
Design, Docking Analysis, and Structure–Activity Relationship of Ferrocene-Modified Tyrosine Kinase Inhibitors: Insights into BCR-ABL Interactions

 , , ,
, , ,  ,
,  and
and 
Abstract

1. Introduction


2. Results and Discussion
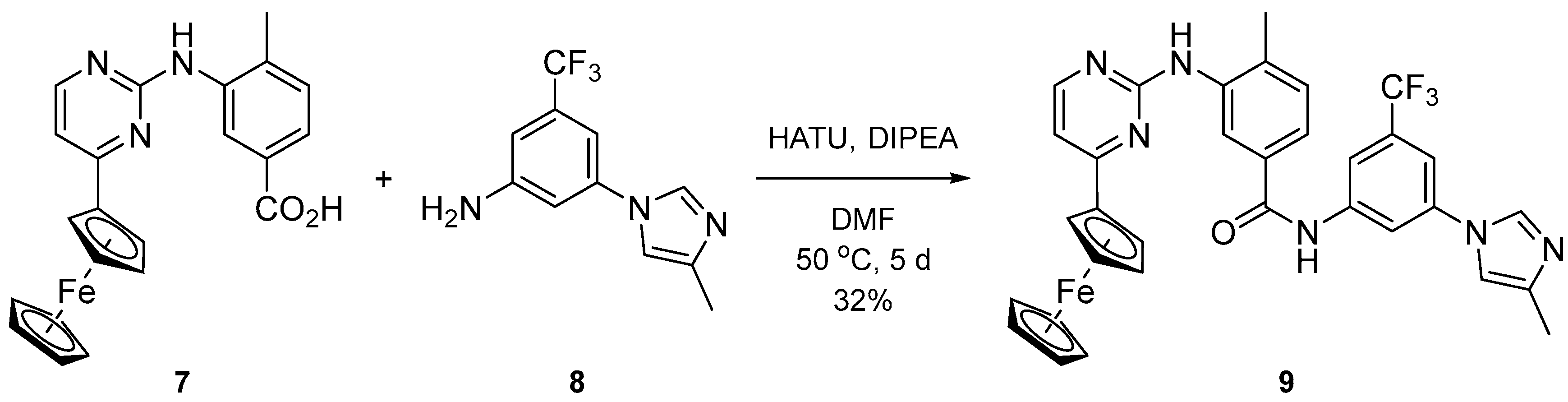
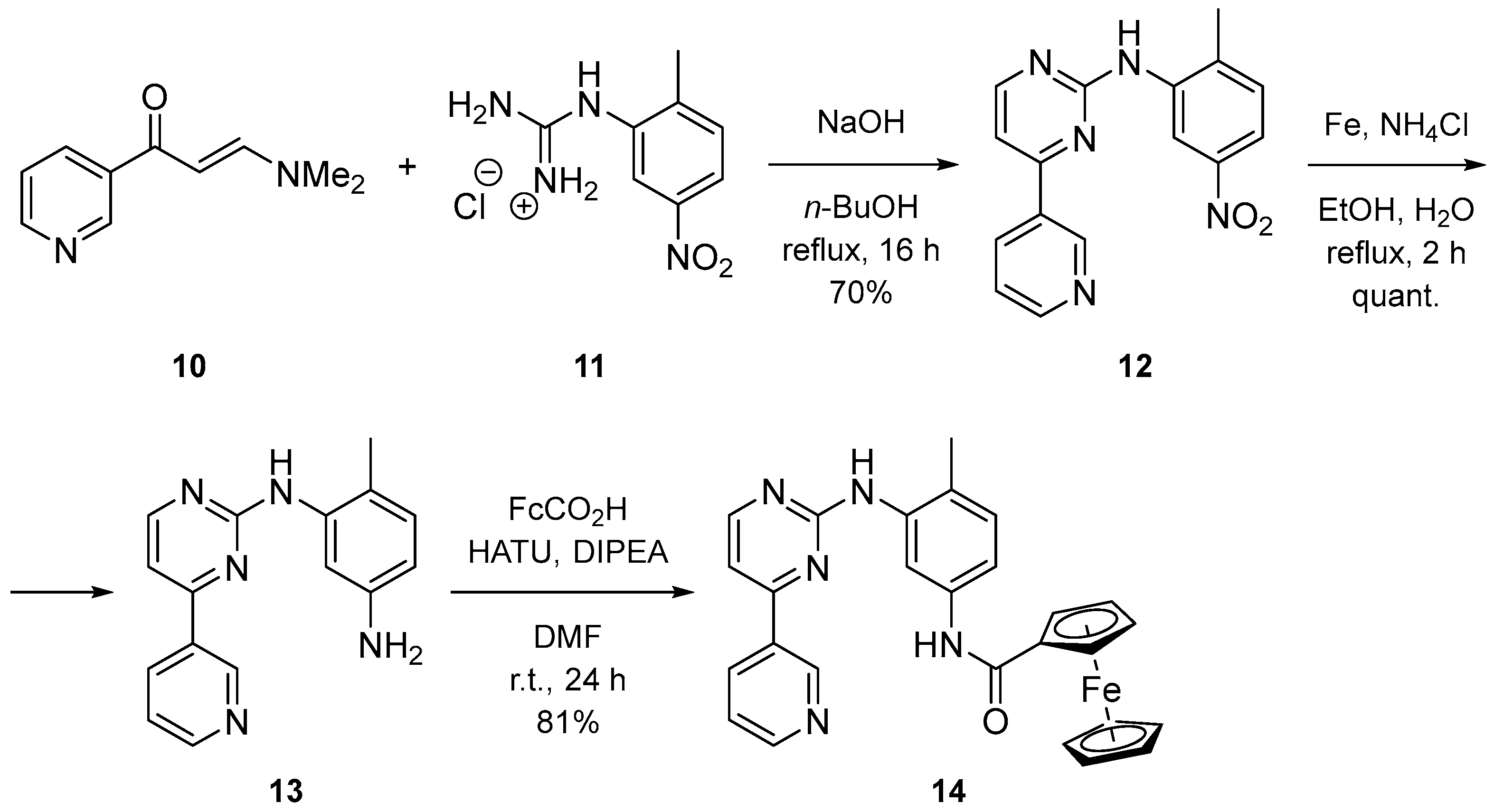
2.1. Syntesis
2.2. Results from In Vitro Cytotoxicity Studies
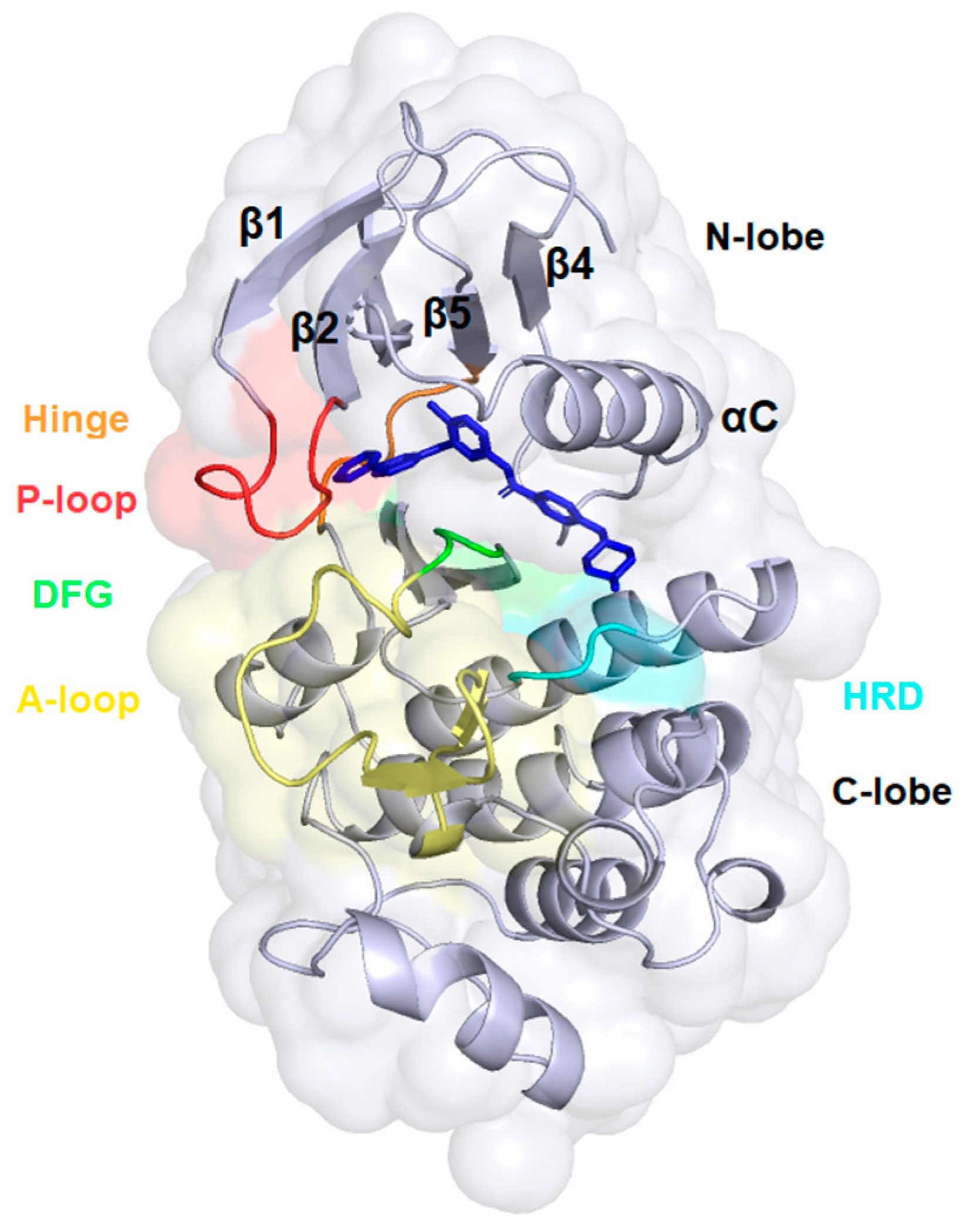
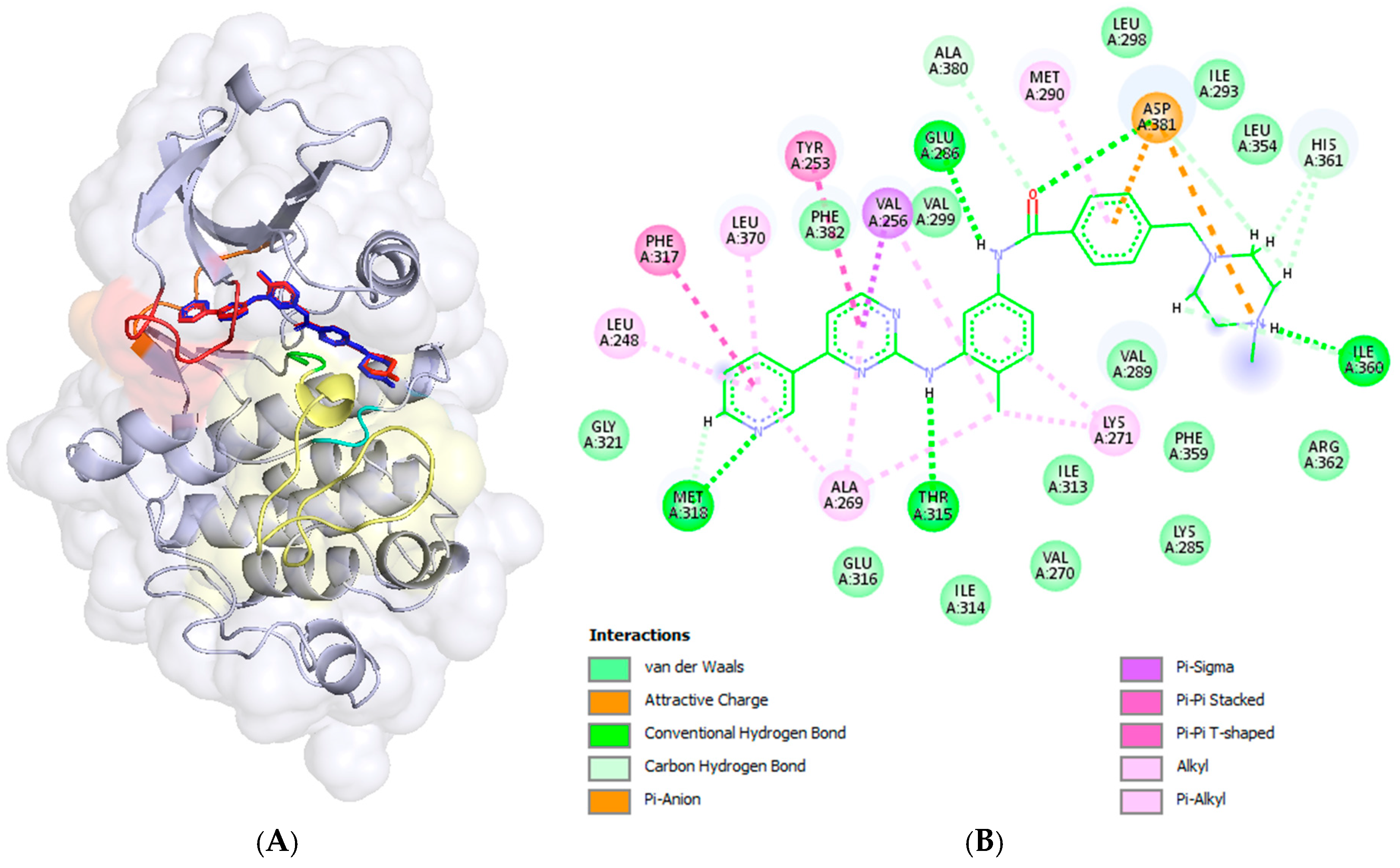
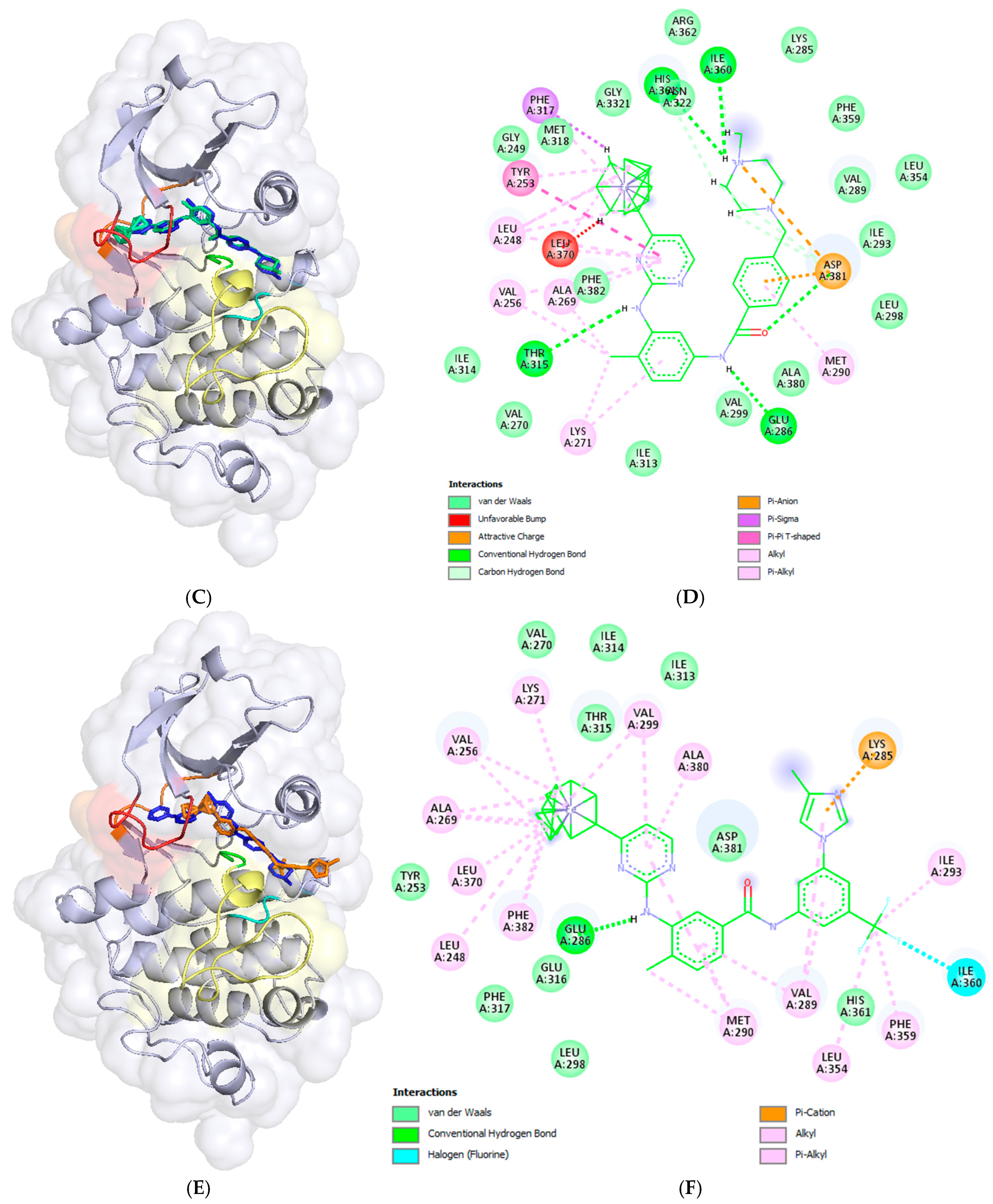
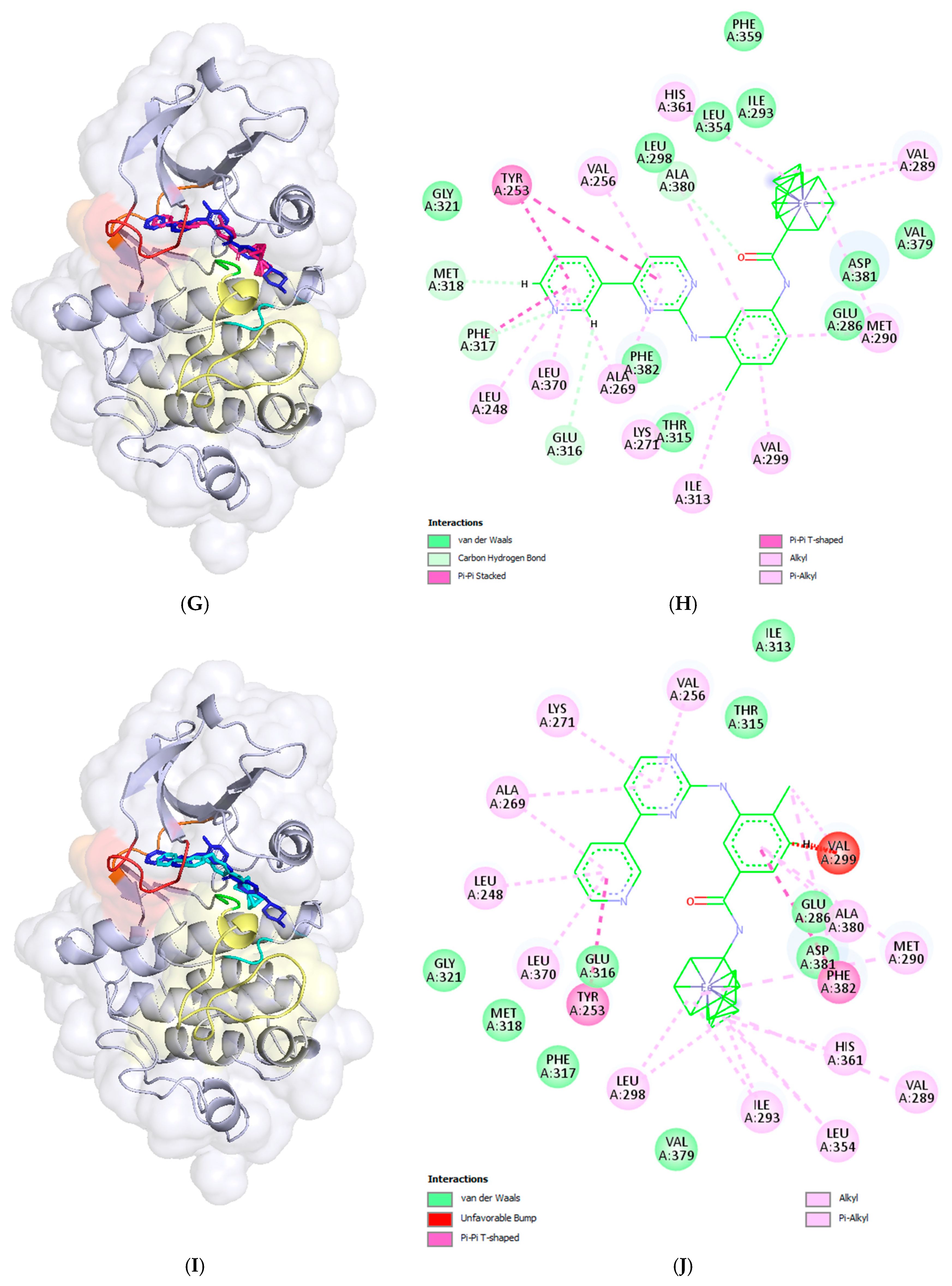
2.3. Docking Analysis—In Silico Modelling of the Interactions Between the Novel Ferrocene Derivatives and c-Abl Tyrosine Kinase
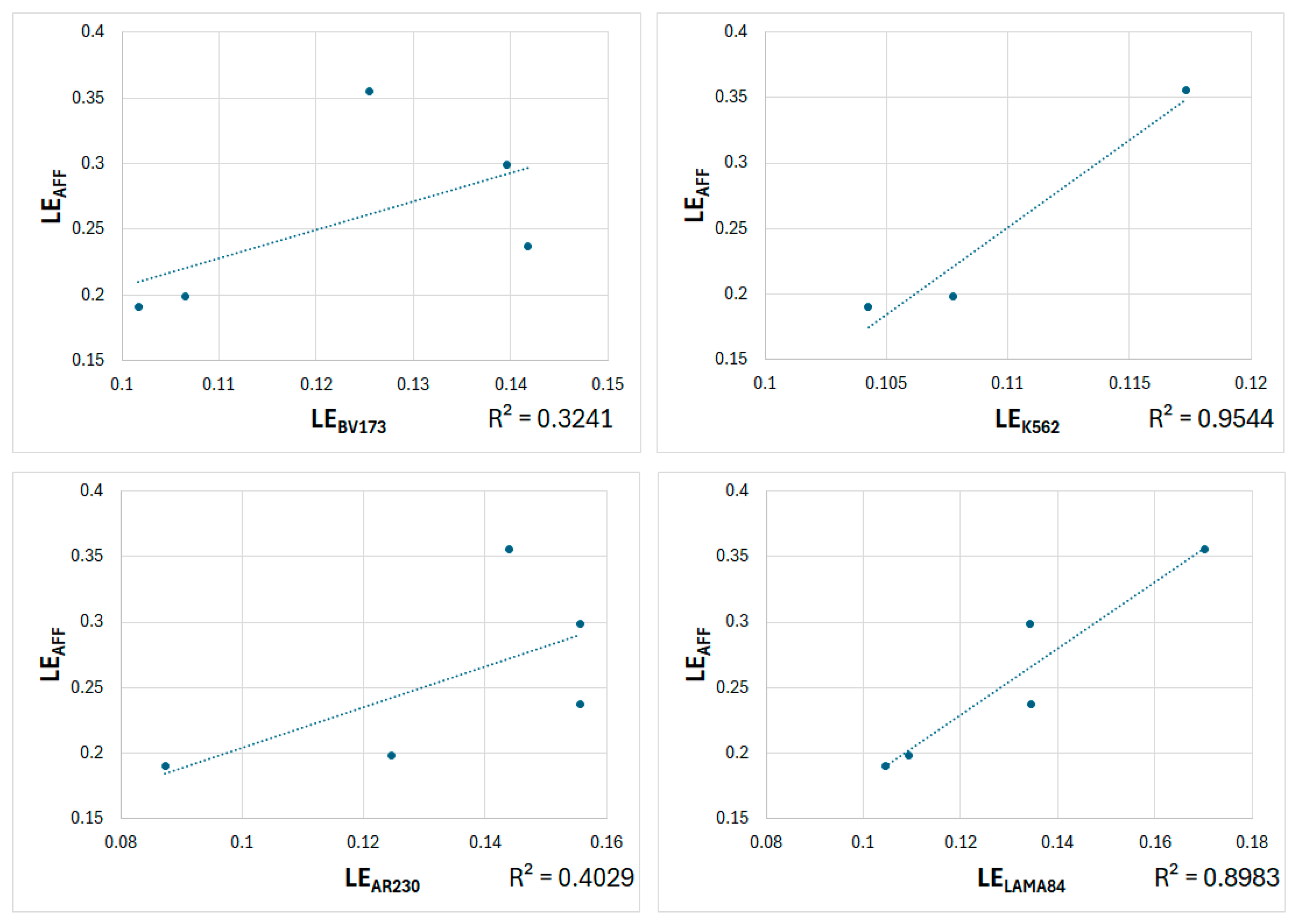
2.4. Comparative Analysis Between Experimental and Predicted Ligand Efficiencies
3. Materials and Methods
3.1. Synthesis
3.1.1. General
3.1.2. Benzyl (4-Methyl-3-nitrophenyl)carbamate (1a)
3.1.3. Benzyl (3-Amino-4-methylphenyl)carbamate (2a)
3.1.4. Benzyl (3-Guanidino-4-methylphenyl)carbamate Hydrochloride (3a)
3.1.5. Benzyl (4-Methyl-3-((4-ferrocenylpyrimidin-2-yl)amino)phenyl)carbamate (4a)
3.1.6. N-(4-Methyl-3-nitrophenyl)acetamide (1b)
3.1.7. N-(3-Amino-4-methylphenyl)acetamide (2b)
3.1.8. N-(3-Guanidino-4-methylphenyl)acetamide Hydrochloride (3b)
