
Ribosomally Synthesized and Post-Translationally Modified Peptides Assembled by ThiF-like Adenylyltransferases: Recent Advances and Future Perspectives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
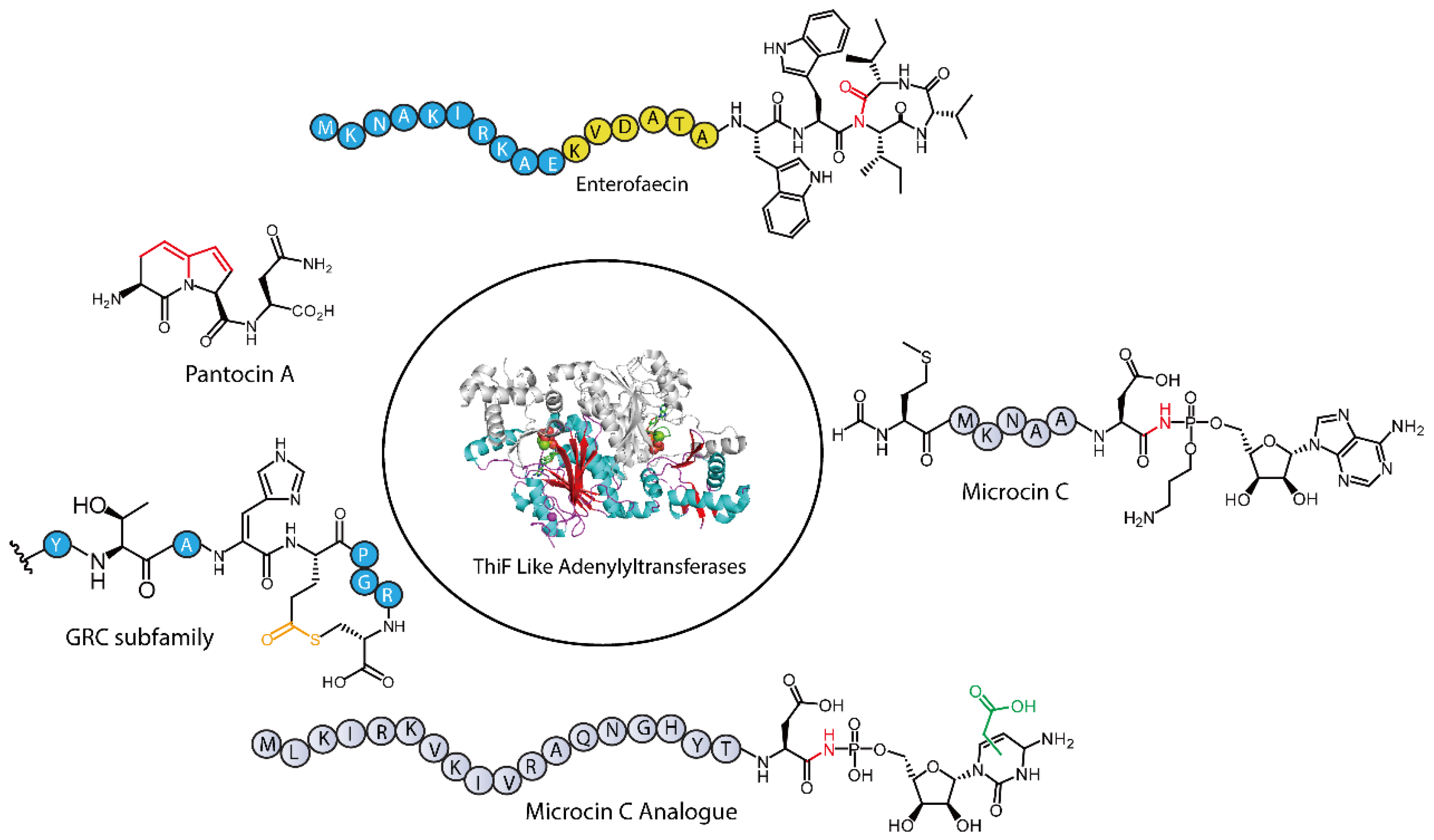
2. TLATs in Microcin C-Type RiPPs Biosynthesis
2.1. Functions of Microcin C-Type RiPPs
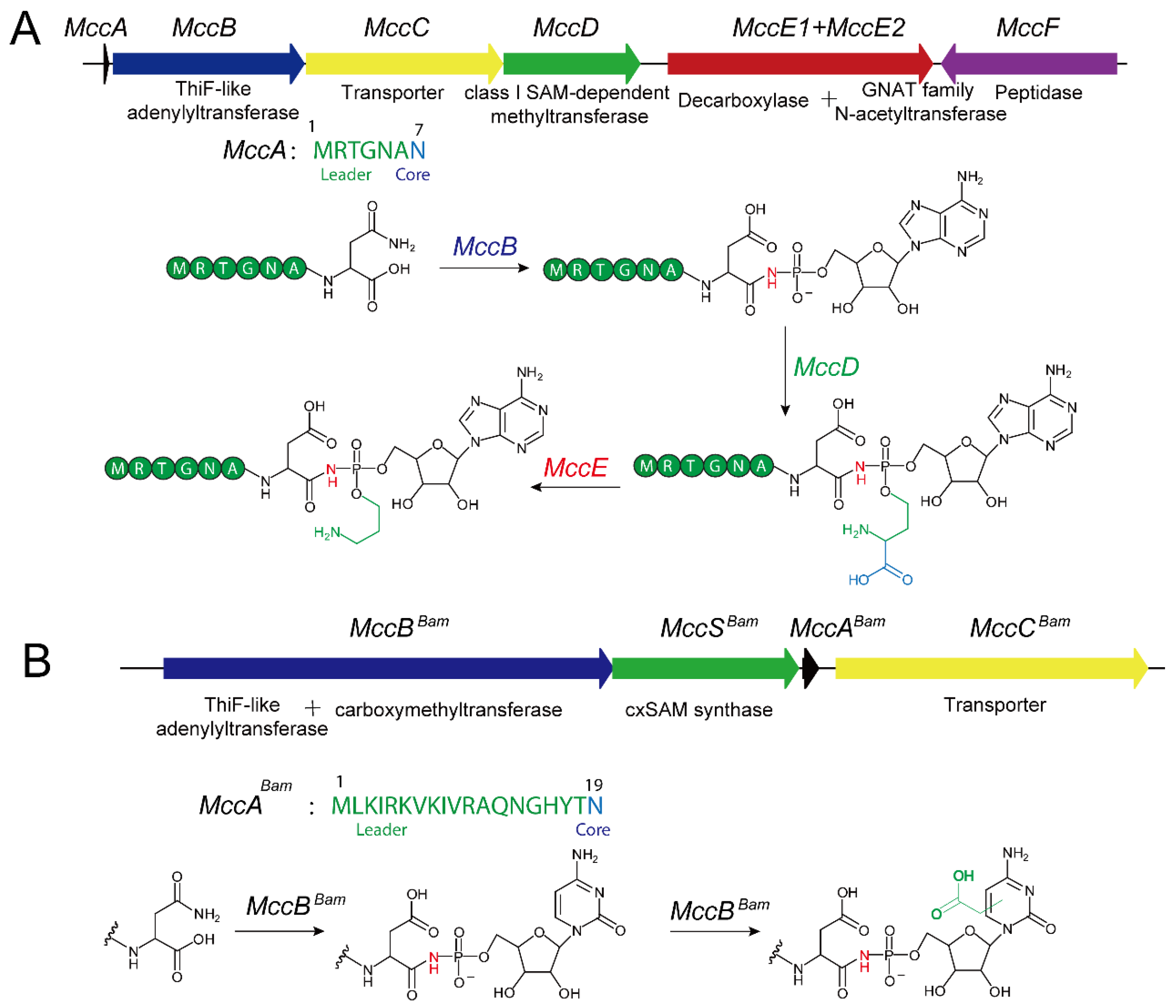
2.2. Molecular Structure and Biosynthetic Pathway of Microcin C-Class Natural Products
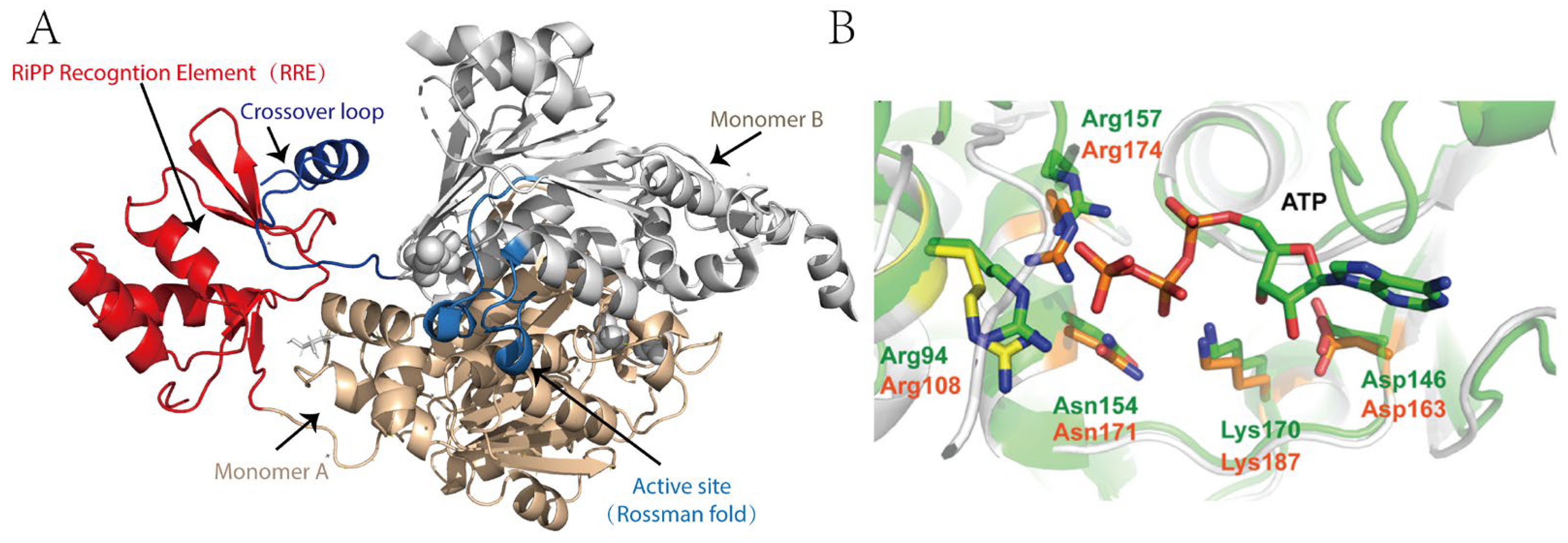
2.3. Structure and Catalytic Mechanism of TLATs in Microcin C-Type RiPPs Biosynthesis
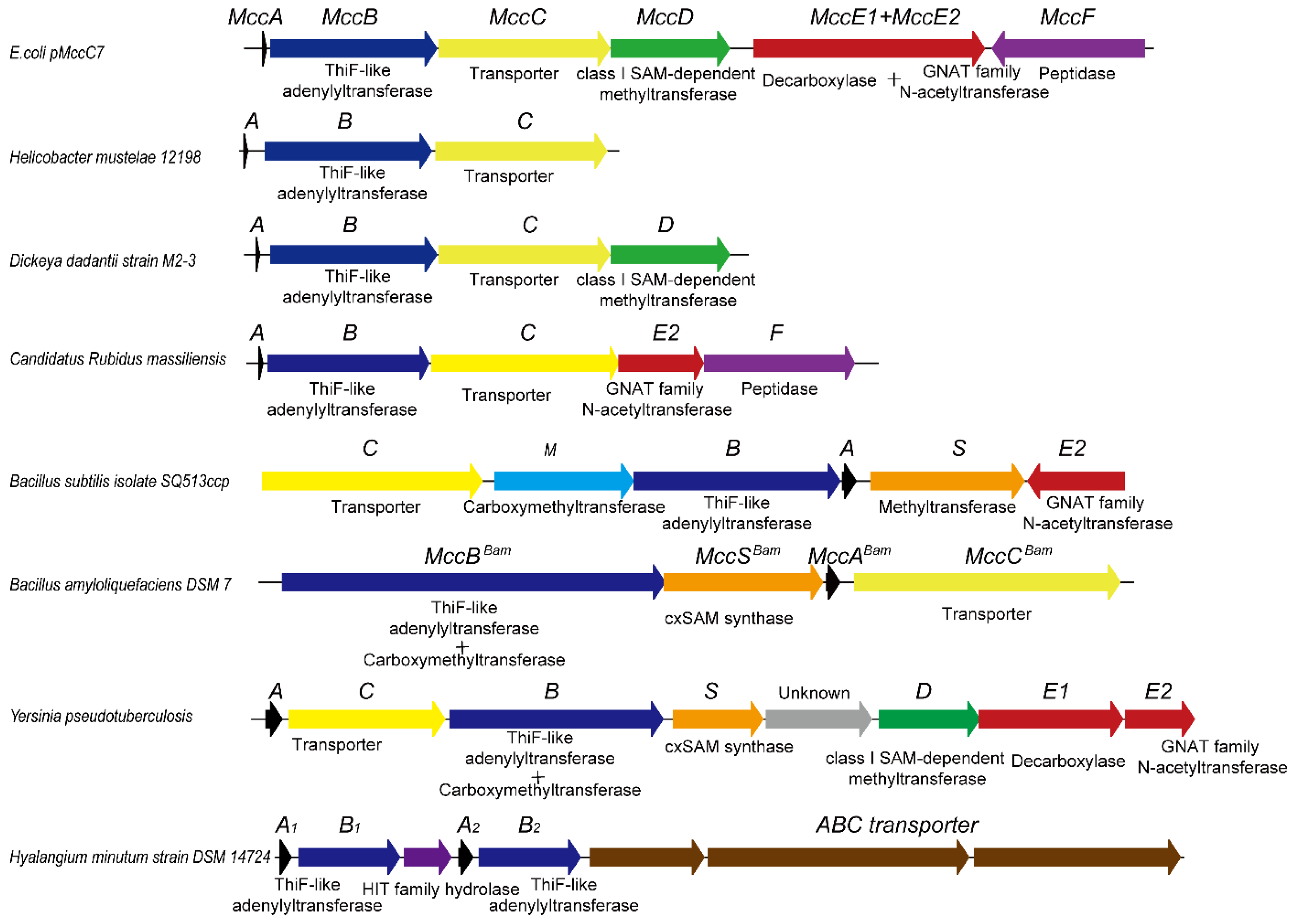
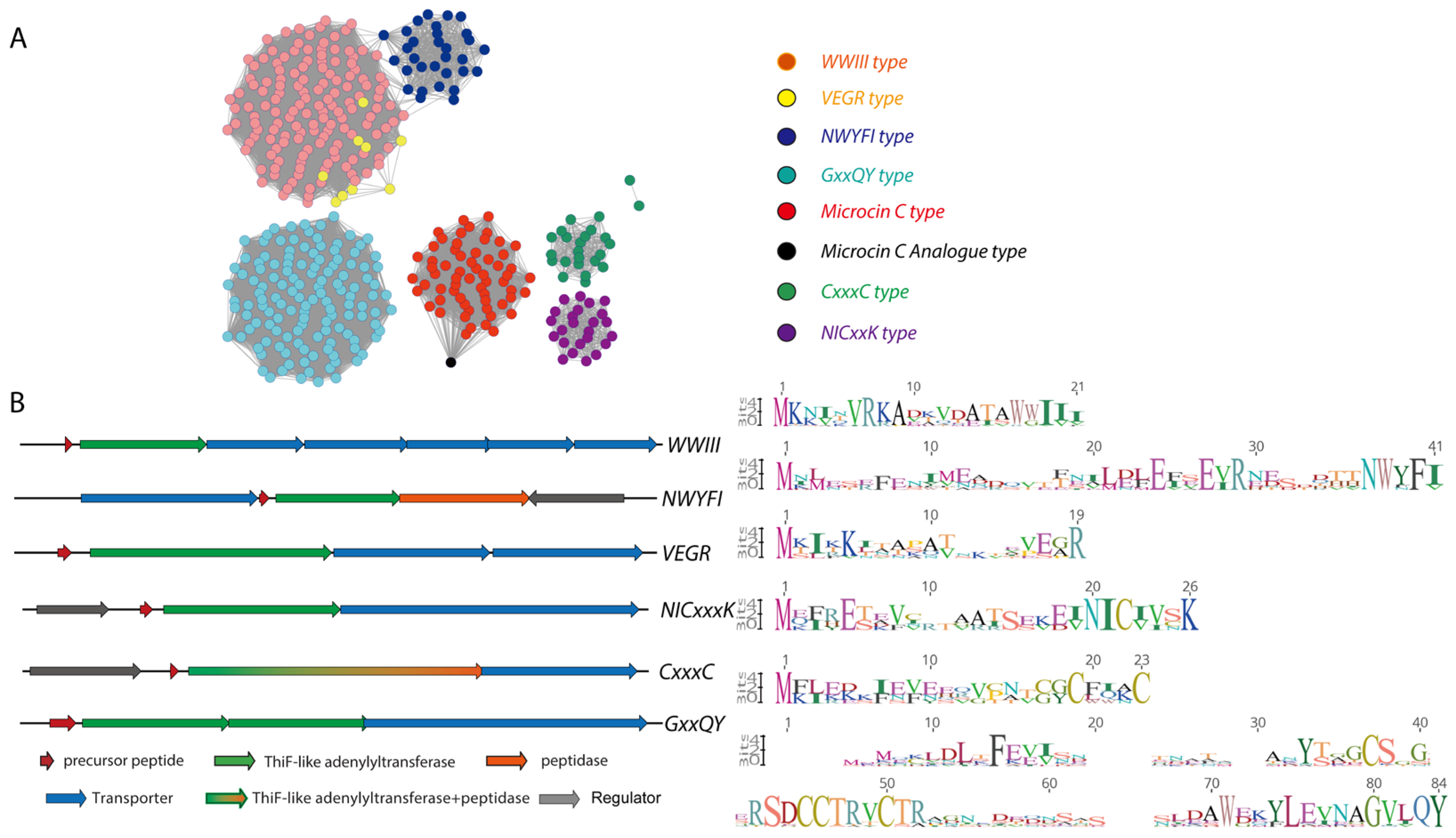
2.4. Genome Mining for Microcin C-Type RiPPs
3. TLATs in Pantocin A-Type RiPPs Biosynthesis
3.1. Biological Function of Pantocin A
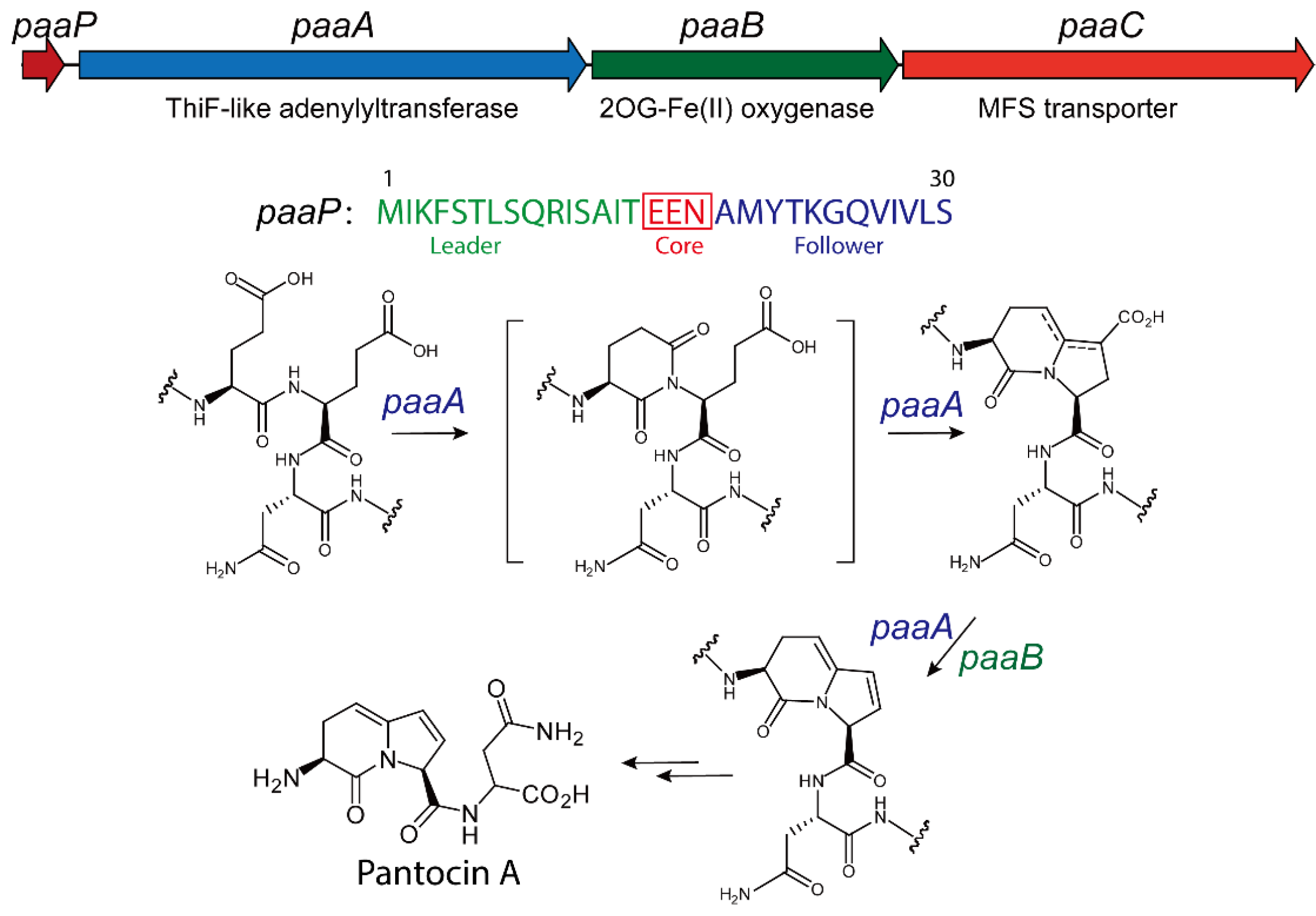
3.2. Biosynthesis of Pantocin A
3.3. Structure and Catalytic Mechanism of TLATs in Pantocin A Biosynthesis
3.4. Genome Mining and Synthetic Biology for PantocinA-Type RiPPs
4. TLATs in GRC-Type RiPPs Biosynthesis
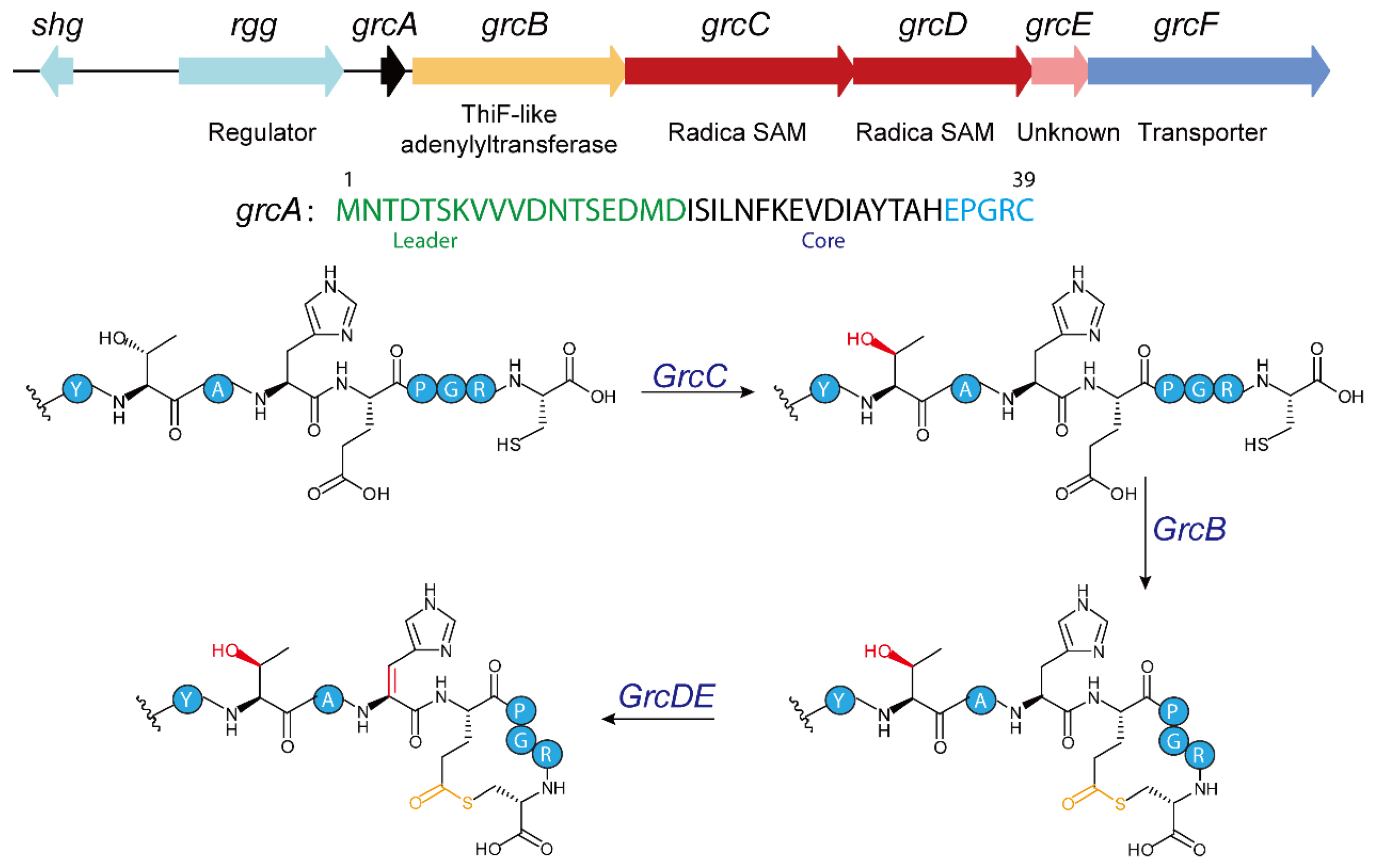
4.1. Biosynthesis of GRC-Type RiPPs
4.2. Genome Mining for GRC-Type RiPPs
5. TLATs in Enterofaecin-Type RiPPs Biosynthesis
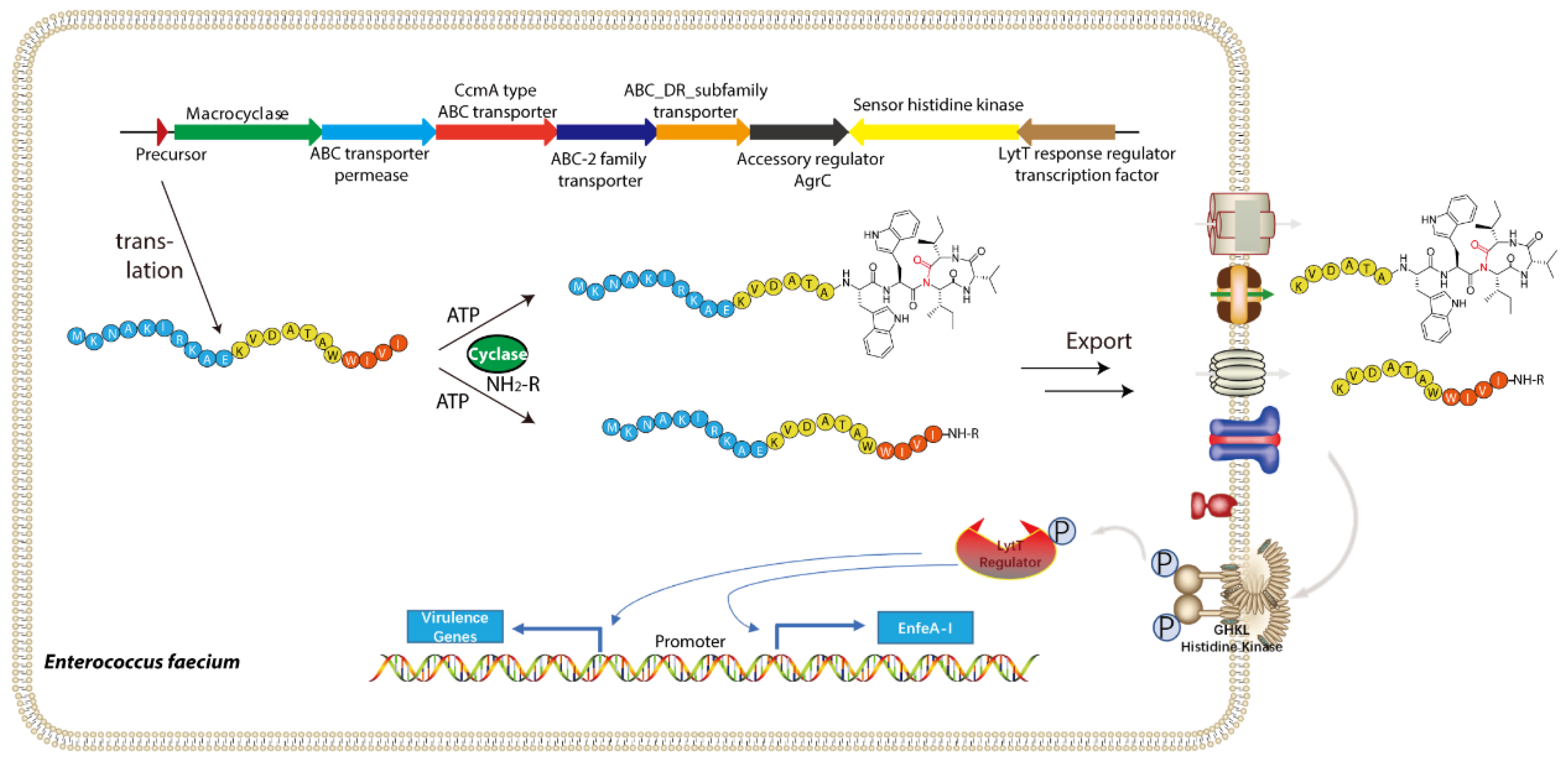
5.1. Biosynthesis of Enterofaecin-Type RiPPs
5.2. Genome Mining for Enterofaecin-Type RiPPs
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Sherry, S.T.; Yankie, L.; Karsch-Mizrachi, I. GenBank 2024 Update. Nucleic Acids Res. 2023, 52, D134–D137. [Google Scholar] [CrossRef] [PubMed]
- Gough, J.; Chothia, C. SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments. Nucleic Acids Res. 2002, 30, 268–272. [Google Scholar] [CrossRef]
- Kaushik, S.; Nair, A.G.; Mutt, E.; Subramanian, H.P.; Sowdhamini, R. Rapid and enhanced remote homology detection by cascading hidden Markov model searches in sequence space. Bioinformatics 2015, 32, 338–344. [Google Scholar] [CrossRef]
- Rutledge, P.J.; Challis, G.L. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol. 2015, 13, 509–523. [Google Scholar] [CrossRef]
- Jensen, P.R. Natural Products and the Gene Cluster Revolution. Trends Microbiol. 2016, 24, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.-M.; Chang, S.-L.; Oakley, B.R.; Wang, C.C.C. Recent advances in awakening silent biosynthetic gene clusters and linking orphan clusters to natural products in microorganisms. Curr. Opin. Chem. Biol. 2011, 15, 137–143. [Google Scholar] [CrossRef]
- Corre, C.; Challis, G.L. New natural product biosynthetic chemistry discovered by genome mining. Nat. Prod. Rep. 2009, 26, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, J.E.; van der Donk, W.A. Genome mining for ribosomally synthesized natural products. Curr. Opin. Chem. Biol. 2011, 15, 11–21. [Google Scholar] [CrossRef]
