

Identification of Anticancer Target Combinations to Treat Pancreatic Cancer and Its Associated Cachexia Using Constraint-Based Modeling

Abstract

1. Introduction
2. Results and Discussion
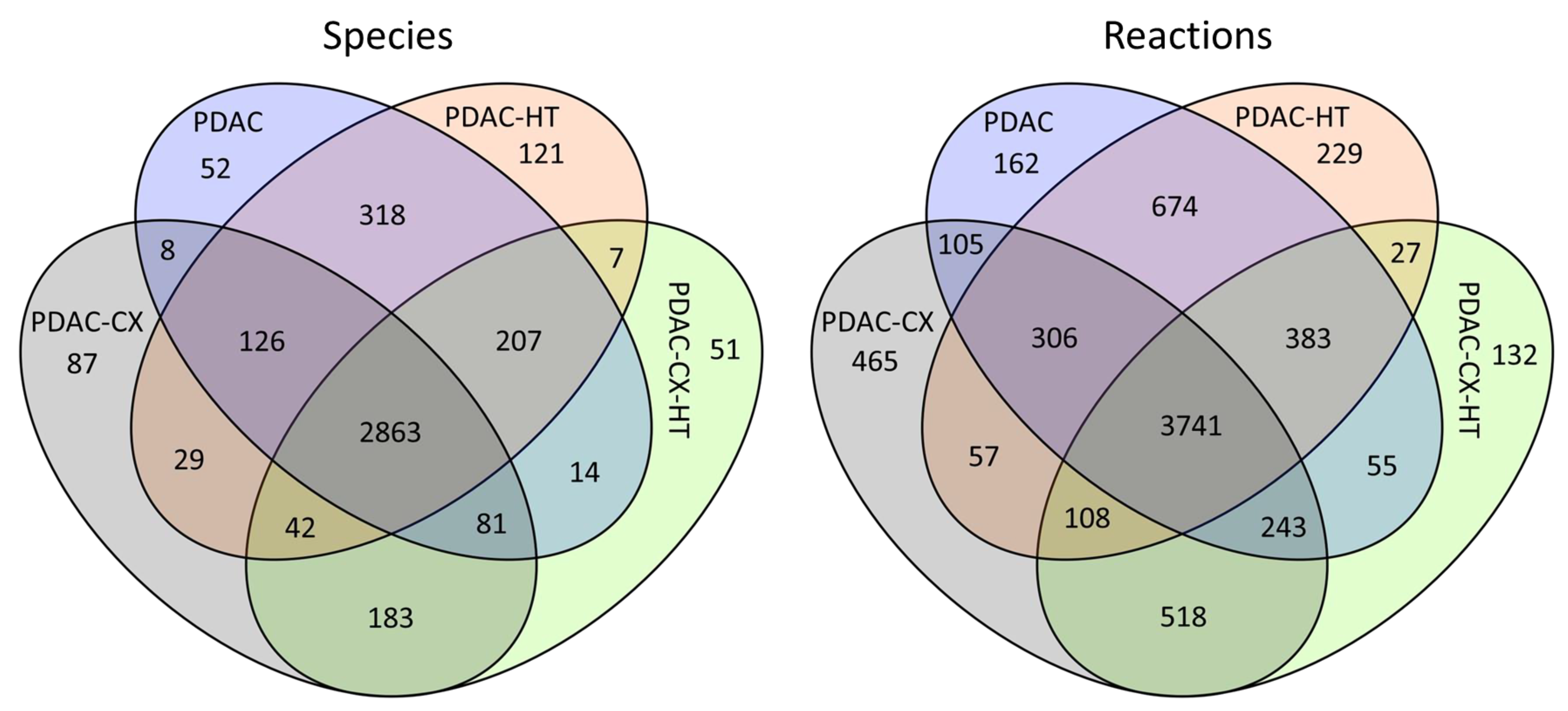
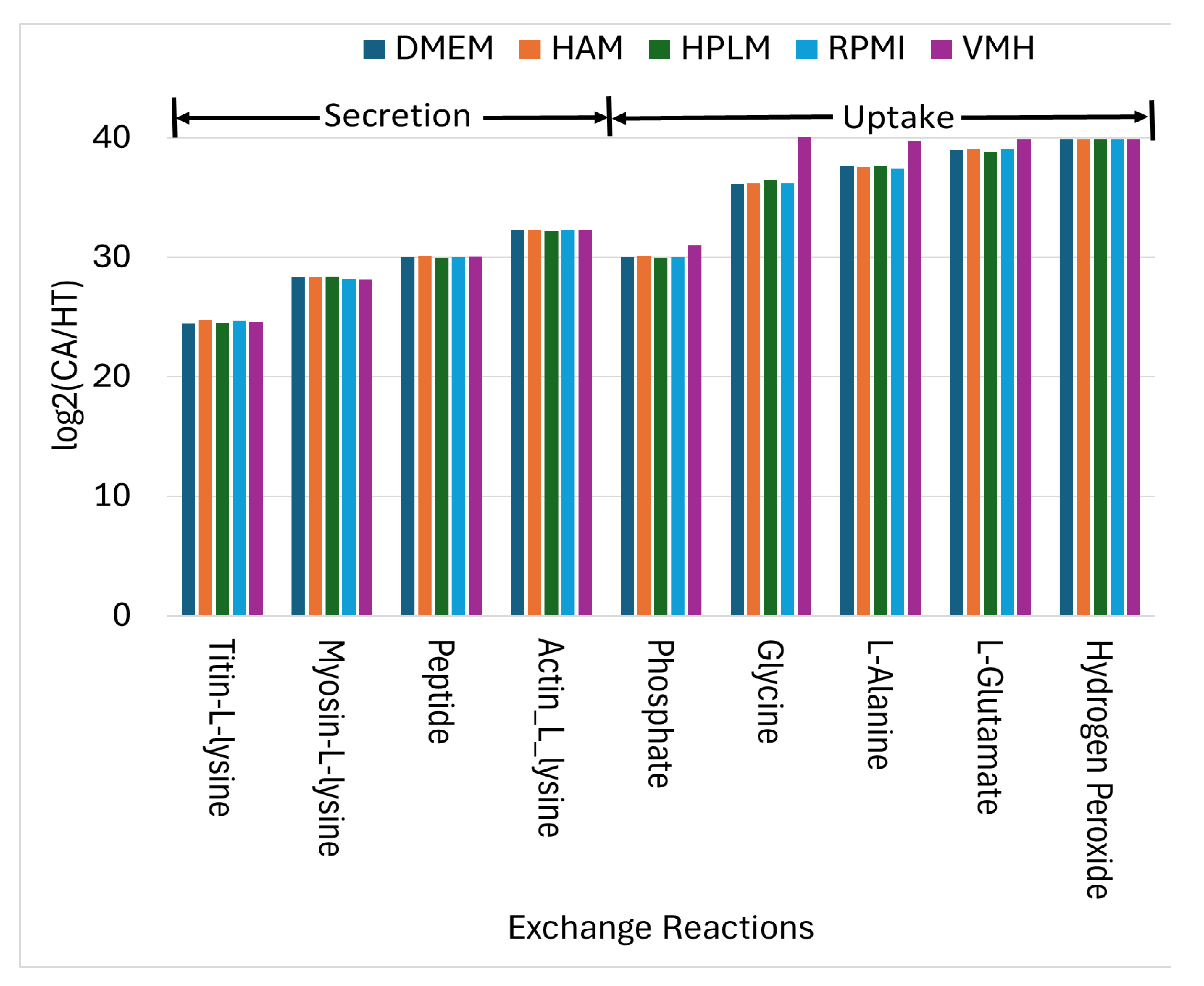
2.1. Cell-Specific Genome-Scale Metabolic Models
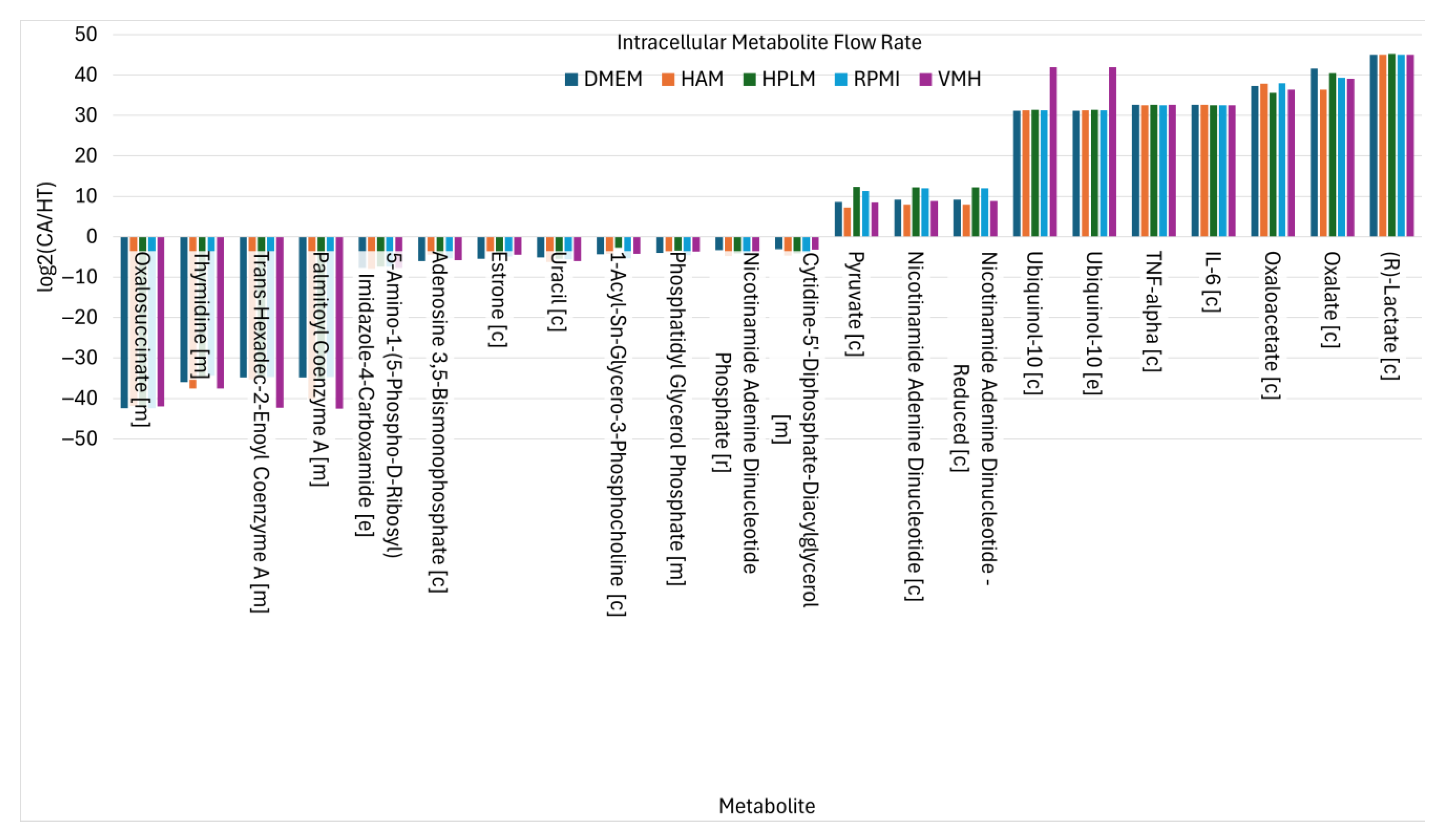
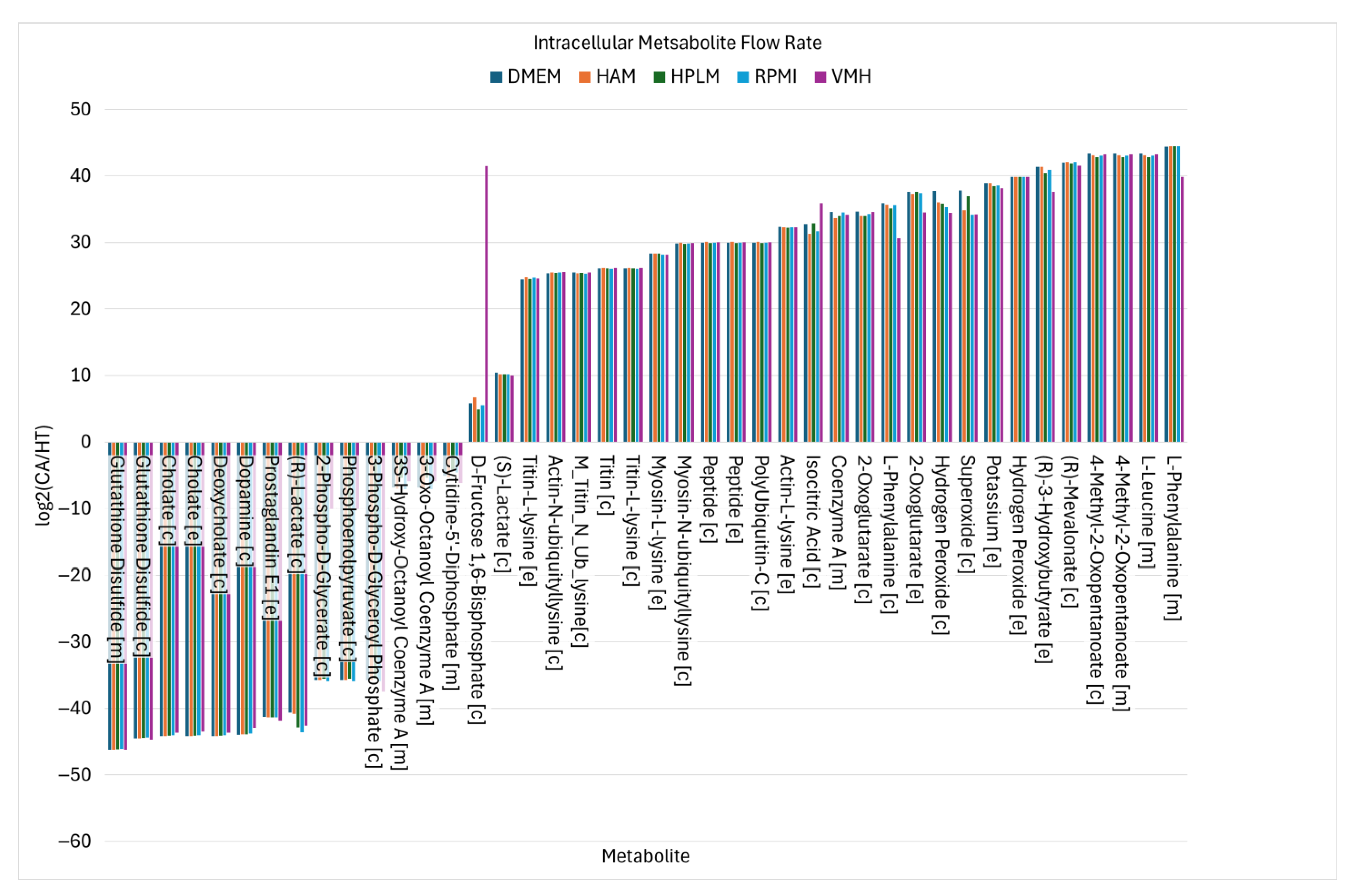
2.2. Identified Potential Biomarkers
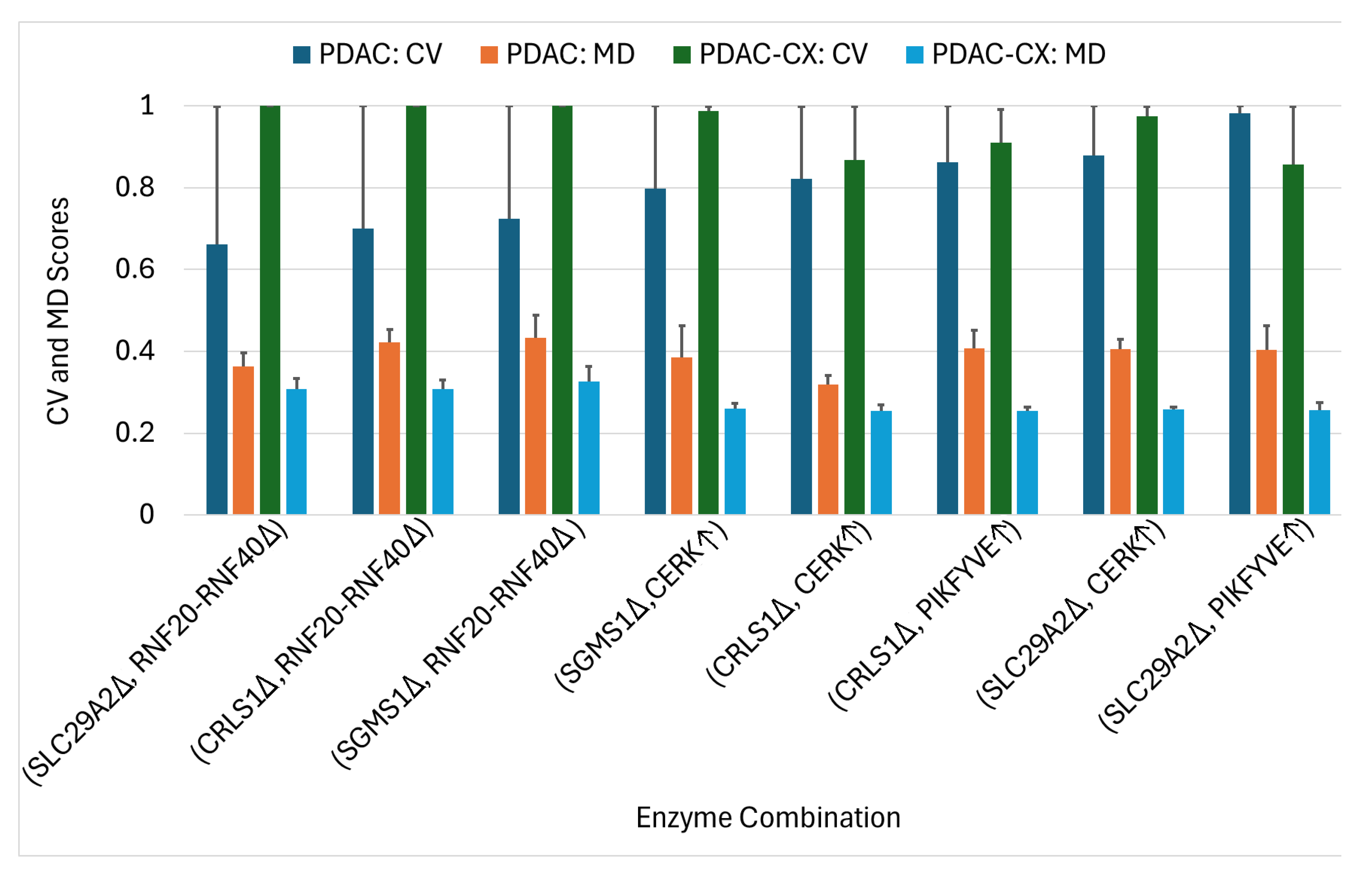
2.3. Enzyme Targets Predicted Using Constraint-Based Modeling
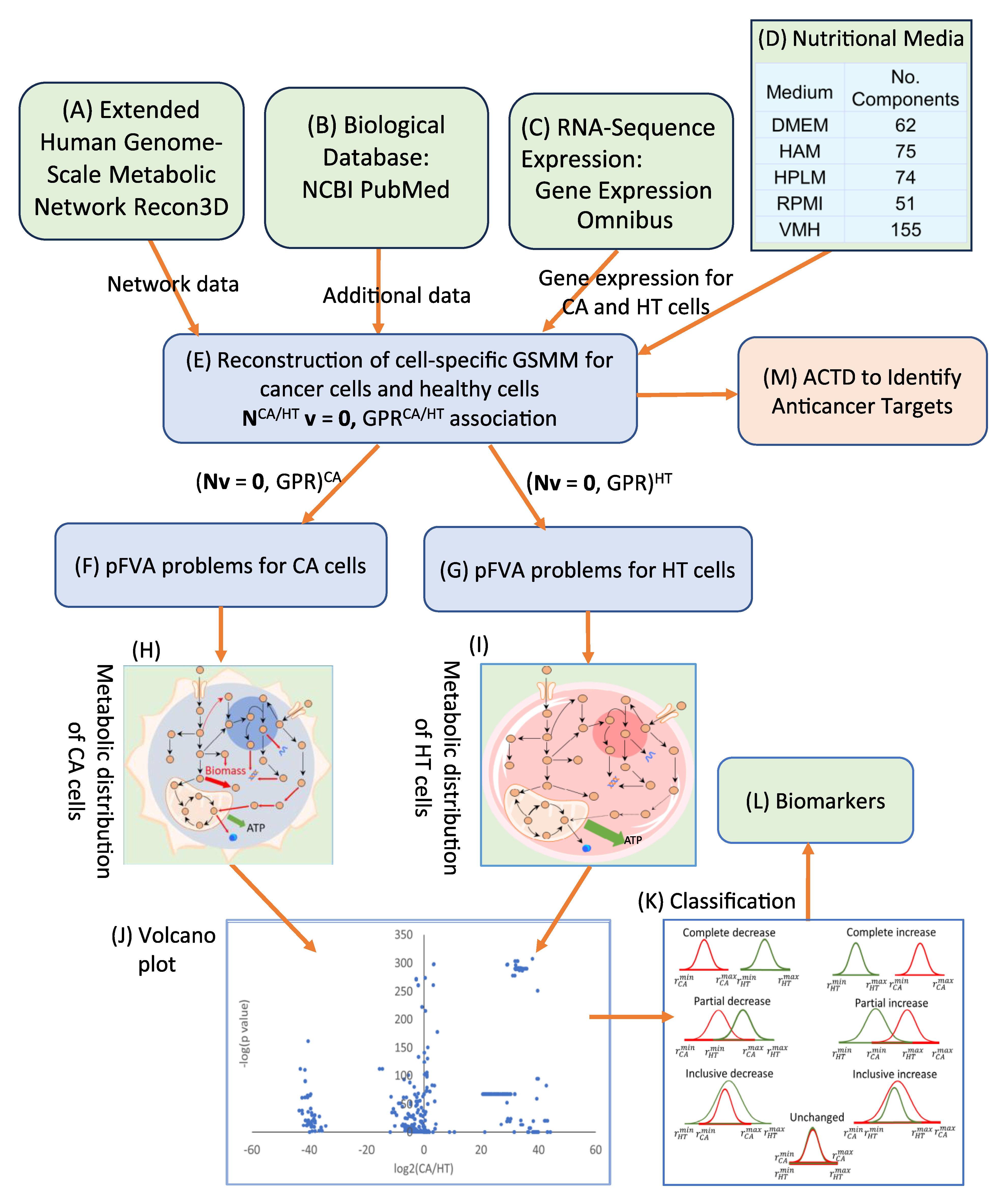
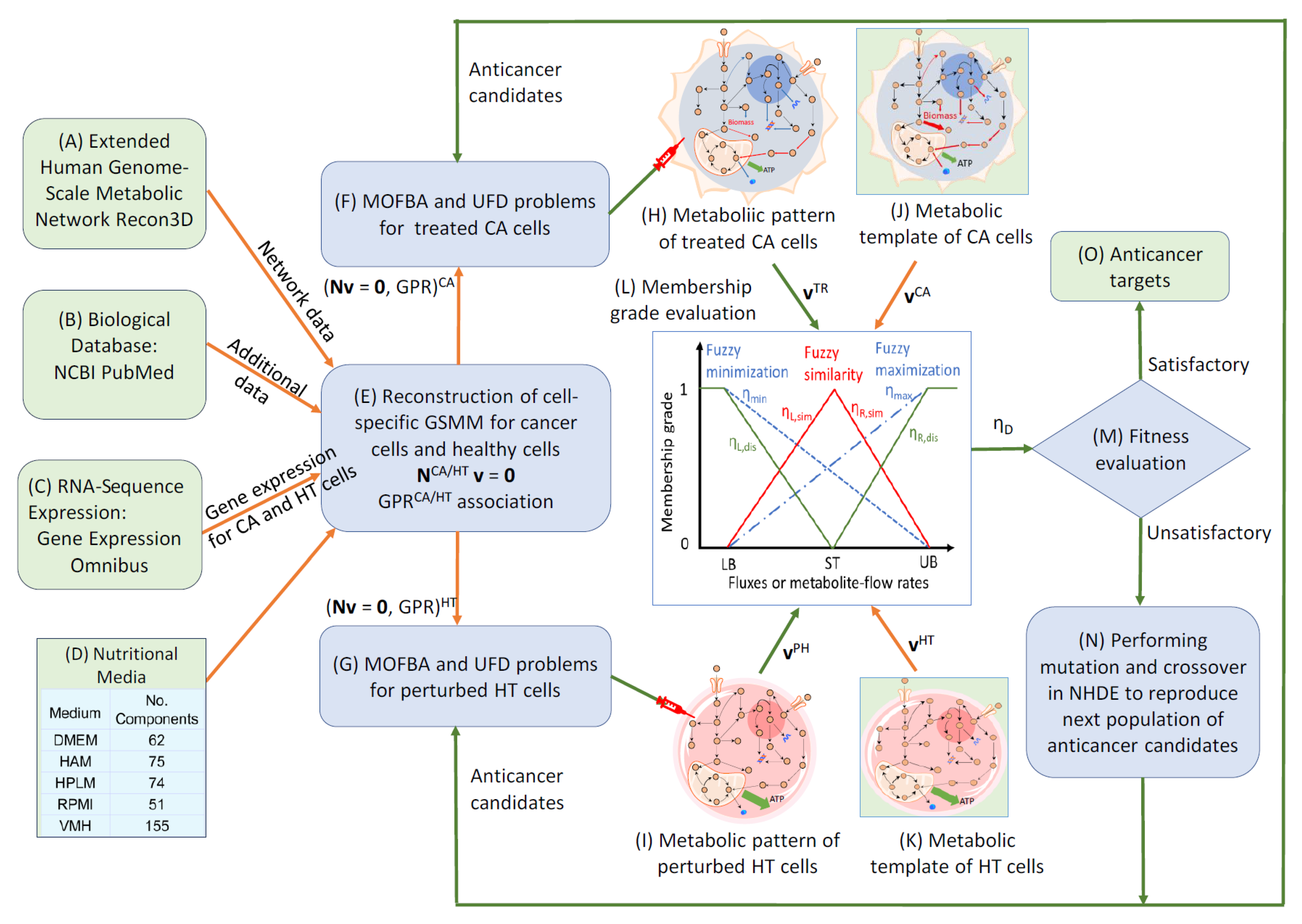
3. Methods
3.1. Extension of Human Genome-Scale Metabolic Network
3.2. Parsimonious Multi-Objective Flux Balance Analysis
3.3. Parsimonious Metabolite Flow Variability Analysis
3.4. Anticancer Target Discovery Framework
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Kang, Y.P.; Ward, N.P.; DeNicola, G.M. Recent advances in cancer metabolism: A technological perspective. Exp. Mol. Med. 2018, 50, 31. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Johnen, H.; Lin, S.; Kuffner, T.; Brown, D.A.; Tsai, V.W.W.; Bauskin, A.R.; Wu, L.; Pankhurst, G.; Jiang, L.; Junankar, S.; et al. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat. Med. 2007, 13, 1333–1340. [Google Scholar] [CrossRef]