3.1.9. N-(4-Methyl-3-((4-ferrocenylpyrimidin-2-yl)amino)phenyl)acetamide (4b)
3.1.10. 6-Methyl-N1-(4-ferrocenylpyrimidin-2-yl)benzene-1,3-diamine (5)
3.1.11. N-(4-Methyl-3-((4-ferrocenylpyrimidin-2-yl)amino)phenyl)-4-((4-methylpiperazin-1-yl)methyl)benzamide (6)
3.1.12. 4-Methyl-N-(3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-ferrocenylpyrimidin-2-yl)amino)benzamide (9)
3.1.13. 1-(2-Methyl-5-nitrophenyl)guanidine Hydrochloride (11)
3.1.14. N-(2-Methyl-5-nitrophenyl)-4-(pyridin-3-yl)pyrimidin-2-amine (12)
3.1.15. 6-Methyl-N1-(4-(pyridin-3-yl)pyrimidin-2-yl)benzene-1,3-diamine (13)
3.1.16. N-(4-Methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)ferrocenecarboxamide (14)
3.1.17. 4-Methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)benzoic Acid (17)
3.1.18. N-Ferrocenyl-4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)benzamide (18)
3.2. Cell Lines and Culture Conditions
3.3. Cell Viability Assay
3.4. Docking Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jaouen, G.; Vessieres, A.; Butler, I.S. Bioorganometallic Chemistry: A Future Direction for Transition Metal Organometallic Chemistry? Acc. Chem. Res. 1993, 26, 361–369. [Google Scholar] [CrossRef]