- Bauman, K.D.; Butler, K.S.; Moore, B.S.; Chekan, J.R. Genome mining methods to discover bioactive natural products. Nat. Prod. Rep. 2021, 38, 2100–2129. [Google Scholar] [CrossRef]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar]
- Ortega, M.A.; van der Donk, W.A. New Insights into the Biosynthetic Logic of Ribosomally Synthesized and Post-translationally Modified Peptide Natural Products. Cell Chem. Biol. 2016, 23, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Finking, R.; Marahiel, M.A. Biosynthesis of Nonribosomal Peptides1. Annu. Rev. Microbiol. 2004, 58, 453–488. [Google Scholar] [CrossRef]
- Hegemann, J.D.; Zimmermann, M.; Xie, X.; Marahiel, M.A. Lasso Peptides: An Intriguing Class of Bacterial Natural Products. Acc. Chem. Res. 2015, 48, 1909–1919. [Google Scholar] [CrossRef]
- Maksimov, M.O.; Pan, S.J.; James Link, A. Lasso peptides: Structure, function, biosynthesis, and engineering. Nat. Prod. Rep. 2012, 29, 996–1006. [Google Scholar] [CrossRef]
- Repka, L.M.; Chekan, J.R.; Nair, S.K.; van der Donk, W.A. Mechanistic Understanding of Lanthipeptide Biosynthetic Enzymes. Chem. Rev. 2017, 117, 5457–5520. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.H.; Truman, A.W. Genome mining strategies for ribosomally synthesised and post-translationally modified peptides. Comput. Struct. Biotechnol. J. 2020, 18, 1838–1851. [Google Scholar] [CrossRef]
- Clark, K.A.; Bushin, L.B.; Seyedsayamdost, M.R. RaS-RiPPs in Streptococci and the Human Microbiome. ACS Bio. Med. Chem. Au. 2022, 2, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.A.; Seyedsayamdost, M.R. Bioinformatic Atlas of Radical SAM Enzyme-Modified RiPP Natural Products Reveals an Isoleucine–Tryptophan Crosslink. J. Am. Chem. Soc. 2022, 144, 17876–17888. [Google Scholar] [CrossRef]
- Johnson, B.A.; Clark, K.A.; Bushin, L.B.; Spolar, C.N.; Seyedsayamdost, M.R. Expanding the Landscape of Noncanonical Amino Acids in RiPP Biosynthesis. J. Am. Chem. Soc. 2024, 146, 3805–3815. [Google Scholar] [CrossRef]
- Bushin, L.B.; Clark, K.A.; Pelczer, I.; Seyedsayamdost, M.R. Charting an Unexplored Streptococcal Biosynthetic Landscape Reveals a Unique Peptide Cyclization Motif. J. Am. Chem. Soc. 2018, 140, 17674–17684. [Google Scholar] [CrossRef]
- He, Y.; Fan, A.; Han, M.; Li, H.; Li, M.; Fan, H.; An, X.; Song, L.; Zhu, S.; Tong, Y. Mammalian Commensal Streptococci Utilize a Rare Family of Class VI Lanthipeptide Synthetases to Synthesize Miniature Lanthipeptide-type Ribosomal Peptide Natural Products. Biochemistry 2023, 62, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, M.; Han, M.; Niu, X.; Fan, A.; Zhu, S.; Tong, Y. Mining the Biosynthetic Landscape of Lactic Acid Bacteria Unearths a New Family of RiPPs Assembled by a Novel Type of ThiF-like Adenylyltransferases. ACS Omega 2024, 9, 30891–30903. [Google Scholar] [CrossRef]