- Suriben, R.; Chen, M.; Higbee, J.; Oeffinger, J.; Ventura, R.; Li, B.; Mondal, K.; Gao, Z.; Ayupova, D.; Taskar, P.; et al. Antibody-mediated inhibition of GDF15–GFRAL activity reverses cancer cachexia in mice. Nat. Med. 2020, 26, 1264–1270. [Google Scholar] [CrossRef]
- Rohm, M.; Herzig, S. An antibody attack against body wasting in cancer. Cell Metab. 2020, 32, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Aydogdu, T.; Kunzevitzky, N.; Guttridge, D.C.; Khuri, S.; Koniaris, L.G.; Zimmers, T.A.; Parise, G. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 2011, 6, e22538. [Google Scholar] [CrossRef] [PubMed]
- Talbert, E.E.; Cuitiño, M.C.; Ladner, K.J.; Rajasekerea, P.V.; Siebert, M.; Shakya, R.; Leone, G.W.; Ostrowski, M.C.; Paleo, B.; Weisleder, N.; et al. Modeling human cancer-induced cachexia. Cell Rep. 2019, 28, 1612–1622.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, W.; Gu, X.; Miao, C.; Feng, L.; Shen, Q.; Liu, X.; Zhang, X. GDF-15 in tumor-derived exosomes promotes muscle atrophy via Bcl-2/caspase-3 pathway. Cell Death Discov. 2022, 8, 162. [Google Scholar] [CrossRef] [PubMed]
