Abstract
A group of sulfonium and selenonium salts bearing diverse benzylidene acetal substituents on their side chain moiety were designed and synthesized. Compared with our previous study, structural modifications in this study focused on multi-substitution of the phenyl ring and bioisosteric replacements at the sulfonium cation center. In vitro biological evaluation showed that selenonium replacement could significantly improve their α-glucosidase inhibitory activity. The most potent inhibitor 20c (10.0 mg/kg) reduced postprandial blood glucose by 48.6% (15 min), 52.8% (30 min), and 48.1% (60 min) in sucrose-loaded mice, outperforming acarbose (20.0 mg/kg). Docking studies of 20c with ntMGAM presented a new binding mode. In addition to conventional hydrogen bonding and electrostatic interaction, amino residue Ala-576 was first identified to contribute to binding affinity through π-alkyl and alkyl interactions with the chlorinated substituent and aromatic ring. The selected compounds exhibited a high degree of safety in cytotoxicity tests against normal cells. Kinetic characterization of α-glucosidase inhibition confirmed a fully competitive inhibitory mode of action for these sulfonium salts.
1. Introduction
Diabetes mellitus typically arises due to either insufficient insulin secretion resulting from the destruction of β cells or the body’s ineffective utilization of normally produced insulin. It is projected that by 2045, the number of individuals suffering from diabetes will reach 783 million [1]. Elevated blood glucose levels are closely associated with a range of complications, including vascular damage, kidney failure, nerve damage, and severe foot infections, among others [2]. The most frequently prescribed medications for T2DM, accounting for over 90% of all diabetes cases, are oral antidiabetic agents, encompassing insulin secretagogues (such as sulfonylureas), insulin sensitizers (like metformin), PPARγ agonists (thiazolidinediones), DPP-4 inhibitors (e.g., sitagliptin) and GLP-1 mimetics (e.g., exenatide) [3,4], sodium-glucose cotransporter 2 (SGLT2) inhibitors [5,6], and α-glucosidase inhibitors (e.g., acarbose) [7,8]. Among the various therapeutic strategies available, one particularly effective method involves delaying intestinal glucose absorption by interfering with the breakdown of polysaccharides [9]. This can be accomplished by inhibiting different α-glucosidases located at the brush border of the intestine, a strategy that has garnered significant attention in medicinal chemistry due to its lack of severe side effects. This approach is especially beneficial for Asian patients, whose diets predominantly consist of carbohydrates [10,11], a dietary pattern that contributes to the high prevalence of diabetes in densely populated Asian countries, thereby driving the global diabetes epidemic. The three currently approved α-glucosidase inhibitors for hyperglycemia—acarbose, voglibose, and miglitol—all belong to the azasugar or iminosugar derivative classes [12,13]. While these agents offer moderate therapeutic benefits for T2DM, they are sometimes associated with gastrointestinal disturbances, such as bloating, diarrhea, nausea, liver dysfunction, and skin allergies, among other adverse effects [14]. Consequently, the identification of potent α-glucosidase inhibitors with high safety profiles remains a focal area of research for medicinal chemists [15,16,17,18,19,20,21,22,23,24].
Since 1997, a group of sulfonium salts characterized by polyhydroxylated side chain structure have been isolated from plants belonging to the genus Salacia [25,26,27]. These compounds, featuring distinct five-membered-ring sulfonium derivatives, can be categorized into two groups. Compounds 1–4 [28] present a unique zwitterionic sulfonium-sulfate salt structure, while compounds 5–8 [29,30,31,32] are identified as the de-O-sulfonated derivatives of 1–4, respectively (Figure 1). In vitro biological assessments have demonstrated that these sulfonium salts exhibit potent α-glucosidase inhibitory activity comparable to clinically prescribed anti-diabetic drugs such as acarbose and voglibose. Their unique structural character, coupled with promising biological activities, has established them as a novel class of drug candidates, specifically as potent α-glucosidase inhibitors. Consequently, significant efforts have been directed towards the total synthesis [29,33,34,35,36,37,38,39,40,41,42,43] and structural modification [44,45,46,47,48,49,50,51,52] of this series of compounds.
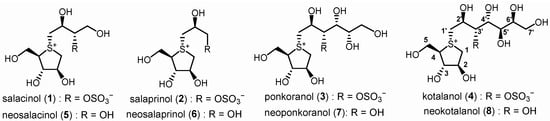
Figure 1.
Sulfonium type α-glucosidase inhibitors isolated from Genus Salacia.
Natural products. Previous structure–activity relationship studies have unequivocally demonstrated that the five-membered sulfonium ring fragment represents the most structurally conserved region responsible for potent bioactivity, with the cooperative effect of the 2′S-OH and 4′R-OH groups on the side chain being crucial for achieving strong inhibitory effects [29] (Figure 1 and Figure 2). Polyhydroxylated side chains longer than four carbons, such as those found in compounds 7 and 8, do not significantly enhance the enzyme inhibitory potency [29]. Additionally, the hydrophilic 3′-O-sulfate moiety in compounds 1–4 is constrained by adjacent hydrophobic residues of the target enzymes [53,54], explaining why de-O-sulfonated analogs 5–8 (Figure 1) typically exhibit superior inhibitory potency compared to their sulfated counterparts 1–4. As part of a rational drug design approach, a series of hydrophobic substituents were deliberately introduced at the 3′-O position of neosalacinol by us [48]. It was discovered that sulfonium salt 9 (Figure 2), which features a substituted benzyl group at the 3′-O position, exhibited significantly increased α-glucosidase inhibitory activities. In silico docking studies revealed an effective π-π stacking interaction between the phenyl ring of the amino acid residue Phe450 and the benzyl group. Recently, we reported a group of sulfonium derivatives (10) with a five-carbon side chain structure on which 3′-OH and 5′-OH diol were protected by a benzylidene acetal ring [54]. Mono-substitution on the benzene ring resulted in different sulfonium salts with different enzyme inhibitory profiles. Specifically, the introduction of a strong electron withdrawing group at the ortho position was most beneficial to present potent α-glucosidase inhibitory activities. Two sulfonium salts with a nitro- or trifluromethyl group attached at the ortho position on the benzene ring exhibited potent in vitro and in vivo bioactivities. Based on previous investigation, we believe that there remains a potential for further structural modification of the benzylidene acetal moiety. For example, an increased dihedral angle between the phenyl ring and acetal ring, as well as a stronger electron-deficient effect on the phenyl ring, may be more beneficial for further enhancing α-glucosidase inhibitory potency. As a continuous SAR study, this paper describes a more comprehensive structural modification focusing on the benzene ring within the benzylidene group. Multiple substituents were introduced onto the benzene ring. Furthermore, utilizing the principle of “bioisosterism”, we replaced the sulfonium cation core moiety with a selenonium cation structure, and a series of target compounds were designed and synthesized. In vitro enzymatic inhibitory activity against maltase and sucrase was evaluated. The most potent inhibitor was selected for in vivo antihyperglycemic efficacy investigation.
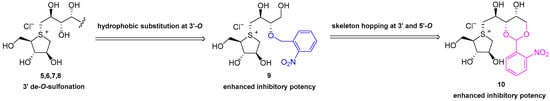
Figure 2.
Our previous structural modification at the 3′-O and 5′-O positions of Salacia-derived Sulfonium type α-glucosidase inhibitors.
2. Results and Discussion
2.1. Chemistry
Starting from compound 11 [49], selective removal of the benzylidene protecting group [55] using 30% trifluoroacetic acid (TFA) solution yielded 12 in 73% yield, serving as a pivotal intermediate for diverse downstream chemical reactions (Scheme 1). Compound 12 was subjected to acid-catalyzed acetalization with different substituted benzaldehydes to give 13a–r in 41–60% yield. 13a–r underwent direct vicinal cyclization [56] to produce epoxide 14a–r with a yield of 85–96%. Subsequently, a coupling reaction with the five-membered thiosugar 15 [35] or selenosugar 16 [45] was conducted, following a previously reported procedure [29] but in a modified manner. In this step, 14a–r was coupled with thiosugar 15 and selenosugar 16 in the presence of a catalytic amount of trifluoroacetic acid (TFA), resulting in the formation of sulfonium salts 17a–r and 18a–r. Under these reaction conditions, the acid-sensitive benzylidene moiety remained. Target compounds were obtained in satisfactory yield without any observation of any degradation products. Following a direct ion exchange reaction with IRA 400J (in its chloride form), sulfonium chloride 19a–r and selenonium chloride 20a–r were obtained, respectively.
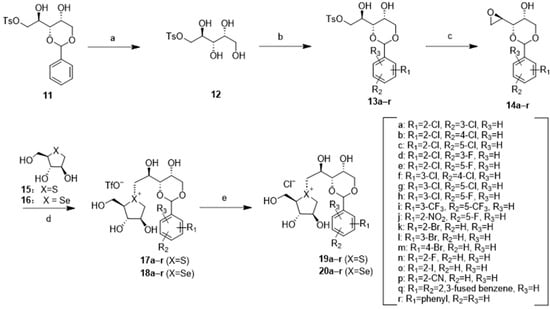
Scheme 1.
Reagents and conditions: (a) 30% aq TFA, DCM, r.t, 4 h, 73%; (b) R-benzaldehyde, p-TSA, MeCN, r.t, 24 h, 41–60%; (c) DBU, DCM, 0 °C, 2 h, 85–96%; (d) TFA, MeCN, −20 °C-r.t, 8 h; (e) IRA400J (Cl− form), MeOH, r.t, 4 h, 45–62%.
2.2. α-Glucosidase Inhibitory Activity Evaluation
The α-glucosidase inhibitory activity of compounds 19a–r and 20a–r was evaluated in vitro against rat intestinal sucrase and maltase, in comparison with the natural sulfonium salt neoponkoranol (7) and the clinical anti-diabetic drugs voglibose and acarbose. IC50 values are shown in Table 1 and Table 2. Our previous investigation revealed that an electron withdrawing group attached at the ortho-position on the phenyl ring is the most beneficial way to increase enzyme inhibitory activity of the resulting sulfonium salts. Thus, most of the designed products maintained an electron withdrawing substituent at ortho-position on phenyl ring. Compounds 19a–19e are sulfonium salts featuring a second halogen substitution at either the meta- or para-position on the phenyl ring structure.

Table 1.
IC50 Values for synthetic sulfonium salts 19a–r, acarbose, and voglibose against maltase and sucrase.
Table 2.
IC50 values for synthetic selenonium salts 20a–r, acarbose, and voglibose against maltase and sucrase.
Generally, the introduction of a second halogen enhanced the maltase inhibitory activity of sulfonium salts compared to acarbose. Specifically, compounds 19a, 19c–e exhibited IC50 values ranging from 0.50 to 1.0 μM, demonstrating approximately seven-fold greater potency than that of acarbose (IC50 value of 3.5 against maltase). For compounds 19f–h, chlorine substituent was moved from the ortho- to the meta-position. The introduction of a second chlorine did not further increase the inhibitory efficacy of the resulting sulfonium salts, which clearly demonstrated the critical role of ortho-substitution by an electron withdrawing group for strong bioactivity. When two strong electron-withdrawing group (EWG) trifluoromethyl groups were introduced at two meta-positions, the resulting sulfonium salt 19i also showed modest inhibitory potency (IC50 = 8.9 μM) against maltase. For sulfonium salt 19j, a strong EWG nitro group was placed at the ortho-position again and the second halogen (fluorine) atom introduced at the para-position yielded compound 19j with increased maltase inhibition (IC50 = 0.75 μM). In this study, we also synthesized several sulfonium salts with mono-substitution on the benzene ring as a supplementary investigation to our previous study. When bromine was introduced, sulfonium salt 19k with ortho-Br attachment showed stronger maltase inhibitory activity than that of 19l and 19m. It is interesting that 19m with bromine attached at the para-position is more potent than 19l, a meta-bromo-substituted sulfonium analog, which is inconsistent with findings from our previous study [57]. The known sulfonium salt 19n also showed potent α-glucosidase inhibitory activity in the present study. Iodine is a halogen atom with relatively weak electron-withdrawing capacity. Unsurprisingly, compound 19o with ortho-iodine substitution exhibited slightly decreased inhibitory activity. When ortho-iodine was replaced by an ortho-cyano group, the resulting sulfonium salt 19p exhibited increased maltase inhibitory activity comparable to that of 19n.
In this study, we also attempted to replace the phenyl ring with other aromatic functional groups, such as naphthyl and biphenyl groups, resulting in compounds 19q and 19r. Unfortunately, both compounds showed decreased inhibitory efficacy when compared with the benzaldehyde analog. In a sucrase enzymatic assay, a similar trend to that discussed above was observed. 19n presented the strongest inhibitory activity against both maltase and sucrase.
By applying the principle of “bioisosterism”, a group of selenonium analogs (20a–r) were also synthesized. Their inhibitory activity against maltase and sucrase was evaluated. To our delight, the biological activities of selenonium salts were increased to some extent when compared with their sulfonium counterparts. Especially, selenonium salt 20c could effectively inhibit maltase with an IC50 value of 0.14 μM and sucrase with an IC50 value of 0.05 μM, representing 25-fold and 44-fold greater potency than acarbose, respectively. This inhibitory potency is also much superior to that of voglibose, a clinically used strong α-glucosidase inhibitor. Unlike sulfonium salts 19f–i, which only showed modest enzymatic inhibitory activities, the inhibitory efficacy of selenonium salts 20f–i against maltase and sucrase increased from 7-fold to 25-fold. It should be noted that selenonium analog 20m with para-bromo substitution even showed superior α-glucosidase inhibitory activities compared to its ortho-bromo and meta-bormo counterparts 20k and 20l. Equipped with the selenonium core structure, compounds 20q and 20r, featuring naphthyl and biphenyl substituents, also showed improved inhibitory activities against both maltase and sucrase. Especially, 20q was shown to be a stronger maltase inhibitor even compared to voglibose.
2.3. Kinetic Enzymatic Studies
In order to explore the inhibitory mechanism of the α-glucosidase by this family of compounds, enzyme kinetic assays were conducted on 20a, 20c, and 10, followed by a detailed kinetic analysis using the Lineweaver–Burk plots. (Figure 3) The small intestinal brush border membrane vesicles were incubated with varying concentrations of maltose (3.27 mg/mL) for maltase assays and sucrose (52.37 mg/mL) for sucrase assays. The plots of 1/V against 1/[S] revealed a series of linear relationships with varying slopes, indicating that this family of compounds competitively inhibits the enzymes by binding to their active sites and competing with the natural substrates. The inhibition constants (Ki) of all selected inhibitors are listed in Table 3. Compound 10 displayed the highest binding affinity for maltase, as indicated by its Ki value of 0.42 μg/mL. Similarly, compound 20c exhibited the highest affinity for sucrase, with a Ki of 0.20 μg/mL.
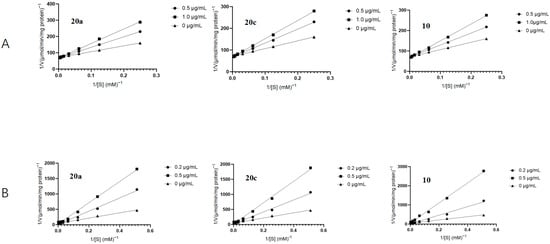
Figure 3.
Lineweaver–Burk plots of the inhibition of rat intestinal maltase (A) and sucrase (B) activities by compounds 20a, 20c, and 10 (Maltase concentration: 0.25 mg/mL; Sucrase concentration: 1.31 mg/mL).
Table 3.
Ki values of compounds 20a, 20c, and 10 for small intestinal α-glucosidase.
2.4. Cytotoxicity
The cytotoxicity of selected sulfonium salts (20a, 20c, and 10) was then evaluated via the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) assay against different cell lines. All compounds showed non-toxic behavior, indicating a high level of safety profile.
2.5. Hypoglycemic Effect in Normal Rats
Research has shown a strong association between the 2 h postprandial blood glucose level and the risk of cardiovascular events, diabetic microvascular complications such as retinopathy, and the progression of macrovascular diseases [57,58]. Recent clinical data suggest that postprandial hyperglycemia may exacerbate cognitive decline in elderly individuals with type 2 diabetes mellitus (T2DM) [59]. Considering this underlying pathological process, α-glucosidase inhibitors, which efficiently regulate postprandial glucose fluctuations, hold significant therapeutic promise for alleviating diabetes-related complications. In this investigation, selenonium salt derivatives 20c, identified through rigorous screening as the most potent inhibitor of α-glucosidase, was chosen for in vivo assessment of their modulatory effects on endogenous glucose homeostasis mechanisms. In the sucrose challenge model (2.5 g/kg p.o.), fasting ICR mice experienced a swift surge in blood glucose levels, with rapid elevation peaking at 30 min post-administration (Tmax) followed by a gradual return to baseline by 120 min (Figure 4). Pharmacological intervention with acarbose (20 mg/kg) demonstrated dose-dependent suppression across all measured time points (15, 30, 60, and 120 min). Relative to acarbose, naturally derived neoponkoranol (10.0 mg/kg) showed a similar trend of hypoglycemic effects but with relatively lower efficacy (Table 4). Candidate compound 20c was initially dosed at 10.0 mg/kg, exhibiting a robust and early-phase suppression of postprandial glucose levels comparable to acarbose but with greater potency (Figure 4). Compound 20c (10 mg/kg) exhibited superior early-phase glucose modulation compared to acarbose (20 mg/kg), achieving 48.6% reduction at 15 min (vs. 45.7% for acarbose) and peak efficacy of 52.8% at 30 min (vs. 48.4% control) (Table 4). The 46.4% decrease in the AUC for 20c (10 mg/kg) was also greater than the 40.5% decrease observed for acarbose (20 mg/kg). Sulfonium salt 10, the best inhibitor identified in our previous investigation, was also tested for hypoglycemic effect assay. Although 20c showed a slightly higher blood glucose reduction rate than 10 at 15 min post-administration, compound 10 exhibited slightly stronger glucose-lowering efficacy at 30 min and 60 min. In terms of the AUC reduction, both compounds demonstrated almost the same effects. Among all administered compounds, naturally occurring neoponkoranol yielded the least favorable results, further validating our strategy of bridging 3′ and 5′-hydroxyls via benzylidene acetal cyclization of the side chain moiety of these naturally derived sulfonium salts. Subsequently, doses were reduced to 1.0 mg/kg. As expected, the three administered compounds, neoponkoranol, 20c, and 10, exhibited decreased blood glucose reduction rates at 15 min, 30 min, and 60 min, respectively. Although compounds 20c and 10 (1 mg/kg) exhibited lower glucose-lowering efficacy compared to acarbose (20 mg/kg), both derivatives maintained statistically significant attenuation of postprandial hyperglycemia (Table 5 & Figure 5). At this low dose, 20c still demonstrated antihyperglycemic potency comparable to compound 10 across all time points (15–60 min).
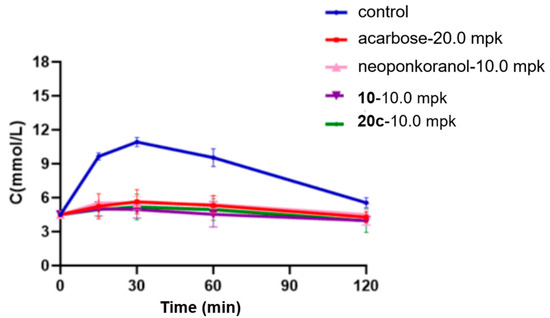
Figure 4.
Effects of 20c (10.0 mpk), 10 (1.0 mpk), neoponkoranol (10.0 mpk), and acarbose (20.0 mpk) on blood glucose levels in the sucrose loading test. The experiments were performed in three independent experiments. Each value represents the mean ± SD (n = 8).
Table 4.
Declined rates of blood glucose levels and AUC caused by 20c (10 mpk), 10, neoponkoranol (10 mpi), and acarbose (20 mpk) in sucrose loading test.
Table 5.
Declined rates of blood glucose levels and AUC caused by 20c (1 mpk), 10, neoponkoranol (1 mpk), and acarbose (20 mpk) in sucrose loading test.
