

Isolation and Identification of Secondary Metabolites in Rheum tataricum L.fil. Growing in Kazakhstan and Surveying of Its Anticancer Potential

 , , , ,
, , , , 
Abstract

1. Introduction
2. Results and Discussion
2.1. Analysis of Components of TBME and EtOAc Fractions of Ethanol Extract of R. Tataricum by HLPC Method
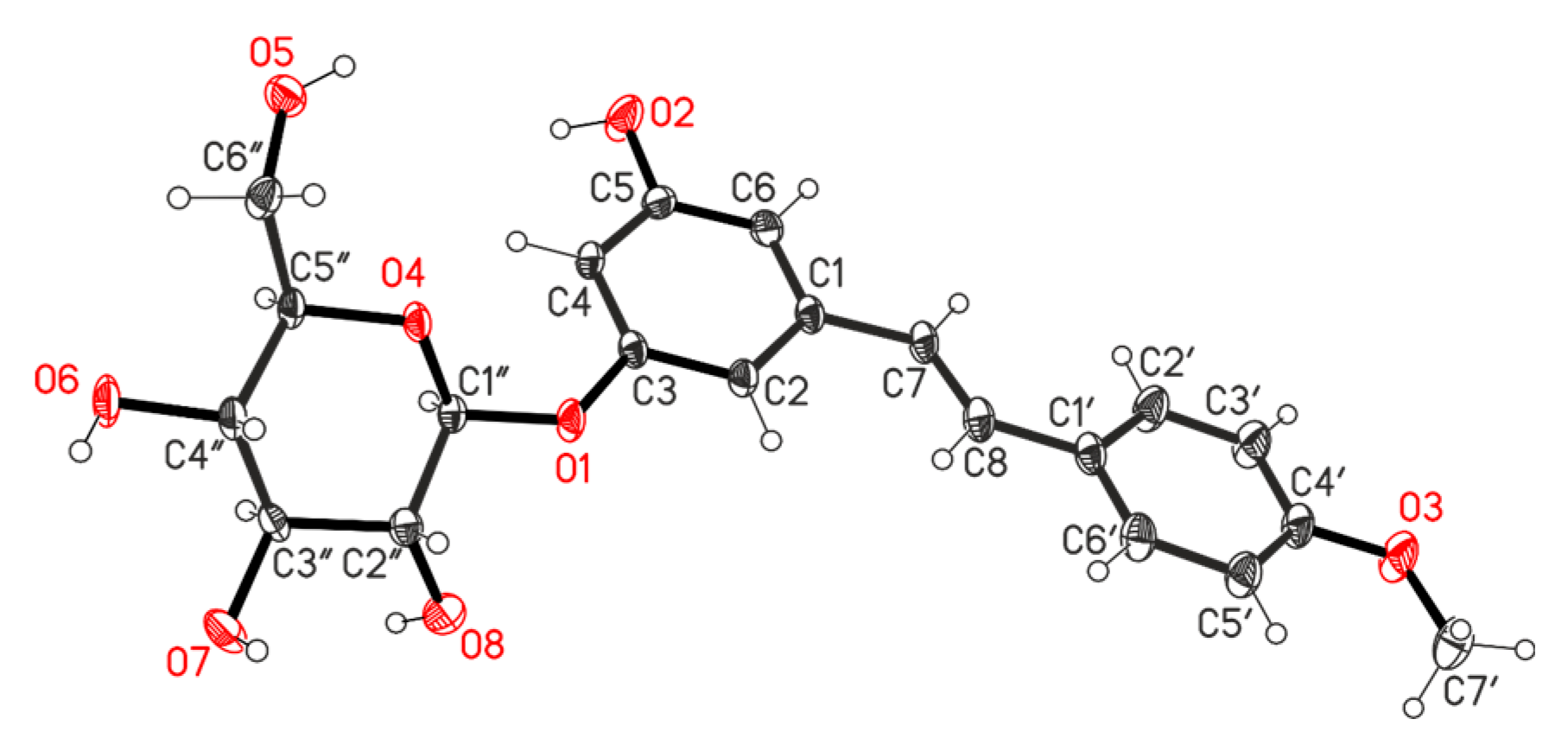
2.2. Separation of TBME and EtOAc Tractions of Ethanol Extract of R. tataricum and Structural Elucidation of Isolated Compounds
2.2.1. Separation of TBME Fraction
2.2.2. Separation of EtOAc Fraction
2.3. Cytotoxicity Assay
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Plant Material
3.1.3. Preparation of Extracts of R. tataricum
3.1.4. Acetylation of Glycosides (1 + 3) (From TBME Fraction of Ethanol Extract of R. tataricum)
3.1.5. Hydrolysis of Glycosides (1 + 3)
3.1.6. Structure Determination of Isolated Compounds
- (R)-4-(4-Hydroxyphenyl)-2-butanol 2-β-D-glucopyranoside (Rhododendrin) (1). White solid. M.p. 183–186 °C (ether). [α]D26 −42.7 (c 0.5, EtOH); (lit.: M.p. 188–189 °C. [α]D −46 (c 1.0, EtOH) [20]; M.p. 189 °C. [α]D −38 (c 0.5, MeOH) [40]); 1H NMR (400 MHz, (CD3)2SO, δ, ppm): 1.19 (3H, d, J = 6.8 Hz, CH3-1), 1.55–1.60 (1H, ddd, J = 13.2, 6.7, 2.0 Hz, H-3), 1.50–1.75 (1H, ddd, J = 13.2, 7.0, 2.0 Hz, H-3), 2.84 (2H, m, H-4), 2.93 (1H, dd, J = 8.0, 8.2 Hz, H-2′), 3.04 (1H, m, H-5′), 3.14 (1H, m, H-4′), 3.34 (3H, br.s, OH), 3.34 (1H, dd, J = 12.0, 5.2 Hz, H-6′), 3.41 (1H, dd, J = 12.0, 2.2 Hz, H-6′), 3.64 (1H, dd, J = 12.0, 8.2 Hz, H-3′), 3.79 (1H, m, H-2), 4.16 (1H, d, J = 8.2 Hz, H-1′), 5.03 (2H, br.s, OH), 6.63 (2H, d, J = 7.8 Hz, H-7,9), 6.98 (2H, d, J = 7.8 Hz, H-6,10); 13C NMR (101 MHz, (CD3)2SO, δ, ppm): 21.74 (C-1), 29.77 (C-4), 38.50 (C-3), 61.12 (C-6′), 70.13 (C-4′), 73.66 (C-2′), 74.62 (C-2), 76.68 (C-3′), 76.80 (C-5′), 102.79 (C-1′), 115.08 (C-7,9), 129.17 (C-6,10), 131.93 (C-5), 155.64 (C-8); IR (KBr, ν, cm−1): 3430, 3225 (OH), 2988, 1605, 1505, 1490 (C=C), 1158 C-O), 1110, 1062, 1043, 1006 (glycosidic C-O); UV (MeOH) λmax (lgε): 223 (4.79), 279 (4.56) nm; HR-MS, m/z (Irel., %): 328 (3, M+), 219 (10), 208 (5), 207 (17), 190 (23), 177 (27), 166 (28), 148 (32), 133 (22), 107 (100); calcd for C16H24O7: 328.1517; found [M − H]+ m/z: 328.1113.
