


Synthesis, Structure, Electrochemistry, and In Vitro Anticancer and Anti-Migratory Activities of (Z)- and (E)-2-Substituted-3-Ferrocene-Acrylonitrile Hybrids and Their Derivatives

 , , , ,
, , , ,  ,
, 
Abstract

1. Introduction
2. Results and Discussion
2.1. Synthesis and Structural Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
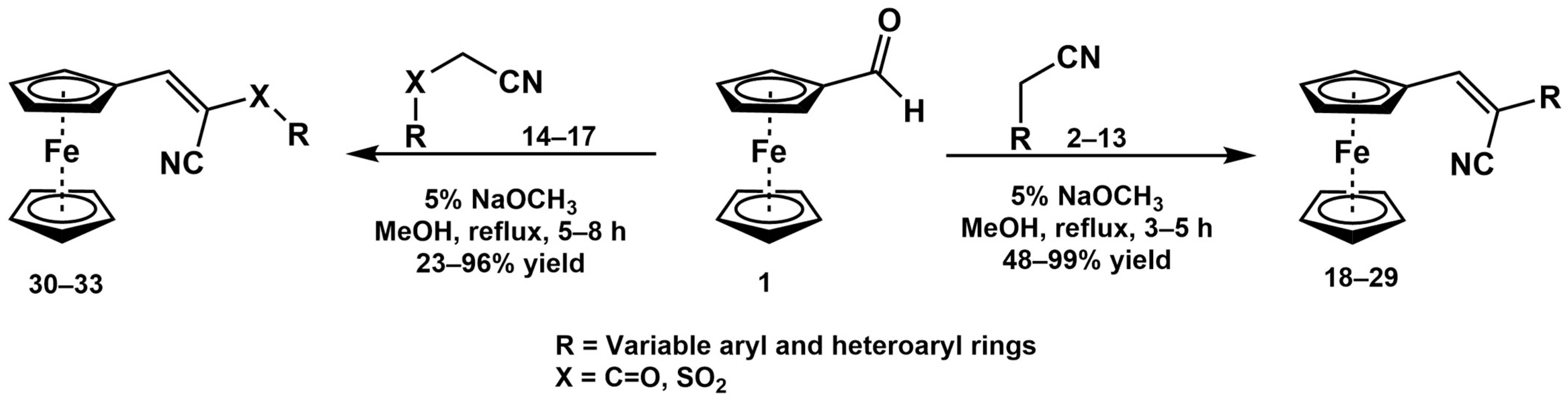
| Substrate | 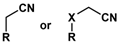 R = Aryl and Heteroaryl Rings X = C=O, SO2 | Product | 2-Substituted-3-Ferroceneacrylonitrile | Yield (%) |
|---|---|---|---|---|
| 2 | 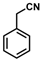 | (Z)-18 | 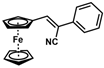 | 98 |
| 3 |  | (Z)-19 |  | 77 |
| 4 | 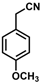 | (Z)-20 | 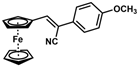 | 59 b |
| 5 | 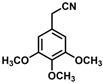 | (Z)-21 |  | 88 |
| 6 |  | (Z)-22 | 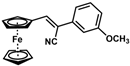 | 61 |
| 7 | 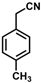 | (Z)-23 | 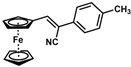 | 89 |
| 8 | 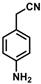 | (Z)-24 |  | 88 |
| 9 | 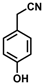 | (Z)-25 | 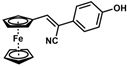 | 48 |
| 10 |  | (Z)-26 a | 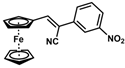 | 70 |
| 11 | 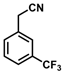 | (Z)-27 | 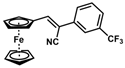 | 99 |
| 12 | 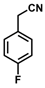 | (Z)-28 a | 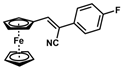 | 87 b |
| 13 | 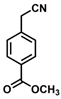 | (Z)-29 |  | 75 |
| 14 | 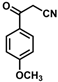 | (E)-30 |  | 23 |
| 15 | 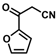 | (E)-31 | 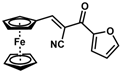 | 57 |
| 16 | 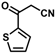 | (E)-32 | 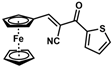 | 96 |
| 17 | 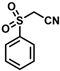 | (E)-33 | 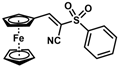 | 73 |
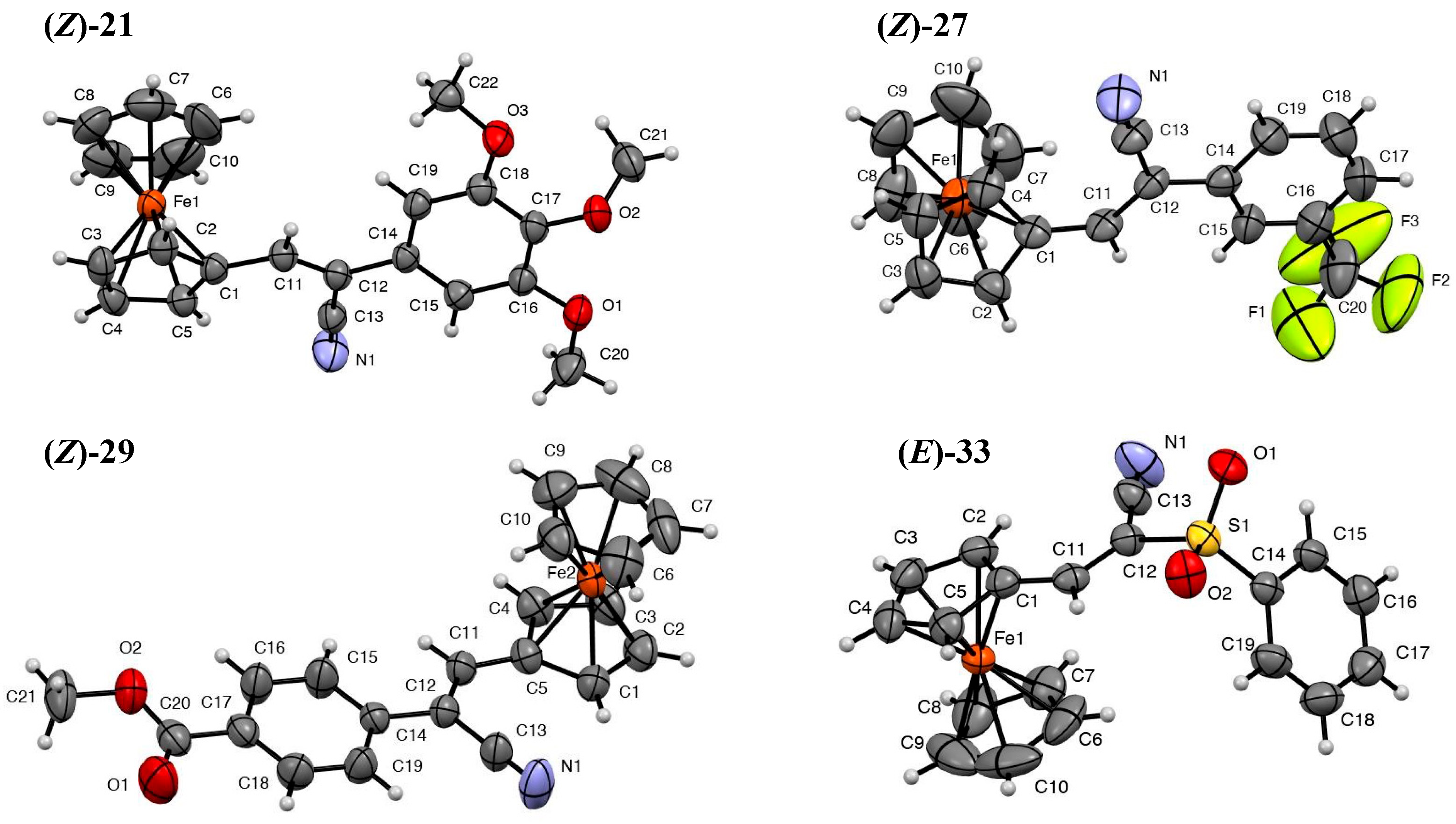
2.1.1. Structure Determination
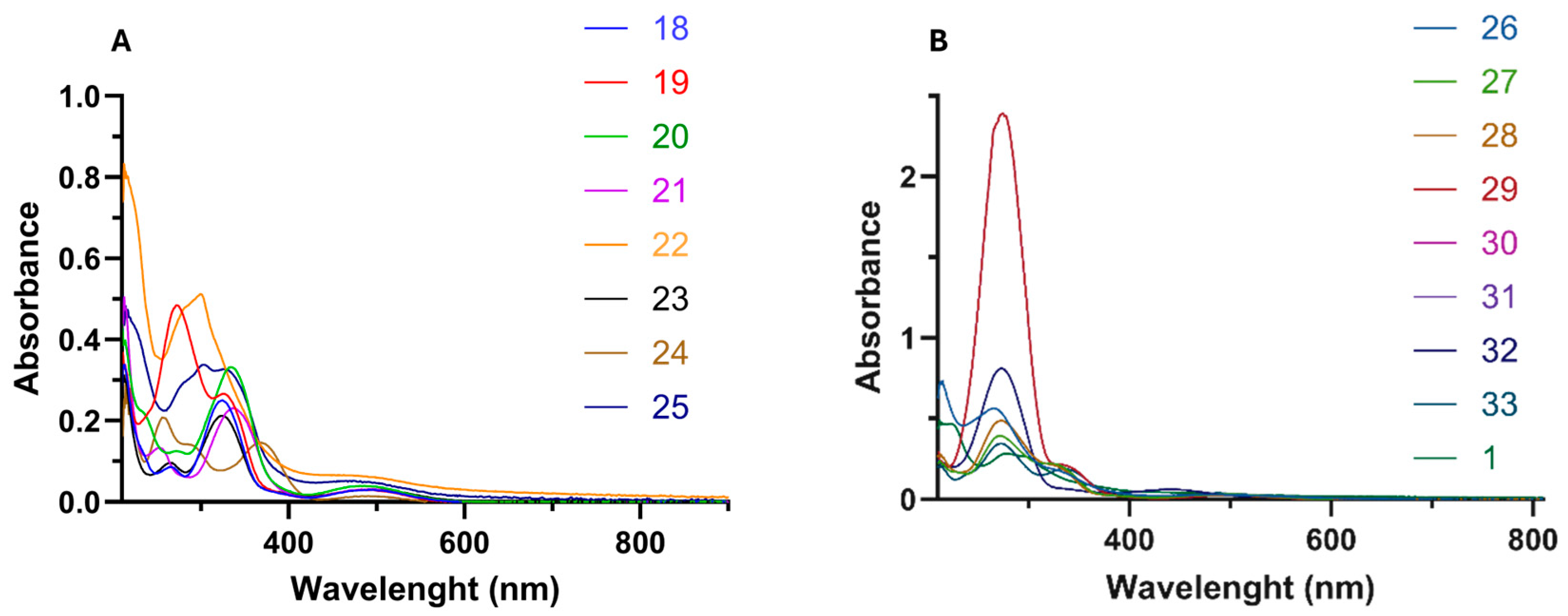
2.1.2. UV/Vis Absorption
| Comp. | π–π* Transition 1 | n–π* Transition 2 | d–d* Transition 3 | |||