- Fish, R.H.; Jaouen, G. Bioorganometallic Chemistry: Structural Diversity of Organometallic Complexes with Bioligands and Molecular Recognition Studies of Several Supramolecular Hosts with Biomolecules, Alkali-Metal Ions, and Organometallic Pharmaceuticals. Organometallics 2003, 22, 2166–2177. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Dyson, P.J. Bioorganometallic Chemistry—From Teaching Paradigms to Medicinal Applications. Chem. Soc. Rev. 2009, 38, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Fouda, M.F.R.; Abd-Elzaher, M.M.; Abdelsamaia, R.A.; Labib, A.A. On the Medicinal Chemistry of Ferrocene. Appl. Organomet. Chem. 2007, 21, 613–625. [Google Scholar] [CrossRef]
- Patra, M.; Gasser, G. The Medicinal Chemistry of Ferrocene and Its Derivatives. Nat. Rev. Chem. 2017, 1, 0066. [Google Scholar] [CrossRef]
- Van Staveren, D.R.; Metzler-Nolte, N. Bioorganometallic Chemistry of Ferrocene. Chem. Rev. 2004, 104, 5931–5986. [Google Scholar] [CrossRef]
- Singh, A.; Lumb, I.; Mehra, V.; Kumar, V. Ferrocene-Appended Pharmacophores: An Exciting Approach for Modulating the Biological Potential of Organic Scaffolds. Dalton Trans. 2019, 48, 2840–2860. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen Type Anti Cancer Drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef]