- Park, J.-H.; Dorrestein, P.C.; Zhai, H.; Kinsland, C.; McLafferty, F.W.; Begley, T.P. Biosynthesis of the Thiazole Moiety of Thiamin Pyrophosphate (Vitamin B1). Biochemistry 2003, 42, 12430–12438. [Google Scholar] [CrossRef]
- Lehmann, C.; Begley, T.P.; Ealick, S.E. Structure of the Escherichia coli ThiS−ThiF Complex, a Key Component of the Sulfur Transfer System in Thiamin Biosynthesis. Biochemistry 2006, 45, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Settembre, E.; Begley, T.P.; Ealick, S.E. Structural biology of enzymes of the thiamin biosynthesis pathway. Curr. Opin. Struct. Biol. 2003, 13, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.J.; Wuebbens, M.M.; Rajagopalan, K.V.; Schindelin, H. Crystal structure of molybdopterin synthase and its evolutionary relationship to ubiquitin activation. Nat. Struct. Biol. 2001, 8, 42–46. [Google Scholar] [CrossRef]
- Leimkühler, S.; Wuebbens, M.M.; Rajagopalan, K.V. Characterization of Escherichia coli MoeB and Its Involvement in the Activation of Molybdopterin Synthase for the Biosynthesis of the Molybdenum Cofactor *. J. Biol. Chem. 2001, 276, 34695–34701. [Google Scholar] [CrossRef]
- Regni, C.A.; Roush, R.F.; Miller, D.J.; Nourse, A.; Walsh, C.T.; Schulman, B.A. How the MccB bacterial ancestor of ubiquitin E1 initiates biosynthesis of the microcin C7 antibiotic. EMBO J. 2009, 28, 1953–1964. [Google Scholar] [CrossRef]
- Dong, S.-H.; Kulikovsky, A.; Zukher, I.; Estrada, P.; Dubiley, S.; Severinov, K.; Nair, S.K. Biosynthesis of the RiPP trojan horse nucleotide antibiotic microcin C is directed by the N-formyl of the peptide precursor. Chem. Sci. 2019, 10, 2391–2395. [Google Scholar] [CrossRef]
- Severinov, K.; Nair, S.K. Microcin C: Biosynthesis and Mechanisms of Bacterial Resistance. Future Microbiol. 2012, 7, 281–289. [Google Scholar] [CrossRef]
- Vondenhoff, G.H.M.; Van Aerschot, A. Microcin C: Biosynthesis, Mode of Action, and Potential as a Lead in Antibiotics Development. Nucleosides Nucleotides Nucleic Acids 2011, 30, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Rebuffat, S. Microcins in action: Amazing defence strategies of Enterobacteria. Biochem. Soc. Trans. 2012, 40, 1456–1462. [Google Scholar] [CrossRef]
- Travin, D.Y.; Severinov, K.; Dubiley, S. Natural Trojan horse inhibitors of aminoacyl-tRNA synthetases. RSC Chem. Biol. 2021, 2, 468–485. [Google Scholar] [CrossRef] [PubMed]
- Kurepina, N.E.; Basyuk, E.I.; Metlitskaya, A.Z.; Zaitsev, D.A.; Khmel, I.A. Cloning and mapping of the genetic determinants for microcin C51 production and immunity. Mol. General. Genet. MGG 1993, 241, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Bouanchaud, D.; Martinez-Perez, M.C.; Fernandez, C. Microcin plasmids: A group of extrachromosomal elements coding for low-molecular-weight antibiotics in Escherichia coli. J. Bacteriol. 1978, 135, 342–347. [Google Scholar] [CrossRef]