|---|---|---|---|---|---|---|
| λ (nm) | ε (×104 cm−1 M−1) | λ (nm) | ε (×104 cm−1 M−1) | λ (nm) | ε (×103 cm−1 M−1) | |
| (Z)-18 | 214 | 3.49 | 326 | 4.90 | 492 | 3.05 |
| (Z)-19 | 211 | 3.64 | 329 | 2.61 | 489 | 2.93 |
| (Z)-20 | 214 | 4.39 | 336 | 3.29 | 486 | 3.83 |
| (Z)-21 | 213 | 5.00 | 338 | 2.28 | 492 | 3.08 |
| (Z)-22 | 213 | 8.30 | 301 | 3.11 | 485 | 6.25 |
| (Z)-23 | 212 | 3.02 | 335 | 2.05 | 498 | 2.99 |
| (Z)-24 | 247 | 2.06 | 360 | 1.44 | 480 | 2.41 |
| (Z)-25 | 215 | 4.80 | 329 | 3.26 | 476 | 5.23 |
| (Z)-26 | 215 | 7.38 | 332 | 1.75 | 483 | 2.85 |
| (Z)-27 | 212 | 2.36 | 325 | 2.19 | 503 | 2.98 |
| (Z)-28 | 213 | 2.56 | 325 | 1.97 | 491 | 2.47 |
| (Z)-29 | 211 | 2.95 | 338 | 2.09 | 509 | 2.40 |
| (E)-30 | 213 | 2.19 | 337 | 0.44 | 426 | 4.99 |
| (E)-31 | 215 | 1.62 | 340 | 0.41 | 442 | 2.55 |
| (E)-32 | 213 | 1.80 | 336 | 0.51 | 442 | 4.77 |
| (E)-33 | 215 | 1.97 | 338 | 1.77 | 490 | 3.54 |
| 1 | 227 | 4.51 | 304 | 2.53 | 460 | 2.18 |
2.1.3. Electrochemical Properties
| Comp. | E½ (mV) a | ΔEp (mV) b |
|---|---|---|
| (Z)-18 | 554 | 73 |
| (Z)-19 | 578 | 76 |
| (Z)-20 | 556 | 77 |
| (Z)-21 | 548 | 88 |
| (Z)-22 | 544 | 86 |
| (Z)-23 | 506 | 67 |
| (Z)-24 | 499 | 77 |
| (Z)-25 | 619 | 115 |
| (Z)-26 | 585 | 64 |
| (Z)-27 | 585 | 73 |
| (Z)-28 | 560 | 75 |
| (Z)-29 | 584 | 63 |
| (E)-30 | 655 | 87 |
| (E)-31 | 678 | 75 |
| (E)-32 | 676 | 72 |
| (E)-33 | 586 | 61 |
2.2. Biological Activity
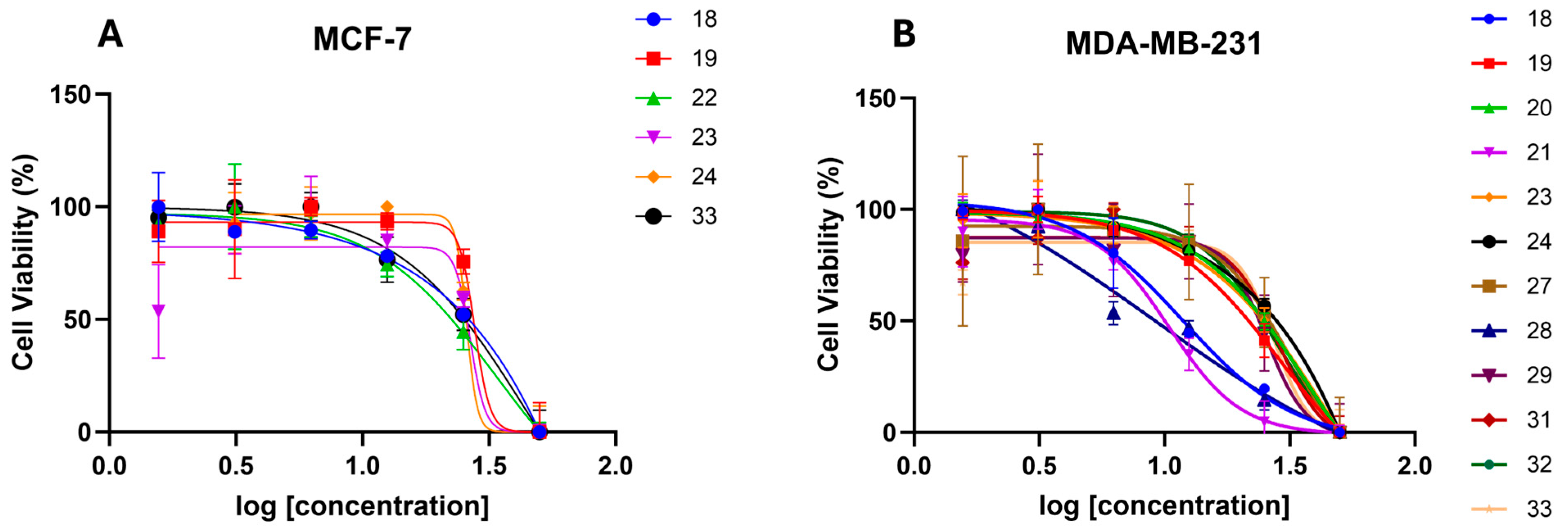
2.2.1. In Vitro Anticancer Activity
| Comp. | GI50 (µM) a | |
|---|---|---|
| MCF-7 | MDA-MB-231 | |
| (Z)-18 | 32.8 | 11.9 |
| (Z)-19 | 27.7 | 29.6 |
| (Z)-20 | >50 | >50 |
| (Z)-21 | >50 | 38.8 |
| (Z)-22 | 33.9 | >50 |
| (Z)-23 | 26.6 | 32.6 |
| (Z)-24 | 25.8 | 33.3 |
| (Z)-25 | >50 | >50 |
| (Z)-26 | >50 | 41.4 |
| (E)-26 | >50 | >50 |
| (Z)-27 | >50 | 31.4 |
| (Z)-28 | >50 | 9.1 |
| (E)-28 | >50 | >50 |
| (Z)-29 | >50 | 25.4 |
| (E)-30 | >50 | >50 |
| (E)-31 | >50 | 28.4 |
| (E)-32 | >50 | 27.3 |
| (E)-33 | 44 | 27.3 |
| (Z)-35 | >50 | >50 |
2.2.2. In Vitro Anti-Migration Activity
| Comp. | Migration (%) a,b | Concentration (µM) c |