- Top, S.; Tang, J.; Vessières, A.; Carrez, D.; Provot, C.; Jaouen, G. Ferrocenyl Hydroxytamoxifen: A Prototype for a New Range of Oestradiol Receptor Site-Directed Cytotoxics. Chem. Commun. 1996, 955–956. [Google Scholar] [CrossRef]
- Nguyen, A.; Vessières, A.; Hillard, E.A.; Top, S.; Pigeon, P.; Jaouen, G. Ferrocifens and Ferrocifenols as New Potential Weapons against Breast Cancer. Chimia 2007, 61, 716. [Google Scholar] [CrossRef]
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic Anticancer Compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef]
- Schatzschneider, U.; Metzler-Nolte, N. New Principles in Medicinal Organometallic Chemistry. Angew. Chem. Int. Ed. 2006, 45, 1504–1507. [Google Scholar] [CrossRef]
- Vessières, A.; Corbet, C.; Heldt, J.M.; Lories, N.; Jouy, N.; Laïos, I.; Leclercq, G.; Jaouen, G.; Toillon, R.-A. A Ferrocenyl Derivative of Hydroxytamoxifen Elicits an Estrogen Receptor-Independent Mechanism of Action in Breast Cancer Cell Lines. J. Inorg. Biochem. 2010, 104, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Zeh, G.; Haines, P.; Miehlich, M.E.; Kienz, T.; Neidlinger, A.; Friedrich, R.P.; Özkan, H.G.; Alexiou, C.; Hampel, F.; Guldi, D.M.; et al. Anticancer Effect of an Electronically Coupled Oligoferrocene. Organometallics 2020, 39, 3112–3120. [Google Scholar] [CrossRef]