- Garcia-Bustos, J.F.; Pezzi, N.; Mendez, E. Structure and mode of action of microcin 7, an antibacterial peptide produced by Escherichia coli. Antimicrob. Agents Chemother. 1985, 27, 791–797. [Google Scholar] [CrossRef]
- Piskunova, J.; Maisonneuve, E.; Germain, E.; Gerdes, K.; Severinov, K. Peptide-nucleotide antibiotic Microcin C is a potent inducer of stringent response and persistence in both sensitive and producing cells. Mol. Microbiol. 2017, 104, 463–471. [Google Scholar] [CrossRef]
- Paz-Yepes, J.; Brahamsha, B.; Palenik, B. Role of a Microcin-C-like biosynthetic gene cluster in allelopathic interactions in marine Synechococcus. Proc. Natl. Acad. Sci. USA 2013, 110, 12030–12035. [Google Scholar] [CrossRef]
- Cursino, L.; Šmajs, D.; Šmarda, J.; Nardi, R.M.D.; Nicoli, J.R.; Chartone-Souza, E.; Nascimento, A.M.A. Exoproducts of the Escherichia coli strain H22 inhibiting some enteric pathogens both in vitro and in vivo. J. Appl. Microbiol. 2006, 100, 821–829. [Google Scholar] [CrossRef]
- Dai, Z.; Shang, L.; Wang, F.; Zeng, X.; Yu, H.; Liu, L.; Zhou, J.; Qiao, S. Effects of Antimicrobial Peptide Microcin C7 on Growth Performance, Immune and Intestinal Barrier Functions, and Cecal Microbiota of Broilers. Front. Vet. Sci. 2022, 8, 813629. [Google Scholar] [CrossRef]
- Yang, G.; Shang, L.; Liu, L.; Li, Z.; Zeng, X.; Ding, X.; Huang, J.; Qiao, S.; Yu, H. Engineering and Purification of Microcin C7 Variants Resistant to Trypsin and Analysis of Their Biological Activity. Antibiotics 2023, 12, 1346. [Google Scholar] [CrossRef]
- Yang, F.; Yang, F.; Huang, J.; Yu, H.; Qiao, S. Microcin C7 as a Potential Antibacterial-Immunomodulatory Agent in the Postantibiotic Era: Overview of Its Bioactivity Aspects Applications. Int. J. Mol. Sci. 2024, 25, 7213. [Google Scholar] [CrossRef] [PubMed]
- Vondenhoff, G.H.M.; Dubiley, S.; Severinov, K.; Lescrinier, E.; Rozenski, J.; Van Aerschot, A. Extended targeting potential and improved synthesis of Microcin C analogs as antibacterials. Bioorg. Med. Chem. 2011, 19, 5462–5467. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, J.I.; González-Pastor, J.E.; Baleux, F.; Millán, J.L.S.; Castilla, M.A.; Rico, M.; Moreno, F.; Delepierre, M. Chemical Structure and Translation Inhibition Studies of the Antibiotic Microcin C7 *. J. Biol. Chem. 1995, 270, 23520–23532. [Google Scholar] [CrossRef] [PubMed]
- Metlitskaya, A.; Kazakov, T.; Kommer, A.; Pavlova, O.; Praetorius-Ibba, M.; Ibba, M.; Krasheninnikov, I.; Kolb, V.; Khmel, I.; Severinov, K. Aspartyl-tRNASynthetase Is the Target of Peptide Nucleotide Antibiotic Microcin C *. J. Biol. Chem. 2006, 281, 18033–18042. [Google Scholar] [CrossRef]
- Vondenhoff, G.H.M.; Blanchaert, B.; Geboers, S.; Kazakov, T.; Datsenko, K.A.; Wanner, B.L.; Rozenski, J.; Severinov, K.; Aerschot, A.V. Characterization of Peptide Chain Length and Constituency Requirements for YejABEF-Mediated Uptake of Microcin C Analogues. J. Bacteriol. 2011, 193, 3618–3623. [Google Scholar] [CrossRef]
