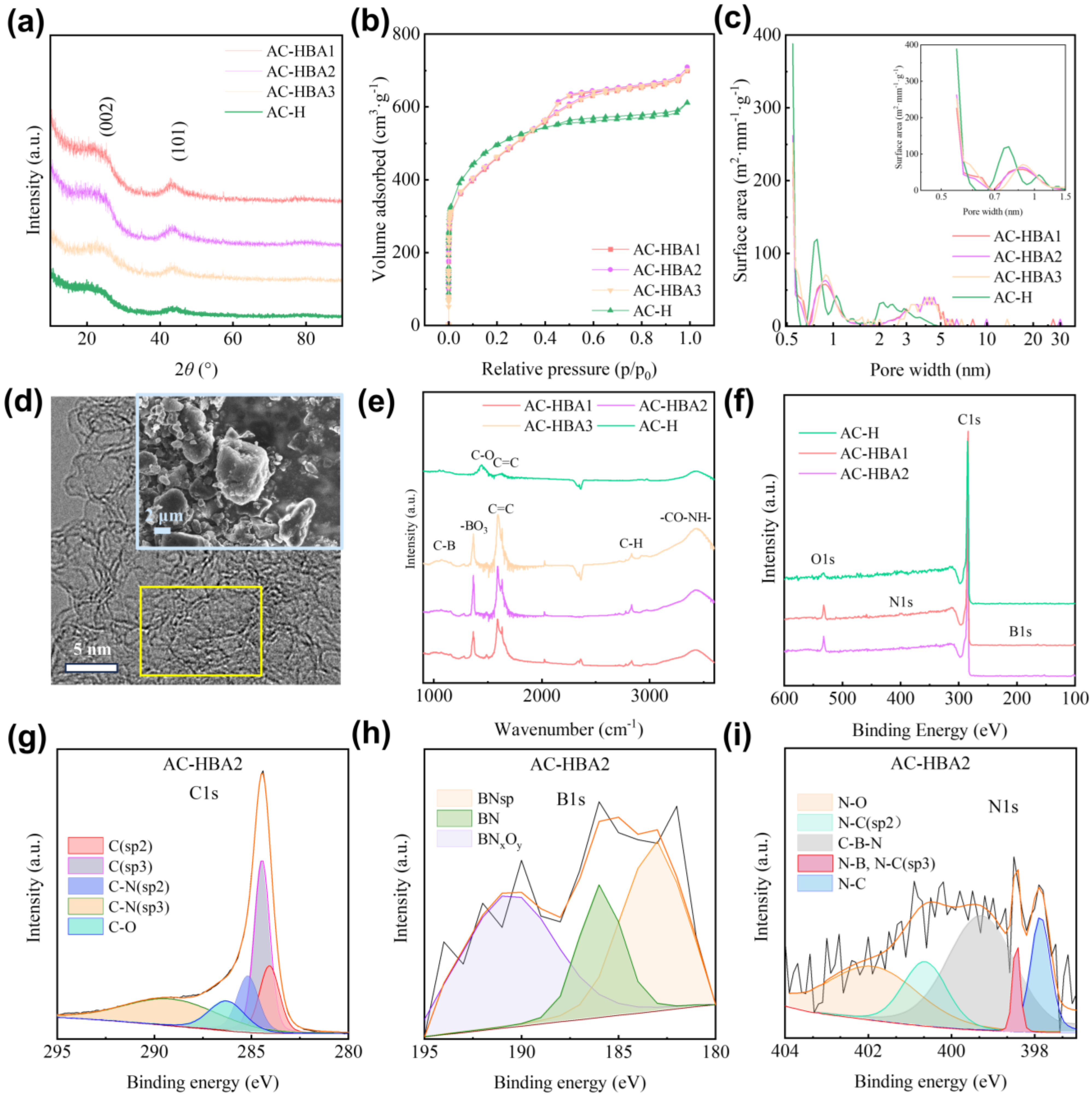
2.2.1. Structural Analysis of Co-Modification
2.2.2. Microscopic Morphology Analysis of Co-Modification
2.2.3. Surface Property Analysis of Co-Modification
2.2.4. Capacitance Performance Analysis of Co-Modification
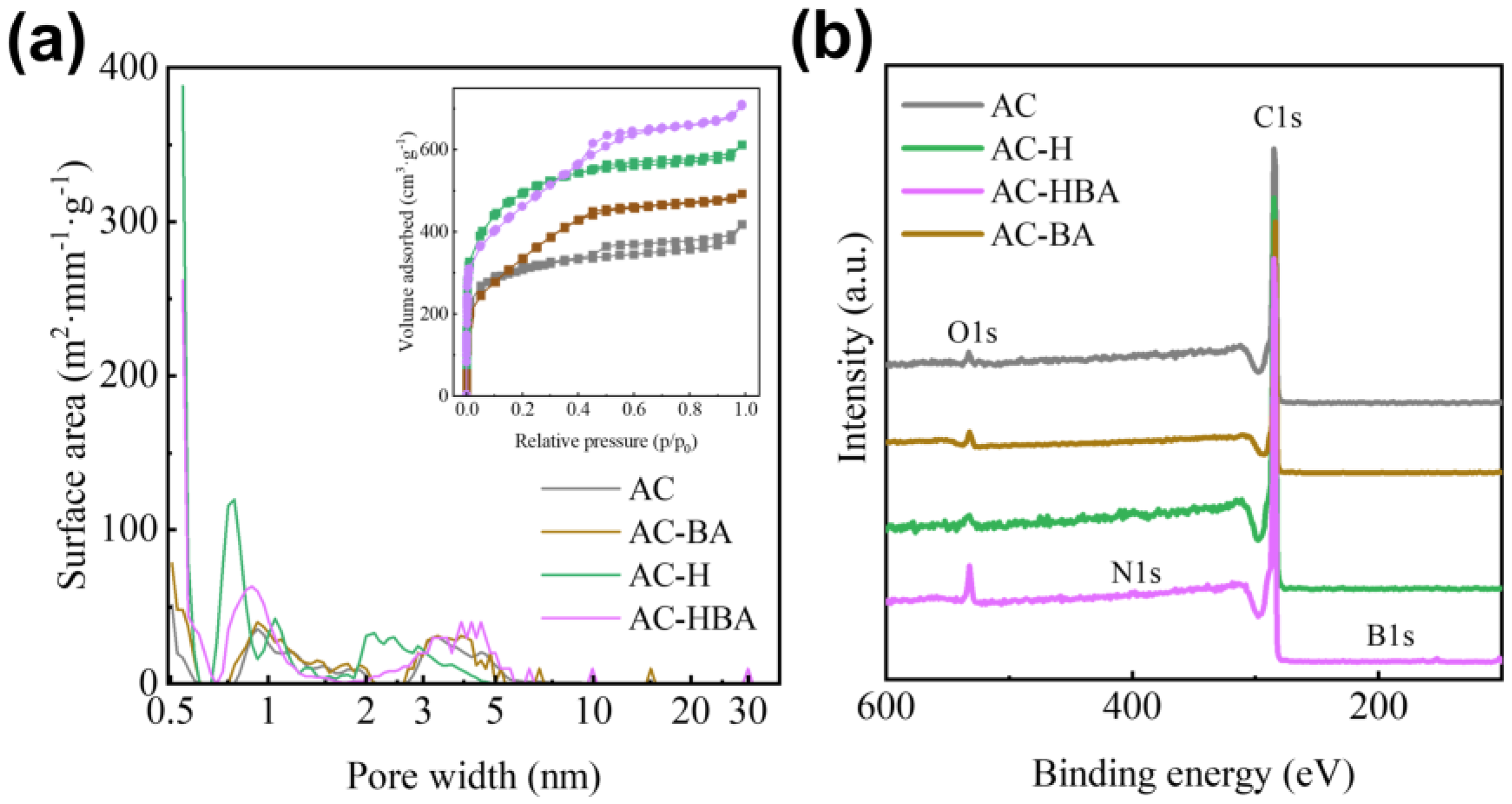
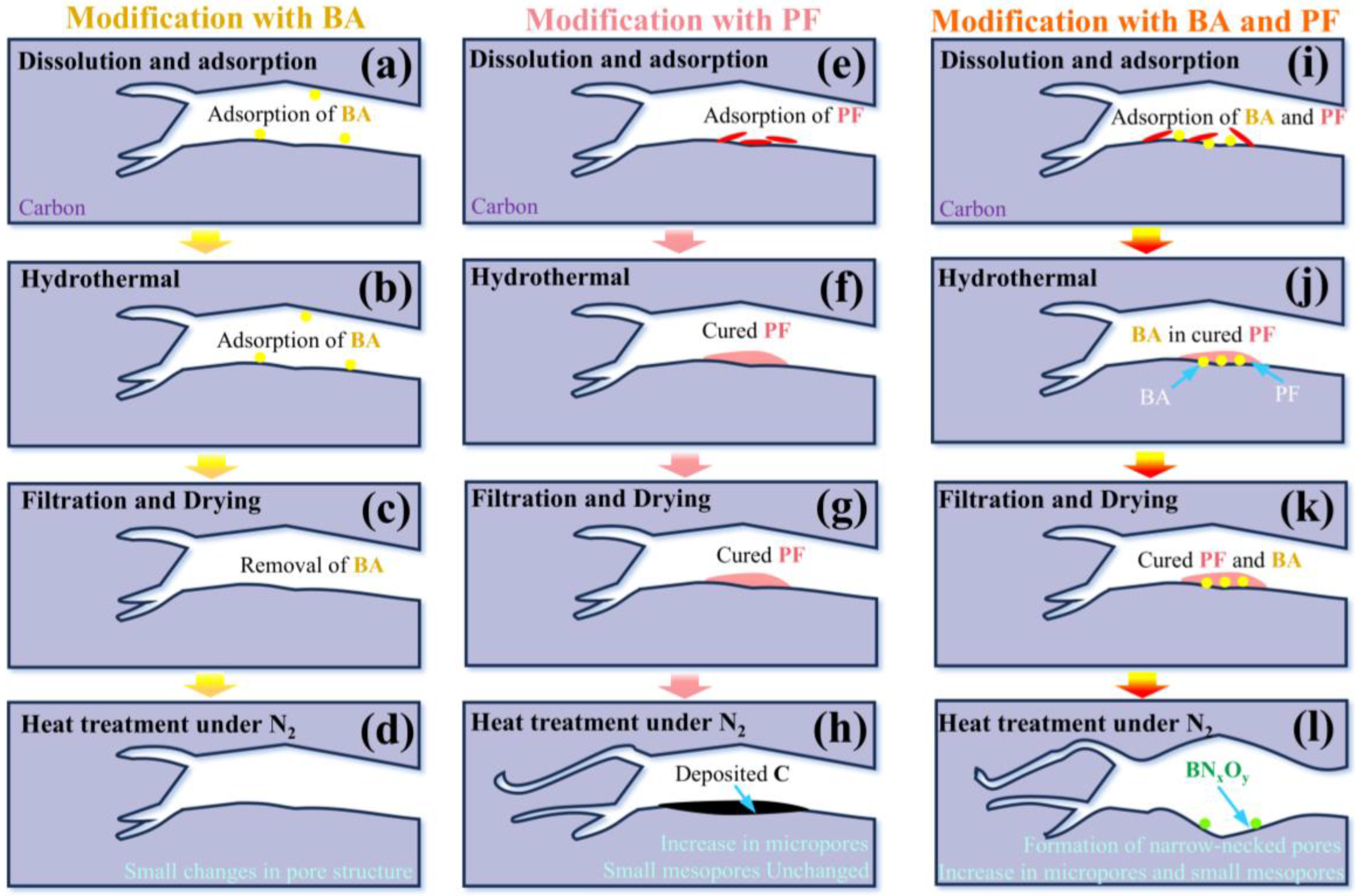
2.3. Synergistic Pore-Tuning Mechanism of PF and BA
3. Experiment
3.1. Materials
3.2. Preparation of Samples
3.2.1. Modification with PF
3.2.2. Modification with BA
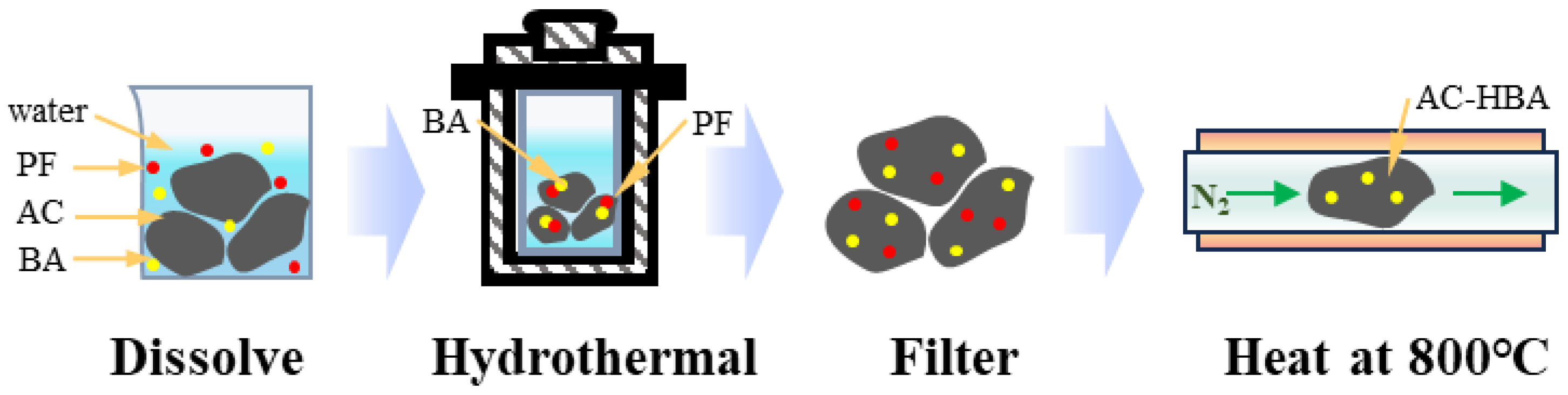
3.2.3. Modification with Both BA and PF
3.3. Analysis and Characterization
3.4. Electrochemical Performance Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jagadale, A.; Zhou, X.; Xiong, R.; Dubal, D.P.; Xu, J.; Yang, S. Lithium ion capacitors (LICs): Development of the materials. Energy Storage Mater. 2019, 19, 314–329. [Google Scholar] [CrossRef]
- Yuan, S.; Lai, Q.; Duan, X.; Wang, Q. Carbon-based materials as anode materials for lithium-ion batteries and lithium-ion capacitors: A review. J. Energy Storage 2023, 61, 106716. [Google Scholar] [CrossRef]
- Li, B.; Zheng, J.; Zhang, H.; Jin, L.; Yang, D.; Lv, H.; Shen, C.; Shellikeri, A.; Zheng, Y.; Gong, R.; et al. Electrode materials, electrolytes, and challenges in nonaqueous lithium-ion capacitors. Adv. Mater. 2018, 30, 1705670. [Google Scholar] [CrossRef] [PubMed]
- Soltani, M.; Beheshti, S.H. A comprehensive review of lithium ion capacitor: Development, modelling, thermal management and applications. J. Energy Storage 2021, 34, 102019. [Google Scholar] [CrossRef]