- (S)-4-(4-Hydroxyphenyl)-2-butanol 2-β-D-glucopyranoside (epi-rhododendrin) (3). White solid. M.p. 83–86 °C (ethylacetate), [α]D26 −13.6 (c 0.5, EtOH); (lit.: [α]D20 −15.5 (c 0.1, EtOH) [19]); 1H NMR (400 MHz, (CD3)2SO, δ, ppm): 1.24 (3H, d, J = 7.0 Hz, CH3-1), 1.59 (1H, ddd, J = 13.5, 7.0, 2.0 Hz, H-3), 1.83 (1H, ddd, J = 13.5, 7.0, 2.0 Hz, H-3), 2.64 (2H, m, H-4), 3.03 (1H, dd, J = 8.0, 8.2 Hz, H-2′), 3.14 (1H, m, H-5′), 3.25 (1H, m, H-4′), 3.49 (1H, dd, J = 12.0, 5.2 Hz, H-6′), 3.66 (1H, dd, J = 12.0, 2.2 Hz, H-6′), 3.71 (1H, dd, J = 12.0, 8.2 Hz, H-3′), 3.74 (1H, m, H-2), 4.22 (1H, d, J = 8.2 Hz, H-1′), 6.61 (2H, d, J = 7.8 Hz, H-7,9), 7.02 (2H, d, J = 7.8 Hz, H-6,10); 13C NMR (101 MHz, CDCl3, δ, ppm): 22.43 (C-1), 30.57 (C-4), 38.06 (C-3), 61.18 (C-6′), 72.07 (C-4′), 75.64 (C-2′), 76.46 (C-3′), 77.51 (C-2), 78.16 (C-5′), 104.08 (C-1′), 115.12 (C-7,9), 129.35 (C-6,10), 131.87 (C-5), 156.71 (C-8); IR KBr, ν, cm−1): 3419 (OH), 1612, 1518, 1500 (C=C), 1178 C-O), 1110, 1076, 1050, 1010 (glycosidic C-O); UV (MeOH) λmax (lgε): 224 (4.81), 278 (4.11) nm; HR-MS, m/z (Irel., %): 328 (1, M+), 219 (10), 207 (16), 191 (4), 190 (20), 178 (9), 177 (32), 166 (41), 149 (15), 148 (48), 133 (28), 121 (17), 108 (23), 107 (100); calcd for C16H24O7: 328.1517; found [M − H]+ m/z: 328.1125.
- (R)-4-(4-Hydroxyphenyl)-2-butanol (-)-Rhododendrol) (4). White needles. M.p. 81–84 °C (EtOAc), [α]D26 −16.2 (c 0.5, EtOH); (lit.: M.p. 81–83 °C, [α]D −17.1 (c 2.0, EtOH,) [40]); 1H NMR (500 MHz, CD3OD+CDCl3, δ, ppm): 1.15 (3H, d, J = 6.5 Hz, CH3-1), 1.51 (1H, m, H-3), 1.65 (1H, ddd, J = 13.5, 6.8, 1.8 Hz, H-3), 2.51(1H, m, H-4), 2.60 (1H, m, H-4), 3.65 (1H, ddd, J = 12.0, 6.5, 5.0 Hz, H-2), 6.59 (2H, d, J = 7.8 Hz, H-7,9), 6.95 (2H, d, J = 7.8 Hz, H-6,10); 13C NMR (125 MHz, CDCl3+CDCl3, δ, ppm): 22.56 (C-1), 27.51 (C-4), 39.23 (C-3), 69.32 (C-2), 114.61 (C-7,9), 128.32 (C-6,10), 131.15 (C-5), 155.34 (C-8); IR (KBr, ν, cm−1): 3323 (OH), 1610, 1500 (C=C); UV (MeOH) λmax (lgε): 221 (4.26), 278 (4.09) nm; HR-MS, m/z (Irel., %): 167 (1), (166 (9, M+), 148 (6), 133 (15), 108 (7), 107 (20), 46 (31), 45 (53), 31 (100); calcd for C10H14O2: 166.0988; found [M − H]+ m/z: 166.0989.