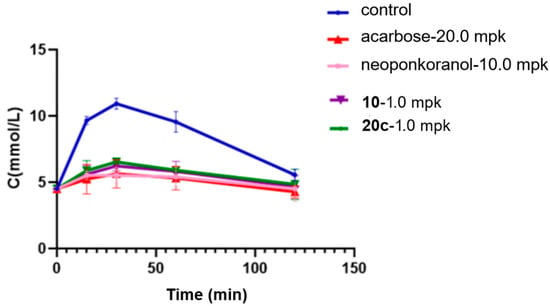
Figure 5.
Effects of 20c (1.0 mpk), 10 (1.0 mpk), neoponkoranol (1.0 mpk), and acarbose (20.0 mpk) on blood glucose levels in the sucrose loading test. The experiments were performed three times independently. Each value represents the mean ± SD (n = 8).
A maltose loading test on compounds 10, 20c, neoponkoranol, and positive control (voglibose) was conducted to further evaluate their in vivo hypoglycemic effects against maltase. As shown in Figure 6, when fasted rats were administered maltose at a dose of 2.0 g/kg, their blood glucose levels swiftly rose from 4.75 mmol/L to a peak of 14.62 mmol/L within 30 min post-ingestion. Subsequently, these levels returned to the pretreatment baseline at 120 min. The clinically used strong α-glucosidase inhibitor voglibose (1.0 mg/kg) showed an evident antihyperglycemic effect across all measured time points. At an equimolar dose, compound 10 presented the strongest capability to delay the hydrolysis of maltose. As shown in Table 6, compound 10 achieved blood glucose reduction rates of 52.8%, 55.0%, and 47.2% at 15, 30, and 60 min, respectively, which are superior to those of voglibose. Compound 20c also showed evident hypoglycemic effects. Although less potent than 10, compound 20c achieved a 43.5% blood glucose reduction rate at 60 min (vs. 40.1% for voglibose). Notably, administration of 20c led to a 40.2% decrease in AUC, which was also greater than that of voglibose (a 39.8% decrease in AUC). Naturally occurring neoponkoranol showed reduction rates of 42.3%, 33.9%, and 19.4% at 15, 30, and 60 min and a 21.5% decrease in AUC, making it the least effective among the four tested compounds.
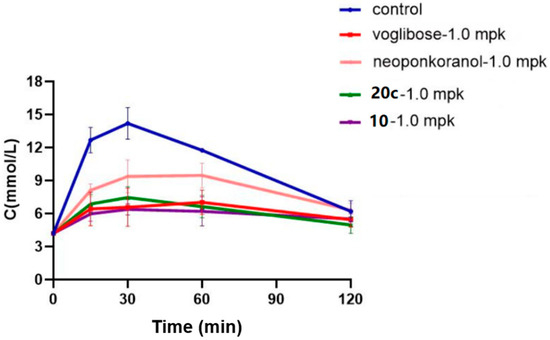
Figure 6.
Effects of 20c (1.0 mpk), 10 (1.0 mpk), neoponkoranol (1.0 mpk), and voglibose (1.0 mpk) on blood glucose levels in the maltose loading test. The experiments were performed in three independent experiments. Each value represents the mean ± SD (n = 8).
Table 6.
Declined rates of blood glucose levels and AUC resulted by 20c (1 mpk), 10, neoponkoranol (1 mpk), and voglibose (1.0 mpk) in the maltose loading test.
2.6. Docking Studies on Compound 20c with ntMGAM
In molecular docking studies, binding energy serves as a critical parameter for evaluating the binding stability between ligands and receptors. Generally, a binding energy below 0 kcal/mol indicates spontaneous binding, while values lower than −5 kcal/mol typically indicate strong binding affinity. In this study, molecular docking revealed a binding energy of −6.8 kcal/mol for the ligand–target protein complex, suggesting a thermodynamically favorable and high-affinity interaction. Analysis of the binding mode demonstrated that Asp-203 (2.5 Å), His-600 (2.1 Å), and Asp-443 (2.4 Å and 2.6 Å) stabilize the complex through hydrogen bonding interactions. Notably, Asp-203 also forms a carbon–hydrogen bond with the hydroxyl-bearing carbon atom in the ligand’s dioxane ring. Furthermore, the selenium atom participates in attractive charge interactions with Asp-443 and Asp-542, while Asp-542 engages in a π-anion interaction with the aromatic ring of the ligand, collectively enhancing binding affinity and specificity. Concurrently, Ala-576 contributes to hydrophobic stabilization via π-alkyl and alkyl interactions with the chlorinated substituent and aromatic ring. These synergistic interactions reinforce complex stability. Collectively, the results suggest that this ligand exhibits both favorable binding energetics and a pharmacologically relevant interaction pattern within the target protein’s active site (Figure 7).
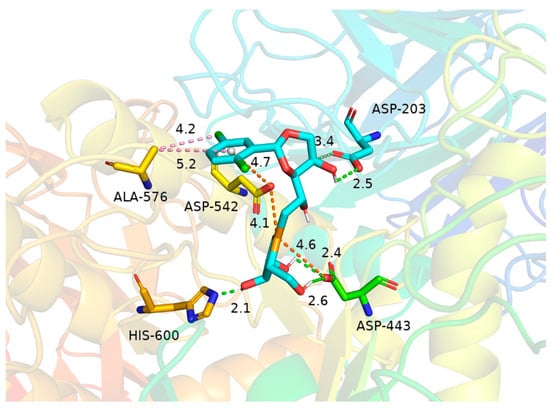
Figure 7.
Interactions of 20c with human N-terminal maltase-glucoamylase (hNtMGAM) (PDB ID: 3L4U). Dotted lines show hydrogen bonding (light green); dotted lines with yellow color show salt bridges. The hydrophobic interaction between ALA-576 and chlorinated substituent and aromatic ring shown in the pink dotted lines were calculated to be 4.2 Å. The electrostatic interaction between two chloride atoms of 20c and the amino acid residues Asp 542 and ASP 443 was calculated to be 4.1 Å and 4.6 Å, respectively.
3. Experimental Section
3.1. Materials and Instruments
Reagents and Solvents: All solvents were purified and dried according to standard methods. PE refers to petroleum ether (b.p. 60–90 °C), EA refers to ethyl acetate, DCM refers to methylene dichloride, and DMF refers to dimethyl formamide.
Chromatography: Flash column chromatography was carried out using commercially available 200–300 mesh under pressure and conducted by eluting with PE/EA, which are listed as volume/volume ratios.
Data Collection: The melting point (m.p.) was measured on a microscopic melting point apparatus. 1H NMR spectra were collected on a BRUKER AV-300 (Billerica, MA, USA, 300 MHz) spectrometer using CDCl3 or DMSO-d6 or methanol-d4 as solvent. Chemical shifts of 1H NMR were recorded in parts per million (ppm, δ) relative to tetramethylsilane (δ = 0.00 ppm) with the solvent resonance as an internal standard (CDCl3, δ = 7.26 ppm, DMSO-d6, δ = 2.50 ppm, methanol-d4, δ = 3.31 ppm). Data are reported as follows: chemical shift in ppm (δ), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, brs = broad singlet, m = multiplet), coupling constant (Hz), and integration. 13C NMR spectra were collected on a BRUKER AV-300 (75 MHz) and BRUKER AVANCE500 (125 MHz) spectrometer using CDCl3 or DMSO-d6 or methanol-d4 as solvent. Chemical shifts of 13C NMR were reported in ppm with the solvent as the internal standard (chloroform-d6, δ = 77.0 ppm, DMSO-d6, δ = 39.5 ppm, methanol-d4, δ = 47.1 ppm). Low-resolution mass spectra (MS) were measured on Finnigan MAT 95 spectrometer (Finnigan, München, Germany). Highresolution mass measurement was performed on Agilent QTOF 6520 mass spectrometer (Santa Clara, CA, USA) with electron spray ionization (ESI) as the ion source. All compounds selected for in vivo hypoglycemic effect investigation were >95% pure according to HPLC (SHIMADZU Labsolutions, Kyoto, Japan, UV detection at λ = 230 nm) analysis on the Agilent C18 column (4.6 × 150 mm2, 5 μm) eluting at 1 mL/min of 90% methanol/10% water.
3.2. Synthesis
3.2.1. Preparation of 5-O-Tosyl-d-arabinitol (12)
To a solution of 11 (20.0 g, 50.75 mmol) in DCM (100 mL) was added 30% aq TFA (500 mL), and the reaction mixture was stirred at room temperature for 4 h. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (DCM/MeOH = 20:1) on silica gel to afford the pure product 12 (11.34 g, 73%) as a white solid, m.p. 285–286 °C. = −37.2 (c = 1.00, MeOH). 1H NMR (300 MHz, DMSO-d6) δ 7.78 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 8.0 Hz, 2H), 4.19 (dd, J = 9.6, 2.2 Hz, 1H), 4.04 (br,4H), 3.93 (dd, J = 9.6, 6.6 Hz, 1H), 3.65 (m, J = 17.5, 6.6, 1.8 Hz, 2H), 3.34 (dd, J = 6.6, 2.3 Hz, 2H), 3.31−3.25 (m, 1H), 2.42 (s, 3H). 13C NMR (75 MHz, DMSO-d6) 144.8, 132.5, 130.2, 127.7, 73.8, 69.8, 69.5, 68.4, 62.6, 21.2. MS (ESI) 307.1 (M + 1).
3.2.2. General Procedure for the Synthesis of Acetals (13a–r)
To a solution of 12 (612 mg, 2.0 mmol) in MeCN (10 mL) were added the appropriate benzaldehyde (3.0 mmol) and p-toluenesulfonic acid (50 mg); the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was neutralized by triethylamine. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (PE/EtOAc = 2/1) on silica gel to afford the pure products 13a–r as pale-yellow oils with yields from 41 to 60%.
- 5-O-tosyl-1,3-O-(2,3-Dichlorobenzylidene)-d-arabinitol (13a). 13a can be obtained from 12 with a yield of 49%. Pale-yellow oils. = −14.5 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.74 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 7.6 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 7.23 (t, J = 5.9 Hz, 2H), 7.19–7.11 (m, 1H), 5.64 (s, 1H), 4.23 (t, J = 9.7 Hz, 2H), 4.09–4.02 (m, 3H), 3.85 (s, 2H), 3.74 (b r, 1H), 3.62 (br,1H), 2.35 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.2, 136.8, 133.1, 132.2, 131.2, 131.1, 129.9, 128.0, 127.5, 125.8, 98.5, 78.2, 72.6, 71.3, 67.2, 62.5, 21.7. MS (ESI) 463.0 (m + 1).
- 5-O-Tosyl-1,3-O-(2,4-dichlorobenzylidene)-d-arabinitol (13b). 13b can be obtained from 12 with a yield of 54%. Pale-yellow oils. = −23.6 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.73 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 8.4 Hz, 1H), 7.36–7.32 (m, 1H), 7.25–7.18 (m, 3H), 5.61 (s, 1H), 4.27–4.18 (m, 2H), 4.06 (t, J = 4.4 Hz, 3H), 4.00 (br, 1H), 3.89–3.81 (m, 2H), 3.50 (br, 1H), 2.36 (s, 3H), 13C NMR (75 MHz, CDCl3) δ 145.1, 135.5, 133.5, 133.3, 132.3, 129.9, 129.3, 128.7, 128.0, 127.2, 97.9, 78.1, 72.6, 71.3, 67.2, 62.4, 21.7. MS (ESI) 463.0 (M + 1).
- 5-O-Tosyl-1,3-O-(2,5-dichlorobenzylidene)-d-arabinitol (13c). 13c can be obtained from 12 with a yield of 44%. Pale-yellow oils. = −11.6 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.75 (d, J = 8.2 Hz, 2H), 7.55 (s, 1H), 7.29–7.21 (m, 4H), 5.61 (s, 1H), 4.31–4.19 (m, 2H), 4.12–4.04 (m, 3H), 3.88–3.81 (m, 2H), 3.49 (s, 1H), 2.34 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.0, 136.2, 132.8, 132.2, 131.1, 130.7, 130.3, 130.0, 129.9, 128.0, 127.8, 97.7, 78.1, 72.6, 71.2, 67.1, 62.4, 21.7. MS (ESI) 463.0 (M + 1).
- 5-O-Tosyl-1,3-O-(2-chloro-3-fluorobenzylidene)-d-arabinitol (13d). 13d can be obtained from 12 with a yield of 43%. Pale-yellow oils. = −22.3(c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.75 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 7.7 Hz, 1H), 7.33–7.28 (m, 1H), 7.25 (d, J = 8.1 Hz, 2H), 7.16 (d, J = 8.4 Hz, 1H), 5.65 (s, 1H), 4.27–4.21 (m, 2H), 4.12–4.04 (m, 3H), 3.89–3.84 (m, 2H), 3.35 (br, 1H), 2.37 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 158.0 (d, J = 247.9 Hz), 145.1, 136.8, 132.3, 130.0, 129.9, 128.1, 128.0, 127.7 (d, J = 7.8 Hz), 122.7 (d, J = 3.4 Hz), 120.0 (d, J = 18.9 Hz), 119.9, 117.1 (d, J = 21.3 Hz), 98.0 (d, J = 3.0 Hz), 78.2, 72.6, 71.3, 67.2, 62.5, 21.6. MS (ESI) 447.1 (M + 1).
- 5-O-Tosyl-1,3-O-(2-chloro-5-fluorobenzylidene)-d-arabinitol (13e). 13e can be obtained from 12 with a yield of 43%. Pale-yellow oils. = −24.2(c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.70 (dd, J = 8.4, 2.0 Hz, 2H), 7.26–7.19 (m, 4H), 6.98–6.91 (m, 1H), 5.57 (s, 1H), 4.23–4.13 (m, 2H), 4.08–4.00 (m, 4H), 3.82–3.77 (m, 2H), 3.17 (s, 1H), 2.92 (s, 1H), 2.32 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.2, 136.5 (d, J = 7.6 Hz), 132.3, 131.0 (d, J = 8.1 Hz), 130.1, 130.0, 128.1, 127.7 (d, J = 3.0 Hz), 117.5 (d, J = 23.0 Hz), 114.9 (d, J = 24.9 Hz), 98.0, 78.3, 72.7, 71.1, 67.5, 62.6, 21.7. MS (ESI) 447.1 (M + 1).
- 5-O-Tosyl-1,3-O-(3,4-Dichlorobenzylidene)-d-arabinitol (13f). 13f can be obtained from 12 with a yield of 42%. Pale-yellow oils. = −33.7 (c = 0.50,MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.80–7.75 (m, 2H), 7.44–7.42 (m, 1H), 7.39 (d, J = 8.3 Hz, 1H), 7.27 (d, J = 8.2 Hz, 2H), 7.18 (dd, J = 8.3, 2.1 Hz, 1H), 5.40 (s, 1H), 4.22–4.18 (m, 2H), 4.10–4.02 (m, 2H), 3.90–3.87 (m, 1H), 3.87–3.80 (m, 2H), 3.65 (br, 2H), 2.36 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.3, 137.5, 133.1, 132.4, 132.3, 130.3, 130.0, 128.1, 128.0, 125.6, 99.3, 77.7, 72.4, 70.9, 67.3, 62.5, 21.7. MS (ESI) 463.0 (M + 1).
- 5-O-Tosyl-1,3-O-(3,5-Dichlorobenzylidene)-d-arabinitol (13g). 13g can be obtained from 12 with a yield of 55%. Pale-yellow oils. = −33.7 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.80 (d, J = 8.3 Hz, 2H), 7.35–7.33 (m, 2H), 7.31 (s, 1H), 7.26 (d, J = 2.0 Hz, 2H), 5.41 (s, 1H), 4.24 (d, J = 3.0 Hz, 2H), 4.10–4.03 (m, 2H), 3.91–3.82 (m, 3H), 3.09 (br, 1H), 2.86 (br, 1H), 2.39 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.4, 140.4, 135.0, 132.4, 132.4, 130.1, 129.2, 128.1, 124.7, 99.2, 77.9, 72.5, 70.8, 67.6, 62.6, 21.8. MS (ESI) 463.0 (M + 1).
- 5-O-Tosyl-1,3-O-(3-Chloro-5-fluorobenzylidene)-d-arabinitol (13h). 13h can be obtained from 12 with a yield of 41%. Pale-yellow oils. = −13.7 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.81 (d, J = 8.1 Hz, 2H), 7.33 (d, J = 8.1 Hz, 3H), 7.09 (dq, J = 8.3, 2.8, 2.2 Hz, 1H), 6.98 (dt, J = 8.9, 1.9 Hz, 1H), 5.43 (s, 1H), 4.30–4.20 (m, 3H), 4.07 (d, J = 11.5 Hz, 2H), 3.94–3.86 (m, 3H), 3.19 (d, J = 6.4 Hz, 1H), 2.92 (d, J = 10.3 Hz, 1H), 2.41 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.4, 140.9(d, J = 8.5 Hz), 135.1 (d, J = 10.6 Hz), 132.5, 130.1, 128.2, 128.1, 122.3 (d, J = 3.15 Hz), 116.8 (d, J = 24.6 Hz), 111.9 (d, J = 23.0 Hz), 99.2, 77.9, 72.5, 70.8, 67.6, 62.6, 21.7. MS (ESI) 447.1 (M + 1).
- 5-O-Tosyl-1,3-O-(3,5-bis(trifluoromethyl)benzylidene)-d-arabinitol (13i). 13i can be obtained from 12 with a yield of 56%. Pale-yellow oils. = −55.4 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.88 (d, J = 5.4 Hz, 3H), 7.81–7.78 (m, 2H), 7.30 (d, J = 8.1 Hz, 2H), 5.59 (s, 1H), 4.35–4.29 (m, 1H), 4.24 (d, J = 3.7 Hz, 2H), 4.14 (d, J = 12.1 Hz, 2H), 3.95 (d, J = 13.4 Hz, 2H), 3.46 (br, 2H), 2.38 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.5, 139.9, 132.4, 131.7 (q, J = 33.6 Hz), 130.3, 130.1, 128.1, 126.7, 124.07 (q, J = 161.5 Hz), 99.0, 78.1, 72.6, 71.0, 67.5, 62.6. MS (ESI) 531.0 (M + 1).
- 5-O-Tosyl-1,3-O-(5-fluoro-2-nitrobenzylidene)-d-arabinitol (13j). 13j can be obtained from 12 with a yield of 51%. Pale-yellow oils. = −38.7 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.94–7.90 (m, 1H), 7.81 (d, J = 6.4 Hz, 2H), 7.45 (d, J = 8.6 Hz, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.19–7.14 (m, 1H), 6.04 (s, 1H), 4.22 (d, J = 10.8 Hz, 2H), 4.07 (t, J = 8.8 Hz, 3H), 3.86 (d, J = 8.6 Hz, 2H), 3.31 (br, 2H), 2.39 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 166.2, 162.8, 145.2, 144.2, 134.78 (d, J = 9.0Hz), 132.4, 130.0, 128.1, 127.24 (J = 9.5Hz), 116.60 (d, J = 23.5 Hz), 115.18 (d, J = 26.1 Hz), 96.3, 78.6, 72.8, 71.3, 67.4, 62.4, 21.7. MS (ESI) 458.1 (M + 1).
- 5-O-Tosyl-1,3-O-(2-bromobenzylidene)-d-arabinitol (13k). 13k can be obtained from 12 with a yield of 54%. Pale-yellow oils. = −43.2 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.73 (d, J = 8.3 Hz, 2H), 7.58–7.52 (m, 1H), 7.50 (d, J = 1.4 Hz, 1H), 7.29–7.22 (m, 1H), 7.19 (d, J = 8.5 Hz, 3H), 5.56 (s, 1H), 4.31–4.22 (m, 1H), 4.18 (d, J = 12.9 Hz, 1H), 4.10–3.97 (m, 3H), 3.83 (d, J = 5.5 Hz, 2H), 3.74 (br, 2H), 2.31 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 144.9, 136.1, 132.7, 132.3, 130.6, 129.8, 127.9, 127.8, 127.4, 122.3, 100.2, 78.0, 72.5, 71.3, 67.0, 62.3, 21.6. MS (ESI) 473.0 (M + 1).