- Wang, X.; Liu, L.; Niu, Z. Carbon-based materials for lithium-ion capacitors. Mater. Chem. Front. 2019, 3, 1265–1279. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Hou, H.; Xu, W.; Duan, G.; He, S.; Liu, K.; Jiang, S. Recent progress in carbon-based materials for supercapacitor electrodes: A review. J. Mater. Sci. 2021, 56, 173–200. [Google Scholar] [CrossRef]
- Barbieri, O.; Hahn, M.; Herzog, A.; Kötz, R. Capacitance limits of high surface area activated carbons for double layer capacitors. Carbon 2005, 43, 1303–1310. [Google Scholar] [CrossRef]
- Xu, B.; Wu, F.; Chen, R.; Cao, G.; Chen, S.; Zhou, Z.; Yang, Y. Highly mesoporous and high surface area carbon: A high capacitance electrode material for EDLCs with various electrolytes. Electrochem. Commun. 2008, 10, 795–797. [Google Scholar] [CrossRef]
- Chmiola, J.; Yushin, G.; Gogotsi, Y.; Portet, C.; Simon, P.; Taberna, P.L. Anomalous increase in carbon capacitance at pore sizes less than 1 nanometer. Science 2006, 313, 1760–1763. [Google Scholar] [CrossRef]
- Kondrat, S.; Wu, P.; Qiao, R.; Kornyshev, A.A. Accelerating charging dynamics in subnanometre pores. Nat. Mater. 2014, 13, 387–393. [Google Scholar] [CrossRef]
- Xu, Z.; Ma, R.; Wang, X. Ultrafast, long-life, high-loading, and wide-temperature zinc ion supercapacitors. Energy Storage Mater. 2022, 46, 233–242. [Google Scholar] [CrossRef]
- Yin, J.; Zhang, W.; Alhebshi, N.A.; Salah, N.; Alsharee, H.N. Synthesis strategies of porous carbon for supercapacitor applications. Small Methods 2020, 4, 1900853. [Google Scholar] [CrossRef]
- Guo, Z.; Han, X.; Zhang, C.; He, S.; Liu, K.; Hu, J.; Yang, W.; Jian, S.; Jiang, S.; Duan, G. Activation of biomass-derived porous carbon for supercapacitors: A review. Chin. Chem. Lett. 2024, 35, 109007. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.; Hyeon, T. Recent progress in the synthesis of porous carbon materials. Adv. Mater. 2006, 18, 2073–2094. [Google Scholar] [CrossRef]