- (S)-4-(4-Hydroxyphenyl)-2-butanol (+)-Rhododendrol (5). White solid. M.p. 75–78 °C (ether), [α]D26 +12.8 (c 0.5, EtOH); (lit.: M.p. 80–81 °C. [α]D +13.6 (c 1.0, EtOH) [41]; [α]D +15.5 (c 0.36, MeOH) [42]); 1H NMR (400 MHz, (CD3OD+CDCl3, δ, ppm): 1.20 (3H, d, J = 7.0 Hz, CH3-1), 1.55 (1H, m, H-3), 1.67 (1H, ddd, J = 13.5, 7.0, 1.8 Hz, H-3), 2.58–2.65 (2H, m, H-4), 3.61–3.74 (1H, m, H-2), 6.64 (2H, d, J = 8.2 Hz, H-7,9), 7.03 (2H, d, J = 8.2 Hz, H-6,10); 13C NMR (101 MHz, CDCl3, δ, ppm): 22.56 (C-1), 27.51 (C-4), 39.23 (C-3), 69.32 (C-2), 114.61 (C-7,9), 128.32 (C-6,10), 131.15 (C-5), 155.34 (C-8); IR (KBr, ν, cm−1): 3430 (OH), 1605, 1505, 1490 (C=C), 1185 C-O); UV (MeOH) λmax (lgε): 223 (4.79), 279 (4.14) nm; HR-MS, m/z (Irel., %): 166 (20, M+), 148 (18), 133 (61), 108 (14), 107 (100); calcd for C10H14O2: 166.0988; found [M − H]+ m/z: 166.0991.
- 1-O-Galloyl-β-D-glucopyranose (β-glucogallin) (6). White powder. M.p. 210–212 °C (ether), [α]D26 −24.2 (c 0.2, CHCl3); [lit.: M.p. 214 °C (ethanol) [43]; [α]D −23.4 (c 1.0, EtOH) [44]; [α]D −8 (c 0.1, MeOH) [45]), 1H NMR (400 MHz, CDCl3+CD3OD, δ, ppm): 3.25–3.41 (4H, m, H-2′,3′,4′,5′), 3.64 (1H, dd, J = 12.4, 5.8 Hz, H-6′), 4.08 (1H, dd, J = 12.4, 1.5 Hz, H-6′), 4.61 (1H, d, J = 7.8 Hz, H-1′), 7.11 (2H, s, H-2,6); 13C NMR (101 MHz, CDCl3+CD3OD, δ, ppm): 61.09 (C-6′), 66.30 (C-4′), 73.60 (C-2′), 76.46 (C-3′), 77.14 (C-5′), 103.87(C-1′), 104.80 (C-5), 125.53 (C-6), 138.29 (C-3), 152.01 (C-4), 166.35 (C-7).
- 3,5-Di-O-Caffeoylquinic Acid (7). Amorphous white powder. 1H NMR (400 MHz, (CDCl3+CD3OD, δ, ppm): 2.08 (4H, br.s, (H-2,6), 3.94 (1H, br.s, H-4), 5.32 (2H, br.s, (H-3,5), 6.27 (2H, d, J = 15.9 Hz, 2-H-2′), 6.89 (2H, d, J = 7.8 Hz, 2-H-8′), 7.01 (2H, d, J = 1.8 Hz, 2-H-5′), 7.05 (2H, dd, J = 7.8, 1.8 Hz, 2-H-9′), 7.58 (2H, d, J = 15.9 Hz, 2-H-3′) (OH protons not observed); 13C NMR (101 MHz, CDCl3+CD3OD, δ, ppm): 31.61 (C-6), 34.03 (C-2), 63.19 (C-4), 65.04 (C-3), 65.09 (C-5), 70.14 (C-1), 109.15 (2C-5′), 115.46 (2C-8′), 115.50 (2C-2′), 122.95 (2C-9′), 129.81 (2C-4′), 144.57 (2C-3′), 146.61 (2C-6′), 147.76 (2C-7′), 167.36 (2C-1′), 174.29 (C-7); IR (KBr, ν, cm−1): 3420, 3380 (OH), 1695 (C=O), 1605, 1525, 812, 745 (C=C); UV (MeOH) λmax (lgε): 228 (4.05), 243 (3.41), 300 sh (3.05), 325 (3.91) nm. NMR, IR and UV spectral data match well with those reported previously [43]. Anal. calcd for C25H24O12, %: C, 58.14; H, 4.68; found, %: C, 58.45; H, 4.79.