- 5-O-Tosyl-1,3-O-(m-bromobenzylidene)-d-arabinitol (13l). 13l can be obtained from 12 with a yield of 53%. Pale-yellow oils. = −30.2 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.78 (d, J = 7.8 Hz, 2H), 7.48 (s, 1H), 7.32–7.22 (m, 4H), 7.22–7.17 (m, 1H), 5.38 (s, 1H), 4.19 (q, J = 11.8, 11.0 Hz, 3H), 4.10–3.99 (m, 2H), 3.87–3.80 (m, 2H), 3.71 (br, 1H), 3.28 (br, 1H), 2.34 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.2, 139.5, 132.4, 132.1, 130.0, 129.9, 129.1, 128.0, 124.8, 122.3, 99.8, 77.7, 72.4, 71.0, 67.3, 62.5, 21.7. MS (ESI) 473.0 (M + 1).
- 5-O-Tosyl-1,3-O-(p-Bromobenzylidene)-d-arabinitol (13m). 13m can be obtained from 12 with a yield of 60%. Pale-yellow oils. = −42.2 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.79 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 7.9 Hz, 2H), 7.31 (d, 1H), 7.29–7.27 (d, 2H), 7.24 (d, 1H), 5.44 (s, 1H), 4.29–4.20 (m, 3H), 4.14–4.03 (m, 2H), 3.90–3.82 (m, 2H), 3.11 (br, 1H), 2.96 (br, 1H), 2.41 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.3, 136.4, 132.5, 131.7, 131.5, 130.1, 128.1, 127.8, 123.3, 100.3, 78.0, 72.5, 70.9, 67.6, 62.7, 21.8. MS (ESI) 473.0 (M + 1).
- 5-O-Tosyl-1,3-O-(2-fluorophenylmethylene)-d-arabinitol (13n). 13n can be obtained from 12 with a yield of 58%. Colorless oil. = −9.9 (c = 0.32, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.75 (d, J = 8.2 Hz, 2H), 7.51 (tdd, J = 7.5, 5.3, 1.9 Hz, 1H), 7.34 (tdd, J = 7.5, 5.3, 1.9 Hz, 1H), 7.24 (d, J = 8.2 Hz, 2H), 7.13 (td, J = 7.5, 1.1 Hz, 1H), 7.04 (ddd, J = 9.8, 8.3, 1.1 Hz, 1H), 5.68 (s, 1H), 4.29–4.23 (m, 1H), 4.23–4.17 (m, 1H), 4.13–4.02 (m, 3H), 3.87–3.89 (m, 2H), 3.19 (br., 2H), 2.36 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 161.7, 145.1, 131.0 (d, JC-F = 8.1 Hz), 130.0, 129.9, 128.1, 128.0, 127.6 (d, JC-F = 3.5 Hz), 124.1 (d, JC-F = 3.6 Hz), 115.5 (d, JC-F = 21.3 Hz), 96.6, 78.2, 72.6, 71.2, 67.5, 62.6, 21.7. MS (ESI) 413.1 (M + 1).
- 5-O-Tosyl-1,3-O-(2-iodobenzylidene)-d-arabinitol (13o). 13o can be obtained from 12 with a yield of 57%. Colorless oil. = −27.4 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.82 (d, J = 7.8 Hz, 1H), 7.77 (d, J = 8.1 Hz, 2H), 7.54 (dd, J = 7.8, 1.7 Hz, 1H), 7.33 (t, J = 7.4 Hz, 1H), 7.24 (d, J = 8.0 Hz, 2H), 7.06 (td, J = 7.6, 1.8 Hz, 1H), 5.43 (s, 1H), 4.33 (d, J = 8.8 Hz, 1H), 4.27–4.19 (m, 1H), 4.16 (d, J = 5.4 Hz, 1H), 4.08 (t, J = 10.0 Hz, 2H), 3.87 (d, J = 8.1 Hz, 2H), 3.39 (br, 2H), 2.36 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.0, 139.4, 138.9, 132.4, 130.8, 129.9, 128.2, 128.0, 127.6, 103.8, 97.0, 78.1, 72.5, 71.5, 67.2, 62.4, 21.7. MS (ESI) 521.0 (M + 1).
- 5-O-Tosyl-1,3-O-(2-cyanobenzylidene)-d-arabinitol (13p). 13p can be obtained from 12 with a yield of 55%. Pale-yellow oils. = −28.7 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d)) δ 7.77–7.74 (m, 2H), 7.67 (d, J = 7.5 Hz, 1H), 7.60–7.55 (m, 2H), 7.48–7.44 (m, 1H), 7.24 (d, J = 8.3Hz, 2H), 5.63 (s, 1H), 4.30–4.22 (m, 3H), 4.14–4.10 (m, 3H), 3.90 (d, J = 8.9 Hz, 2H), 3.62 (br, 1H), 2.35 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.0, 140.4, 133.3, 132.9, 132.4, 129.9, 129.7, 128.1, 128.0, 127.7, 118.2, 110.6, 99.6, 78.2, 72.5, 71.4, 67.2, 62.6, 21.7. MS (ESI) 420.1 (M + 1).
- 5-O-Tosyl-1,3-O-(1-naphthalenebenzenemethylene)-d-arabinitol (13q). 13q can be obtained from 12 with a yield of 52%. Pale-yellow oils. = −24.7 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.95 (d, J = 7.5 Hz, 1H), 7.83 (t, J = 8.1 Hz, 2H), 7.69 (d, J = 8.0 Hz, 2H), 7.64 (d, J = 7.4 Hz, 1H), 7.51 (dt, J = 9.5, 5.8 Hz, 2H), 7.39 (t, J = 7.7 Hz, 1H), 7.05 (d, J = 8.0 Hz, 2H), 5.91 (s, 1H), 4.21 (d, J = 12.2 Hz, 1H), 4.17–4.12 (m, 2H), 4.07 (dd, J = 10.6, 5.0 Hz, 2H), 3.94 (d, J = 8.9 Hz, 1H), 3.84 (s, 1H), 3.29 (br, 2H), 2.15 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.0, 133.7, 132.6, 132.3, 130.4, 129.9, 129.8, 128.7, 128.0, 126.6, 125.9, 125.1, 123.8, 123.7, 99.3, 77.9, 72.5, 71.1, 67.3, 62.7, 21.5. MS (ESI) 445.1 (M + 1).
- 5-O-Tosyl-1,3-O-(2-phenylbenzylidene)-d-arabinitol (13r). 13r can be obtained from 12 with a yield of 57%. Pale-yellow oils. = −20.9 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.81 (d, J = 8.1 Hz, 2H), 7.76 (dd, J = 7.3, 1.9 Hz, 1H), 7.51–7.42 (m, 6H), 7.41–7.38 (m, 2H), 7.35–7.30 (m, 2H), 5.45 (s, 1H), 4.24–4.16 (m, 2H), 4.13–3.99 (m, 2H), 3.87 (d, J = 12.1 Hz, 1H), 3.78 (s, 1H), 3.67 (d, J = 8.3 Hz, 1H), 3.43 (br, 2H), 2.44 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 145.0, 141.1, 140.2, 134.7, 132.5, 130.1, 129.9, 129.3, 129.2, 128.2, 128.0, 127.6, 127.5, 126.6, 100.0, 78.1, 72.3, 71.4, 67.3, 62.5, 21.7. MS (ESI) 471.1 (M + 1).
3.2.3. General Procedure for the Synthesis of Epoxides (14a–r)
To a solution of 13a–r (2.0 mmol) in DCM (10 mL) under an argon atmosphere was added DBU (2.5 mL) at 0 °C. The mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (PE/EtOAc, 5:1) on silica gel to afford the pure products 14a–r as colorless oil with yields from 85 to 93%.
- 4,5-Anhydro-1,3-O-(2,3-dichlorobenzylidene)-d-arabinitol (14a). 14a can be obtained from 13a with a yield of 85%. Colorless oil. = –31.6 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.61 (d, J = 7.7 Hz, 1H), 7.46 (d, J = 8.0 Hz, 1H), 7.27–7.21 (m, 1H), 5.82 (s, 1H), 4.23 (dt, J = 12.0, 1.8 Hz, 1H), 4.08 (d, J = 12.1 Hz, 1H), 3.76 (d, J = 5.3 Hz, 2H), 3.33–3.29 (m, 1H), 2.98 (br, 1H), 2.91–2.88 (m, 1H), 2.82 (dt, J = 4.6, 2.0 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 136.8, 133.3, 131.3, 127.6, 126.0, 99.1, 80.2, 72.6, 64.3, 50.7, 45.9. MS (ESI) 291.0 (M + 1).
- 4,5-Anhydro-1,3-O-(2,4-dichlorobenzylidene)-d-arabinitol (14b). 14b can be obtained from 13b with a yield of 89%. Colorless oil. = –26.4 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.64 (d, J = 8.4 Hz, 1H), 7.39 (d, J = 2.1 Hz, 1H), 7.29 (dd, J = 8.3, 2.0 Hz, 1H), 5.78 (s, 1H), 4.24 (dd, J = 12.1, 1.8 Hz, 1H), 4.08 (dd, J = 12.1, 1.4 Hz, 1H), 3.78–3.74 (m, 2H), 3.32 (ddd, J = 5.4, 3.9, 2.6 Hz, 1H), 3.06 (d, J = 9.5 Hz, 1H), 2.91 (t, J = 4.5 Hz, 1H), 2.84–2.81 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 135.8, 133.5, 133.3, 129.5, 128.9, 127.4, 98.5, 80.1, 72.6, 64.3, 50.7, 45.9. MS (ESI) 291.0 (M + 1).
- 4,5-Anhydro-1,3-O-(2,5-dichlorobenzylidene)-d-arabinitol (14c). 14c can be obtained from 13c with a yield of 88%. Colorless oil. = –16.3 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.70 (d, J = 2.1 Hz, 1H), 7.27 (d, J = 4.2 Hz, 2H), 5.79 (s, 1H), 4.25 (dd, J = 12.2, 1.9 Hz, 1H), 4.09 (d, J = 12.1 Hz, 1H), 3.80–3.75 (m, 2H), 3.34 (p, J = 2.9 Hz, 1H), 3.03 (br, 1H), 2.93 (t, J = 4.5 Hz, 1H), 2.85–2.83 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 136.1, 133.1, 131.1, 130.8, 130.6, 128.1, 98.4, 80.2, 72.6, 64.3, 50.7, 46.0. MS (ESI) 291.0 (M + 1).
- 4,5-Anhydro-1,3-O-(2-chloro-3-fluorobenzylidene)-d-arabinitol (14d). 14d can be obtained from 13d with a yield of 88%. Colorless oil. = –23.1 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.43 (d, J = 7.9 Hz, 1H), 7.23–7.17 (m, 1H), 7.09 (t, J = 8.6 Hz, 1H), 5.75 (s, 1H), 4.17 (d, J = 12.1 Hz, 1H), 4.02 (d, J = 1 2.1 Hz, 1H), 3.70 (d, J = 5.3 Hz, 2H), 3.27–3.23 (m, 1H), 2.99 (d, J = 9.1 Hz, 1H), 2.85–2.82 (m, 1H), 2.76 (dd, J = 5.1, 2.6 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 158.1 (d, J = 248.1 Hz), 136.8, 127.9 (d, J = 7.7 Hz), 123.0 (d, J = 3.7 Hz), 120.1 (d, J = 18.6 Hz)117.3 (d, J = 21.1 Hz), 98.5 (d, J = 3.9 Hz). 80.1, 72.6, 64.3, 50.7, 45.9. MS (ESI) 275.0 (M + 1).
- 4,5-Anhydro-1,3-O-(2-chloro-5-fluorobenzylidene)-d-arabinitol (14e). 14e can be obtained from 13e with a yield of 91%. Colorless oil. = –26.3 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.43 (dd, J = 9.1, 3.0 Hz, 1H), 7.32 (dd, J = 8.9, 5.0 Hz, 1H), 7.02 (tdd, J = 7.5, 3.1, 1.2 Hz, 1H), 5.78 (s, 1H), 4.25 (d, J = 12.1 Hz, 1H), 4.09 (d, J = 12.2 Hz, 1H), 3.80–3.76 (m, 2H), 3.33 (ddd, J = 5.1, 3.9, 2.4 Hz, 1H), 2.99 (d, J = 10.1 Hz, 1H), 2.92 (td, J = 4.5, 3.9, 1.3 Hz, 1H), 2.85–2.82 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 161.4 (d, J = 246.8 Hz), 136.5 (d, J = 7.6 Hz), 131.0 (d, J = 8.1 Hz), 127.6 (d, J = 3.3 Hz), 117.6 (d, J = 22.9 Hz), 115.2 (d, J = 24.8 Hz). 98.4, 80.2, 72.6, 64.3, 50.7, 45.9. MS (ESI) 275.0 (M + 1).
- 4,5-Anhydro-1,3-O-(3,4-Dichlorobenzylidene)-d-arabinitol (14f). 14f can be obtained from 13f with a yield of 94%. Colorless oil. = –53.2 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.61 (d, J = 2.0 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.32 (dd, J = 8.3, 2.0 Hz, 1H), 5.50 (s, 1H), 4.24 (dd, J = 12.1, 1.9 Hz, 1H), 4.05 (dd, J = 12.1, 1.4 Hz, 1H), 3.75 (t, J = 2.4 Hz, 2H), 3.31 (ddd, J = 5.0, 4.0, 2.6 Hz, 1H), 3.02 (br, 1H), 2.92 (dd, J = 5.1, 4.0 Hz, 1H), 2.85 (dd, J = 5.0, 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 137.5, 133.2, 132.5, 130.4, 128.3, 125.6, 99.7, 79.5, 72.3, 64.2, 50.9, 45.9. MS (ESI) 291.0 (M + 1).
- 4,5-Anhydro-1,3-O-(3,5-dichlorobenzylidene)-d-arabinitol (14g). 14g can be obtained from 13g with a yield of 93%. Colorless oil. = –38.4 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.39 (d, J = 2.0 Hz, 2H), 7.35 (t, J = 2.0 Hz, 1H), 5.49 (s, 1H), 4.24 (dd, J = 12.1, 1.8 Hz, 1H), 4.04 (dd, J = 12.1, 1.4 Hz, 1H), 3.78–3.75 (m, 2H), 3.32 (ddd, J = 5.0, 4.0, 2.6 Hz, 1H), 3.05 (d, J = 10.0 Hz, 1H), 2.93 (dd, J = 5.1, 3.9 Hz, 1H), 2.85 (dd, J = 5.0, 2.6 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 140.4, 135.0, 129.2, 124.9, 99.5, 79.6, 72.3, 64.2, 50.8, 45.9. MS (ESI) 291.0 (M + 1).
- 4,5-Anhydro-1,3-O-(3-chloro-5-fluorobenzylidene)-d-arabinitol (14h). 14h can be obtained from 13h with a yield of 84%. Colorless oil. = –40.6 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.26 (d, J = 1.9 Hz, 1H), 7.07 (ddt, J = 12.8, 8.4, 2.1 Hz, 2H), 5.46 (s, 1H), 4.21 (dd, J = 12.1, 1.8 Hz, 1H), 4.01 (dd, J = 12.1, 1.4 Hz, 1H), 3.73 (d, J = 6.0 Hz, 2H), 3.28 (td, J = 4.4, 2.5 Hz, 1H), 2.98 (d, J = 10.0 Hz, 1H), 2.89 (t, J = 4.6 Hz, 1H), 2.82 (dd, J = 5.0, 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 158.5 (d, J = 250.0 Hz), 134.6 (d, J = 3.8 Hz),128.7, 126.2 (d, J = 7.4 Hz), 121.0 (d, J = 18.0 Hz), 116.5 (d, J = 21.3 Hz). 99.9, 79.6, 72.3, 64.2, 50.9, 45.9. MS (ESI) 275.0 (M + 1).
- 4,5-Anhydro-1,3-O-(3,5-bis(trifluoromethyl)benzylidene)-d-arabinitol (14i). 14i can be obtained from 13i with a yield of 87%. Colorless oil. = –18.1 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.97 (s, 2H), 7.87 (s, 1H), 5.65 (s, 1H), 4.29 (d, J = 12.3 Hz, 1H), 4.10 (d, J = 12.2 Hz, 1H), 3.80 (d, J = 8.8 Hz, 2H), 3.37–3.33 (m, 1H), 3.00–2.93 (m, 2H), 2.87 (dd, J = 5.1, 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 139.8, 131.8 (q, J = 33.7 Hz), 126.7 (d, J = 2.61 Hz), 125.1, 123.1 (dq, J = 7.6, 3.9 Hz), 121.5, 99.2, 79.7, 72.4, 64.2, 50.8, 45.9. MS (ESI) 359.1 (M + 1).
- 4,5-Anhydro-1,3-O-(5-fluoro-2-nitrobenzylidene)-d-arabinitol (14j). 14j can be obtained from 13j with a yield of 86%. Colorless oil. = –11.4 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.96 (dd, J = 9.0, 4.9 Hz, 1H), 7.59 (dd, J = 9.2, 2.9 Hz, 1H), 7.17 (ddd, J = 9.0, 7.1, 2.9 Hz, 1H), 6.17 (s, 1H), 4.23 (dd, J = 12.1, 1.8 Hz, 1H), 4.09 (dd, J = 12.2, 1.4 Hz, 1H), 3.79–3.75 (m, 2H), 3.29 (ddd, J = 5.1, 4.0, 2.6 Hz, 1H), 2.93–2.89 (m, 2H), 2.82–2.80 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 164.7 (d, J = 256.5 Hz), 135.0 (d, J = 8.9 Hz), 127.5 (d, J = 9.6 Hz), 116.7 (d, J = 23.3 Hz), 115.3 (d, J = 26.0 Hz), 99.7, 96.7, 80.3, 72.7, 64.3, 50.6, 45.7. MS (ESI) 286.1 (M + 1).
- 4,5-Anhydro-1,3-O-(o-bromobenzylidene)-d-arabinitol (14k). 14k can be obtained from 13k with a yield of 92%. Colorless oil. = –34.7 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.68 (d, J = 7.8 Hz, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.35 (t, J = 7.6 Hz, 1H), 7.22 (t, J = 7.7 Hz, 1H), 5.77 (s, 1H), 4.24 (d, J = 12.1 Hz, 1H), 4.08 (d, J = 12.0 Hz, 1H), 3.75 (t, J = 7.1 Hz, 2H), 3.35–3.31 (m, 1H), 3.07 (d, J = 10.0 Hz, 1H), 2.87 (dt, J = 18.0, 4.6 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 136.1, 132.9, 130.9, 128.1, 127.6, 122.4, 101.0, 80.2, 72.6, 64.3, 50.7, 46.0. MS (ESI) 301.0 (M + 1).
- 4,5-Anhydro-1,3-O-(m-bromobenzylidene)-d-arabinitol (14l). 14l can be obtained from 13l with a yield of 85%. Colorless oil. = –28.9 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.66 (s, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.24 (t, J = 7.8 Hz, 1H), 5.51 (s, 1H), 4.23 (dd, J = 12.1, 1.9 Hz, 1H), 4.04 (dd, J = 12.1, 1.4 Hz, 1H), 3.77–3.73 (m, 2H), 3.31 (ddd, J = 5.2, 4.0, 2.6 Hz, 1H), 3.02 (br, 1H), 2.91 (dd, J = 5.1, 3.9 Hz, 1H), 2.85 (dd, J = 5.0, 2.7 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 139.5, 132.3, 130.0, 129.3, 124.8, 122.4, 100.3, 79.6, 72.3, 64.3, 50.9, 46.0. MS (ESI) 301.0 (M + 1).