- Kim, M.; Leong, K.K.; Amiralian, N.; Bando, Y.; Ahamad, T.; Alshehri, S.M.; Yamauchi, Y. Nanoarchitectured MOF-derived porous carbons: Road to future carbon materials. Appl. Phys. Rev. 2024, 11, 041317. [Google Scholar] [CrossRef]
- Pavlenko, V.; Żółtowska, S.; Haruna, A.B.; Zahid, M.; Mansurov, Z.; Supiyeva, Z.; Galal, A.; Ozoemena, K.I.; Abbas, Q.; Jesionowski, T.; et al. A comprehensive review of template-assisted porous carbons: Modern preparation methods and advanced applications. Mater. Sci. Eng. R Rep. 2022, 149, 100682. [Google Scholar] [CrossRef]
- Sevilla, M.; Díez, N.; Fuertes, A.B. More sustainable chemical activation strategies for the production of porous carbons. ChemSusChem 2021, 14, 94–117. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zha, Q.; Qiu, J.; Han, X.; Guo, Y.; Li, Z.; Yuan, A.; Sun, X. Preparation of porous carbons from petroleum coke by different activation methods. Fuel 2005, 84, 1992–1997. [Google Scholar] [CrossRef]
- Bae, Y.S.; Yazaydın, A.O.; Snurr, R.Q. Evaluation of the BET method for determining surface areas of MOFs and zeolites that contain ultra-micropores. Langmuir 2010, 26, 5475–5483. [Google Scholar] [CrossRef]
- Khani, H.; Moradi, O. Influence of surface oxidation on the morphological and crystallographic structure of multi-walled carbon nanotubes via different oxidants. J. Nanostructure Chem. 2013, 3, 73. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing Kenneth, S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Levi, G.; Senneca, O.; Causa, M.; Salatino, P.; Lacovig, P.; Lizzit, S. Probing the chemical nature of surface oxides during coal char oxidation by high-resolution XPS. Carbon 2015, 90, 181–196. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; López-Ramón, M.V.; Carrasco-Marın, F. Changes in surface chemistry of activated carbons by wet oxidation. Carbon 2000, 38, 1995–2001. [Google Scholar] [CrossRef]