- 5-Caffeoyl-3-O-(p-coumaroyl)quinic Acid (8). Amorphous yellowish powder. 1H NMR (400 MHz, (CDCl3+CD3OD, δ, ppm): 2.06 (4H, br.s, (H-2,6), 3.95 (1H, br.s, H-4), 5.16 (1H, br.s, (H-5), 5.26 (1H, br.s, (H-5), 6.27 (1H, d, J = 15.9 Hz, H-2′), 6.28 (1H, d, J = 15.9 Hz, H-2″), 6.82 (2H, d, J = 8.2 Hz, H-6″,8′′), 6.90 (1H, d, J = 7.8 Hz, H-8′), 7.01 (H, d, J = 1.8 Hz, H-5′), 7.05 (H, dd, J = 7.8, 1.8 Hz, H-9′), 7.40 (2H, d, J = 8.2 Hz, H-5″,9″), 7.60 (H, d, J = 15.9 Hz, H-3′), 7.62 (H, d, J = 15.9 Hz, H-3″) (OH protons not observed); 13C NMR (101 MHz, CDCl3+CD3OD, δ, ppm): 31.63 (C-6), 34.05 (C-2), 63.21 (C-4), 65.01 (C-3), 65.18 (C-5), 70.14 (C-1), 109.57 (C-5′), 115.46 (C-8′), 115.62 (C-2″),115.50 (C-2′), 115.74 (C-6″,8″), 122.95 (C-9′), 126.99 (C-4″), 129.80 (C-4′), 129.81 (C-5″,9″), 144.22 (C-3″), 144.57 (2C-3′), 146.61 (2C-6′), 147.76 (2C-7′), 157.76 (C-7″) 167.35 (C-1′), 167.51 (C-1″), 174.29 (C-7); IR (KBr, ν, cm−1): 3432 (OH), 1695 (C=O), 3080, 1600, 1523, 812, 763 (C=C); UV (MeOH) λmax (lgε): 218 (4.32), 237 (3.53), 300 sh (3.15), 326 (3.75) nm. Anal. calcd for C25H24O11, %: C, 60.00; H, 4.83; found, %: C, 60.18; H, 4.67.
- 4-(4-Hydroxyphenyl)butan-2-one (9). White powder. M.p. 82–85 °C (ether); (lit.: M.p. 83.5–84.5 °C [46]); 1H NMR (500 MHz, CDCl3, δ, ppm): 2.11 (3H, s, CH3), 2.72, 2.82 (both 2H, A2B2 system, J = 7.6 Hz, CH2CH2-H-3,4), 5.35 (1H, br.s, OH), 6.73 (2H, d, J = 8.4 Hz, H-7,9), 7.01(2H, d, J = 8.4 Hz, H-6,10). 13C- NMR (125 MHz, CDCl3, δ, ppm): 28.81 (C-4), 30.29 (CH3), 45.51 (C-3), 115.34 (C-7,9), 130.22 (C-6,10), 132.46(C-5), 154.19 (C-8), 209.89 (C=O); HR-MS, m/z (Irel., %): 164 (38, M+), 149 (6, [M-CH3]+), 121 (15 [M-Ac]+), 107 (100 [M-CH2Ac]+), 77 (51); calcd for C10H12O2: 164.0872; found [M − H]+ m/z: 164.0879.
- Rhaponticin (3,3′,5-Trihydroxy-4′-methoxystilbene-3-O-β-D-glucopyranoside) (10). Yellowish needles; M.p. 248–250 °C (EtOH), [α]D −55.2 (c 0.5; EtOH); (lit.: M.p. 246–248 °C (dil. acetone), [α]D −56.3 (c 0.88, aq. acetone, 1:1) [30]); 1H NMR (400 MHz, CDCl3+CD3OD, δ, ppm): 3.26 (1H, m, H-2″), 3.38–3.46 (1H, m, H-3″,4″), 3.58 (1H, m, H-5″), 3.71 (1H, dd, J = 7.0, 11.8 Hz, H-6″), 3.78 (3H, s, 4′-OCH3), 3.93 (1H, dd, J = 3.8, 11.8 Hz, H-6″), 4.85 (1H, d, J = 7.0 Hz, H-1″), 6.35 (1H, t, J = 1.8 Hz, H-4), 6.58, 6.77 (each 1H, br.s, H-2,6), 6.85 (1H, d, J = 16.2 Hz, H-7), 7.02 (1H, d, J = 16.2 Hz, H-8), 6.84 (1H, d, J = 8.2 Hz, H-5′), 6.98 (1H, dd, J = 2,0, 8.2 Hz, H-6′), 7.08 (1H, d, J = 2.0 Hz, H-2′) (OH protons not observed); 13C NMR (101 MHz, CDCl3 +CD3OD, δ, ppm): 55.63 (OCH3), 60.91 (C-6″), 71.05 (C-4″), 74.31 (C-2″), 76.23 (C-5″), 77.48 (C-3″), 100.51 (C-1″), 102.84 (C-4), 105.02 (C-2), 107.21 (C-6), 112.11 (C-5′), 112.72 (C-2′), 118.61 (C-6′), 126.21 (C-7), 128.45 (C-8), 129.78 (C-1′), 139.14 (C-1), 146.43 (C-3′), 147.41 (C-4′), 158.08 (C-5), 158.71 (C-3); IR (KBr, ν, cm−1): 3456, 3340 (OH), 1610, 1583, 1510, 815, 723 (C=C), 1085, 1015 (sugar); UV (MeOH) λmax (lgε): 220 (4.36), 302 (4.09), 324 (4.45); HRMS: (Irel., %): 420 (M+, 1), 258 (100, M+- Glc), 257 (32), 256 (18), 197 (26), 129 (16), 115 (16); calcd for C21H24O9: 420.1466; found [M − H]+ m/z: 420.1461.