- 4,5-Anhydro-1,3-O-(p-bromobenzylidene)-d-arabinitol (14m). 14m can be obtained from 13m with a yield of 85%. Colorless oil. = −42.4 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.56 (d, J = 8.5 Hz, 2H), 7.43 (d, J = 8.5 Hz, 2H), 5.56 (s, 1H), 4.28 (dd, J = 12.0, 1.9 Hz, 1H), 4.10 (dd, J = 12.0, 1.4 Hz, 1H), 3.81–3.78 (m, 2H), 3.35 (dt, J = 4.7, 2.3 Hz, 1H), 3.11 (br, 1H), 2.97–2.88 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 136.5, 131.5, 127.9, 123.4, 100.6, 79.6, 72.3, 64.3, 50.9, 46.0. MS (ESI) 301.0 (M + 1).
- 4,5-Anhydro-1,3-O-(o-fluorophenylmethylene)-d-arabinitol (14n). 14n can be obtained from 13n with a yield of 91%. Pale-yellow oils. = −33.1 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.62 (td, J = 7.5, 1.9 Hz, 1H), 7.38–7.33 (m, 1H), 7.16 (td, J = 7.5, 1.3 Hz, 1H), 7.09–7.03 (m, 1H), 5.83 (s, 1H), 4.24 (dd, J = 12.1, 2.0 Hz, 1H), 4.08 (dd, J = 12.1, 1.3 Hz, 1H), 3.79–3.74 (m, 2H), 3.32 (ddd, J = 5.2, 4.0, 2.6 Hz, 1H), 2.96–2.89 (m, 2H), 2.83 (dd, J = 5.2, 2.6 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 131.2 (d, JC-F = 8.5 Hz), 127.8 (d, JC-F = 3.6 Hz), 124.3 (d, JC-F = 3.6 Hz), 115.7 (d, JC-F = 21.1 Hz), 97.0 (d, JC-F = 4.2 Hz), 80.1, 72.6, 64.5, 50.8, 46.0. MS (ESI) 241.1 (M + 1).
- 4,5-Anhydro-1,3-O-(o-iodobenzylidene)-d-arabinitol (14o). 14o can be obtained from 13o with a yield of 90%. Colorless oil. = –29.6 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.85 (dd, J = 7.9, 1.2 Hz, 1H), 7.65 (dd, J = 7.8, 1.7 Hz, 1H), 7.40 (td, J = 7.6, 1.2 Hz, 1H), 7.08 (td, J = 7.6, 1.8 Hz, 1H), 5.60 (s, 1H), 4.26 (dd, J = 12.1, 1.9 Hz, 1H), 4.11 (dd, J = 12.1, 1.4 Hz, 1H), 3.80 (s, 1H), 3.76 (dd, J = 5.5, 1.2 Hz, 1H), 3.35 (ddd, J = 5.4, 3.9, 2.6 Hz, 1H), 3.11 (d, J = 10.4 Hz, 1H), 2.92 (dd, J = 5.1, 4.0 Hz, 1H), 2.88 (dd, J = 5.1, 2.6 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 139.6, 138.9, 131.1, 128.4, 127.8, 104.6, 97.0, 80.3, 72.5, 64.3, 50.6, 46.0. MS (ESI) 349.0 (M + 1).
- 4,5-Anhydro-1,3-O-(o-cyanobenzylidene)-d-arabinitol (14p). 14p can be obtained from 13p with a yield of 91%. Colorless oil. = –49.1 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.71 (d, J = 7.4 Hz, 1H), 7.64–7.57 (m, 1H), 7.51–7.46 (m, 1H), 5.72 (s, 1H), 4.29 (dd, J = 12.1, 2.0 Hz, 1H), 4.10 (d, J = 12.1 Hz, 1H), 3.88 (d, J = 4.7 Hz, 1H), 3.81 (br, 1H), 3.42–3.28 (m, 2H), 2.91 (s, 2H). 13C NMR (75 MHz, CDCl3) δ 140.3, 133.6, 132.9, 129.9, 127.9, 118.5, 110.9, 100.4, 79.4, 72.6, 64.5, 62.5, 50.9, 45.6. MS (ESI) 248.1 (M + 1).
- 4,5-Anhydro-1,3-O-(1-naphthalene)-d-arabinitol (14q). 14q can be obtained from 13q with a yield of 85%. Colorless oil. = –35.2 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 8.12 (dd, J = 8.4, 1.6 Hz, 1H), 7.89–7.88 (m, 1H), 7.81 (dd, J = 7.2, 1.3 Hz, 1H), 7.51 (dtd, J = 11.3, 8.1, 4.4 Hz, 3H), 6.10 (s, 1H), 4.30 (dd, J = 12.0, 2.0 Hz, 1H), 4.15 (dd, J = 12.1, 1.4 Hz, 1H), 3.87–3.79 (m, 2H), 3.38–3.36 (m, 1H), 3.09 (br, 1H), 2.91–2.87 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 133.8, 132.6, 130.4, 130.0, 128.8, 126.5, 125.8, 125.1, 124.2, 123.8, 100.3, 79.9, 72.5, 64.5, 51.0, 46.0. MS (ESI) 273.1 (M + 1).
- 4,5-Anhydro-1,3-O-(2-phenylbenzylidene)-d-arabinitol (14r). 14r can be obtained from 13r with a yield of 87%. Colorless oil. = –13.0 (c = 0.50, MeOH). 1H NMR (300 MHz, chloroform-d) δ 7.70–7.63 (m, 1H), 7.36–7.32 (m, 7H), 7.22–7.17 (m, 1H), 5.32 (s, 1H), 4.06 (t, J = 10.0 Hz, 1H), 3.78 (d, J = 11.8 Hz, 1H), 3.57 (d, J = 10.6 Hz, 1H), 3.41 (d, J = 5.5 Hz, 1H), 3.17 (t, J = 4.3 Hz, 1H), 2.78–2.71 (m, 2H), 2.63–2.58 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 141.1, 140.4, 134.7, 130.3, 129.6, 129.3, 128.1, 127.8, 127.5, 126.5, 100.4, 79.9, 72.3, 64.4, 50.7, 45.9. MS (ESI) 299.1 (M + 1).
3.2.4. General Procedure for Coupling Reactions
Coupling reaction to construct sulfonium salts: To a solution of 14a–r (0.4 mmol) and 15 (0.36 mmol) in MeCN (4.0 mL) under an argon atmosphere was added trifluoroacetic acid (0.36 mmol) at −20 °C; after stirring for 10 min, the reaction temperature was warmed up to room temperature and stirred for another 8 h. The reaction mixture was concentrated under reduced pressure. The resulting residue was resolved in MeOH, and ion exchange reagent IRA (Cl− form, 2.0 g) was added; the reaction mixture was stirred at room temperature for 4 h, filtered through celite, and washed with MeOH. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (DCM/MeOH, 15:1) on silica gel to afford the pure products as pale-yellow oils with yields from 38 to 56%.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(2,3-dichlorobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19a). 19a was obtained from 14a and 15 with a yield of 46%. Pale-yellow oils. = –15.6 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.77 (dd, J = 7.8, 1.6 Hz, 1H), 7.55 (dd, J = 8.0, 1.7 Hz, 1H), 7.34 (t, J = 7.9 Hz, 1H), 5.93 (s, 1H), 4.64 (q, J = 2.6 Hz, 1H), 4.42–4.35 (m, 2H), 4.22–4.17 (m, 2H), 4.11–4.05 (m, 1H), 4.03–4.02 (m, 2H), 3.96 (s, 1H), 3.92 (d, J = 2.1 Hz, 1H), 3.86 (dd, J = 3.2, 1.7 Hz, 3H), 3.74 (dd, J = 13.2, 8.8 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 138.8, 133.7, 132.0, 132.0, 128.8, 127.8, 99.6, 82.8, 79.5, 73.9, 73.8, 67.1, 63.1, 61.0, 52.0, 51.9. HRMS (ESI) calcd for C17H23Cl2O7S+ [M]+ 441.0536 found 441.0528.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(2,4-dichlorobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19b). 19b was obtained from 14b and 15 with a yield of 46%. Pale-yellow oils. = –15.2 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.80 (d, J = 8.3 Hz, 1H), 7.45 (d, J = 2.1 Hz, 1H), 7.36 (dd, J = 8.4, 2.1 Hz, 1H), 5.88 (s, 1H), 4.65 (q, J = 2.6 Hz, 1H), 4.41–4.35 (m, 2H), 4.25–4.17 (m, 2H), 4.11–4.00 (m, 3H), 3.98–3.96 (m, 1H), 3.92–3.91 (m, 1H), 3.88–3.83 (m, 3H), 3.74 (dd, J = 13.1, 9.3 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 136.4, 135.4, 134.6, 130.7, 129.9, 128.3, 99.0, 82.6, 79.4, 73.8, 73.7, 67.1, 63.0, 60.9, 51.9, 51.8. HRMS (ESI) calcd for C17H23Cl2O7S+ [M]+ 441.0536 found 441.0537.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(2,5-dichlorobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19c). 19c was obtained from 14c and 15 with a yield of 46%. Pale-yellow oils. = –24.3 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.85–7.82 (m, 1H), 7.41–7.35 (m, 2H), 5.90 (s, 1H), 4.63 (d, J = 2.5 Hz, 1H), 4.38 (q, J = 5.4 Hz, 2H), 4.23–4.17 (m, 2H), 4.09–3.96 (m, 4H), 3.92 (q, J = 3.9, 3.2 Hz, 1H), 3.87–3.84 (m, 3H), 3.73 (dd, J = 13.1, 8.7 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 138.2, 134.0, 132.2, 131.8, 131.4, 129.6, 98.9, 82.7, 79.5, 73.9, 73.8, 67.1, 63.1, 60.9, 52.0, 51.9. HRMS (ESI) calcd for C17H23Cl2O7S+ [M]+ 441.0536 found 441.0526.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(2-chloro-3-fluorobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19d). 19d was obtained from 14d and 15 with a yield of 55%. Pale-yellow oils. = –26.3 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.64 (dt, J = 7.8, 1.4 Hz, 1H), 7.36 (td, J = 8.1, 5.1 Hz, 1H), 7.25 (td, J = 8.7, 1.7 Hz, 1H), 5.92 (s, 1H), 4.64 (q, J = 2.6 Hz, 1H), 4.42–4.35 (m, 2H), 4.22 (dd, J = 3.8, 1.7 Hz, 2H), 4.11–4.00 (m, 4H), 3.96–3.92 (m, 1H), 3.88–3.86 (m, 3H), 3.74 (dd, J = 13.2, 8.8 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 159.1 (d, J = 246.4 Hz), 138.7, 129.1 (d, J = 7.8 Hz), 124.7 (d, J = 3.3 Hz), 120.8 (d, J = 18.6 Hz), 117.9 (d, J = 21.3 Hz), 99.0 (d, J = 3.6 Hz), 82.7, 79.5, 73.9, 73.8, 67.1, 63.1, 61.0, 52.0, 51.9. HRMS (ESI) calcd for C17H23ClFO7S+ [M]+ 425.0832 found 425.0816.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(2-chloro-5-fluorobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19e). 19e was obtained from 14e and 15 with a yield of 51%. Pale-yellow oils. = –24.6 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.61 (dd, J = 9.4, 3.1 Hz, 1H), 7.48–7.42 (m, 1H), 7.16 (td, J = 8.3, 3.1 Hz, 1H), 5.92 (s, 1H), 4.69–4.61 (m, 1H), 4.45–4.39 (m, 2H), 4.24 (dd, J = 5.0, 1.8 Hz, 2H), 4.14–4.09 (m, 2H), 3.95 (d, J = 2.2 Hz, 1H), 3.90 (t, J = 3.3 Hz, 4H), 3.81–3.74 (m, 1H). 13C NMR (75 MHz, methanol-d4) δ 162.7 (d, J = 245.2 Hz), 138.6 (d, J = 7.7 Hz), 132.0 (d, J = 8.3 Hz), 128.7, 118.4 (d, J = 23.4 Hz), 116.4 (d, J = 25.2 Hz), 99.0, 82.7, 79.5, 73.9, 73.8, 67.1, 63.1, 60.9, 52.0, 51.8. HRMS (ESI) calcd for C17H23ClFO7S+ [M]+ 425.0832 found 425.0811.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(3,4-dichlorobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19f). 19f was obtained from 14f and 15 with a yield of 43%. Pale-yellow oils. = –26.4 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.74–7.72 (m, 1H), 7.54–7.44 (m, 2H), 5.65 (d, J = 4.6 Hz, 1H), 4.64 (q, 1H), 4.42–4.33 (m, 2H), 4.25–4.12 (m, 3H), 4.11–4.02 (m, 3H), 3.98–3.92 (m, 2H), 3.87–3.83 (m, 2H), 3.79–3.71 (m, 1H). 13C NMR (75 MHz, methanol-d4) δ 140.0, 131.3, 129.8, 127.5, 100.9, 82.6, 79.5, 73.8, 73.6, 67.3, 63.2, 61.0, 52.0, 51.9. HRMS (ESI) calcd for C17H23Cl2O7S+ [M]+ 441.0536 found 441.0519.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(3,5-dichlorobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19g). 19g was obtained from 14g and 15 with a yield of 47%. Pale-yellow oils. = –19.9 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.54–7.52 (m, 2H), 7.43 (t, J = 2.0 Hz, 1H), 5.64 (s, 1H), 4.64 (q, J = 2.6 Hz, 1H), 4.41–4.33 (m, 2H), 4.24–4.13 (m, 3H), 4.04 (dd, J = 8.2, 4.4 Hz, 2H), 3.98–3.88 (m, 3H), 3.87 (d, J = 2.6 Hz, 2H), 3.76 (dd, J = 13.2, 9.1 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 143.0, 135.9, 129.8, 126.5, 126.3, 100.7, 82.6, 79.5, 73.7, 73.6, 67.3, 63.2, 61.0, 52.1, 51.9. HRMS (ESI) calcd for C17H23Cl2O7S+ [M]+ 441.0536 found 441.0522.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(3-chloro-5-fluorobenzylidene)-d-arabinitol5-yl) episulfoniumylidene]-d-arabinitol chloride (19h). 19h was obtained from 14h and 15 with a yield of 41%. Pale-yellow oils. = –23.9 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 5.86 (d, J = 1.8 Hz, 1H), 5.75–5.66 (m, 1H), 5.62 (dh, J = 9.2, 2.1 Hz, 1H), 4.10 (d, J = 3.9 Hz, 1H), 3.11–3.04 (m, 1H), 2.87–2.70 (m, 2H), 2.63 (qd, J = 11.5, 10.9, 5.6 Hz, 3H), 2.55–2.47 (m, 2H), 2.44–2.36 (m, 2H), 2.33–2.24 (m, 3H), 2.24–2.18 (m, 1H). 13C NMR (75 MHz, methanol-d4) δ 162.6 (d, J = 248.1 Hz), 142.0 (d, J = 8.3 Hz), 134.5 (d, J = 10.5 Hz), 122.6 (d, J = 3.0 Hz), 116.0 (d, J = 25.3 Hz), 112.1 (d, J = 23.0 Hz), 99.4, 81.3, 78.2, 72.4, 72.3, 66.0, 61.9, 59.7. HRMS (ESI) calcd for C17H23ClFO7S+ [M]+ 425.0832 found 425.0866.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(3,5-bis(trifluoromethyl)benzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19i). 19i was obtained from 14i and 15 with a yield of 38%. Pale-yellow oils. = –26.1 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 8.19–8.16 (m, 2H), 7.96 (s, 1H), 5.85 (d, J = 1.9 Hz, 1H), 4.62 (dq, J = 14.0, 2.9 Hz, 1H), 4.45–4.36 (m, 2H), 4.29–4.23 (m, 2H), 4.12–4.04 (m, 3H), 3.96–3.95 (m, 2H), 3.92–3.87 (m, 3H), 3.77 (dd, J = 13.2, 8.9 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 142.4, 130.2, 132.6 (q, J = 33.4 Hz), 126.6, 128.6 (d, J = 3.2 Hz), 123.6 (dq, J = 8.3, 4.8 Hz), 122.9, 100.7, 82.7, 79.5, 73.7, 67.3, 63.2, 61.0, 52.0, 51.9. HRMS (ESI) calcd for C19H23F6O7S+ [M]+ 509.1063 found 509.1085.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(5-fluoro-2-nitrobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19j). 19j was obtained from 14j and 15 with a yield of 39%. Pale-yellow oils. = –29.7 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 8.02–7.97 (m, 2H), 7.67 (dd, J = 9.5, 2.8 Hz, 1H), 7.37–7.31 (m, 1H), 6.18 (s, 1H), 4.68–4.58 (m, 1H), 4.43 (d, J = 2.5 Hz, 1H), 4.39–4.33 (m, 1H),4.25–4.15 (m, 3H),4.10 (d, J = 7.2 Hz, 3H),3.99–3.94 (m, 2H), 3.88–3.83 (m, 2H),3.65–3.58 (m, 1H). 13C NMR (75 MHz, methanol-d4) δ 165.7 (d, J = 253.6 Hz), 146.0 (d, J = 3.2 Hz), 136.3 (d, J = 9.2 Hz), 128.3 (d, J = 10.0 Hz), 117.7 (d, J = 23.6 Hz), 116.1 (d, J = 26.1 Hz), 97.5, 82.4, 79.6, 79.4, 73.9, 73.8, 66.8, 62.9, 60.9, 51.8, 51.5. HRMS (ESI) calcd for C17H23FNO9S+ [M]+ 436.1072 found 436.1035.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(o-bromobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19k). 19k was obtained from 14k and 15 with a yield of 51%. Pale-yellow oils. = –13.7 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.83 (dd, J = 7.8, 1.8 Hz, 1H), 7.60 (dd, J = 7.9, 1.3 Hz, 1H), 7.44–7.39 (m, 1H), 7.31 (qd, J = 7.2, 2.6 Hz, 1H), 5.87 (s, 1H), 4.67 (q, J = 2.4 Hz, 1H), 4.45–4.38 (m, 2H), 4.29–4.20 (m, 2H), 4.16–4.11 (m, 1H), 4.08–4.00 (m, 2H), 4.00–3.92 (m, 2H), 3.92–3.88 (m, 3H), 3.79 (dt, J = 13.1, 7.1 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 138.0, 133.6, 131.8, 129.6, 128.5, 123.4, 101.7, 82.7, 79.4, 73.9, 73.7, 67.1, 63.1, 60.9, 51.9. HRMS (ESI) calcd for C17H24BrO7S+ [M]+ 451.0421 found 451.0411.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(m-bromobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19l). 19l was obtained from 14l and 15 with a yield of 54%. Pale-yellow oils. = –13.9 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.75 (t, J = 1.9 Hz, 1H), 7.50 (dd, J = 7.8, 1.8 Hz, 2H), 7.32–7.25 (m, 1H), 5.63 (s, 1H), 4.64–4.58 (m, 1H), 4.42–4.34 (m, 2H), 4.23–4.16 (m, 2H), 4.13–4.06 (m, 2H), 3.98–3.88 (m, 3H), 3.86 (d, J = 2.6 Hz, 2H), 3.83–3.82 (m, 1H), 3.77 (dd, J = 9.0, 4.1 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 141.8, 133.0, 131.0, 130.6, 126.5, 123.0, 101.6, 82.6, 79.5, 73.8, 73.6, 67.3, 63.2, 61.5, 61.0, 52.0, 51.9. HRMS (ESI) calcd for C17H24BrO7S+ [M]+ 451.0421 found 451.0429.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(p-bromobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19m). 19m was obtained from 14m and 15 with a yield of 56%. Pale-yellow oils. = –18.7 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.54–7.45 (m, 4H), 5.63 (s, 1H), 4.63 (q, J = 2.6 Hz, 1H), 4.42–4.34 (m, 2H), 4.22–4.16 (m, 2H), 4.08–4.00 (m, 3H), 3.98–3.95 (m, 1H), 3.92–3.90 (m, 1H), 3.87–3.82 (m, 3H), 3.79–3.72 (m, 1H). 13C NMR (75 MHz, methanol-d4) δ 138.7, 132.2, 129.5, 123.8, 101.7, 82.5, 79.5, 73.8, 73.5, 67.3, 63.2, 61.5, 60.9, 52.0, 51.9. HRMS (ESI) calcd for C17H24BrO7S+ [M]+ 451.0421 found 451.0433.