|---|---|---|
| (Z)-18 | 98.14 ± 4.31 | 2.4 |
| (Z)-19 | 97.99 ± 2.82 | 5.9 |
| (Z)-20 | 96.98 ± 3.54 | 10 |
| (Z)-21 | 96.91 ± 3.74 | 7.8 |
| (Z)-22 | 99.47 ± 1.32 | 10 |
| (Z)-23 | 98.32 ± 3.17 | 6.5 |
| (Z)-24 | 98.46 ± 4.17 | 6.7 |
| (Z)-25 | 87.46 ± 4.11 | 10 |
| (Z)-26 | 99.18 ± 0.47 | 10 |
| (E)-26 | 99.04 ± 2.16 | 10 |
| (Z)-27 | 99.37 ± 0.53 | 6.3 |
| (Z)-28 | 97.35 ± 4.65 | 1.8 |
| (E)-28 | 98.79 ± 0.03 | 10 |
| (Z)-29 | 99.38 ± 1.10 | 5.1 |
| (E)-30 | 99.02 ± 0.03 | 5.4 |
| (E)-31 | 98.85 ± 0.03 | 5.1 |
| (E)-32 | 99.28 ± 0.03 | 5.4 |
| (E)-33 | 98.98 ± 2.78 | 10 |
| (Z)-35 | 99.10 ± 0.03 | 10 |
3. Materials and Methods
3.1. General Methods
3.2. Synthesis Methods
General Procedure for the Synthesis of 2-Substituted-3-Ferrocene-Acrylonitrile Derivatives 18–33
- (Z)-3-Ferrocenyl-2-phenylacrylonitrile (18)
- (Z)-3-Ferrocenyl-2-(3-pyridyl)acrylonitrile (19)
- (Z)-3-Ferrocenyl-2-(4-methoxyphenyl)acrylonitrile (20)
- (Z)-3-Ferrocenyl-2-(3,4,5-trimethoxyphenyl)acrylonitrile (21)
- (Z)-3-Ferrocenyl-2-(3-methoxyphenyl)acrylonitrile (22)
- (Z)-3-Ferrocene-2-(4-methylphenyl)acrylonitrile (23)
- (Z)-3-Ferrocenyl-2-(4-aminophenyl)acrylonitrile (24)
- (Z)-3-Ferrocenyl-2-(4-hydroxyphenyl)acrylonitrile (25)
- (Z)-3-Ferrocenyl-2-(3-nitrophenyl)acrylonitrile (26)
- (E)-3-Ferrocenyl-2-(3-nitrophenyl)acrylonitrile (26)
- (Z)-3-Ferrocenyl-2-[3-(trifluoromethyl)phenyl]acrylonitrile (27)
- (Z)-3-Ferrocenyl-2-(4-fluorophenyl)acrylonitrile (28)
- (E)-3-Ferrocenyl-2-(-(4-fluorophenyl)acrylonitrile (E-28)
- (Z)-3-Ferrocenyl-2-(4-methylbenzoate)acrylonitrile (29)
- (E)-3-Ferrocenyl-2-(4-methoxybenzoyl)acrylonitrile (30)
- (E)-3-Ferrocenyl-2-(2-furanoyl)acrylonitrile (31)
- (E)-3-Ferrocene-2-(2-thienylketone)acrylonitrile (32)
- (E)-3-Ferrocenyl-2-(phenylsulfonyl)acrylonitrile (33)
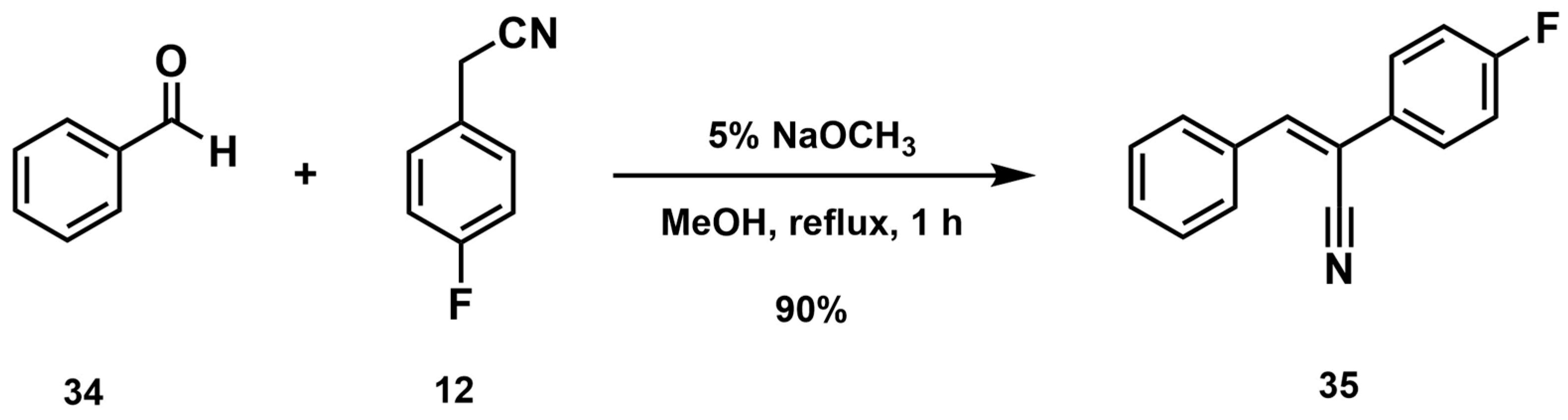
- Synthesis of (Z)-2-(4-Fluorophenyl)-3-phenylacrylonitrile (35)
3.3. Single-Crystal X-Ray Diffraction Experiments
3.4. Cyclic Voltammetry
3.5. Biological Evaluation
3.5.1. Cell Culture
3.5.2. Sulforhodamine B (SRB) Assay