- Novikova, M.; Metlitskaya, A.; Datsenko, K.; Kazakov, T.; Kazakov, A.; Wanner, B.; Severinov, K. The Escherichia coli Yej Transporter Is Required for the Uptake of Translation Inhibitor Microcin C. J. Bacteriol. 2007, 189, 8361–8365. [Google Scholar] [CrossRef]
- Ran, R.; Zeng, H.; Zhao, D.; Liu, R.; Xu, X. The Novel Property of Heptapeptide of Microcin C7 in Affecting the Cell Growth of Escherichia coli. Molecules 2017, 22, 432. [Google Scholar] [CrossRef]
- Kazakov, T.; Vondenhoff, G.H.; Datsenko, K.A.; Novikova, M.; Metlitskaya, A.; Wanner, B.L.; Severinov, K. Escherichia coli Peptidase A, B, or N Can Process Translation Inhibitor Microcin C. J. Bacteriol. 2008, 190, 2607–2610. [Google Scholar] [CrossRef]
- González-Pastor, J.E.; Millán, J.L.S.; Castilla, M.A.; Moreno, F. Structure and organization of plasmid genes required to produce the translation inhibitor microcin C7. J. Bacteriol. 1995, 177, 7131–7140. [Google Scholar] [CrossRef]
- Metlitskaya, A.; Kazakov, T.; Vondenhoff, G.H.; Novikova, M.; Shashkov, A.; Zatsepin, T.; Semenova, E.; Zaitseva, N.; Ramensky, V.; Aerschot, A.V.; et al. Maturation of the Translation Inhibitor Microcin C. J. Bacteriol. 2009, 191, 2380–2387. [Google Scholar] [CrossRef] [PubMed]
- Kulikovsky, A.; Serebryakova, M.; Bantysh, O.; Metlitskaya, A.; Borukhov, S.; Severinov, K.; Dubiley, S. The Molecular Mechanism of Aminopropylation of Peptide-Nucleotide Antibiotic Microcin C. J. Am. Chem. Soc. 2014, 136, 11168–11175. [Google Scholar] [CrossRef] [PubMed]
- Yagmurov, E.; Tsibulskaya, D.; Livenskyi, A.; Serebryakova, M.; Wolf, Y.I.; Borukhov, S.; Severinov, K.; Dubiley, S. Histidine-Triad Hydrolases Provide Resistance to Peptide-Nucleotide Antibiotics. mBio 2020, 11, e00497-20. [Google Scholar] [CrossRef] [PubMed]
- Serebryakova, M.; Tsibulskaya, D.; Mokina, O.; Kulikovsky, A.; Nautiyal, M.; Van Aerschot, A.; Severinov, K.; Dubiley, S. A Trojan-Horse Peptide-Carboxymethyl-Cytidine Antibiotic from Bacillus amyloliquefaciens. J. Am. Chem. Soc. 2016, 138, 15690–15698. [Google Scholar] [CrossRef]
- Kazakov, T.; Kuznedelov, K.; Semenova, E.; Mukhamedyarov, D.; Datsenko, K.A.; Metlitskaya, A.; Vondenhoff, G.H.; Tikhonov, A.; Agarwal, V.; Nair, S.; et al. The RimL Transacetylase Provides Resistance to Translation Inhibitor Microcin C. J. Bacteriol. 2014, 196, 3377–3385. [Google Scholar] [CrossRef] [PubMed]
- Novikova, M.; Kazakov, T.; Vondenhoff, G.H.; Semenova, E.; Rozenski, J.; Metlytskaya, A.; Zukher, I.; Tikhonov, A.; Van Aerschot, A.; Severinov, K. MccE Provides Resistance to Protein Synthesis Inhibitor Microcin C by Acetylating the Processed Form of the Antibiotic *. J. Biol. Chem. 2010, 285, 12662–12669. [Google Scholar] [CrossRef]