- 1,4-Dideoxy-1,4-{(R)-[5-deoxy-1,3-O-(o-fluorophenylmethylene)-d-arabinitol-5-yl] episulfoniumylidene}-d-arabinitol chloride (19n). 19n was obtained from 14n and 15 with a yield of 45%. Pale-yellow oils. = −31.6 (c = 0.542, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.65 (td, J = 7.5, 1.8 Hz, 1H), 7.28 (tdd, J = 7.5, 5.3, 1.8 Hz, 1H), 7.09 (td, J = 7.5, 1.1 Hz, 1H), 6.98 (ddd, J = 10.5, 8.3, 1.1 Hz, 1H), 5.82 (s, 1H), 4.53 (q, J = 2.6 Hz, 1H), 4.32–4.26 (m, 2H), 4.09 (qd, J = 12.3, 1.8 Hz, 2H), 3.93 (dd, J = 7.3, 5.5 Hz, 2H), 3.88 (dd, J = 8.0, 1.6 Hz, 1H), 3.85–3.79 (m, 1H), 3.75 (td, J = 8.0, 7.3, 2.5 Hz, 4H), 3.63 (dd, J = 13.2, 8.7 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 163.1, 159.8, 132.0 (d, JC-F = 8.4 Hz), 129.3 (d, J = 3.3 Hz), 129.3 (d, JC-F = 3.3 Hz), 125.2 (d, JC-F = 3.6 Hz), 116.1 (d, JC-F = 21.5 Hz), 97.3 (d, JC-F = 4.8 Hz), 82.7, 79.5, 74.0, 73.7, 67.2, 63.3, 61.0, 51.9. HRMS (ESI) calcd for C17H24FO7S+ [M]+ 391.1221, found 391.1214.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(o-iodobenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19o). 19o was obtained from 14o and 15 with a yield of 50%. Pale-yellow oils. = –29.2 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.84 (dd, J = 8.1, 3.9 Hz, 1H), 7.73 (dd, J = 8.4, 4.1 Hz, 1H), 7.40 (td, J = 7.6, 3.7 Hz, 1H), 7.12–7.06 (m, 1H), 5.64 (d, J = 4.2 Hz, 1H), 4.64–4.56 (m, 1H), 4.42–4.34 (m, 2H), 4.27–4.19 (m, 2H), 4.08–3.97 (m, 3H), 3.93–3.85 (m, 3H), 3.79–3.64 (m, 1H). 13C NMR (75 MHz, methanol-d4) δ 139.6, 139.1, 130.6, 127.9, 104.2, 96.8, 81.4, 78.2, 72.6, 72.5, 65.8, 61.7, 59.7, 50.8, 50.6, 47.5, 47.2, 46.9. HRMS (ESI) calcd for C17H24IO7S+ [M]+ 499.0282 found 499.0295.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(o-cyanobenzyliden)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19p). 19p was obtained from 14p and 15 with a yield of 48%. Pale-yellow oils. = –32.5 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.86 (d, J = 7.6 Hz, 1H), 7.73 (d, J = 11.3 Hz, 2H), 7.56 (d, J = 7.6 Hz, 1H), 5.90 (s, 1H), 4.65 (s, 1H), 4.41 (s, 2H), 4.26 (d, J = 7.2 Hz, 2H), 4.08 (t, J = 12.8 Hz, 4H), 3.89 (s, 4H), 3.78 (d, J = 9.4 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 142.0, 134.2, 130.8, 128.2, 118.9, 111.7, 100.0, 82.3, 79.6, 79.2, 73.7, 73.6, 67.0, 63.1, 60.9, 51.7, 51.6. HRMS (ESI) calcd for C18H24NO7S+ [M]+ 398.1268 found 398.1273.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(1-naphthalene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19q). 19q was obtained from 14q and 15 with a yield of 56%. Pale-yellow oils. = –63.2 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 8.33 (d, J = 8.3 Hz, 1H), 7.91–7.85 (m, 2H), 7.80 (dt, J = 7.4, 1.9 Hz, 1H), 7.50 (ddt, J = 14.4, 6.6, 1.6 Hz, 3H), 6.20 (s, 1H), 4.58 (q, J = 2.5 Hz, 1H), 4.48–4.40 (m, 1H), 4.34–4.33 (m, 1H), 4.28 (t, J = 2.4 Hz, 2H), 4.16–4.06 (m, 4H), 3.98–3.96 (m, 1H), 3.93–3.87 (m, 2H), 3.85–3.83 (m, 1H), 3.79 (d, J = 2.4 Hz, 2H), 3.76–3.70 (m, 1H). 13C NMR (75 MHz, methanol-d4) δ 135.2, 134.6, 131.9, 130.6, 129.4, 127.2, 126.8, 126.0, 125.7, 125.6, 101.7, 82.8, 79.4, 73.8, 73.7, 67.4, 63.4, 60.9, 54.8, 52.0. HRMS (ESI) calcd for C21H27O7S+ [M]+ 423.1472 found 423.1477.
- 1,4-Dideoxy-1,4-[(R)-(5-deoxy-1,3-O-(2-phenylbenzylidene)-d-arabinitol-5-yl) episulfoniumylidene]-d-arabinitol chloride (19r). 19r was obtained from 14r and 15 with a yield of 41%. Pale-yellow oils. = –37.6 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.90–7.87 (m, 1H), 7.42–7.39 (m, 7H), 7.27–7.24 (m, 1H), 5.51 (s, 1H), 4.62 (d, J = 2.7 Hz, 1H), 4.39–4.36 (m, 2H), 4.30–4.15 (m, 3H),4.11–4.05 (m, 3H), 3.94–3.90 (m, 3H), 3.71 (s, 2H), 3.60 (d, J = 5.1 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 142.4, 141.8, 136.4, 130.7, 130.3, 130.0, 129.4, 129.1, 128.5, 128.2, 100.9, 82.9, 79.4, 73.8, 73.5, 67.2, 63.1, 61.0, 52.2, 51.9. HRMS (ESI) calcd for C23H29O7S+ [M]+ 449.1629 found 449.1663.
Coupling reaction to construct selenonium salts: To a solution of 14a–r (0.4 mmol) and 16 (0.36 mmol) in MeCN (4.0 mL) under an argon atmosphere was added trifluoroacetic acid (0.36 mmol) at −20 °C; after stirring for 10 min, the reaction temperature was warmed up to room temperature and stirred for another 8 h. The reaction mixture was concentrated under reduced pressure. The resulting residue was resolved in MeOH, and ion exchange reagent IRA (Cl− form, 2.0 g) was added; the reaction mixture was stirred at room temperature for 4 h, filtered through celite, and washed with MeOH. The filtrate was concentrated under reduced pressure, and the resulting residue was purified by flash column chromatography (DCM/MeOH, 15:1) on silica gel to afford the pure products as colorless solid with yields from 39 to 53%.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(2,3-Dichlorophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20a). 20a was obtained from 14a and 16 with a yield of 43%. Colorless solid. = –24.7 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.79–7.75 (m, 1H), 7.56 (dd, J = 8.1, 1.6 Hz, 1H), 7.34 (t, J = 7.9 Hz, 1H), 5.94 (s, 1H), 4.74 (q, J = 2.5 Hz, 1H), 4.47–4.43 (m, 1H), 4.36 (m, J = 8.1, 3.8 Hz, 1H), 4.21 (dd, J = 3.0, 1.6 Hz, 2H), 4.19–4.15 (m, 1H), 4.01–3.95 (m, 2H), 3.95–3.85 (m, 2H), 3.85–3.77 (m, 2H), 3.76 (d, J = 2.5 Hz, 2H). 13C NMR (75 MHz, methanol-d4) δ 138.9, 133.8, 132.1, 128.8, 127.8, 99.7, 83.5, 80.4, 80.3, 74.2, 73.8, 67.1, 63.4, 61.0, 49.8. HRMS (ESI) calcd for C17H23Cl2O7Se+ [M]+ 488.9981 found 488.9987.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(2,4-Dichlorophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20b). 20b was obtained from 14b and 16 with a yield of 43%. Colorless solid. = –23.8 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.80 (dd, J = 8.4, 3.9 Hz, 1H), 7.47 (d, J = 2.1 Hz, 1H), 7.37 (dd, J = 8.5, 2.2 Hz, 1H), 5.89 (s, 1H), 4.79–4.60 (m, 1H), 4.46 (dd, J = 2.9, 1.3 Hz, 1H), 4.35 (m, J = 8.0, 3.8 Hz, 1H), 4.21–4.13 (m, 3H), 4.11–4.06 (m, 1H), 4.03–3.93 (m, 3H), 3.91–3.87 (m, 1H), 3.82 (d, J = 8.4 Hz, 2H), 3.75 (d, J = 2.5 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 136.6, 135.5, 134.7, 130.7, 130.0, 128.3, 99.1, 83.4, 80.4, 80.3, 74.2, 73.8, 67.1, 63.3, 61.0, 49.7,49.5. HRMS (ESI) calcd for C17H23Cl2O7Se+ [M]+ 488.9981 found 488.9979.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(2,5-Dichlorophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20c). 20c was obtained from 14c and 16 with a yield of 43%. Colorless solid. = –33.6 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.85 (s, 1H), 7.41–7.35 (m, 2H), 5.91 (s, 1H), 4.74 (s, 1H), 4.47 (d, J = 2.8 Hz, 1H), 4.36 (td, J = 8.2, 3.8 Hz, 1H), 4.20 (dd, J = 5.0, 2.1 Hz, 2H), 4.10 (q, J = 7.2 Hz, 2H), 4.06–3.88 (m, J = 6.9, 6.2 Hz, 4H), 3.84–3.83 (m, 1H), 3.78 (dd, J = 10.3, 3.0 Hz, 2H). 13C NMR (75 MHz, methanol-d4) δ 138.2, 134.0, 132.2, 131.8, 131.5, 129.6, 99.0, 83.4, 80.4, 80.3, 74.1, 73.8, 67.0, 63.3, 61.0, 49.8, 49.3. HRMS (ESI) calcd for C17H23Cl2O7Se+ [M]+ 488.9981 found 488.9984.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(2-Chloro-3-fluorophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20d). 20d was obtained from 14d and 16 with a yield of 52%. Colorless solid. = –36.2 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.64 (d, J = 7.7 Hz, 1H), 7.37 (m, J = 7.9, 5.3 Hz, 1H), 7.29–7.23 (m, 1H), 4.74 (s, 1H), 4.47 (d, J = 2.9 Hz, 1H), 4.36 (dt, J = 8.2, 4.1 Hz, 1H), 4.22–4.17 (m, 3H), 4.06–3.90 (m, 4H), 3.83 (d, J = 8.5 Hz, 2H), 3.76 (d, J = 2.6 Hz, 2H). 13C NMR (75 MHz, methanol-d4) δ 160.8, 157.5, 138.8, 129.2, 129.1, 124.7, 124.6, 120.7, 118.1, 117.8, 99.1, 99.1, 83.5, 80.4, 80.2, 74.2, 73.8, 67.1, 63.4, 61.0, 49.7,49.6. HRMS (ESI) calcd for C17H23ClFO7Se+ [M]+ 473.0276 found 473.0298.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(2-Chloro-5-fluorophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20e). 20e was obtained from 14e and 16 with a yield of 45%. Colorless solid. = –33.9 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.58 (dd, J = 9.4, 3.2 Hz, 1H), 7.41 (dd, J = 8.8, 4.9 Hz, 1H), 7.13 (m, J = 8.3, 3.1 Hz, 1H), 5.90 (s, 1H), 4.74 (s, 1H), 4.46 (d, J = 2.7 Hz, 1H), 4.39–4.33 (m, 1H), 4.33–4.13 (m, 3H), 4.10 (q, J = 7.2 Hz, 1H), 4.03–3.91 (m, 3H), 3.91–3.81 (m, 2H), 3.78 (dd, J = 12.6, 3.1 Hz, 2H). 13C NMR (75 MHz, methanol-d4) δ 132.0, 131.9, 118.6, 118.3, 116.6, 116.3, 99.0, 83.4, 80.4, 80.3, 74.2, 73.8, 67.0, 63.3, 61.5, 61.0, 49.8. HRMS (ESI) calcd for C17H23ClFO7Se+ [M]+ 473.0276 found 473.0277.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(3,4-Dichlorophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20f). 20f was obtained from 14f and 16 with a yield of 52%. Colorless solid. = –34.7 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.75 (d, J = 2.1 Hz, 1H), 7.54–7.45 (m, 2H), 5.65 (s, 1H), 4.74 (d, J = 2.8 Hz, 1H), 4.47 (d, J = 3.1 Hz, 1H), 4.35 (m, J = 7.7, 3.9 Hz, 1H), 4.19 (m, J = 6.9, 5.8, 3.1 Hz, 3H), 4.13–4.04 (m, 1H), 3.97 (m, J = 11.5, 6.1, 3.7 Hz, 3H), 3.89–3.78 (m, 2H), 3.78–3.72 (m, 1H). 13C NMR (75 MHz, methanol-d4) δ 140.0, 133.1, 131.4, 129.8, 127.5, 100.9, 83.1, 80.4, 80.2, 74.0, 73.6, 67.3, 63.4, 61.0, 49.0,48.7. HRMS (ESI) calcd for C17H23Cl2O7Se+ [M]+ 488.9981 found 488.9986.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(3,5-Dichlorophenyl)-5-hydroxy-1,3–dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium Chloride (20g). 20g was obtained from 14g and 16 with a yield of 46%. Colorless solid. = –29.8 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.54 (t, J = 2.3 Hz, 2H), 7.43 (t, J = 1.9 Hz, 1H), 5.65 (d, J = 3.7 Hz, 1H), 4.75 (p, J = 2.9 Hz, 1H), 4.47 (m, J = 4.6, 2.8, 1.5 Hz, 1H), 4.35 (m, J = 7.9, 3.8 Hz, 1H), 4.20 (m, J = 13.1, 6.2, 4.4 Hz, 3H), 4.13–4.06 (m, 1H), 4.01–3.92 (m, 3H), 3.89–3.81 (m, 2H), 3.78–3.73 (m, 2H). 13C NMR (75 MHz, methanol-d4) δ 143.0, 135.9, 129.8, 126.4, 126.4, 100.7, 83.2, 80.4, 80.2, 73.9, 73.6, 67.2, 63.4, 61.1, 49.7,49.6. HRMS (ESI) calcd for C17H23Cl2O7Se+ [M]+ 488.9981 found 488.9984.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(3-Chloro-5-fluorophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20h). 20h was obtained from 14h and 16 with a yield of 43%. Colorless solid. = –32.5 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.43 (s, 1H), 7.28 (d, J = 9.6 Hz, 1H), 7.20 (s, 1H), 5.65 (s, 1H), 4.74 (s, 1H), 4.47 (dd, J = 4.6, 2.2 Hz, 1H), 4.36 (td, J = 7.8, 7.1, 3.0 Hz, 1H), 4.18 (dt, J = 6.9, 2.6 Hz, 3H), 4.02–3.92 (m, 4H), 3.86–3.81 (m, 2H), 3.78–3.72 (m, 2H). 13C NMR (75 MHz, methanol-d4) δ 165.5, 143.4, 143.3, 136.0, 135.8, 123.9, 123.8, 117.5, 117.2, 113.5, 113.2, 100.7, 83.2, 82.6, 81.4, 80.4, 80.2, 74.0, 73.6, 67.3, 66.6, 63.4, 61.1, 49.7. HRMS (ESI) calcd for C17H23ClFO7Se+ [M]+ 473.0276 found 473.0297.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(3,5-Bis(trifluoromethyl)phenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20i). 20i was obtained from 14i and 16 with a yield of 40%. Colorless solid. = –42.3 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.72 (d, J = 6.6 Hz, 2H), 7.52 (s, 1H), 4.29 (s, 1H), 4.02 (d, J = 3.0 Hz, 1H), 3.92 (q, J = 5.8, 4.1 Hz, 1H), 3.89–3.78 (m, 2H), 3.74–3.71 (m, 1H), 3.65–3.48 (m, 3H), 3.42 (d, J = 5.2 Hz, 2H), 3.36–3.28 (m, 2H).13C NMR (75 MHz, methanol-d4) δ 142.4, 142.4, 132.8, 132.4, 128.5, 126.6, 123.0, 100.7, 83.3, 82.7, 80.4, 80.2, 73.9, 73.7, 67.2, 63.4, 61.1, 54.8, 49.7,49.6. HRMS (ESI) calcd for C19H23F6O7Se+ [M]+ 557.0508 found 557.0525.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(5-Fluoro-2-nitrophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20j). 20j was obtained from 14j and 16 with a yield of 39%. Colorless solid. = –43.2 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.99 (dd, J = 8.9, 4.9 Hz, 1H), 7.67 (dd, J = 9.6, 2.9 Hz, 1H), 7.34 (td, J = 9.3, 8.5, 2.7 Hz, 1H), 6.20 (d, J = 7.4 Hz, 1H), 4.78 (s, 1H), 4.51 (s, 1H), 4.33 (m, J = 7.8, 5.1 Hz, 1H), 4.28–4.18 (m, 3H), 4.10 (q, J = 7.2 Hz, 1H), 4.01 (m, J = 14.1, 13.0, 7.9 Hz, 2H), 3.94–3.85 (m, 3H), 3.77 (dd, J = 11.9, 3.2 Hz, 1H), 3.68 (dd, J = 11.9, 7.7 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 167.4, 164.1, 136.4, 136.3, 128.4, 128.3, 117.9, 117.5, 116.2, 115.9, 97.6, 83.2, 80.5, 80.2, 74.4, 73.9, 66.6, 63.1, 61.5, 61.1,49.5,49.3. HRMS (ESI) calcd for C17H23FNO9Se+ [M]+ 484.0517 found 484.0536.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(2-Bromophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20k). 20k was obtained from 14k and 16 with a yield of 53%. Colorless solid. = –24.3 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.82–7.79 (m, 1H), 7.57 (dd, J = 7.9, 1.3 Hz, 1H), 7.41–7.36 (m, 1H), 7.27 (td, J = 7.7, 1.8 Hz, 1H), 5.85 (s, 1H), 4.73 (q, J = 2.8 Hz, 1H), 4.46 (pd, J = 3.7, 2.9, 1.5 Hz, 1H), 4.36 (td, J = 8.0, 3.8 Hz, 1H), 4.25–4.15 (m, 3H), 4.10 (q, J = 7.1 Hz, 1H), 3.98–3.94 (m, 2H), 3.91 (dd, J = 4.3, 3.1 Hz, 1H), 3.87–3.82 (m, 2H), 3.74 (d, J = 2.5 Hz, 2H). 13C NMR (75 MHz, methanol-d4) δ 138.0, 133.6, 131.8, 129.6, 128.6, 123.4, 101.7, 83.4, 80.4, 80.2, 74.2, 73.8, 67.1, 63.4, 61.5, 61.0, 48.7. HRMS (ESI) calcd for C17H24BrO7Se+ [M]+ 498.9865 found 498.9867.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(3-Bromophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20l). 20l was obtained from 14l and 16 with a yield of 51%. Colorless solid. = –24.7 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.76 (s, 1H), 7.51 (d, J = 7.8 Hz, 2H), 7.32–7.27 (m, 1H), 5.65 (d, J = 3.4 Hz, 1H), 4.73 (s, 1H), 4.47 (m, 1H), 4.36 (td, J = 7.6, 3.9 Hz, 1H), 4.29–4.16 (m, 3H), 4.14–4.00 (m, 2H), 3.99–3.95 (m, 2H), 3.87–3.80 (m, 2H), 3.77–3.69 (m, 2H). 13C NMR (75 MHz, methanol-d4) δ 141.8, 133.0, 131.0, 130.6, 126.5, 123.1, 101.6, 83.2, 80.4, 80.2, 74.0, 73.6, 67.3, 63.5, 61.1, 49.7,49.6. HRMS (ESI) calcd for C17H24BrO7Se+ [M]+ 498.9865 found 498.9869.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(4-Bromophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20m). 20m was obtained from 14m and 16 with a yield of 53%. Colorless solid. = –29.2 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.50 (q, J = 8.6 Hz, 4H), 5.64 (s, 1H), 4.73 (d, J = 2.6 Hz, 1H), 4.60 (br, 1H), 4.46 (d, J = 2.5 Hz, 1H), 4.36 (td, J = 7.4, 4.2 Hz, 1H), 4.18–4.12 (m, 3H), 3.98 (d, J = 5.2 Hz, 1H), 3.96–3.90 (m, 2H), 3.87–3.79 (m, 2H), 3.77–3.68 (m, 2H). 13C NMR (75 MHz, methanol-d4) δ 138.7, 132.3, 129.5, 123.9, 101.8, 83.2, 80.4, 80.2, 74.1, 73.6, 67.4, 63.5, 61.1, 49.7. HRMS (ESI) calcd for C17H24BrO7Se+ [M]+ 498.9865 found 498.9868.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(2-Fluorophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20n). 20n was obtained from 14n and 16 with a yield of 47%. Colorless solid. = –43.7 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.76 (td, J = 7.4, 2.0 Hz, 1H), 7.43–7.36 (m, 1H), 7.20 (t, J = 7.5 Hz, 1H), 7.09 (dd, J = 10.5, 8.3 Hz, 1H), 5.93 (s, 1H), 4.72 (d, J = 2.7 Hz, 1H), 4.45 (d, J = 2.9 Hz, 1H), 4.37 (m, J = 7.8, 4.0 Hz, 1H), 4.23–4.15 (m, 3H), 4.12–4.00 (m, 1H), 3.98–3.92 (m, 2H), 3.89 (t, J = 3.8 Hz, 1H), 3.86–3.78 (m, 2H), 3.75 (d, J = 2.6 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 132.1, 132.0, 129.3, 129.3, 126.6, 126.4, 125.2, 125.2, 116.3, 116.0, 97.3, 97.2, 83.3, 80.3, 80.2, 74.1, 73.7, 67.1, 63.4, 61.0, 49.7, 49.5. HRMS (ESI) calcd for C17H24FO7Se+ [M]+ 439.0666 found 439.0672.