- Bloch, S.A.; Lee, J.Y.; Syburra, T.; Rosendahl, U.; Griffiths, M.J.; Kemp, P.R.; Polkey, M.I. Increased expression of GDF-15 may mediate ICU-acquired weakness by down-regulating muscle microRNAs. Thorax 2015, 70, 219–228. [Google Scholar] [CrossRef]
- Garfield, B.E.; Crosby, A.; Shao, D.; Yang, P.; Read, C.; Sawiak, S.; Moore, S.; Parfitt, L.; Harries, C.; Rice, M.; et al. Growth/differentiation factor 15 causes TGFβ-activated kinase 1-dependent muscle atrophy in pulmonary arterial hypertension. Thorax 2019, 74, 164–176. [Google Scholar] [CrossRef]
- Cheng, C.T.; Wang, T.Y.; Chen, P.R.; Wu, W.H.; Lai, J.M.; Sawiak, S.; Moore, S.; Parfitt, L.; Harries, C.; Rice, M.; et al. Computer-aided design for identifying anticancer targets in genome-scale metabolic models of colon cancer. Biology 2021, 10, 1115. [Google Scholar] [CrossRef]
- Wang, Y.T.; Lin, M.R.; Chen, W.C.; Wu, W.H.; Wang, F.S. Optimization of a modeling platform to predict oncogenes from genome-scale metabolic networks of non-small-cell lung cancers. FEBS Openbio 2021, 11, 2078–2094. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.S.; Zhang, H.X. Identification of anticancer enzymes and biomarkers for hepatocellular carcinoma through constraint-based modeling. Molecules 2024, 29, 2594. [Google Scholar] [CrossRef]
- Brunk, E.; Sahoo, S.; Zielinski, D.C.; Altunkaya, A.; Dräger, A.; Mih, N.; Gatto, F.; Nilsson, A.; Preciat Gonzalez, G.A.; Aurich, M.K.; et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 2018, 36, 272–281. [Google Scholar] [CrossRef]
- Yang, S.; Tang, W.; Azizian, A.; Gaedcke, J.; Ströbel, P.; Wang, L.; Cawley, H.; Ohara, Y.; Valenzuela, P.; Zhang, L.; et al. Dysregulation of HNF1B/Clusterin axis enhances disease progression in a highly aggressive subset of pancreatic cancer patients. Carcinogenesis 2022, 43, 1198–1210. [Google Scholar] [CrossRef]
- Narasimhan, A.; Zhong, X.; Au, E.P.; Ceppa, E.P.; Nakeeb, A.; House, M.G.; Zyromski, N.J.; Schmidt, C.M.; Schloss, K.N.H.; Schloss, D.E.I.; et al. Profiling of adipose and skeletal muscle in human pancreatic cancer cachexia reveals distinct gene profiles with convergent pathways. Cancers 2021, 13, 1975. [Google Scholar] [CrossRef]