- Nocek, B.; Tikhonov, A.; Babnigg, G.; Gu, M.; Zhou, M.; Makarova, K.S.; Vondenhoff, G.; Aerschot, A.V.; Kwon, K.; Anderson, W.F.; et al. Structural and Functional Characterization of Microcin C Resistance Peptidase MccF from Bacillus anthracis. J. Mol. Biol. 2012, 420, 366–383. [Google Scholar] [CrossRef]
- Tikhonov, A.; Kazakov, T.; Semenova, E.; Serebryakova, M.; Vondenhoff, G.; Van Aerschot, A.; Reader, J.S.; Govorun, V.M.; Severinov, K. The Mechanism of Microcin C Resistance Provided by the MccF Peptidase *. J. Biol. Chem. 2010, 285, 37944–37952. [Google Scholar] [CrossRef]
- Roush, R.F.; Nolan, E.M.; Löhr, F.; Walsh, C.T. Maturation of an Escherichia coli Ribosomal Peptide Antibiotic by ATP-Consuming N−P Bond Formation in Microcin C7. J. Am. Chem. Soc. 2008, 130, 3603–3609. [Google Scholar] [CrossRef]
- Kazakov, T.; Metlitskaya, A.; Severinov, K. Amino Acid Residues Required for Maturation Cell Uptake Processing of Translation Inhibitor Microcin C. J. Bacteriol. 2007, 189, 2114–2118. [Google Scholar] [CrossRef]
- Bantysh, O.; Serebryakova, M.; Makarova, K.S.; Dubiley, S.; Datsenko, K.A.; Severinov, K. Enzymatic Synthesis of Bioinformatically Predicted Microcin C-like Compounds Encoded by Diverse Bacteria. mBio 2014, 5, e01059-14. [Google Scholar] [CrossRef]
- Tsibulskaya, D.; Mokina, O.; Kulikovsky, A.; Piskunova, J.; Severinov, K.; Serebryakova, M.; Dubiley, S. The Product of Yersinia pseudotuberculosis mcc Operon Is a Peptide-Cytidine Antibiotic Activated Inside Producing Cells by the TldD/E Protease. J. Am. Chem. Soc. 2017, 139, 16178–16187. [Google Scholar] [CrossRef] [PubMed]
- Francés, J.; Bonaterra, A.; Moreno, M.C.; Cabrefiga, J.; Badosa, E.; Montesinos, E. Pathogen aggressiveness and postharvest biocontrol efficiency in Pantoea agglomerans. Postharvest Biol. Technol. 2006, 39, 299–307. [Google Scholar] [CrossRef]
- Costa, E.; Teixidó, N.; Usall, J.; Atarés, E.; Viñas, I. Production of the biocontrol agent Pantoea agglomerans strain CPA-2 using commercial products and by-products. Appl. Microbiol. Biotechnol. 2001, 56, 367–371. [Google Scholar] [CrossRef]
- Pusey, P.L.; Stockwell, V.O.; Reardon, C.L.; Smits, T.H.M.; Duffy, B. Antibiosis Activity of Pantoea agglomerans Biocontrol Strain E325 Against Erwinia amylovora on Apple Flower Stigmas. Phytopathology 2011, 101, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Jeong, J.C.; Lee, J.-S.; Park, J.M.; Yang, J.-W.; Lee, M.H.; Choi, S.H.; Kim, C.Y.; Kim, D.-H.; Kim, S.W.; et al. Potential of Pantoea dispersa as an effective biocontrol agent for black rot in sweet potato. Sci. Rep. 2019, 9, 16354. [Google Scholar] [CrossRef] [PubMed]
- Smits, T.H.M.; Duffy, B.; Blom, J.; Ishimaru, C.A.; Stockwell, V.O. Pantocin A, a peptide-derived antibiotic involved in biological control by plant-associated Pantoea species. Arch. Microbiol. 2019, 201, 713–722. [Google Scholar] [CrossRef]