- Desoxyrhaponticin (3,5-Dihydroxy-4′-methoxystilbene-3-O-β-D-glucopyranoside) (11). Colorless plate. M.p. 227–230 °C (EtOH), [α]D −56.6 (c 0.3, EtOH); (lit.: M.p. 226–228 °C (EtOH), [α]D −50 ± 2 (c 0.2; aq. acetone, 1:1) [47]). 1H NMR (500 MHz, CDCl3+CD3OD, δ, ppm): 3.38–3.43 (1H, m, H-2″), 3.45–3.51 (2H, m, H-3″,4″), 3.50–3.53 (1H, m, H-5″), 3.78 (1H, ddd, J = 11.6, 7.2, 3.2 Hz, H-6″), 3.82 (3H, s, 4′-OCH3), 3.93 (1H, dd, J = 11.6, 3.6 Hz, H-6″), 4.92 (1H, d, J = 7.2 Hz, H-1″), 6.48 (1H, t, J = 2.0 Hz, H-4), 6.66 (1H, t, J = 1.6 Hz, H-6), 6.78 (1H, t, J = 1.6 Hz, H-2), 6.89 (1H, d, J = 16.2 Hz, H-7), 6.95 (2H, brd, J = 8.8 Hz, H-3′, 5′), 7.03 (1H, d, J = 16.2 Hz, H-8), 7.44 (2H, br. d, J = 8.8 Hz, H-2′, 6′) (OH protons not observed); 13C NMR (125 MHz, CDCl3 +CD3OD, δ, ppm): 54.61 (4′-OCH3), 61.08 (C-6″), 69.62 (C-4″), 72.89 (C-2″), 76.04 (C-5″), 77.18 (C-3″), 100.49 (C-1″), 102.57 (C-4), 105.69 (C-6), 107.02 (C-1′), 113.51 (C3′, 5′), 125.73 (C-2), 127.19 (C-2′,6′), 128.05 (C-7), 129.61 (C-8), 139.38 (C-1), 157.63 (C-5), 158.26 (C-3), 159.84 (C-4′); IR (KBr, ν, cm−1): 3380 (OH), 1600, 1592, 1512, 832, 710 (C=C), 1086, 1012 (sugar); UV (MeOH) λmax (lgε): 222 (3.72), 307 (4.84), 321 (4.65); HRMS: calcd for C21H24O8: 404.1471; found {M − H]+ m/z: 404.1468.
3.2. Cell Culture and Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, X.; Dai, L.; Yan, F.; Ma, Y.; Guo, X.; Jenis, J.; Wang, Y.; Zhang, J.; Miao, X.; Shang, X. The phytochemistry and pharmacology of three Rheum species: A comprehensive review with future perspectives. Phytomedicine 2024, 131, 155772. [Google Scholar] [CrossRef]
- Xiang, H.; Zuo, J.; Guo, F.; Dong, D. What we already know about rhubarb: A comprehensive review. Chin. Med. 2020, 15, 88. [Google Scholar] [CrossRef]
- Cao, Y.-J.; Pu, Z.-J.; Tang, Y.-P.; Shen, J.; Chen, Y.-Y.; Kang, A.; Zhou, G.-S.; Duan, J.-A. Advances in bioactive constituents, pharmacology and clinical applications of rhubarb. Chin. Med. 2017, 12, 36. [Google Scholar] [CrossRef]
- Mohtashami, L.; Amiri, M.S.; Ayati, Z.; Ramezani, M.; Jamialahmadi, T. Ethnobotanical uses, phytochemistry and pharmacology of different Rheum species (Polygonaceae): A review. Adv. Exp. Med. Biol. 2021, 1308, 309–352. [Google Scholar] [CrossRef]
- Krafczyk, N.; Kötke, M.; Lehnert, N.; Glomb, M.A. Phenolic composition of rhubarb. Eur. Food Res. Technol. 2008, 228, 187–196. [Google Scholar] [CrossRef]
- Ren, G.; Li, L.; Hu, H.; Li, Y.; Liu, C.; Wei, S. Influence of the environmental factors on the accumulation of the bioactive ingredients in Chinese rhubarb products. PLoS ONE 2016, 11, e0154649. [Google Scholar] [CrossRef]
- Han, J.; Ye, M.; Xu, M.; Qiao, X.; Chen, H.; Wang, B.; Zheng, J.; Guo, D. Comparison of phenolic compounds of rhubarbs in the section deserticola with Rheum palmatum by HPLC-DAD-ESI-MS. Planta Medica 2008, 74, 873–879. [Google Scholar] [CrossRef]
- Golubkina, N.; Kharchenko, V.; Bogachuk, M.; Koshevarov, A.; Sheshnitsan, S.; Kosheleva, O.; Pirogov, N.; Cariso, G. Biochemical characteristics and elemental composition peculiarities of Rheum tataricum L. in semi-desert conditions and of European garden rhubarb. Int. J. Plant Biol. 2022, 13, 368–380. [Google Scholar] [CrossRef]
- Chumbalov, P.K.; Nurgalieva, G.M. Flavonoids of Rheum tataricum. Chem. Nat. Compd. 1967, 3, 291. [Google Scholar] [CrossRef]
- Chumbalov, P.K.; Nurgalieva, G.M. Catechins of Rheum tataricum. Chem. Nat. Compd. 1967, 3, 236–237. [Google Scholar] [CrossRef]
- Samappito, S.; Page, J.E.; Schmidt, J.; De-Eknamkul, W.; Kutchan, T.M. Aromatic and pyrone polyketides synthesized by a stilbene synthase from Rheum tataricum. Phytochemistry 2003, 62, 313–323. [Google Scholar] [CrossRef]