- Murillo-Acevedo, Y.; Giraldo, L.; Moreno-Piraján, J.C. Nanoparticles size distribution and phenol photodegradation with TiO2/C support obtained by phosphoric acid activation of palm kernel shell. Microporous Mesoporous Mater. 2020, 304, 109325. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R.; White, H.S. Electrochemical Methods: Fundamentals and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2022. [Google Scholar]
- Chen, J.; Li, K.; Luo, Y.; Guo, X.; Li, D.; Deng, M.; Huang, S.; Meng, Q. A flexible carbon counter electrode for dye-sensitized solar cells. Carbon 2009, 47, 2704–2708. [Google Scholar] [CrossRef]
- Wang, L.; Geng, M.; Ding, X.; Fang, C.; Zhang, Y.; Shi, S.; Zheng, Y.; Yang, K.; Zhan, C.; Wang, X. Research progress of the electrochemical impedance technique applied to the high-capacity lithium-ion battery. Int. J. Miner. Metall. Mater. 2021, 28, 538–552. [Google Scholar] [CrossRef]
- Su, C.; Suarez, D.L. Coordination of adsorbed boron: A FTIR spectroscopic study. Environ. Sci. Technol. 1995, 29, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Valle, C.; Reynard, B.; Daniel, I.; Lecuyer, C.; Martinez, I.; Chervin, J.C. Boron isotopic fractionation between minerals and fluids: New insights from in situ high pressure-high temperature vibrational spectroscopic data. Geochim. Cosmochim. Acta 2005, 69, 4301–4313. [Google Scholar] [CrossRef]
- Peak, D.; Luther IIIG, W.; Sparks, D.L. ATR-FTIR spectroscopic studies of boric acid adsorption on hydrous ferric oxide. Geochim. Cosmochim. Acta 2003, 67, 2551–2560. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Feng, K.; Li, Y.; Li, W.; Zhao, C.; Zhao, Y. Preparation of boron carbon nitride thin films by radio frequency magnetron sputtering. Appl. Surf. Sci. 2006, 252, 4185–4189. [Google Scholar] [CrossRef]