- (1S,2R,3S,4S)-3,4-Dihydroxy-1-((2S)-2-hydroxy-2-((4S,5R)-5-hydroxy-2-(2-iodophenyl)-1,3-dioxan-4-yl)ethyl)-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20o). 20o was obtained from 14o and 16 with a yield of 52%. Colorless solid. = –19.7 (c = 0.165, MeOH). = (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.87–7.84 (m, 1H), 7.73 (m, J = 7.7, 2.1 Hz, 1H), 7.43–7.38 (m, 1H), 7.11 (m, J = 7.7, 5.1, 2.5 Hz, 1H), 5.66 (s, 1H), 4.73 (s, 1H), 4.47–4.46 (m, 1H), 4.36 (q, J = 4.1 Hz, 1H), 4.23–4.19 (m, 2H), 4.11–4.06 (m, 1H), 3.99–3.94 (m, 3H), 3.90 (d, J = 8.1 Hz, 1H), 3.86–3.82 (m, 2H), 3.75 (d, J = 2.5 Hz, 2H). 13C NMR (75 MHz, methanol-d4) δ 140.9, 140.4, 131.9, 129.2, 105.6, 98.0, 83.4, 80.4, 80.2, 74.3, 73.8, 67.1, 63.3, 61.5, 61.0, 50.0. HRMS (ESI) calcd for C17H24IO7Se+ [M]+ 546.9726 found 546.9736.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-(2-Cyanophenyl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20p). 20p was obtained from 14p and 16 with a yield of 50%. Colorless solid. = –35.2 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.86 (d, J = 7.8 Hz, 1H), 7.79–7.68 (m, 2H), 7.56 (m, J = 8.4, 4.2 Hz, 1H), 5.91 (s, 1H), 4.74 (s, 1H), 4.48 (s, 1H), 4.35 (d, J = 20.7 Hz, 2H), 4.27–4.24 (m, 2H), 4.03 (m, J = 8.3, 5.8, 3.8 Hz, 2H), 3.97 (d, J = 2.8 Hz, 1H), 3.94–3.85 (m, 3H), 3.79–3.70 (m, 2H). 13C NMR (75 MHz, methanol-d4) δ 142.2, 134.2, 130.8, 128.2, 118.9, 111.8, 100.1, 83.0, 80.5, 80.0, 74.1, 73.8, 66.9, 63.3, 61.0. HRMS (ESI) calcd for C18H24NO7Se+ [M]+ 446.0713 found 446.0726.
- (1S,2R,3S,4S)-3,4-dihydroxy-1-((2S)-2-hydroxy-2-((4S,5R)-5-hydroxy-2-(naphthalen-1-yl)-1,3-dioxan-4-yl)ethyl)-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20q). 20q was obtained from 14q and 16 with a yield of 45%. Colorless solid. = –80.3 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 8.34 (d, J = 8.2 Hz, 1H), 7.88 (d, J = 8.1 Hz, 2H), 7.80 (d, J = 7.1 Hz, 1H), 7.55–7.47 (m, 3H), 6.22 (d, J = 2.0 Hz, 1H), 4.65 (s, 1H), 4.41 (s, 2H), 4.29 (s, 2H), 4.13 (d, J = 8.1 Hz, 2H), 3.96–3.93 (m, 2H), 3.88 (dd, J = 10.3, 3.9 Hz, 3H), 3.72–3.62 (m, 1H), 3.55 (d, J = 11.9 Hz, 1H). 13C NMR (75 MHz, methanol-d4) δ 135.2, 131.9, 130.7, 129.5, 127.3, 126.8, 126.0, 125.7, 125.6, 101.8, 83.4, 80.5, 74.0, 73.7, 67.5, 63.8, 61.0. HRMS (ESI) calcd for C21H27O7Se+ [M]+ 471.0917 found 471.0930.
- (1S,2R,3S,4S)-1-((2S)-2-((4S,5R)-2-([1,1′-biphenyl]-2-yl)-5-hydroxy-1,3-dioxan-4-yl)-2-hydroxyethyl)-3,4-dihydroxy-2-(hydroxymethyl)tetrahydro-1H-selenophen-1-ium chloride (20r). 20r was obtained from 14r and 16 with a yield of 49%. Colorless solid. = –51.2 (c = 0.165, MeOH). 1H NMR (300 MHz, methanol-d4) δ 7.86 (dd, J = 6.7, 3.1 Hz, 1H), 7.36 (q, J = 7.8, 7.3 Hz, 7H), 7.23–7.20 (m, 1H), 5.46 (s, 1H), 4.67 (d, J = 15.3 Hz, 1H), 4.42 (s, 1H), 4.25 (s, 1H), 4.12–4.05 (m, 3H), 3.93–3.84 (m, 3H), 3.65 (d, J = 16.4 Hz, 4H), 3.59–3.54 (m, 1H). 13C NMR (75 MHz, methanol-d4) δ 142.4, 141.7, 136.4, 130.8, 130.4, 130.0, 129.4, 128.5, 128.4, 128.2, 101.0, 83.6, 80.4, 80.2, 74.0, 73.6, 67.2, 63.4, 61.0, 49.7. HRMS (ESI) calcd for C23H29O7Se+ [M]+ 497.1073 found 497.1091.
3.3. In Vitro α-Glucosidase Inhibition Assay
α-Glucosidase inhibitory activities of the target compounds 19a–r and 20a–r were tested for rat intestinal α-glucosidases such as maltase and sucrase in vitro and compared with those of clinically used anti-diabetic agent voglibose and acarbose. The testing method previously described was modified and used. Thus, rat small intestinal brush border membrane vesicles were first prepared, and its suspension in 0.1 M maleate buffer (pH = 6.0) was used as small intestinal α-glucosidases of maltase and sucrase. A test compound was dissolved in dimethylsulfoxide (DMSO), and the resulting solution was diluted with 0.1 M maleate buffer to prepare the test compound solution (concentration of DMSO: 10%). The substrate solution in maleate buffer (maltose, 74 μM, sucrose, 74 μM, 50 μL), a test compound solution (25 μL), and the enzyme solution (25 μL) were mixed and incubated at 37 °C for 30 min. After incubation, the solution was immediately heated by boiling water for 2 min to stop the reaction, and the solution was mixed with water (150 μL). Glucose concentration was determined by the glucose-oxidase method. Final concentration of DMSO in the test solution was 2.5% and no influence of DMSO was detected on the inhibitory activity. The IC50 value was determined graphically by a plot of percent inhibition versus log of the test compound.
3.4. Cytotoxicity Evaluation of Compounds (20b, 20e and 21b) on Normal HELF Cell Lines
The cytotoxicity of the most potent inhibitors (20a, 20c and 20m) obtained from the present study was evaluated via the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) assay on HELF, a human embryonic lung fibroblast cell line which was a kind gift from KeyGEN BioTECH. Briefly, the HELF-adherent cells were cultured in a 96-well plate overnight in a CO2 environment at 37 °C. Supernatant was removed, and 100 μL serially diluted compounds with complete medium DMEM supplemented with 10% (v/v) fetal bovine serum, penicillin (100 units/mL), and streptomycin (100 μg/mL) were added to each well. After the incubation, the culture medium was aspirated carefully, and 50 μL of MTT solution (2mg/mL PBS) was added to each well and further incubated for 4h. After this, MTT solution was aspirated, and cells were PBS-washed once, and 100 μL of DMSO was added to dissolve the blue insoluble MTT formazan produced by the action of mitochondrial dehydrogenase. The plate was agitated at room temperature for 15 min and then read at 570 nm by using a microplate reader. The percentage of viable cells was calculated as the relative ratio of optical densities.
3.5. Molecular Docking
The crystalline complex of the natural product neokotalanol with ntMGAM (pdbcode: 3L4U) was selected from the PDB database. The .pdb file for 3L4U was downloaded. Proteins (PDB ID:3L4U) were visualized and checked using PyMOL 3.0.3 software to ensure file integrity and docking suitability. Ligands were geometrically optimized by the MMFF94 force field of OpenBabel 2.4.1 software. The optimized molecular structures had the lowest energy and optimal conformation to ensure their reliability for subsequent molecular docking. Receptor proteins and ligand molecules were pre-processed using AutoDock Tools 1.5.6, and molecular docking parameters were set in the Grid module. The molecular docking was performed in semi-flexible docking mode with the docking accuracy parameter exhaustiveness = 25. The chosen docking algorithm was the Lamarckian Genetic Algorithm, and the docking calculations were completed by running AutoDock Vina 1.2.0 software. The docking results were visualized by Pymol.
3.6. Inhibition-Kinetic Studies
For kinetic analyses of maltase and sucrase by compounds (20a, 20c, and 10), the enzyme and test samples (1.0 mg/mL for maltase and 0.2 mg/mL for sucrase) were incubated with increasing concentrations of maltose (2.5, 5, 10, 20, 50, 100 μM) and sucrose (5, 10, 20, 50, 100, 1000 μM) at 37 °C for 30 min. The tube was immediately immersed in boiling water for 2 min to stop the reaction, then cooled with water. The glucose concentration was determined by the glucose-oxidase method. The results plotted according to Lineweaver–Burk revealed a fully competitive type of inhibition on each α-glucosidase. The Lineweaver–Burk equation follows as:
Ki values for each inhibitor were determined by the equation:
3.7. Hypoglycemic Effect in Normal ICR Mice
The animal experimental protocols in this study were approved by the Experimental Animal Center of Shanghai Institute of Materia Medica (Shanghai, China). Male ICR (Institute of Cancer Research) mice (29−33 g) after an overnight fast were used for acute maltose and sucrose loading tests. In the maltose loading test, maltose (2.0 mpk of body weight) and the inhibitors and voglibose were dissolved in distillation−distillation water and administered to the mice via a stomach tube. A control group was loaded with distillation−distillation water. In the sucrose loading test, sucrose (2.5 g/kg of body weight) and the inhibitor miglitol were dissolved in distillation−distillation water and administered to the mice via a stomach tube. A control group was loaded with distillation−distillation water. Blood samples for glucose measurements were obtained from the tail vein at 0, 15, 30, 60, and 120 min after maltose and sucrose loading. The blood glucose levels were measured by a portable kit, StatStrip Xpress (Nova Biomedical Co., Ltd., Waltham, MA, USA). The AUC over was calculated by the trapezoidal method.
4. Conclusions
Based on our previously reported Salacia-derived sulfonium salts featuring a 3′,5′-benzylidene core structure on side chain moiety, a more comprehensive structural modification was conducted in the present investigation. Multiple hydrophobic halogens were attached on the phenyl ring of the benzylidene structure, and an additional phenyl ring was incorporated as either naphthyl or biphenyl groups. Furthermore, applying the principle of “bioisosterism”, the sulfonium cation center was replaced by a selenonium cation. Most of the resulting sulfonium or selenonium salts presented strong inhibitory activity against both maltase and sucrase. Strong electron-withdrawing groups attached at the ortho-position on the phenyl ring, such as nitro or chloro, play a critical and fundamental role in potent α-glucosidase inhibitory activities. Compared to sulfonium salts, their selenonium analogs usually generally exhibited improved in vitro inhibitory potency against both maltase and sucrase. The most potent inhibitor, selenonium 20c (IC50 = 0.05 mM against sucrase and 0.14 mM against maltase), was selected for in vivo hypoglycemic evaluation. Although slightly less potent than compound 10, 20c (10.0 mg/kg) showed significant antihyperglycemic effects superior to those of acarbose (20.0 mg/kg) in the sucrose leading assay. At an equimolar dose, 20c (1.0 mg/kg) demonstrated antihyperglycemic effects comparable to voglibose (1.0 mg/kg) in the maltose loading assay. Enzyme kinetic studies on 20c showed fully competitive inhibition against α-glucosidases. Cytotoxicity evaluation of 20c against multiple cell lines indicated low toxicity. Docking studies of 20c with ntMGAM revealed binding to α-glucosidase via hydrogen bonds, π-alkyl interactions, alkyl interactions, and critical electrostatic interactions contributing to its potent inhibitory activity. This study provides strong evidence that replacing the sulfonium cation core with selenonium may enhance the α-glucosidase inhibitory activity of certain Salacia-derived inhibitors.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules30132856/s1. The supporting information contains 1H and 13C NMR, curves of selected sulfonium and selenonium salts in enzyme assay and IC50 Values of 20a and 20c against different normal cell lines.