3.5.3. Wound-Healing Assay (Scratch Method) Using MDA-MB-231 Cancer Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Statistics Explorer Network. Available online: https://seer.cancer.gov/statistics-network/ (accessed on 12 October 2024).
- Rueth, N.M.; Lin, H.Y.; Bedrosian, I.; Shaitelman, S.F.; Ueno, N.T.; Shen, Y.; Babiera, G. Underuse of Trimodality Treatment Affects Survival for Patients with Inflammatory Breast Cancer: An Analysis of Treatment and Survival Trends from the National Cancer Database. J. Clin. Oncol. 2014, 32, 2018–2024. [Google Scholar] [CrossRef]
- Yih Ching, O.; Gasser, G. Organometallic compounds in drug discovery: Past, present and future. Drug Discov. Today Technol. 2020, 37, 117–124. [Google Scholar]
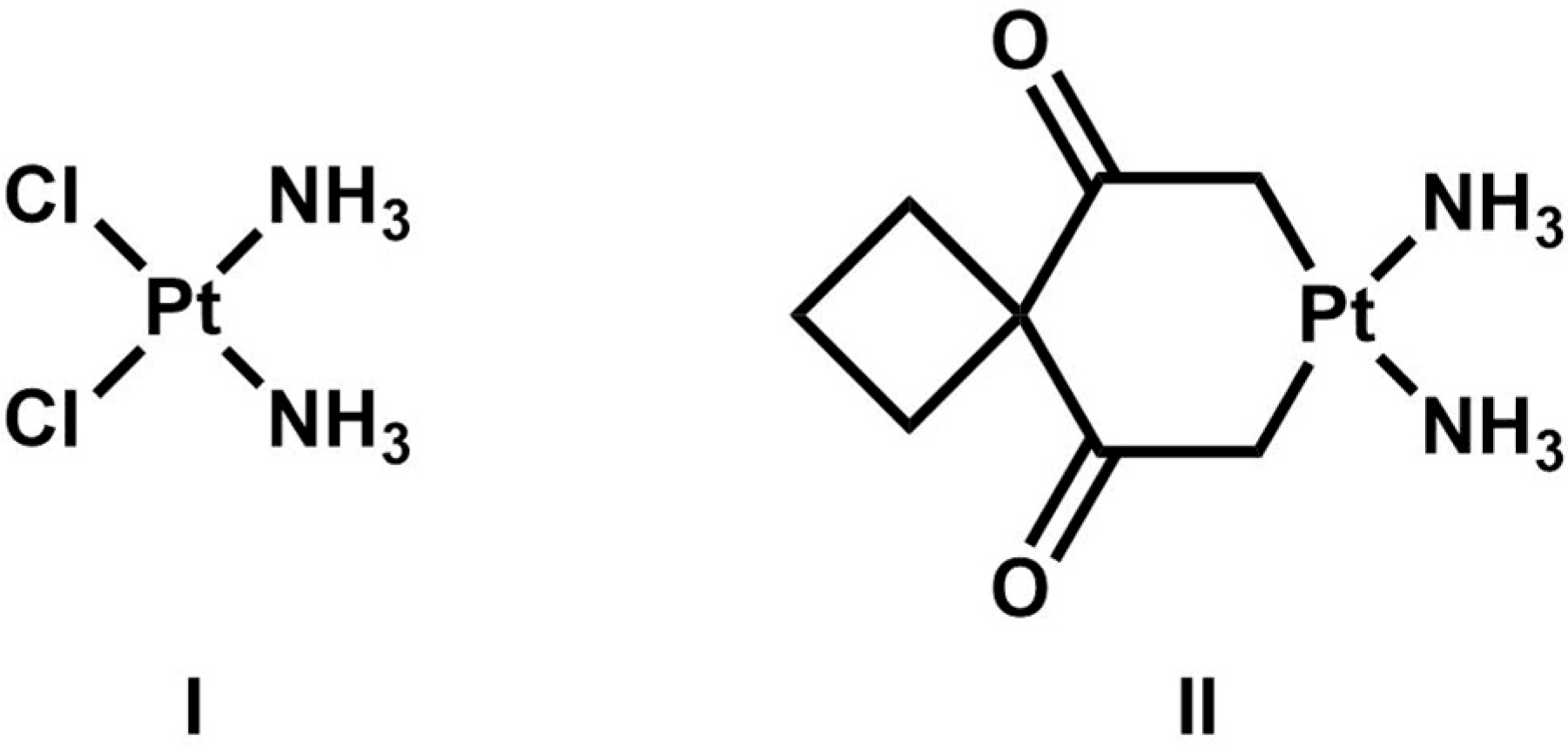
- Rozencweig, M.; Von Hoff, D.D.; Slavik, M.; Muggia, F.M. Cis-diamminedichloroplatinum (II), a new anticancer drug. Ann. Intern. Med. 1977, 86, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Shaloam, D.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar]
- Dong, W.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar]
- Caliskan, B. Radical Mechanisms in the Metallocenes. In Recent Progress in Organometallic Chemistry; IntechOpen: Rijeka, Croatia, 2017; pp. 25–45. [Google Scholar]
- Cullen, W.R.; Woollins, J.D. Ferrocene-containing metal complexes. Coord. Chem. Rev. 1981, 39, 1–30. [Google Scholar] [CrossRef]
- Rausch, M.D. Metallocene chemistry—A decade of progress. Can. J. Chem. 1963, 41, 1289–1314. [Google Scholar] [CrossRef]
- Avishek, P.; Borrelli, R.; Bouyanfif, H.; Gottis, S.; Sauvage, F. Tunable redox potential, optical properties, and enhanced stability of modified ferrocene-based complexes. ACS Omega 2019, 4, 14780–14789. [Google Scholar]
- Patra, M.; Gasser, G. The Medicinal Chemistry of Ferrocene and Its Derivatives. Nat. Rev. Chem. 2017, 1, 0066. [Google Scholar] [CrossRef]
- Li, W.; Yu, J.; Wang, J.; Fan, X.; Xu, X.; Wang, H.; Xiong, Y.; Li, X.; Zhang, X.; Zhang, Q.; et al. How Does Ferrocene Correlate with Ferroptosis? Multiple Approaches to Explore Ferrocene-Appended GPX4 Inhibitors as Anticancer Agents. Chem. Sci. 2024, 15, 10477–10490. [Google Scholar] [CrossRef]