- Chumbalov, P.K.; Nurgalieva, G.M. Carbohydrates of Rheum tataricum. Chem. Nat. Compd. 1966, 2, 230. [Google Scholar] [CrossRef]
- Amangeldinova, M.; Ersatır, M.; Necip, A.; Yilmaz, M.A.; Cimentepe, M.; Kudrina, N.; Terletskaya, N.V.; Ozturk Cimentepe, O.; Yildirim, M. Simultaneous quantitative screening of 53 phytochemicals from Rheum tataricum L. roots: A comparative study of supercritical CO2, subcritical ethanol, and ultrasound-assisted extraction for enhanced antioxidant, antibacterial activities, and molecular docking study. Front. Plant Sci. 2024, 15, 1513875. [Google Scholar] [CrossRef]
- Shults, E.E.; Shakirov, M.M.; Pokrovsky, M.A.; Petrova, T.N.; Pokrovsky, A.G.; Gorovoy, P.G. Bioactive phenolic compounds from Glycyrrhiza pallidiflora and their cytotoxic activity. Nat. Prod. Res. 2017, 31, 445–452. [Google Scholar] [CrossRef]
- Archangelsky, K. Ueber rhododendrol, rhododendrin und andromedotoxin. Arch. Exp. Pathol. Pharmakol. 1901, 46, 313–320. [Google Scholar] [CrossRef]
- Santamour, F.S., Jr.; Vettel, H.E. The distribution of rhododendrin in birch (Betula) species. Biochem. Syst. Ecol. 1978, 6, 107–108. [Google Scholar] [CrossRef]
- Šmite, E.; Lundgren, L.N.; Andersson, R. Arylbutanoid and diarylheptanoid glycosides from inner bark of Betula pendula. Phytochemistry 1993, 32, 365–369. [Google Scholar] [CrossRef]
- Khan, M.S.Y.; Kumar, I.; Prasad, J.S.; Nagarajan, G.R.; Parathasarathy, M.R.; Krishnamurty, H.G. Phenolic constituents of Taxus baceata leaves. Planta Med. 1976, 30, 82–85. [Google Scholar] [CrossRef]
- Inoue, T.; Ishidate, Y.; Fujita, M.; Kubo, M.; Fukushima, M.; Nagai, M. Studies on the constituents of Aceraceae plants. I. Constituents in the leaves and the stem bark of Acer nikoense MAXIM. J. Pharm. Soc. Jpn. 1978, 98, 41–46. [Google Scholar] [CrossRef]
- Pan, H.; Lundgren, L.N. Rhododendrol glycosides and phenyl glucoside esters from inner bark Betula pubescens. Phytochemistry 1994, 36, 79–83. [Google Scholar] [CrossRef]
- Shikishima, Y.; Takaishi, Y.; Honda, G.; Ito, M.; Takeda, Y.; Kodzhimatov, O.K.; Ashurmetov, O. Phenylbutanoids and stilbene derivatives of Rheum maximowiczii. Phytochemistry 2001, 56, 377–381. [Google Scholar] [CrossRef]
- Parmar, V.S.; Vardhan, A.; Taneja, P.; Sinha, R.; Patnaik, G.K.; Tripathi, S.C.; Boll, P.M.; Larsen, S. Absolute configuration of epi-rhododendrin and (-)-rhododendrol [ = (-)- betuligenol] and X-Ray crystal and molecular structure of rhododendrin [ = betuloside], a hepatoprotective constituent of Taxus baccata. J. Chem. Soc. Perkin Trans. 1 1991, 1991, 2687–2690. [Google Scholar] [CrossRef]
- Gao, L.-L.; Guo, T.; Xu, X.-D.; Yang, J.-S. Rapid identification and simultaneous analysis of multiple constituents from Rheum tanguticum Maxim. ex Balf. by UPLC/Q-TOF-MS. Nat. Prod. Res. 2017, 31, 1529–1535. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Nonaka, G.-I.; Nishioka, I.; Yamagishi, T. Galloyl and hydroxycinnamoylglucoses from rhubarb. Phytochemistry 1988, 27, 1473–1477. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Yang, C.-Y. Assessment of ethanolic extraction of chlorogenic acid, cynarin, and polyphenols from burdock (Arctium lappa L.) roots under ultrasound. Molecules 2024, 29, 5115. [Google Scholar] [CrossRef]
- Xue, H.Y.; Wei, M.J.; Ji, L.L. Chlorogenic Acids: A pharmacological systematic review on their hepatoprotective effects. Phytomedicine 2023, 118, 154961. [Google Scholar] [CrossRef]
- Gao, L.L.; Xu, X.D.; Nang, H.J.; Yang, J.S.; Chen, S.L. Chemical constituents in Rheum tanguticum. Chin. Trad. Herbal Drugs 2011, 42, 443–446. [Google Scholar]