Author Contributions
Conceptualization, W.X.; methodology, W.X.; software, J.Y. (Jianchen Yang); validation, G.T. and O.M.; formal analysis, J.Y. (Jiahao Yi); investigation, X.H. and J.Y. (Jiahao Yi); resources, W.X.; data curation, X.H.; writing—original draft preparation, W.X.; writing—review and editing, W.X.; visualization, W.X.; project administration, W.X.; funding acquisition, W.X. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to express their appreciation for the National Key R&D Program of China (Grant No. 2022YFE0105400).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of Institutional Animal Care and Use Committee (IACUC) of China Pharmaceutical University (IACYC number: SYXK (SU) 2024-0018).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Magliano, D.J.; Boyko, E.J.; Atlas, I.D. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021. [Google Scholar]
- Bellary, S. The changing character of diabetes complications. Lancet Diabetes Endocrinol. 2022, 10, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Trapp, S.; Stanford, S.C. New developments in the prospects for GLP-1 therapy. Br. J. Pharmacol. 2022, 179, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Min, S.H.; Yoon, J.H.; Hahn, S.; Cho, Y.M. Efficacy and safety of combination therapy with an α-glucosidase inhibitor and a dipeptidyl peptidase-4 inhibitor in patients with type 2 diabetes mellitus: A systematic review with meta-analysis. J. Diabetes Investig. 2018, 9, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Tahrani, A.A. SGLT-2 inhibitors as second-line therapy in type 2 diabetes. Lancet Diabetes Endocrinol. 2014, 2, 678–679. [Google Scholar] [CrossRef]
- Pia, I.L. SGLT2 inhibitors: The story continues to unfold. Eur. Heart J. 2021, 42, 4902–4904. [Google Scholar]
- Levetan, C. Oral antidiabetic agents in type 2 diabetes. Curr. Med. Res. Opin. 2007, 23, 945–952. [Google Scholar] [CrossRef]
- Lebovitz, H.; Bonhomme, Y. Historical Development of Oral Antidiabetic Agents: The Era of Fortuitous Discovery. Front. Diabetes 2020, 29, 115–133. [Google Scholar]
- Hakamata, W.; Kurihara, M.; Okuda, H.; Nishio, T.; Oku, T. Design and screening strategies for α-glucosidase inhibitors based on enzymological information. Curr. Top. Med. Chem. 2009, 9, 3–12. [Google Scholar] [CrossRef]
- Komindr, S.; Ingsriswang, S.; Lerdvuthisopon, N.; Boontawee, A. Effect of long-term intake of Asian food with different glycemic indices on diabetic control and protein conservation in type 2 diabetic patients. J. Med. Assoc. Thail. 2001, 84, 85–97. [Google Scholar]
- Godbout, A.; Chiasson, J.L. Who should benefit from the use of α-glucosidase inhibitors? Curr. Diabetes. Rep. 2007, 7, 333–339. [Google Scholar] [CrossRef]
- Warfield, K.L.; Alonzi, D.S.; Hill, J.C.; Caputo, A.T.; Roversi, P.; Kiappes, J.L.; Sheets, N.; Duchars, M.; Dwek, R.A.; Biggins, J.; et al. Targeting Endoplasmic Reticulum α-Glucosidase I with a Single-Dose Iminosugar Treatment Protects against Lethal Influenza and Dengue Virus Infections. J. Med. Chem. 2020, 63, 4205–4214. [Google Scholar] [CrossRef]
- Varrot, A.; Tarling, C.A.; Macdonald, J.M.; Stick, R.V.; Zechel, D.L.; Withers, S.G.; Davies, G.J. Direct Observation of the Protonation State of an Imino Sugar Glycosidase Inhibitor upon Binding. J. Am. Chem. Soc. 2003, 125, 7496–7497. [Google Scholar] [CrossRef] [PubMed]
- Hossain, U.; Das, A.K.; Ghosh, S.; Sil, P.C. An overview on the role of bioactive α-glucosidase inhibitors in ameliorating diabetic complications. Food Chem. Toxicol. 2020, 145, 111738. [Google Scholar] [CrossRef]
- Li, R.F.; Yang, J.X.; Liu, J.; Ai, G.M.; Zhang, H.Y.; Xu, L.Y.; Chen, S.B.; Zhang, H.X.; Li, X.L.; Cao, Z.R.; et al. Positional Isomeric Effects on the Optical Properties, Multivalent Glycosidase Inhibition Effect, and Hypoglycemic Effect of Perylene Bisimide-deoxynojirimycin Conjugates. J. Med. Chem. 2021, 64, 5863–5873. [Google Scholar] [CrossRef] [PubMed]
- Pawar, N.J.; Parihar, V.S.; Khan, A.; Joshi, R.; Dhavale, D.D. Quaternary Indolizidine and Indolizidone Iminosugars as Potential Immunostimulating and Glycosidase Inhibitory Agents: Synthesis, Conformational Analysis, Biological Activity, and Molecular Docking Study. J. Med. Chem. 2015, 58, 7820–7832. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Hayashi, E.; Miyauchi, S.; Adachi, I.; Imahori, T.; Natori, Y.; Yoshimura, Y.; Nash, R.J.; Shimaoka, H.; Nakagom, I.; et al. α-1-C-Butyl-1,4-dideoxy-1,4-imino-l-arabinitol as a Second-Generation Iminosugar-Based Oral α-Glucosidase Inhibitor for Improving Postprandial Hyperglycemia. J. Med. Chem. 2012, 55, 10347–10362. [Google Scholar] [CrossRef]
- Shen, Q.; Shao, J.; Peng, Q.; Zhang, W.; Ma, L.; Chan, A.C.; Gu, L. Hydroxycoumarin Derivatives: Novel and Potent α-Glucosidase Inhibitors. J. Med. Chem. 2010, 53, 8252–8259. [Google Scholar] [CrossRef]
- Howard, E.; Cousido-Siah, A.; Lepage, M.L.; Schneider, J.P.; Bodlenner, A.; Mitschler, A.; Meli, A.; Izzo, I.; Alvarez, H.A.; Podjarny, A.; et al. Structural basis of outstanding multivalent effects in jack bean α-mannosidase inhibition. Angew. Chem. Int. Ed. 2018, 57, 8002–8006. [Google Scholar] [CrossRef]
- Niaz, H.; Kashtoh, H.; Khan, J.A.; Khan, A.; Alam, M.T.; Khan, K.M.; Perveen, S.; Choudhary, M.I. Synthesis of diethyl 4-substituted-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylates as a new series of inhibitors against yeast α-glucosidase. Eur. J. Med. Chem. 2015, 95, 199–209. [Google Scholar] [CrossRef]
- Hati, S.; Madurkar, S.M.; Bathula, C.; Thulluri, C.; Agarwal, R.; Siddiqui, F.A.; Dangi, P.; Adepally, U.; Singh, A.; Singh, S.; et al. Design, synthesis and biological evaluation of small molecules as potent glucosidase inhibitors. Eur. J. Med. Chem. 2015, 100, 188–196. [Google Scholar] [CrossRef]
- Jabeen, F.; Oliferenko, P.V.; Oliferenko, A.A.; Pillai, G.G.; Ansari, F.L.; Hall, C.D.; Katritzky, A.R. Dual inhibition of the α-glucosidase and butyrylcholinesterase studied by molecular field topology analysis. Eur. J. Med. Chem. 2014, 80, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Rattanangkool, E.; Kittikhunnatham, P.; Damsud, T.; Wacharasindhu, S.; Phuwapraisirisan, P. Quercitylcinnamates, a new series of antidiabetic bioconjugates possessing α-glucosidase inhibition and antioxidant. Eur. J. Med. Chem. 2013, 66, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Mellet, C.O.; Nierengarten, J.F.; Fernández, J.M.G. Multivalency as an action principle in multimodal lectin recognition and glycosidase inhibition: A paradigm shift driven by carbon-based glyconanomaterials. J. Mater. Chem. B 2017, 5, 6428–6436. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Morikawa, T.; Matsuda, H.; Tanabe, G.; Muraoka, O. Absolute stereostructure of potent α-glucosidase inhibitor, salacinol, with unique thiosugar sulfonium sulfate inner salt structure from Salacia reticulata. Bioorg. Med. Chem. 2002, 10, 1547–1554. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Murakami, T.; Shimada, H.; Matsuda, H.; Yamahara, J.; Tanabe, G.; Muraoka, O. Salacinol, potent antidiabetic principle with unique thiosugar sulfonium sulfate structure from the Ayurvedic traditional medicine Salacia reticulata in Sri Lanka and India. Tetrahedron Lett. 1997, 38, 8367–8370. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Xu, F.; Nakamura, S.; Wang, T.; Matsuda, H.; Tanabe, G.; Muraoka, O. Salaprinol and ponkoranol with thiosugar sulfonium sulfate structure from Salacia prinoides and α-glucosidase inhibitory activity of ponkoranol and kotalanol disulfate. Heterocycles 2008, 75, 1397–1405. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Murakami, T.; Yashiro, K.; Matsuda, H. Kotalanol, a potent α-glucosidase inhibitor with thiosugar sulfonium sulfate structure, from antidiabetic Ayurvedic medicine Salacia reticulata. Chem. Pharm. Bull. 1998, 46, 1339–1340. [Google Scholar] [CrossRef]
- Lu, L.; Li, X.Y.; Yang, Y.; Xie, W.J. Recent progress in the construction of natural de-O-sulfonated sulfonium sugaers with antidiabetic activities. Chem. Eur. J. 2019, 25, 13458–13471. [Google Scholar] [CrossRef]
- Ozaki, S.; Oe, H.; Kitamura, S. α-Glucosidase inhibitor from Kothala-himbutu (Salacia reticulata WIGHT). J. Nat. Prod. 2008, 71, 981–984. [Google Scholar] [CrossRef]