- Mitsunaga, S.; Ikeda, M.; Nakachi, K.; Ohno, I.; Shimizu, S.; Takahashi, H.; Okuyama, H.; Inagaki, M.; Furuse, J.; Ochiai, A. Use of elevated IL-1 to predict prognosis in patients with advanced pancreatic cancer with high IL-6 and wasting condition. J. Clin. Oncol. 2011, 29, e14604. [Google Scholar] [CrossRef]
- Balsano, R.; Kruize, Z.; Lunardi, M.; Comandatore, A.; Barone, M.; Cavazzoni, A.; Cecconi, A.D.R.; Morelli, L.; Wilmink, H.; Tiseo, M.; et al. Transforming growth factor-beta signaling in cancer-induced cachexia: From molecular pathways to the clinics. Cells 2022, 11, 2671. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Guerrico, A.M.G.; Nin, V.; Camacho-Pereira, J.; Escande, C.; Barbosa, M.T.; Chini, E.N. Targeting of NAD metabolism in pancreatic cancer cells: Potential novel therapy for pancreatic tumors. Clin. Cancer Res. 2013, 20, 120–130. [Google Scholar] [CrossRef]
- Gao, F.; Sun, K.; Wang, S.; Zhang, X.; Ba, X. Lactate metabolism reprogramming in PDAC: Potential for tumor therapy. Biochim. Biophys. Acta 2025, 1880, 189373. [Google Scholar] [CrossRef]
- Eze-Odurukwe, A.; Rehman, A.; Ayinla, L.; Anika, N.N.; Shahid, R.; Ugwuoru, A.L.; Mansoor, M.; Kamran, M. Metabolite biomarkers for early detection of pancreatic ductal adenocarcinoma: A systematic review. Cureus 2024, 16, e74528. [Google Scholar] [CrossRef]