- Hernández, M.F.; Violini, M.A.; Serra, M.F.; Conconi, M.S.; Suarez, G.; Rendtorff, N.M. Boric acid (H3BO3) as flux agent of clay-based ceramics, B2O3 effect in clay thermal behavior and resultant ceramics properties. J. Therm. Anal. Calorim. 2020, 139, 1717–1729. [Google Scholar] [CrossRef]
- Panayiotatos, Y.; Logothetidis, S.; Handrea, M.; Kautek, W. Homogeneous and amorphous sputtered sp3-bonded BN films at RT: A stress, spectroscopic ellipsometry and XPS study. Diam. Relat. Mater. 2003, 12, 1151–1156. [Google Scholar] [CrossRef]
- Schild, D.; Ulrich, S.; Ye, J.; Stüber, M. XPS investigations of thick, oxygen-containing cubic boron nitride coatings. Solid State Sci. 2010, 12, 1903–1906. [Google Scholar] [CrossRef]
- Guimon, C.; Gonbeau, D.; Pfister-Guillouzo, G.; Dugne, O.; Guette, A.; Naslain, R.; Lahaye, M. XPS study of BN thin films deposited by CVD on SiC plane substrates. Surf. Interface Anal. 1990, 16, 440–445. [Google Scholar] [CrossRef]
- Karamanova, B.; Stoyanova, A.; Shipochka, M.; Veleva, S.; Stoyanova, R. Effect of alkaline-basic electrolytes on the capacitance performance of biomass-derived carbonaceous materials. Materials 2020, 13, 2941. [Google Scholar] [CrossRef]
- Shaibani, M.; Smith SJ, D.; Banerjee, P.C.; Konstas, K.; Zafari, A.; Lobo, D.E.; Nazari, M.; Hollenkamp, A.F.; Hill, M.R.; Majumder, M. Framework-mediated synthesis of highly microporous onion-like carbon: Energy enhancement in supercapacitors without compromising power. J. Mater. Chem. A 2017, 5, 2519–2529. [Google Scholar] [CrossRef]