- Moffitt, W. The Electronic Structure of Bis-Cyclopentadienyl Compounds. J. Am. Chem. Soc. 1954, 76, 3386–3392. [Google Scholar] [CrossRef]
- Laskoski, M.; Steffen, W.; Smith, M.D.; Bunz, U.H.F. Is Ferrocene More Aromatic than Benzene? Chem. Commun. 2001, 691–692. [Google Scholar] [CrossRef]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A Rational Approach in Drug Design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef] [PubMed]
- Yeary, R. Chronic Toxicity of Dicyclopentadienyliron (Ferrocene) in Dogs. Toxicol. Appl. Pharmacol. 1969, 15, 666–676. [Google Scholar] [CrossRef]
- Hanzlik, R.P.; Soine, W.H. Enzymic hydroxylation of ferrocene. J. Am. Chem. Soc. 1978, 100, 1290–1291. [Google Scholar] [CrossRef]
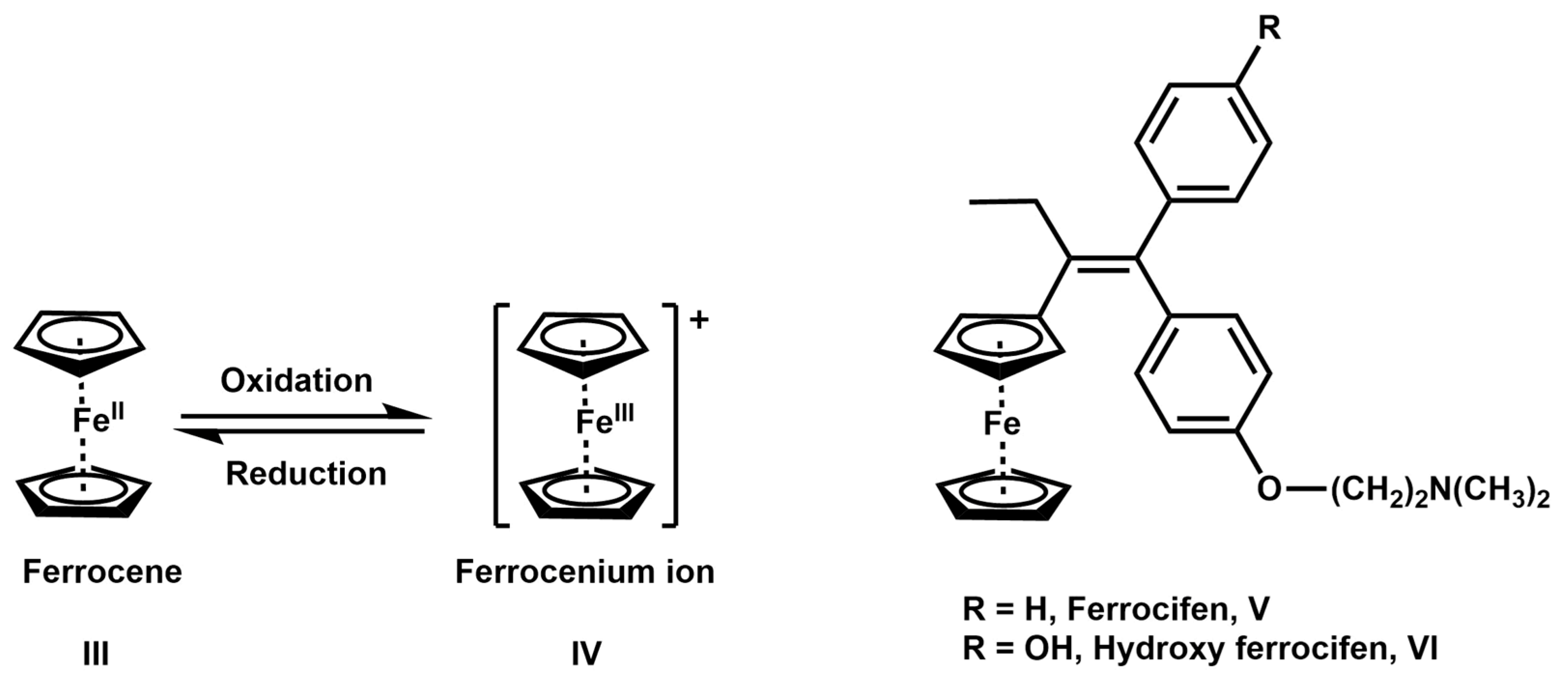
- Fabbrizzi, L. The Ferrocenium/Ferrocene Couple: A Versatile Redox Switch. ChemTexts 2020, 6, 22. [Google Scholar] [CrossRef]
- Fiorina, V.J.; Dubois, R.J.; Brynes, S. Ferrocenyl Polyamines as Agents for the Chemoimmunotherapy of Cancer. J. Med. Chem. 1978, 21, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Top, S.; Pigeon, P.; Vessières, A.; Hillard, E.A.; Plamont, M.-A.; Huché, M.; Rigamonti, C.; Jaouen, G. Synthesis and Structure-Activity Relationships of Ferrocenyl Tamoxifen Derivatives with Modified Side Chains. Chem.-A Eur. J. 2009, 15, 684–696. [Google Scholar] [CrossRef]
- Top, S.; Tang, J.; Vessières, A.; Carrez, D.; Provot, C.; Jaouen, G. Ferrocenyl Hydroxytamoxifen: A Prototype for a New Range of Oestradiol Receptor Site-Directed Cytotoxics. Chem. Commun. 1996, 8, 955–956. [Google Scholar] [CrossRef]
- Richard, M.-A.; Hamels, D.; Pigeon, P.; Top, S.; Dansette, P.M.; Lee, H.Z.S.; Vessières, A.; Mansuy, D.; Jaouen, G. Oxidative Metabolism of Ferrocene Analogues of Tamoxifen: Characterization and Antiproliferative Activities of the Metabolites. ChemMedChem 2015, 10, 981–990. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen Type Anti-Cancer Drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef] [PubMed]
- Köpf-Maier, P.; Köpf, H.; Neuse, E.W. Ferrocenium Salts—The First Antineoplastic Iron Compounds. Angew. Chem. Int. Ed. 1984, 23, 456–457. [Google Scholar] [CrossRef]