- Zhao, R.; Chen, S.; Cui, W.; Xie, C.; Zhang, A. PTPN1 is a prognostic biomarker related to cancer immunity and drug sensitivity: From pan-cancer analysis to validation in breast cancer. Front. Immunol. 2023, 14, 1232047. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Mallidis, C.; Bhasin, S.; Mahabadi, V.; Artaza, J.; Gonzalez-Cadavid, N.; Arias, J.; Salehian, B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am. J. Physiol. Endocrinol. Metab. 2003, 5, E363–E371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Jin, B.; Li, Y.P. C/EBPβ mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 2011, 30, 4323–4335. [Google Scholar] [CrossRef] [PubMed]
- Pierucci, F.; Frati, A.; Battistini, C.; Penna, F.; Costelli, P.; Meacci, E. Control of skeletal muscle atrophy associated to cancer or corticosteroids by ceramide kinase. Cancers 2021, 13, 3285. [Google Scholar] [CrossRef]
- Ikonomov, O.C.; Sbrissa, D.; Delvecchio, K.; Feng, H.Z.; Cartee, G.D.; Jin, J.-P.; Shisheva, A. Muscle-specific Pikfyve gene disruption causes glucose intolerance, insulin resistance, adiposity, and hyperinsulinemia but not muscle fiber-type switching. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E119–E131. [Google Scholar] [CrossRef]