- Bober, P.; Trchová, M.; Morávková, Z.; Kovářová, J.; Vulić, I.; Gavrilov, N.; Pašti, I.A.; Stejskal, J. Phosphorus and nitrogen-containing carbons obtained by the carbonization of conducting polyaniline complex with phosphites. Electrochim. Acta 2017, 246, 443–450. [Google Scholar] [CrossRef]
- Stepniak, I.; Ciszewski, A. New design of electric double layer capacitors with aqueous LiOH electrolyte as alternative to capacitor with KOH solution. J. Power Sources 2010, 195, 2564–2569. [Google Scholar] [CrossRef]
- Krause, A.; Kossyrev, P.; Oljaca, M.; Passerini, S.; Winter, M.; Balducci, A. Electrochemical double layer capacitor and lithium-ion capacitor based on carbon black. J. Power Sources 2011, 196, 8836–8842. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, N.; Sharma, Y.; Leu, J.; Tseng, T.Y. Synthesis of free-standing flexible rGO/MWCNT films for symmetric supercapacitor application. Nanoscale Res. Lett. 2019, 14, 266. [Google Scholar] [CrossRef]
- Wiggins-Camacho, J.D.; Stevenson, K.J. Effect of nitrogen concentration on capacitance, density of states, electronic conductivity, and morphology of N-doped carbon nanotube electrodes. J. Phys. Chem. C 2009, 113, 19082–19090. [Google Scholar] [CrossRef]
- Cohen, Y.; Aizenshtat, Z. Investigation of pyrolytically produced condensates of phenol-formaldehyde resins, in relation to their structure and decomposition mechanism. J. Anal. Appl. Pyrolysis 1992, 22, 153–178. [Google Scholar] [CrossRef]
- Liu, Y.; Bai, X.; Li, S. In-situ preparation of Pd nanoparticles in the pore channel of CMK-3 for Suzuki coupling reaction. Microporous Mesoporous Mater. 2018, 260, 40–44. [Google Scholar] [CrossRef]
- Akiya, N.; Savage, P.E. Roles of water for chemical reactions in high-temperature water. Chem. Rev. 2002, 102, 2725–2750. [Google Scholar] [CrossRef]
