
Inversion Motion of Xanthene and Detection of Its Oxidation Product Xanthone from Gas-Phase Rotational Spectroscopy


Abstract

1. Introduction
2. Results and Discussion
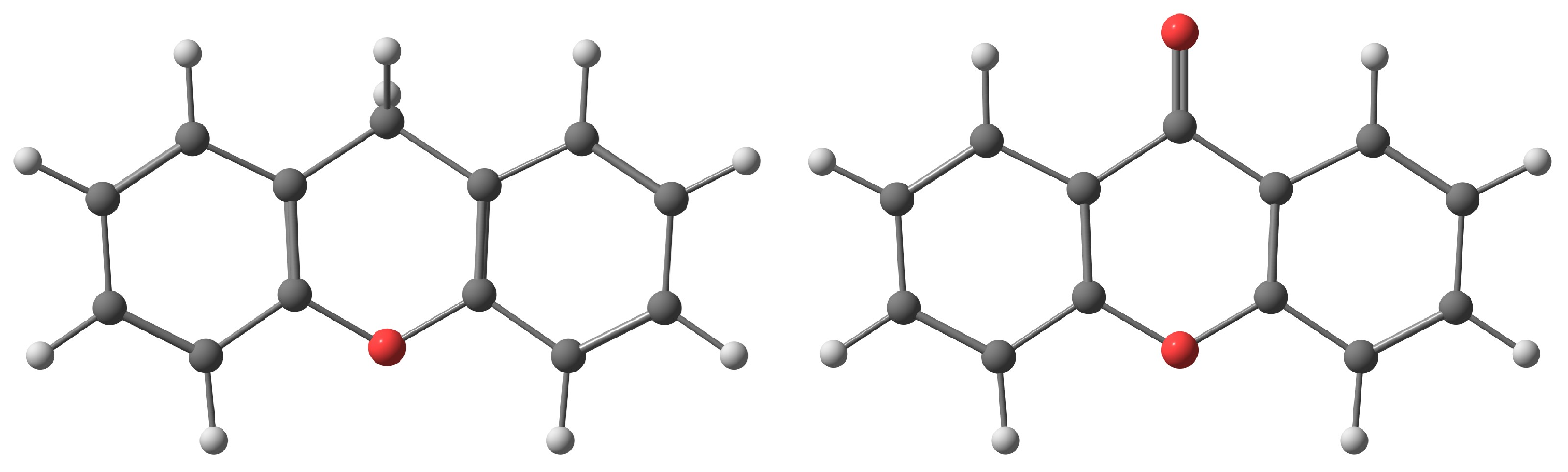
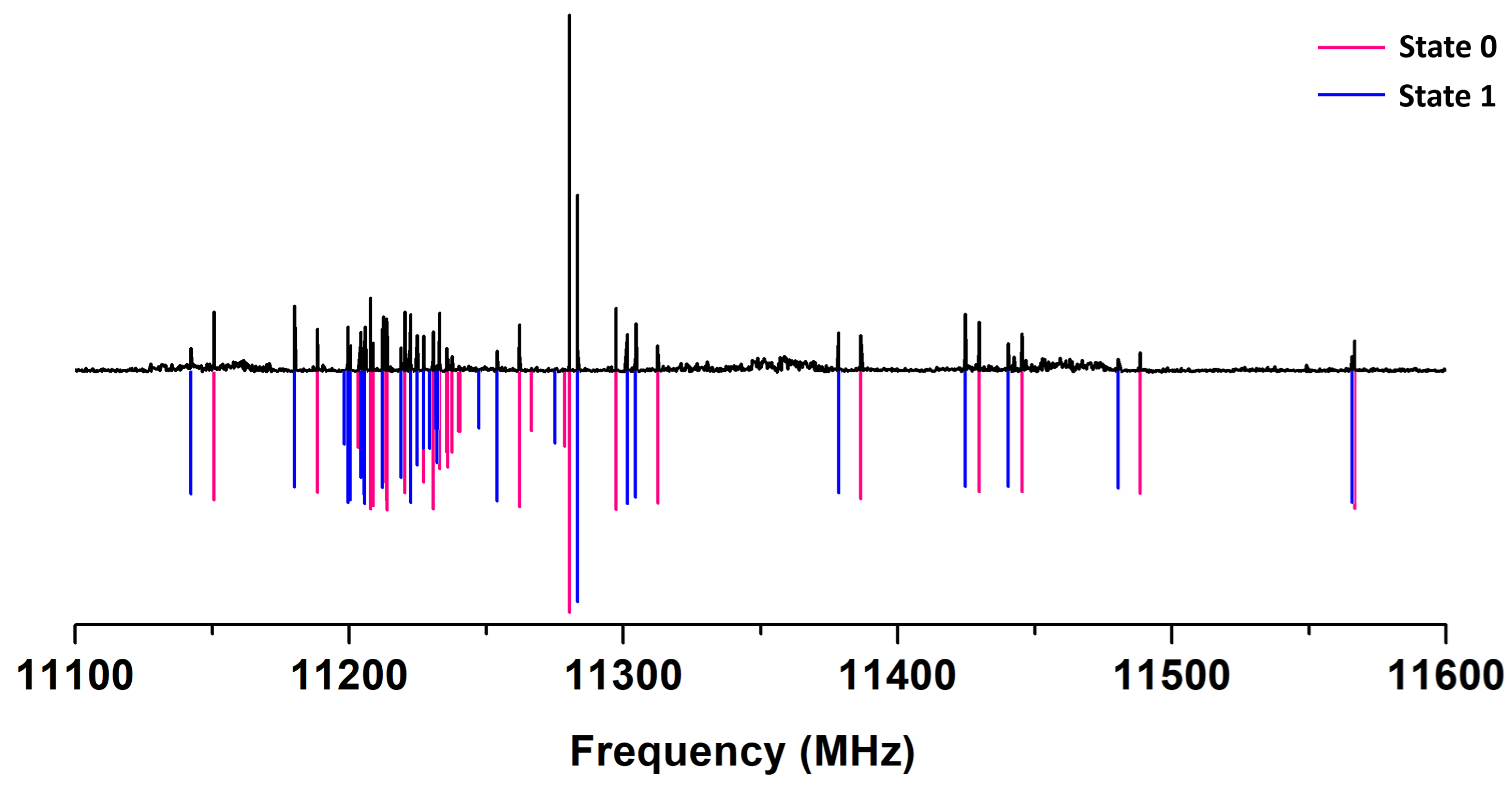