- Haynie, D.T. Biological Thermodynamics, 2nd ed.; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Chu, S.W.; Wang, F.S. Fuzzy optimization for identifying antiviral targets for treating SARS-CoV-2 infection in the heart. BMC Bioinform. 2023, 24, 364. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.S.; Chen, K.L.; Chu, S.W. Human/SARS-CoV-2 genome-scale metabolic modeling to discover potential antiviral targets for COVID-19. J. Taiwan Inst. Chem. Eng. 2023, 133, 10427. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medium | Type | CI | PI | II | ID | PD | CD | Total |
|---|---|---|---|---|---|---|---|---|
| DMEM | PDAC | 97 | 240 | 12 | 26 | 4 | 145 | 524 |
| PDAC-CX | 80 | 134 | 16 | 58 | 24 | 116 | 428 | |
| HAM | PDAC | 132 | 212 | 12 | 20 | 6 | 125 | 507 |
| PDAC-CX | 78 | 130 | 20 | 72 | 20 | 154 | 474 | |
| HPLM | PDAC | 125 | 211 | 17 | 32 | 7 | 91 | 483 |
| PDAC-CX | 88 | 175 | 17 | 112 | 26 | 94 | 512 | |
| RPMI | PDAC | 134 | 222 | 11 | 15 | 6 | 148 | 536 |
| PDAC-CX | 95 | 146 | 20 | 89 | 21 | 125 | 496 | |