- Citta, A.; Folda, A.; Bindoli, A.; Pigeon, P.; Top, S.; Vessières, A.; Salmain, M.; Jaouen, G.; Rigobello, M.P. Evidence for Targeting Thioredoxin Reductases with Ferrocenyl Quinone Methides. A Possible Molecular Basis for the Antiproliferative Effect of Hydroxyferrocifens on Cancer Cells. J. Med. Chem. 2014, 57, 8849–8859. [Google Scholar] [CrossRef]
- Osella, D.; Ferrali, M.; Zanello, P.; Laschi, F.; Fontani, M.; Nervi, C.; Cavigiolio, G. On the Mechanism of the Antitumor Activity of Ferrocenium Derivatives. Inorganica Chim. Acta 2000, 306, 42–48. [Google Scholar] [CrossRef]
- Fatiadi, A.J. Preparation and Synthetic Applications of Cyano Compounds. In The Triple-Bonded Functional Groups; Supplement C; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons, Ltd. eBooks: Hoboken, NJ, USA, 2010; Volume 2, pp. 1057–1303. [Google Scholar]
- Magnus, P.; Scott, D.A.; Fielding, M.R. Direct Conversion of α,β-Unsaturated Nitriles into Cyanohydrins Using Mn(Dpm)3 Catalyst, Dioxygen and Phenylsilane. Tetrahedron Lett. 2001, 42, 4127–4129. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, C.; Tang, S.; Tao, S.; Yuan, M.; Li, R.; Chen, H.; Fu, H.; Zheng, X. Practical Synthesis of (Z)-α,β-Unsaturated Nitriles via a One-Pot Sequential Hydroformylation/Knoevenagel Reaction. J. Org. Chem. 2021, 86, 15413–15422. [Google Scholar] [CrossRef]
- Tan, O.U.; Zengin, M. Insights into the Chemistry and Therapeutic Potential of Acrylonitrile Derivatives. Arch. Der Pharm. 2021, 355, e2100383. [Google Scholar]
- Orellana, E.; Kasinski, A. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio-Protoc. 2016, 6. [Google Scholar] [CrossRef]
- Liang, C.-C.; Park, A.Y.; Guan, J.-L. In Vitro Scratch Assay: A Convenient and Inexpensive Method for Analysis of Cell Migration in Vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef]
- Imrie, C.; Kleyi, P.; Nyamori, V.O.; Gerber, T.I.A.; Levendis, D.C.; Look, J. Further Solvent-Free Reactions of Ferrocenylaldehydes: Synthesis of 1,1′-Ferrocenyldiimines and Ferrocenylacrylonitriles. J. Organomet. Chem. 2007, 692, 3443–3453. [Google Scholar] [CrossRef]