- Lin, V.C.-H.; Ding, H.-Y.; Kuo, S.-Y.; Chin, L.-W.; Wu, J.-Y.; Chang, T.-S. Evaluation of in vitro and in vivo depigmenting activity of raspberry ketone from Rheum officinale. Int. J. Mol. Sci. 2011, 12, 4819–4835. [Google Scholar] [CrossRef]
- Li, X.; Wei, T.; Wu, M.; Chen, F.; Zhang, P.; Deng, Z.; Luo, T. Potential metabolic activities of raspberry ketone. J. Food Biochem. 2022, 46, e14018. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Nonaka, G.I.; Nishioka, I. Studies on rhubarb (Rhei rhizoma). V. Isolation and characterization of stilbenes. Chem. Pharm. Bull. 1984, 32, 3501–3517. [Google Scholar] [CrossRef]
- Zhao, Y.-Y.; Qin, X.-Y.; Chen, S.-P.; Zhang, Y.; Lin, R.-C.; Sun, W.-J. Crystal and molecular structure of rhaponticin from Rheum hotaoense. J. Chem. Cryst. 2011, 41, 409–411. [Google Scholar] [CrossRef]
- Carnat, A.; Heitz, A.; Fraisse, D.; Carnat, A.P.; Lamaison, J.L. Major dicaffeoylquinic acids from Artemisia vulgaris. Fitoterapia 2000, 71, 587–589. [Google Scholar] [CrossRef]
- Teka, T.; Zhang, L.; Ge, X.; Li, Y.; Han, L.; Yan, X. Stilbenes: Source plants, chemistry, biosynthesis, pharmacology, application and problems related to their clinical application—A comprehensive review. Phytochemistry 2022, 197, 113128. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 16, 55–63. [Google Scholar] [CrossRef]
- Aburjai, T.A. Anti-platelet stilbenes from aerial parts of Rheum palaestinum. Phytochemistry 2000, 55, 407–410. [Google Scholar] [CrossRef]
- Li, P.; Tian, W.; Wang, X.; Ma, X. Inhibitory effect of desoxyrhaponticin and rhaponticin, two natural stilbene glycosides from the Tibetan nutritional food Rheum tanguticum Maxim. ex Balf., on fatty acid synthase and human breast cancer cells. Food Funct. 2014, 5, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Kurzava Kendall, L.; Ma, Y.; Yang, T.; Lubecka, K.; Stefanska, B. Epigenetic Effects of Resveratrol on Oncogenic Signaling in Breast Cancer. Nutrients 2024, 16, 699. [Google Scholar] [CrossRef] [PubMed]
- Singaravelan, N.; Tollefsbol, T.O. Polyphenol-Based Prevention and Treatment of Cancer Through Epigenetic and Combinatorial Mechanisms. Nutrients 2025, 17, 616. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatzu, K. Biochemical mechanism of rhododendrol-induced leukoderma. Int. J. Mol. Sci. 2018, 19, 552. [Google Scholar] [CrossRef]
- Kim, M.H.; Nugroho, A.; Choi, J.; Park, J.H.; Park, H.J. Rhododendrin, an analgesic/anti-inflammatory arylbutanoid glycoside, from the leaves of Rhododendron aureum. Arch. Pharm. Res. 2011, 34, 971–978. [Google Scholar] [CrossRef]
- Fushiya, S.; Kabe, Y.; Ikegaya, Y.; Takano, F. (+)-Rhododendrol and epi-rhododendrin suppress the NO production by activated macrophages in vivo. Planta Med. 1998, 64, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Padma Rao, S.; Srinivas, K.V.N.S.; Yadav, J.S. Stereospecific synthesis and absolute configuration of (+)-rhododendrol. Phytochemistry 1993, 33, 1529–1530. [Google Scholar] [CrossRef]
- Schmidt, O.T.; Reuss, H. Über natürliche gerbstoffe, XXX. 2-[p-Hydroxybenzoyl]-glucose und 2-galloyl-glucose. Justus Lieb. Ann. Chem. 1961, 649, 137–148. [Google Scholar] [CrossRef]
- Bikbulatova, T.N.; Beisekova, K.D. Chemical composition of the fruit of Rosa platyacantha. Chem. Nat. Compd. 1979, 15, 372. [Google Scholar] [CrossRef]
- Latte, K.P.; Kolodziej, H. Pelargoniins, new ellagitannins from Pelargonium reniforme. Phytochemistry 2000, 54, 701–708. [Google Scholar] [CrossRef]
- Mannich, C.; Merz, K.W. Über einige vom 1-phenyl-3-amino-butan sich ableitende phenolbasen. Arch. Pharm. 1927, 265, 15–26. [Google Scholar] [CrossRef]