| VMH | PDAC | 63 | 203 | 10 | 31 | 8 | 118 | 433 |
| PDAC-CX | 140 | 171 | 25 | 43 | 29 | 96 | 504 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.-S.; Wu, C.-K.; Huang, K.-T. Identification of Anticancer Target Combinations to Treat Pancreatic Cancer and Its Associated Cachexia Using Constraint-Based Modeling. Molecules 2025, 30, 3200. https://doi.org/10.3390/molecules30153200
Wang F-S, Wu C-K, Huang K-T. Identification of Anticancer Target Combinations to Treat Pancreatic Cancer and Its Associated Cachexia Using Constraint-Based Modeling. Molecules. 2025; 30(15):3200. https://doi.org/10.3390/molecules30153200
Chicago/Turabian StyleWang, Feng-Sheng, Ching-Kai Wu, and Kuang-Tse Huang. 2025. "Identification of Anticancer Target Combinations to Treat Pancreatic Cancer and Its Associated Cachexia Using Constraint-Based Modeling" Molecules 30, no. 15: 3200. https://doi.org/10.3390/molecules30153200
APA StyleWang, F.-S., Wu, C.-K., & Huang, K.-T. (2025). Identification of Anticancer Target Combinations to Treat Pancreatic Cancer and Its Associated Cachexia Using Constraint-Based Modeling. Molecules, 30(15), 3200. https://doi.org/10.3390/molecules30153200