- Giddens, S.R.; Feng, Y.; Mahanty, H.K. Characterization of a novel phenazine antibiotic gene cluster in Erwinia herbicola Eh1087. Mol. Microbiol. 2002, 45, 769–783. [Google Scholar] [CrossRef]
- Giddens, S.R.; Houliston, G.J.; Mahanty, H.K. The influence of antibiotic production and pre-emptive colonization on the population dynamics of Pantoea agglomerans (Erwinia herbicola) Eh1087 and Erwinia amylovora in planta. Environ. Microbiol. 2003, 5, 1016–1021. [Google Scholar] [CrossRef]
- Kamber, T.; Lansdell, T.A.; Stockwell, V.O.; Ishimaru, C.A.; Smits, T.H.M.; Duffy, B. Characterization of the Biosynthetic Operon for the Antibacterial Peptide Herbicolin in Pantoea vagans Biocontrol Strain C9-1 and Incidence in Pantoea Species. Appl. Environ. Microbiol. 2012, 78, 4412–4419. [Google Scholar] [CrossRef]
- Ishimaru, C.A.; Lansdell, T.A.; Clardy, J.; Duffy, B.; Smits, T.H.M. The Histidine-Reversible Antibiotic Herbicolin O Produced by Pantoea Vagans C9-1 Is Pantocin A. J. Plant Pathol. 2017, 99, 91–97. [Google Scholar]
- Smits, T.H.M.; Rezzonico, F.; Kamber, T.; Blom, J.; Goesmann, A.; Ishimaru, C.A.; Frey, J.E.; Stockwell, V.O.; Duffy, B. Metabolic Versatility and Antibacterial Metabolite Biosynthesis Are Distinguishing Genomic Features of the Fire Blight Antagonist Pantoea vagans C9-1. PLoS ONE 2011, 6, e22247. [Google Scholar] [CrossRef]
- Wright, S.A.I.; Zumoff, C.H.; Schneider, L.; Beer, S.V. Pantoea agglomerans Strain EH318 Produces Two Antibiotics That Inhibit Erwinia amylovora In Vitro. Appl. Environ. Microbiol. 2001, 67, 284–292. [Google Scholar] [CrossRef]
- Wodzinski, R.S.; Paulin, J.-P. Frequency and diversity of antibiotic production by putative Erwinia herbicola strains. J. Appl. Bacteriol. 1994, 76, 603–607. [Google Scholar] [CrossRef]
- Jin, M.; Liu, L.; Wright, S.A.I.; Beer, S.V.; Clardy, J. Structural and Functional Analysis of Pantocin A: An Antibiotic from Pantoea agglomerans Discovered by Heterologous Expression of Cloned Genes. Angew. Chem. 2003, 42, 2898–2901. [Google Scholar] [CrossRef]
- Rezzonico, F.; Smits, T.H.M.; Montesinos, E.; Frey, J.E.; Duffy, B. Genotypic comparison of Pantoea agglomeransplant and clinical strains. BMC Microbiol. 2009, 9, 204. [Google Scholar] [CrossRef]
- Davis, L.; Ishimaru, C. Cloning and expression of herbicolin O biosynthesis genes in Escherichia coli. Phytopathology 1993, 83, 1339. [Google Scholar]
- Vanneste, J.; Yu, J.; Cornish, D. Presence of Genes Homologous to Those Necessary for Synthesis of Microcin MccEh252 in Strains of Pantoea agglomerans; Johnson, K.B., Leuven, V.O.S., Eds.; International Society for Horticultural Science (ISHS): Leuven, Belgium, 2008; pp. 391–396. [Google Scholar]
- Jin, M.; Wright, S.A.I.; Beer, S.V.; Clardy, J. The Biosynthetic Gene Cluster of Pantocin A Provides Insights into Biosynthesis and a Tool for Screening. Angew. Chem. Int. Ed. 2003, 42, 2902–2905. [Google Scholar] [CrossRef] [PubMed]