- Banks, H.J.; Cameron, D.W. New natural stilbene glucoside from Rheum rhaponticum (Polygonaceae). Austr. J. Chem. 1971, 24, 2427–2430. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Bruker AXS. SADABS, User Manual; v. 2008-1; Bruker AXS: Madison, WI, USA, 2008. [Google Scholar]
- Spek, A.L. Single-crystal structure validation with the program Platon. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Stree, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Jayatilake, G.S.; Jayasuriya, H.; Lee, E.-S.; Koonchanok, N.M.; Geahlen, R.L.; Ashendel, C.L.; McLaughlin, J.L.; Chang, C.J. Kinase inhibitors from Polygonum cuspidatum. J. Nat. Prod. 1993, 56, 1805–1810. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extract or Compound | Growth Inhibition of Cells (GI50 ± SEM, μM) [a,b] | |||||||
|---|---|---|---|---|---|---|---|---|
| C33 A (HPV- Negative) | CaSki (HPV-16) | HeLa (HPV-18) | MCF-7 | DU-145 | SNB-19 | T98G | VERO | |
| TBME fraction | 41 ± 3.6 | 38 ± 1.2 | 31 ± 2.2 | 39 ± 1.6 | 51 ± 3.1 | 43 ± 5.4 | 66 ± 4.9 | 91 ± 7.8 |
| EtOAc fraction | 61 ± 3.2 | 41 ± 2.8 | 61 ± 6.6 | 43 ± 2.9 | 49 ± 3.4 | 76 ± 6.1 | 49 ± 3.8 | 83 ± 4.6 |
| Ethanol extract | 55 ± 4.2 | 52 ± 4.8 | 65 ± 3.8 | 36 ± 2.8 | 68 ± 5.2 | 52 ± 4.5 | 78 ± 3.1 | 86 ± 6.3 |
| 1 | 27 ± 1.4 | 12 ± 0.7 | 16 ± 0.8 | 10 ± 1.8 | 21 ± 2.3 | 23 ± 4.1 | 29 ± 2.5 | >100 |
| 1a + 3a (5:1) | 25 ± 1.8 | 19 ± 0.5 | 19 ± 1.4 | 17 ± 0.4 | 28 ± 0.7 | 37 ± 1.6 | 45 ± 4.8 | >100 |
| 4 | 28 ± 0.5 | 10 ± 0.6 | 29 ± 0.7 | 11 ± 1.4 | 21 ± 0.8 | 15 ± 0.9 | 18 ± 1.1 | >100 |
| 6 | 27 ± 2.6 | 18 ± 1.4 | 29 ± 2.4 | 31 ± 0.7 | 67 ± 6.4 | 26 ± 1.2 | 21 ± 0.8 | >100 |
| 9 | 19 ± 2.2 | 20 ± 1.5 | 23 ± 3.7 | 21 ± 2.1 | 60 ± 1.7 | 33 ± 1.8 | 19 ± 0.9 | >100 |
| 11 | NT | NT | 17 ± 0.9 | 8 ± 1.8 | NT | 13 ± 1.1 | 18 ± 3.5 | 92 ± 7.9 |
| 12 | NT | NT | 18 ± 1.6 | 7 ± 1.2 | NT | 9.2 ± 0.8 | 21 ± 1.7 | 98 ± 4.8 |
| Doxorubicin | 2.5 ± 0.8 | 10.1 ± 0.7 | 6.1 ± 0.7 | 5.2 ± 0.8 | 15 ± 0.3 | 7.6 ± 0.7 | 13 ± 0.5 | 8 ± 1.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turgunbayeva, A.A.; Sultanova, N.A.; Hamad, M.S.; Savelyev, V.A.; Chernyak, E.I.; Bagryanskaya, I.Y.; Pokrovsky, M.A.; Pokrovsky, A.G.; Gemejiyeva, N.G.; Shults, E.E. Isolation and Identification of Secondary Metabolites in Rheum tataricum L.fil. Growing in Kazakhstan and Surveying of Its Anticancer Potential. Molecules 2025, 30, 2978. https://doi.org/10.3390/molecules30142978
Turgunbayeva AA, Sultanova NA, Hamad MS, Savelyev VA, Chernyak EI, Bagryanskaya IY, Pokrovsky MA, Pokrovsky AG, Gemejiyeva NG, Shults EE. Isolation and Identification of Secondary Metabolites in Rheum tataricum L.fil. Growing in Kazakhstan and Surveying of Its Anticancer Potential. Molecules. 2025; 30(14):2978. https://doi.org/10.3390/molecules30142978
Chicago/Turabian StyleTurgunbayeva, Aiman A., Nurgul A. Sultanova, Mohammad Saleh Hamad, Victor A. Savelyev, Elena I. Chernyak, Irina Yu. Bagryanskaya, Mikhail A. Pokrovsky, Andrey G. Pokrovsky, Nadezhda G. Gemejiyeva, and Elvira E. Shults. 2025. "Isolation and Identification of Secondary Metabolites in Rheum tataricum L.fil. Growing in Kazakhstan and Surveying of Its Anticancer Potential" Molecules 30, no. 14: 2978. https://doi.org/10.3390/molecules30142978
APA StyleTurgunbayeva, A. A., Sultanova, N. A., Hamad, M. S., Savelyev, V. A., Chernyak, E. I., Bagryanskaya, I. Y., Pokrovsky, M. A., Pokrovsky, A. G., Gemejiyeva, N. G., & Shults, E. E. (2025). Isolation and Identification of Secondary Metabolites in Rheum tataricum L.fil. Growing in Kazakhstan and Surveying of Its Anticancer Potential. Molecules, 30(14), 2978. https://doi.org/10.3390/molecules30142978