2.1. Xanthene Analysis
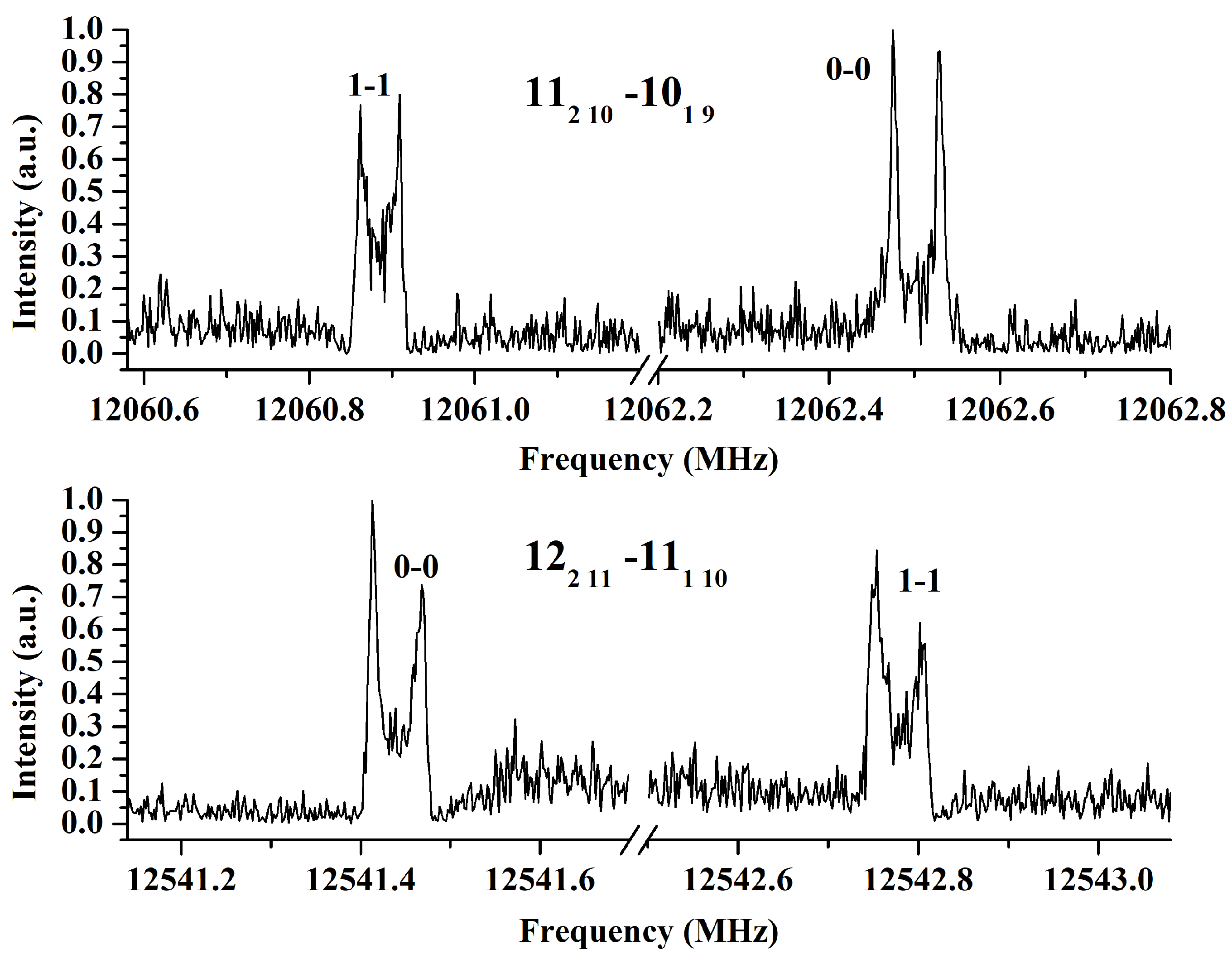
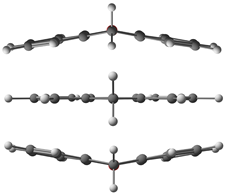