- Wang, L.; Yang, F.H.; Yang, R.T.; Miller, M.A. Effect of surface oxygen groups in carbons on hydrogen storage by spillover. Ind. Eng. Chem. Res. 2009, 48, 2920–2926. [Google Scholar] [CrossRef]
- Li, D.; Oganov, A.R.; Dong, X.; Zhou, X.F.; Zhu, Q.; Qian, G.; Dong, H. Nitrogen oxides under pressure: Stability, ionization, polymerization and superconductivity. Sci. Rep. 2015, 5, 16311. [Google Scholar] [CrossRef]
- Chen, P.; Wang, H.; Qiao, L.; Gu, M.; Luo, K.; Fan, J. Study of the NO formation characteristics and ammonia-N/coal-N transformation mechanism of ammonia-coal co-combustion in O2/CO2 atmosphere. Combust. Flame 2024, 270, 113756. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | d002 (g) | SSABET (m2·g−1) | SSADFT (m2·g−1) | SSA0.5–1.5 (m2·g−1) | SSA2.5–5.5 (m2·g−1) | VDFT (cm3·g−1) | Average Pore Size (nm) | Minimum Pore Size (nm) |
|---|---|---|---|---|---|---|---|---|
| AC | 0.3789 | 1026 | 914 | 394 | 3837 | 0.721 | 0.91 | 0.495 |
| AC-0 | 0.3365 | 983 | 851 | 437 | 370 | 0.679 | 0.89 | 0.504 |
| AC-H | 0.3932 | 1729 | 1699 | 1212 | 263 | 0.862 | 0.54 | 0.545 |
| Sample | d002 (g) | SSABET (m2·g−1) | SSADFT (m2·g−1) | SSA0.5–1.5 (m2·g−1) | SSA2.5–5.5 (m2·g−1) | VDFT (cm3·g−1) | Average Pore Size (nm) | Minimum Pore Size (nm) |
|---|---|---|---|---|---|---|---|---|
| AC-HBA1 | 0.3520 | 1610 | 1624 | 884 | 544 | 1.013 | 0.54 | 0.545 |
| AC-HBA2 | 0.3764 | 1619 | 1640 | 901 | 545 | 1.023 | 0.54 | 0.545 |
| AC-HBA3 | 0.3890 | 1620 | 1632 | 988 | 500 | 1.017 | 0.54 | 0.545 |
| AC-H | 0.3932 | 1729 | 1699 | 1212 | 263 | 0.862 | 0.54 | 0.545 |
| AC | 0.3789 | 1026 | 914 | 394 | 384 | 0.721 | 0.91 | 0.495 |
| Sample | C1A (F·g−1) | C20A/C1A (%) | Rs (Ω) | Rct (Ω) | Diffusion Coefficient (10−16) |