- Özkan, H.G.; Mokhir, A. A Prodrug of 3-(Ferrocenylaminocarbonyloxymethyl)Phenol Activated by Reactive Oxygen Species in Cancer Cells. J. Inorg. Biochem. 2022, 233, 111859. [Google Scholar] [CrossRef]
- Manley, P.W. Investigations into the Potential Role of Metabolites on the Anti-Leukemic Activity of Imatinib, Nilotinib and Midostaurin. Chimia 2019, 73, 561. [Google Scholar] [CrossRef]
- Philipova, I.; Mihaylova, R.; Momekov, G.; Angelova, R.; Stavrakov, G. Ferrocene Modified Analogues of Imatinib and Nilotinib as Potent Anti-Cancer Agents. RSC Med. Chem. 2023, 14, 880–889. [Google Scholar] [CrossRef]
- Huang, W.-S.; Shakespeare, W. An Efficient Synthesis of Nilotinib (AMN107). Synthesis 2007, 2007, 2121–2124. [Google Scholar] [CrossRef]
- Arter, C.; Trask, L.; Ward, S.; Yeoh, S.; Bayliss, R. Structural Features of the Protein Kinase Domain and Targeted Binding by Small-Molecule Inhibitors. J. Biol. Chem. 2022, 298, 102247. [Google Scholar] [CrossRef]
- Kang, Z.-J.; Liu, Y.-F.; Xu, L.-Z.; Long, Z.-J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.-X.; Pan, Y.-J.; Yan, J.-S.; et al. The Philadelphia Chromosome in Leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef]