- Oe, H.; Ozaki, S. Hypoglycemic effect of 13-membered ring thiocyclitol, a novel α-glucosidase inhibitor from Kothala-himbutu (Salacia reticulata). Biosci. Biotechnol. Biochem. 2008, 72, 1962–1964. [Google Scholar] [CrossRef]
- Muraoka, O.; Xie, W.J.; Tanabe, G.; Amer, M.F.A.; Minematsu, T.; Yoshikawa, M. On the structure of the bioactive constituent from ayurvedic medicine Salacia reticulata: Revision of the literature. Tetrahedron Lett. 2008, 49, 7315–7317. [Google Scholar] [CrossRef]
- Yuasa, H.; Takada, J.; Hashimoto, H. Synthesis of salacinol. Tetrahedron Lett. 2000, 41, 6615–6618. [Google Scholar] [CrossRef]
- Ghavami, A.; Johnston, B.D.; Pinto, B.M. A new class of glycosidase inhibitor: Synthesis of salacinol and its stereoisomers. J. Org. Chem. 2001, 66, 2312–2317. [Google Scholar] [CrossRef]
- Johnston, B.D.; Ghavami, A.; Jensen, M.T.; Svensson, B.; Pinto, B.M. Synthesis of selenium analogues of the naturally occurring glycosidase inhibitor salacinol and their evaluation as glycosidase inhibitors. J. Am. Chem. Soc. 2002, 124, 8245–8250. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, A.; Sadalapure, K.S.; Johnston, B.D.; Lobera, M.; Snider, B.B.; Pinto, B.M. Improved syntheses of the naturally occurring glycosidase inhibitor salacinol. Synlett 2003, 34, 1259–1262. [Google Scholar] [CrossRef]
- Johnston, B.D.; Jensen, H.H.; Pinto, B.M. Synthesis of sulfonium sulfate analogues of disaccharides and their conversion to chain-extended homologues of salacinol: New glycosidase inhibitors. J. Org. Chem. 2006, 71, 1111–1118. [Google Scholar] [CrossRef]
- Tanabe, G.; Xie, W.; Ogawa, A.; Cao, C.; Minematsu, T.; Yoshikawa, M.; Muraoka, O. Facile synthesis of de-O-sulfated salacinols: Revision of the structure of neosalacinol, a potent α-glucosidase inhibitor. Bioorg. Med. Chem. Lett. 2009, 19, 2195–2198. [Google Scholar] [CrossRef]
- Jayakanthan, K.; Mohan, S.; Pinto, B.M. Structure proof and synthesis of kotalanol and de-O-sulfonated kotalanol, glycosidase inhibitors isolated from an herbal remedy for the treatment of type-2 diabetes. J. Am. Chem. Soc. 2009, 131, 5621–5626. [Google Scholar] [CrossRef]
- Eskandari, R.; Jayakanthan, K.; Kuntz, D.A.; Rose, D.R.; Pinto, B.M. Synthesis of a biologically active isomer of kotalanol, a naturally occurring glucosidase inhibitor. Bioorg. Med. Chem. 2010, 18, 2829–2835. [Google Scholar] [CrossRef]
- Eskandari, R.; Kuntz, D.A.; Rose, D.R.; Pinto, B.M. Potent glucosidase inhibitors: De-O-sulfonated ponkoranol and its stereo isomer. Org. Lett. 2010, 12, 1632–1635. [Google Scholar] [CrossRef]
- Huang, Y.; Gao, Y.; He, W.; Wang, Z.; Li, W.; Lin, A.; Xu, J.; Tanabe, G.; Muraoka, O.; Wu, X.; et al. Practical Route to Neokotalanol and Its Natural Analogues: Sulfonium Sugars with Antidiabetic Activities. Angew. Chem. Int. Ed. 2019, 58, 6400–6404. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, G.; Ueda, S.; Kurimoto, K.; Sonoda, N.; Marumoto, S.; Ishikawa, F.; Xie, W.; Muraoka, O. Facile Synthesis of Neokotalanol, a Potent α-glycosidase Inhibitor Isolated from the Ayurvedic Traditional Medicine “Salacia”. ACS Omega 2019, 4, 7533–7542. [Google Scholar] [CrossRef]
- Lee, B.H.; Eskandari, R.; Jones, K.; Reddy, K.R.; Quezada Calvillo, R.; Nichols, B.L.; Rose, D.R.; Hamaker, B.R.; Pinto, B.M. Modulation of starch digestion for slow glucose release through “toggling” of activities of mucosal α-glucosidases. J. Biol. Chem. 2012, 287, 31929–31938. [Google Scholar] [CrossRef]
- Liu, H.; Nasi, R.; Jayakanthan, K.; Sim, L.; Heipel, H.; Rose, D.R.; Pinto, B.M. New synthetic routes to chain-extended selenium, sulfur, and nitrogen analogues of the naturally occurring glucosidase inhibitor salacinol and their inhibitory activities against recombinant human maltase glucoamylase. J. Org. Chem. 2007, 72, 6562–6572. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Sim, L.; Rose, D.R.; Pinto, B.M. Synthesis of S-alkylated sulfonium-ions and their glucosidase inhibitory activities against recombinant human maltase glucoamylase. Carbohydr. Res. 2007, 342, 901–912. [Google Scholar] [CrossRef]
- Eskandari, R.; Jones, K.; Rose, D.R.; Pinto, B.M. The effect of heteroatom substitution of sulfur for selenium in glucosidase inhibitors on intestinal α-glucosidase activities. Chem. Commun. 2011, 47, 9134–9136. [Google Scholar] [CrossRef]
- Tanabe, G.; Otani, T.; Cong, W.; Minematsu, T.; Ninomiya, K.; Yoshikawa, M.; Muraoka, O. Biological evaluation of 3′-O alkylated analogs of salacinol, the role of hydrophobic alkyl group at 3′-position in the side chain on the α-glucosidase inhibitory activity. Bioorg. Med. Chem. Lett. 2011, 21, 3159–3162. [Google Scholar] [CrossRef]
- Eskandari, R.; Jones, K.; Reddy, K.R.; Jayakanthan, K.; Chaudet, M.; Rose, D.R.; Pinto, B.M. Probing the intestinal α-glucosidase enzyme specificities of starch-digesting maltase glucoamylase and sucrase-isomaltase: Synthesis and inhibitory properties of 3′- and 5′-maltose-extended de-O-sulfonated ponkoranol. Chem.-Eur. J. 2015, 17, 14817–14825. [Google Scholar] [CrossRef]
- Liu, D.; He, W.; Wang, Z.; Liu, L.; Wang, C.; Zhang, C.; Wang, C.; Wang, Y.; Tanabe, G.; Muraoka, O.; et al. Design, synthesis and biological evaluation of 3′-benzylated analogs of 3′-epi-neoponkoranol as potent α-glucosidase inhibitors. Eur. J. Med. Chem. 2016, 110, 224–236. [Google Scholar] [CrossRef]
- Tanabe, G.; Nakamura, S.; Tsutsui, N.; Balakishan, G.; Xie, W.; Tsuchiya, S.; Akaki, J.; Morikawa, T.; Ninomiya, K.; Nakanishi, I.; et al. In silico design, synthesis and evaluation of 3′-O-benzylated analogs of salacinol, a potent α-glucosidase inhibitor isolated from an Ayurvedic traditional medicine “Salacia”. Chem. Commun. 2012, 48, 8646–8648. [Google Scholar] [CrossRef]
- Lu, L.; Chen, J.Y.; Tao, W.X.; Wang, Z.M.; Liu, D.; Zhou, J.H.; Wu, X.M.; Sun, H.P.; Li, W.; Tanabe, G.; et al. Design and Synthesis of Sulfonium Derivatives: A Novel Class of α-Glucosidase Inhibitors with Potent In Vivo Antihyperglycemic Activities. J. Med. Chem. 2023, 66, 3484–3498. [Google Scholar] [CrossRef] [PubMed]
- Sim, L.; Jayakanthan, K.; Mohan, S.; Nasi, R.; Johnston, B.D.; Pinto, B.M.; Rose, D.R. New glucosidase inhibitors from an ayurvedic herbal treatment for type 2 diabetes: Structures and inhibition of human intestinal maltase-glucoamylase with compounds from Salacia reticulata. Biochemistry 2010, 49, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Takahira, K.; Tanabe, G.; Muraoka, O.; Nakanishi, I. Homology modeling of human alpha-glucosidase catalytic domains and SAR study of salacinol derivatives. Open. J. Med. Chem. 2012, 2, 50–60. [Google Scholar] [CrossRef]
- Yang, Y.; Tong, J.; Xie, X.S.; Cao, H.; Fu, Y.; Luo, Y.; Liu, S.; Chen, W.; Yang, N. Design, synthesis, and biological evaluation of novel chrysin derivatives as poly(ADP-ribose) polymerase 1 (PARP1) inhibitors for the treatment of breast cancer. Chin. J. Nat. Med. 2024, 22, 455–465. [Google Scholar] [CrossRef]
- Tian, H.C.; Zhang, L. Total synthesis of lissodendoric acid A by [4 + 2] cycloaddition of transient cyclic allene. Chin. J. Nat. Med. 2023, 21, 161–162. [Google Scholar] [CrossRef]
- Haller, H. The clinical importance of postprandial glucose. Diabetes Res. Clin. Pract. 1998, 40, S43–S49. [Google Scholar] [CrossRef]
- Rizzo, M.R.; Marfella, R.; Barbieri, M.; Boccardi, V.; Vestini, F.; Lettieri, B.; Canonico, S.; Paolisso, G. Relationships between daily acute glucose fluctuations and cognitive performance among aged type 2 diabetic patients. Diabetes Care 2010, 33, 2169–2174. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, X.Y.; Miao, Y.; Zhu, J.H.; Yan, H.; Wang, B.Y.; Jin, J.; Hu, T.J.; Jia, W.P. The relationship between glucose excursion and cognitive function in aged type 2 diabetes patients. Biomed. Environ. Sci. 2012, 25, 1–7. [Google Scholar]
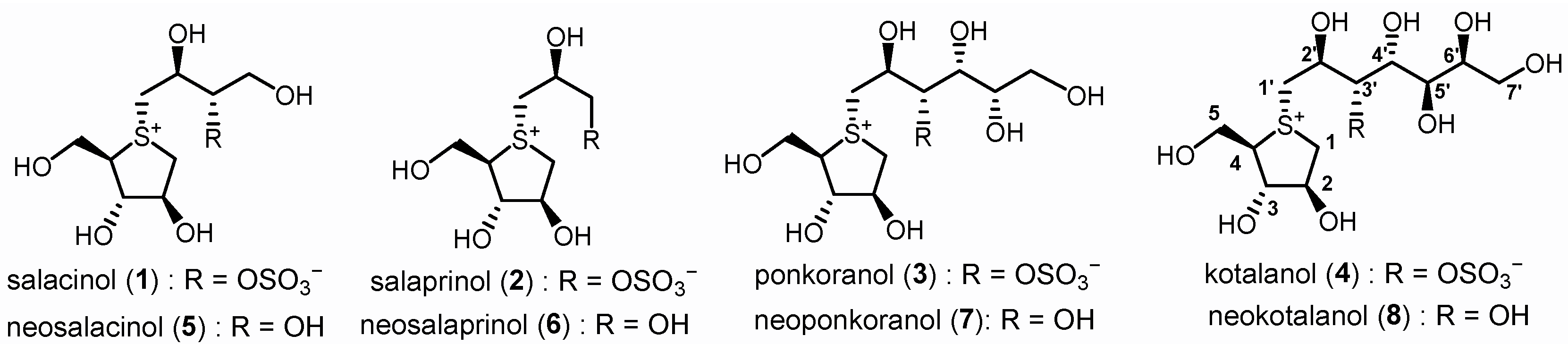
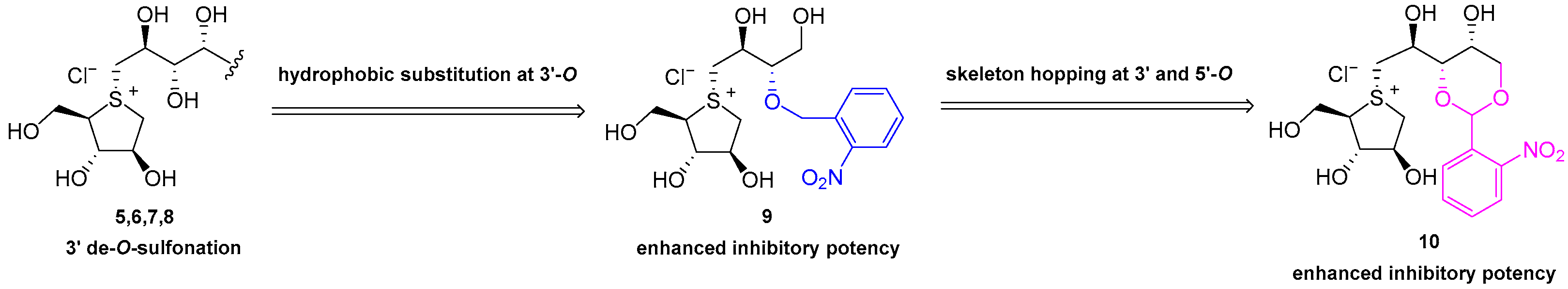
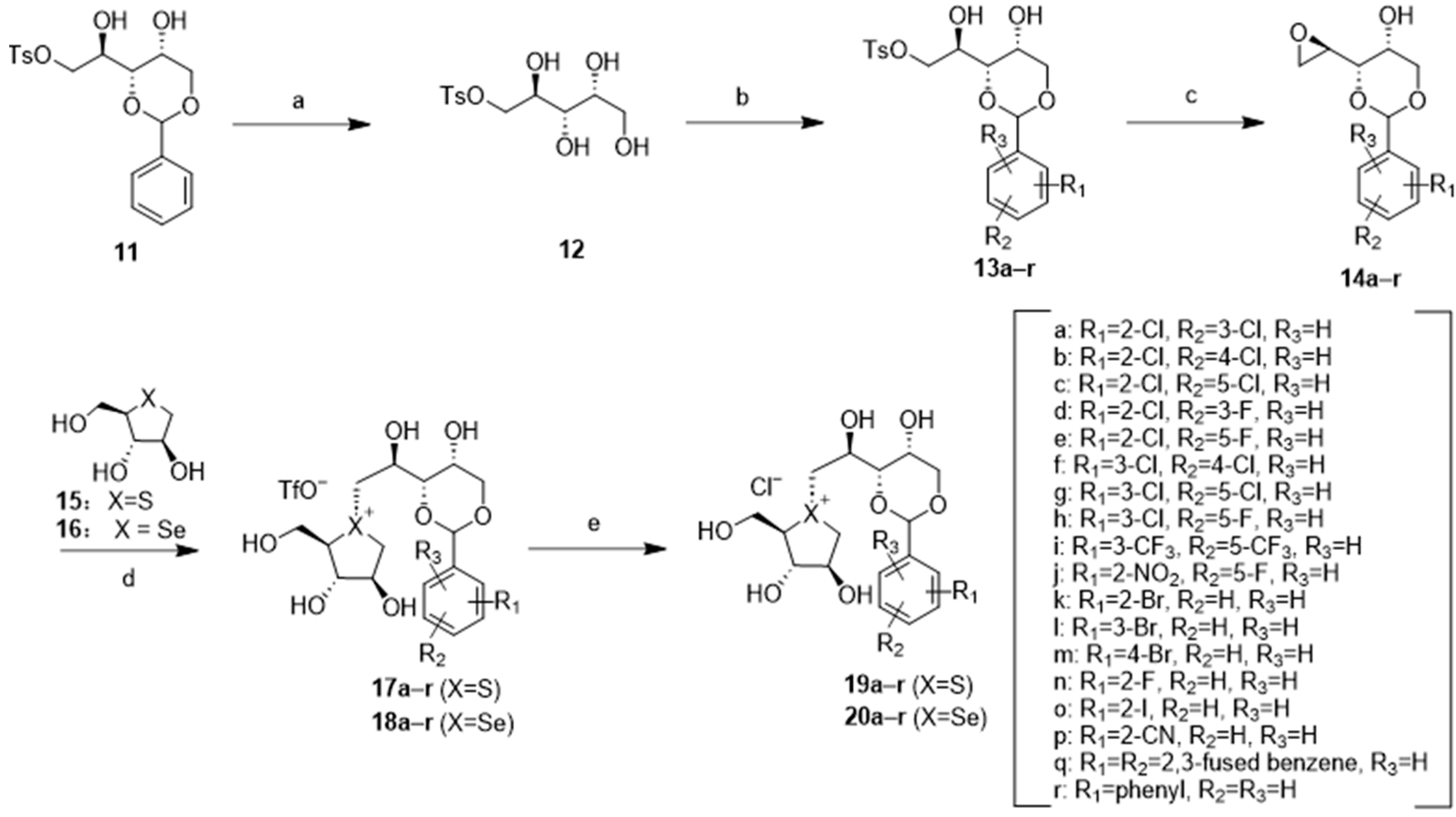
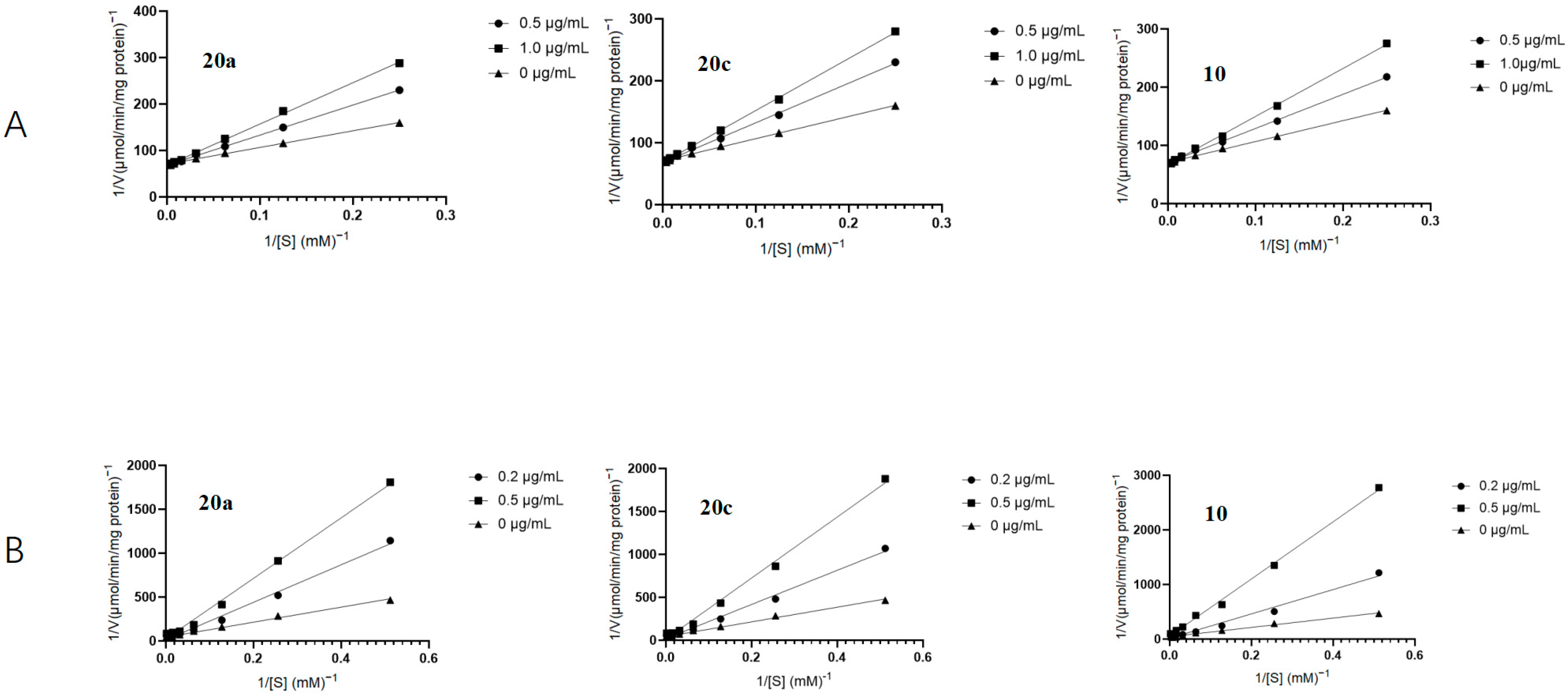
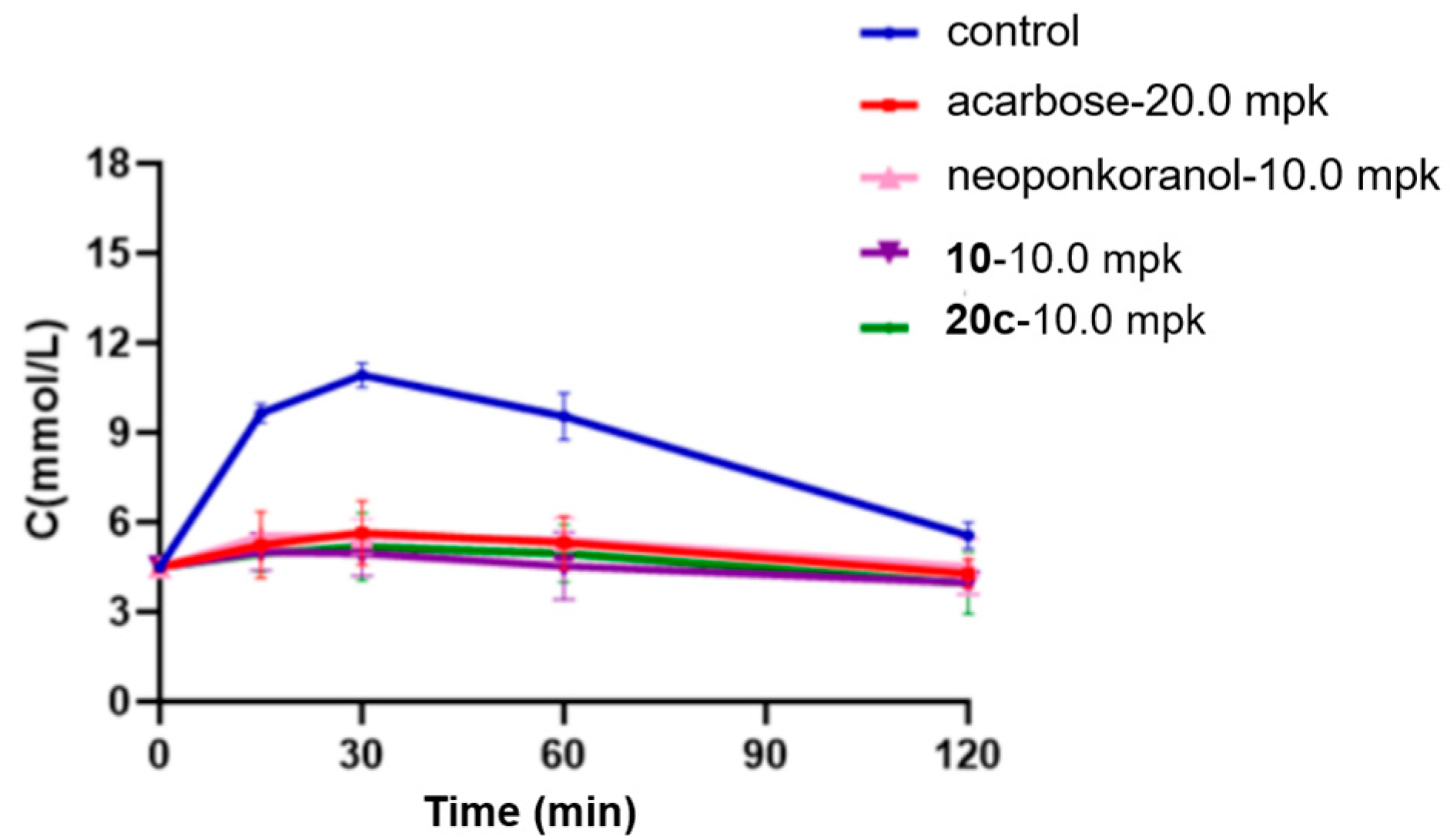
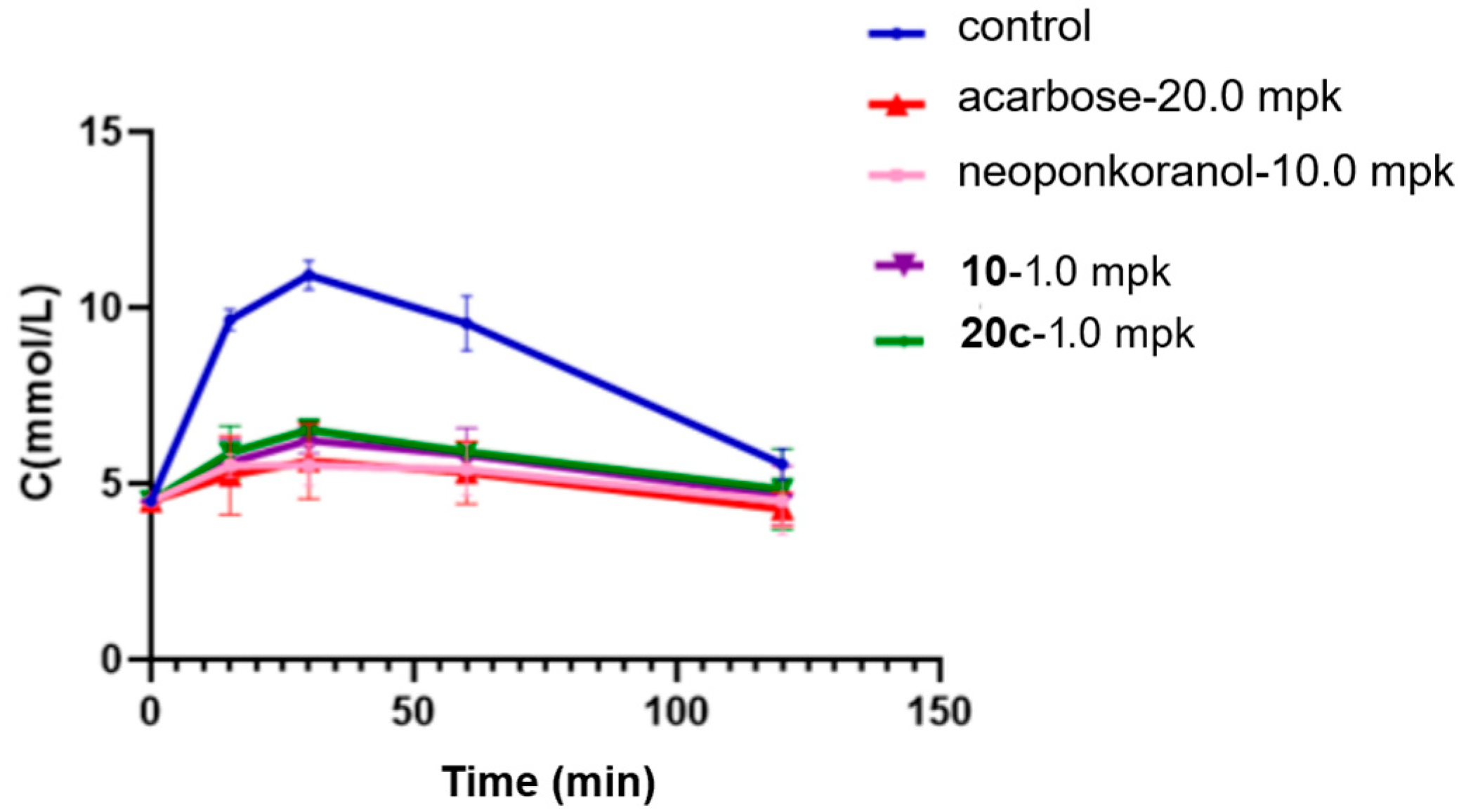
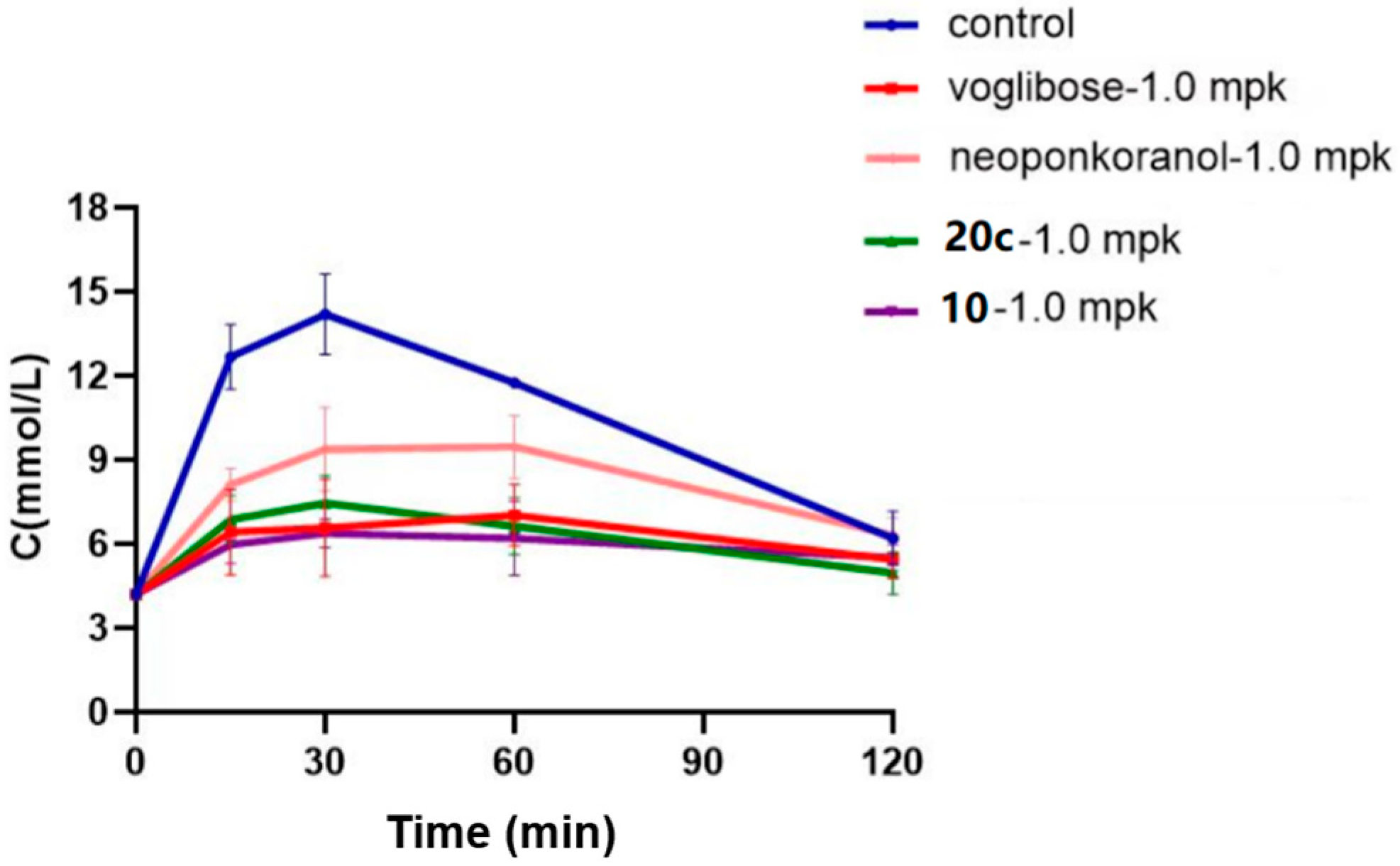
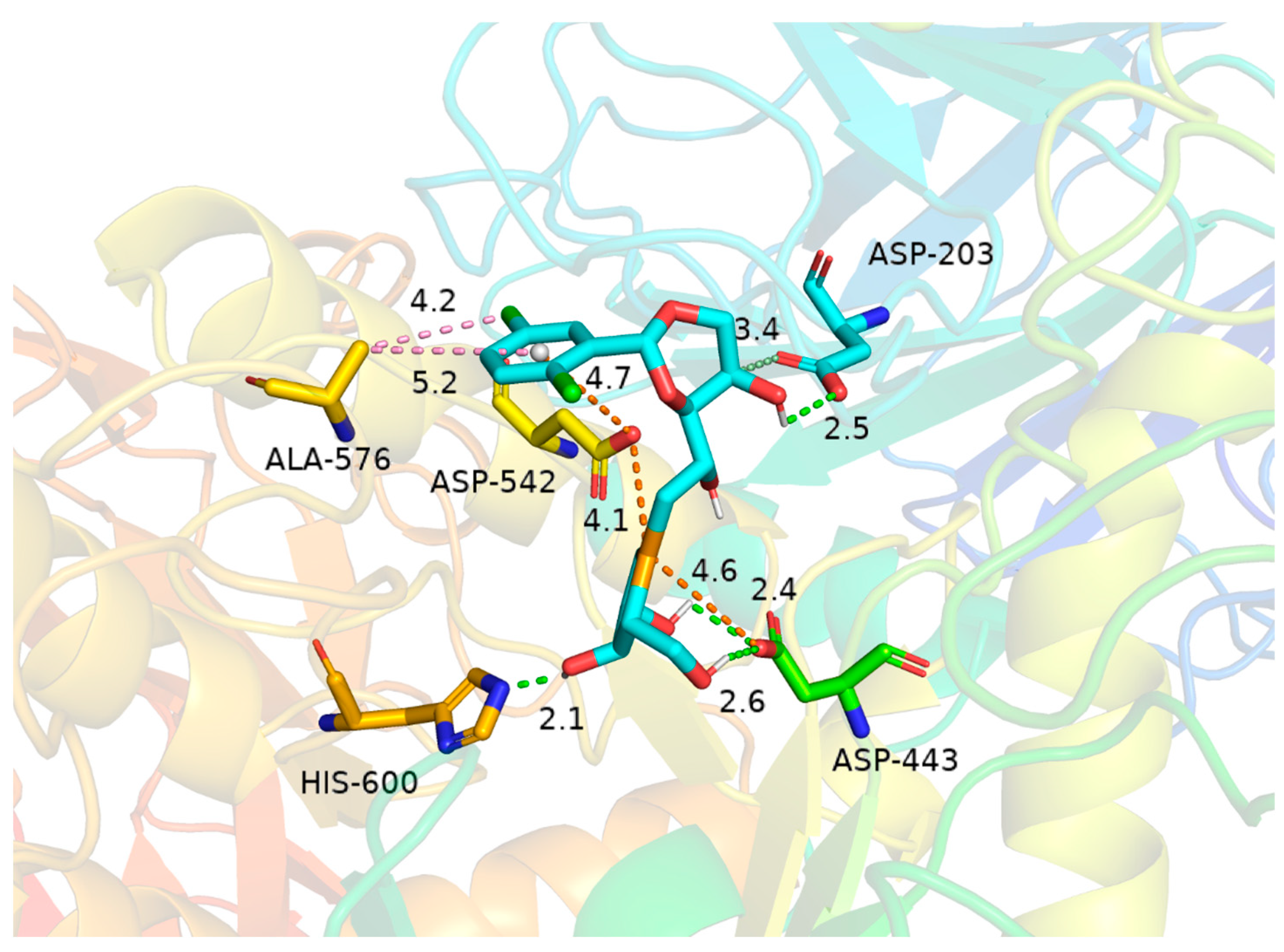
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).