|---|---|---|---|---|---|
| AC-HBA1 | 123 | 18.6 | 1.81 | 0.45 | 7.52 |
| AC-HBA2 | 144 | 30.02 | 1.67 | 0.34 | 21.68 |
| AC-HBA3 | 76 | 11.01 | 1.65 | 0.41 | 12.00 |
| AC-H | 132 | 7.44 | 1.13 | 0.67 | 6.66 |
| AC | 100 | 10.72 | 1.79 | 0.32 | 4.56 |
| Samples | AC | PF | BA | H2O |
|---|---|---|---|---|
| AC-0 | 1 g | 0 | 0 | 20 mL |
| AC-H | 1 g | 0.1 mL | 0 | 20 mL |
| AC-BA | 1 g | 0 | 0.01 g | 20 mL |
| AC-HBA1 | 1 g | 0.1 mL | 0.01 g | 20 mL |
| AC-HBA2 | 1 g | 0.1 mL | 0.001 g | 20 mL |
| AC-HBA3 | 1 g | 0.1 mL | 0.0001 g | 20 mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, Y.; Zhang, F.; Wang, S.; Wei, S.; Zhang, X.; Dong, W.; Shen, D.; Tang, S.; Liu, F.; Chen, Y.; et al. Synergistic Enhancement of Capacitive Performance in Porous Carbon by Phenolic Resin and Boric Acid. Molecules 2025, 30, 1228. https://doi.org/10.3390/molecules30061228
Xia Y, Zhang F, Wang S, Wei S, Zhang X, Dong W, Shen D, Tang S, Liu F, Chen Y, et al. Synergistic Enhancement of Capacitive Performance in Porous Carbon by Phenolic Resin and Boric Acid. Molecules. 2025; 30(6):1228. https://doi.org/10.3390/molecules30061228
Chicago/Turabian StyleXia, Yingkai, Fengzhi Zhang, Shuo Wang, Shuang Wei, Xu Zhang, Wei Dong, Ding Shen, Shuwei Tang, Fengxia Liu, Yuehui Chen, and et al. 2025. "Synergistic Enhancement of Capacitive Performance in Porous Carbon by Phenolic Resin and Boric Acid" Molecules 30, no. 6: 1228. https://doi.org/10.3390/molecules30061228
APA StyleXia, Y., Zhang, F., Wang, S., Wei, S., Zhang, X., Dong, W., Shen, D., Tang, S., Liu, F., Chen, Y., & Yang, S. (2025). Synergistic Enhancement of Capacitive Performance in Porous Carbon by Phenolic Resin and Boric Acid. Molecules, 30(6), 1228. https://doi.org/10.3390/molecules30061228