- Peng, Y.-H.; Shiao, H.-Y.; Tu, C.-H.; Liu, P.-M.; Hsu, J.T.-A.; Amancha, P.K.; Wu, J.-S.; Coumar, M.S.; Chen, C.-H.; Wang, S.-Y.; et al. Protein Kinase Inhibitor Design by Targeting the Asp-Phe-Gly (DFG) Motif: The Role of the DFG Motif in the Design of Epidermal Growth Factor Receptor Inhibitors. J. Med. Chem. 2013, 56, 3889–3903. [Google Scholar] [CrossRef]
- Cowan-Jacob, S.W.; Fendrich, G.; Floersheimer, A.; Furet, P.; Liebetanz, J.; Rummel, G.; Rheinberger, P.; Centeleghe, M.; Fabbro, D.; Manley, P.W. Structural Biology Contributions to the Discovery of Drugs to Treat Chronic Myelogenous Leukaemia. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 80–93. [Google Scholar] [CrossRef]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular Docking with Deep Learning. J. Cheminformatics 2021, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand Efficiency: A Useful Metric for Lead Selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. CP Bioinform. 2006, 15, 5.6.1–5.6.30. [Google Scholar] [CrossRef]
- Aleksandrov, A.; Simonson, T. A Molecular Mechanics Model for Imatinib and Imatinib: Kinase Binding. J. Comput. Chem. 2010, 31, 1550–1560. [Google Scholar] [CrossRef]
- Szakács, Z.; Béni, S.; Varga, Z.; Örfi, L.; Kéri, G.; Noszál, B. Acid−Base Profiling of Imatinib (Gleevec) and Its Fragments. J. Med. Chem. 2005, 48, 249–255. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | BV-173 | LEBV173 | K-562 | LEK562 | AR-230 | LEAR230 | LAMA-84 | LELAMA84 | CCL-1 |