- Ghodge, S.V.; Biernat, K.A.; Bassett, S.J.; Redinbo, M.R.; Bowers, A.A. Post-translational Claisen Condensation and Decarboxylation en Route to the Bicyclic Core of Pantocin A. J. Am. Chem. Soc. 2016, 138, 5487–5490. [Google Scholar] [CrossRef]
- Fleming, S.R.; Himes, P.M.; Ghodge, S.V.; Goto, Y.; Suga, H.; Bowers, A.A. Exploring the Post-translational Enzymology of PaaA by mRNA Display. J. Am. Chem. Soc. 2020, 142, 5024–5028. [Google Scholar] [CrossRef]
- Glassey, E.; Zhang, Z.; King, A.M.; Niquille, D.L.; Voigt, C.A. De novo design of ribosomally synthesized and post-translationally modified peptides. Nat. Chem. 2025, 17, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Morinaka, B.I. Accessing and exploring the unusual chemistry by radical SAM-RiPP enzymes. Curr. Opin. Chem. Biol. 2024, 81, 102483. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.H.; Haedar, J.R.; Kiselov, V.; Romanuks, V.; Smits, G.; Donadio, S.; Phan, C.-S. Radical SAM Enzyme WprB Catalyzes Uniform Cross-Link Topology between Trp-C5 and Arg-Cγ on the Precursor Peptide. ACS Chem. Biol. 2025, 20, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Thoendel, M.; Kavanaugh, J.S.; Flack, C.E.; Horswill, A.R. Peptide Signaling in the Staphylococci. Chem. Rev. 2011, 111, 117–151. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V. Do symbiotic bacteria subvert host immunity? Nat. Rev. Microbiol. 2009, 7, 367–374. [Google Scholar] [CrossRef]
- Ng, W.-L.; Bassler, B.L. Bacterial Quorum-Sensing Network Architectures. Annu. Rev. Genet. 2009, 43, 197–222. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, S.; Liu, Y.; Wang, H.; Sun, J.; Yao, J.; Huang, H. Ribosomally Synthesized and Post-Translationally Modified Peptides Assembled by ThiF-like Adenylyltransferases: Recent Advances and Future Perspectives. Molecules 2025, 30, 2821. https://doi.org/10.3390/molecules30132821
Zhu S, Liu Y, Wang H, Sun J, Yao J, Huang H. Ribosomally Synthesized and Post-Translationally Modified Peptides Assembled by ThiF-like Adenylyltransferases: Recent Advances and Future Perspectives. Molecules. 2025; 30(13):2821. https://doi.org/10.3390/molecules30132821
Chicago/Turabian StyleZhu, Shaozhou, Yan Liu, Hang Wang, Jiabei Sun, Jing Yao, and Haiwei Huang. 2025. "Ribosomally Synthesized and Post-Translationally Modified Peptides Assembled by ThiF-like Adenylyltransferases: Recent Advances and Future Perspectives" Molecules 30, no. 13: 2821. https://doi.org/10.3390/molecules30132821
APA StyleZhu, S., Liu, Y., Wang, H., Sun, J., Yao, J., & Huang, H. (2025). Ribosomally Synthesized and Post-Translationally Modified Peptides Assembled by ThiF-like Adenylyltransferases: Recent Advances and Future Perspectives. Molecules, 30(13), 2821. https://doi.org/10.3390/molecules30132821