- Chen, F.; Ye, H.-Y. (Z)-3-Ferrocenyl-2-(3-Pyridyl)Acrylonitrile. Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64, m1165. [Google Scholar] [CrossRef]
- Cao, L.-Y.; Ye, H.-Y. (Z)-3-Ferrocenyl-2-phenylacrylonitrile. Struct. Rep. Online 2008, 64, m822. [Google Scholar] [CrossRef]
- Alberghina, G.; Amato, M.E.; Corsaro, A.; Fisichella, S.; Scarlata, G. Kinetic Study of the Base-Catalyzed Reactions of Benzaldehyde and Thiophene-2-Carbaldehyde with Acetonitriles. J. Chem. Society. Perkin Trans. II 1985, 353–356. [Google Scholar] [CrossRef]
- Sørensen, T.; Nielsen, M. Synthesis, UV/Vis Spectra and Electrochemical Characterization of Arylthio and Styryl Substituted Ferrocenes. Open Chem. 2011, 9, 610–618. [Google Scholar] [CrossRef]
- Andrews, D.L. Electromagnetic radiation. In Encyclopedia of Spectroscopy and Spectrometry, 2nd ed.; Elsevier Ltd., University of East Anglia: Norwich, UK, 2010; Volume 1, pp. 451–455. [Google Scholar]
- Chataigner, I.; Panel, C.; Gérard, H.; Piettre, S.R. Sulfonyl vs. Carbonyl Group: Which Is More Electron-Withdrawing? Chem. Commun. 2007, 3288–3290. [Google Scholar] [CrossRef]
- Chu, Z.; Yang, J.; Zheng, W.; Sun, J.; Wang, W.; Qian, H. Recent Advances on Modulation of H2O2 in Tumor Microenvironment for Enhanced Cancer Therapeutic Efficacy. Coord. Chem. Rev. 2023, 481, 215049. [Google Scholar] [CrossRef]
- Lin, B.; Chen, H.; Liang, D.; Lin, W.; Qi, X.; Liu, H.; Deng, X. Acidic PH and High-H2O2 Dual Tumor Microenvironment-Responsive Nanocatalytic Graphene Oxide for Cancer Selective Therapy and Recognition. ACS Appl. Mater. Interfaces 2019, 11, 11157–11166. [Google Scholar] [CrossRef]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Transient Exposure to Hydrogen Peroxide Causes an Increase in Mitochondria-Derived Superoxide as a Result of Sustained Alteration in L-Type Ca2+ Channel Function in the Absence of Apoptosis in Ventricular Myocytes. Circ. Res. 2007, 100, 1036–1044. [Google Scholar] [CrossRef]