|---|---|---|---|---|---|---|---|---|---|
| 6 | 33.6 ± 2.1 | 0.107 | 29.9 ± 3.1 | 0.102 | 5.9 ± 0.2 | 0.125 | 25.4 ± 2.9 | 0.109 | 55.7 ± 9.5 |
| 9 | 33.6 ± 2.3 | 0.102 | 25.9 ± 2.9 | 0.104 | 143.8 ± 10.6 | 0.087 | 25.4 ± 2.4 | 0.104 | >200 |
| 14 | 17.9 ± 3.1 | 0.140 | >200 | 5.1 ± 1.3 | 0.156 | 27.3 ± 1.8 | 0.134 | >200 | |
| 18 | 15.1 ± 2.9 | 0.142 | >200 | 5.1 ± 0.9 | 0.156 | 26.8 ± 3.2 | 0.134 | 60.2 ± 8.3 | |
| imatinib | 22.8 ± 4.8 | 0.125 | 45.5 ± 5.0 | 0.117 | 4.7 ± 1.0 | 0.144 | 0.5 ± 0.05 | 0.170 | >200 |
| Compound | Affinity, kcal/mol | CNN Affinity, pK Units | LEAFF | LECNN |
|---|---|---|---|---|
| 6 | −8.35 | 7.552 | 0.200 | 0.180 |
| 9 | −8.39 | 6.946 | 0.191 | 0.158 |
| 14 | −10.17 | 7.337 | 0.299 | 0.216 |
| 18 | −8.06 | 7.023 | 0.237 | 0.207 |
| imatinib | −13.14 | 7.864 | 0.355 | 0.213 |
| LEBV173 | LEK562 | LEAR230 | LELAMA84 | |
|---|---|---|---|---|
| LEAFF | 0.569 | 0.977 | 0.635 | 0.948 |
| LECNN | 0.896 | 0.989 | 0.961 | 0.809 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Philipova, I.; Atanasova, M.; Mihaylova, R.; Dailova-Barzeva, A.; Ivanov, S.M.; Simeonova, R.L.; Stavrakov, G. Design, Docking Analysis, and Structure–Activity Relationship of Ferrocene-Modified Tyrosine Kinase Inhibitors: Insights into BCR-ABL Interactions. Molecules 2025, 30, 3101. https://doi.org/10.3390/molecules30153101
Philipova I, Atanasova M, Mihaylova R, Dailova-Barzeva A, Ivanov SM, Simeonova RL, Stavrakov G. Design, Docking Analysis, and Structure–Activity Relationship of Ferrocene-Modified Tyrosine Kinase Inhibitors: Insights into BCR-ABL Interactions. Molecules. 2025; 30(15):3101. https://doi.org/10.3390/molecules30153101
Chicago/Turabian StylePhilipova, Irena, Mariyana Atanasova, Rositsa Mihaylova, Asine Dailova-Barzeva, Stefan M. Ivanov, Rumyana L. Simeonova, and Georgi Stavrakov. 2025. "Design, Docking Analysis, and Structure–Activity Relationship of Ferrocene-Modified Tyrosine Kinase Inhibitors: Insights into BCR-ABL Interactions" Molecules 30, no. 15: 3101. https://doi.org/10.3390/molecules30153101
APA StylePhilipova, I., Atanasova, M., Mihaylova, R., Dailova-Barzeva, A., Ivanov, S. M., Simeonova, R. L., & Stavrakov, G. (2025). Design, Docking Analysis, and Structure–Activity Relationship of Ferrocene-Modified Tyrosine Kinase Inhibitors: Insights into BCR-ABL Interactions. Molecules, 30(15), 3101. https://doi.org/10.3390/molecules30153101