- Bockris, J.O.; Oldfield, L.F. The Oxidation-Reduction Reactions of Hydrogen Peroxide at Inert Metal Electrodes and Mercury Cathodes. Trans. Faraday Soc. 1955, 51, 249–259. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J. Multivalent Metal Catalysts in Fenton/Fenton-like Oxidation System: A Critical Review. Chem. Eng. J. 2023, 466, 143147. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The Transcription Factor Snail Controls Epithelial–Mesenchymal Transitions by Repressing E-Cadherin Expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Rigaku, O.D. CrysAlis PRO; Rigaku Oxford Diffraction Ltd.: Yarnton, UK, 2019. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendoza-Morales, W.O.; Rodríguez, E.; González, A.; Ramos, Z.; Acosta-Mercado, J.; Piñero-Cruz, D.M.; Ospina, C.A.; Meléndez, E.; Hernández-O’Farrill, E. Synthesis, Structure, Electrochemistry, and In Vitro Anticancer and Anti-Migratory Activities of (Z)- and (E)-2-Substituted-3-Ferrocene-Acrylonitrile Hybrids and Their Derivatives. Molecules 2025, 30, 2835. https://doi.org/10.3390/molecules30132835
Mendoza-Morales WO, Rodríguez E, González A, Ramos Z, Acosta-Mercado J, Piñero-Cruz DM, Ospina CA, Meléndez E, Hernández-O’Farrill E. Synthesis, Structure, Electrochemistry, and In Vitro Anticancer and Anti-Migratory Activities of (Z)- and (E)-2-Substituted-3-Ferrocene-Acrylonitrile Hybrids and Their Derivatives. Molecules. 2025; 30(13):2835. https://doi.org/10.3390/molecules30132835
Chicago/Turabian StyleMendoza-Morales, William O., Esteban Rodríguez, Aliana González, Zulma Ramos, Jemily Acosta-Mercado, Dalice M. Piñero-Cruz, Claudia A. Ospina, Enrique Meléndez, and Eliud Hernández-O’Farrill. 2025. "Synthesis, Structure, Electrochemistry, and In Vitro Anticancer and Anti-Migratory Activities of (Z)- and (E)-2-Substituted-3-Ferrocene-Acrylonitrile Hybrids and Their Derivatives" Molecules 30, no. 13: 2835. https://doi.org/10.3390/molecules30132835
APA StyleMendoza-Morales, W. O., Rodríguez, E., González, A., Ramos, Z., Acosta-Mercado, J., Piñero-Cruz, D. M., Ospina, C. A., Meléndez, E., & Hernández-O’Farrill, E. (2025). Synthesis, Structure, Electrochemistry, and In Vitro Anticancer and Anti-Migratory Activities of (Z)- and (E)-2-Substituted-3-Ferrocene-Acrylonitrile Hybrids and Their Derivatives. Molecules, 30(13), 2835. https://doi.org/10.3390/molecules30132835