2.2. Barrier to Rings Inversion
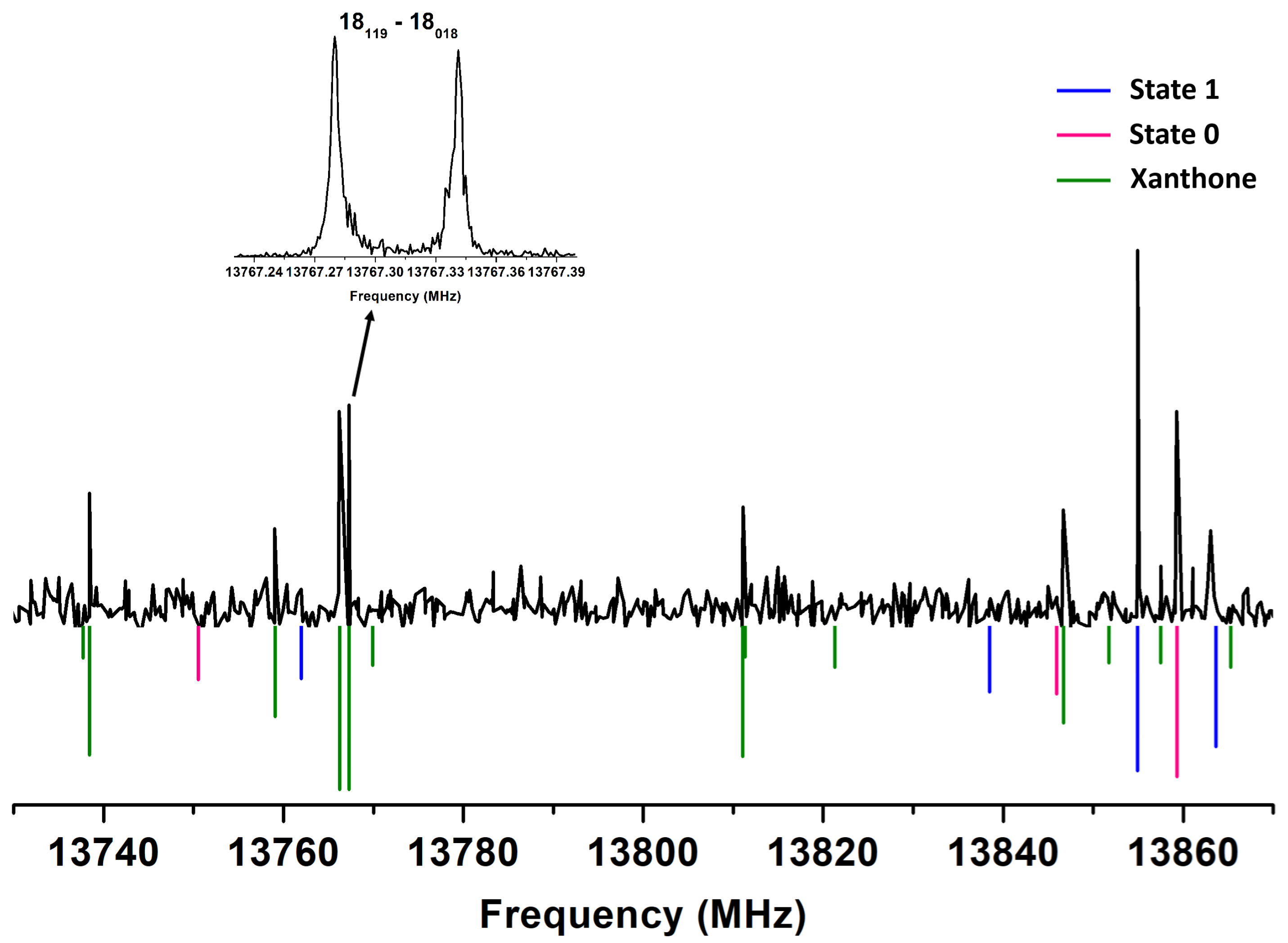
2.3. Xanthone Analysis
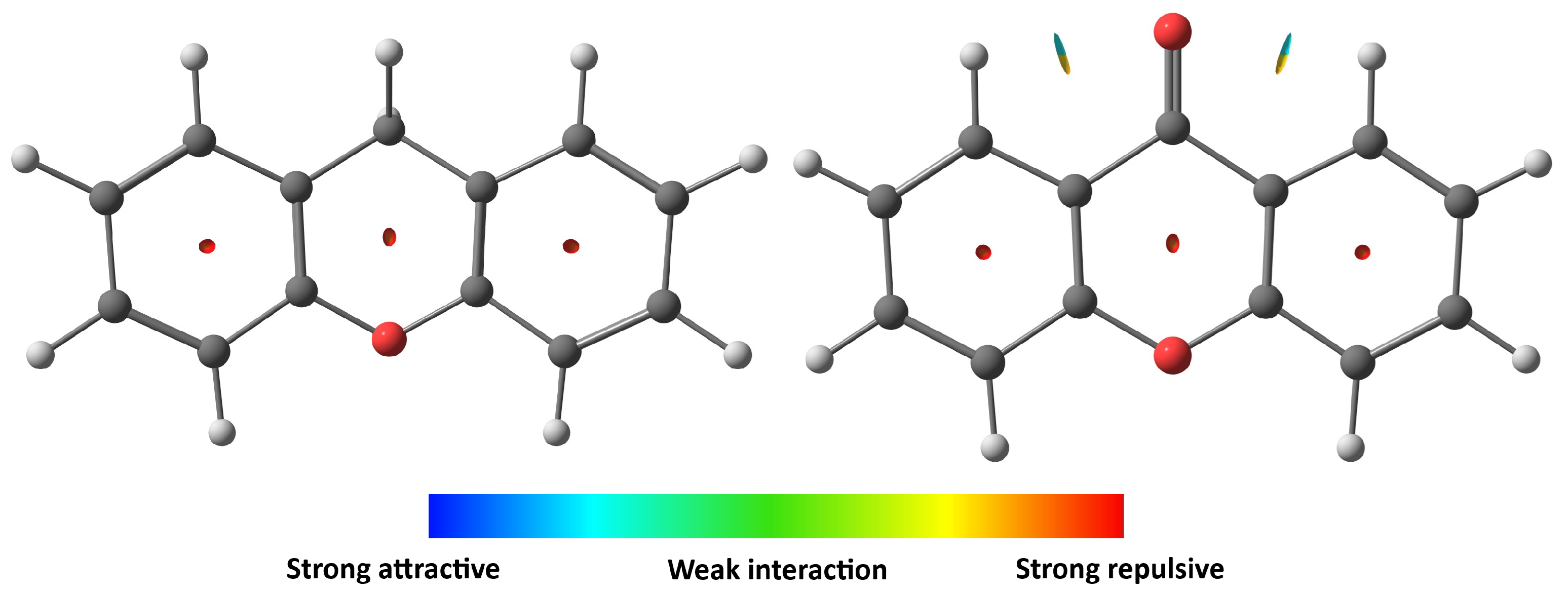
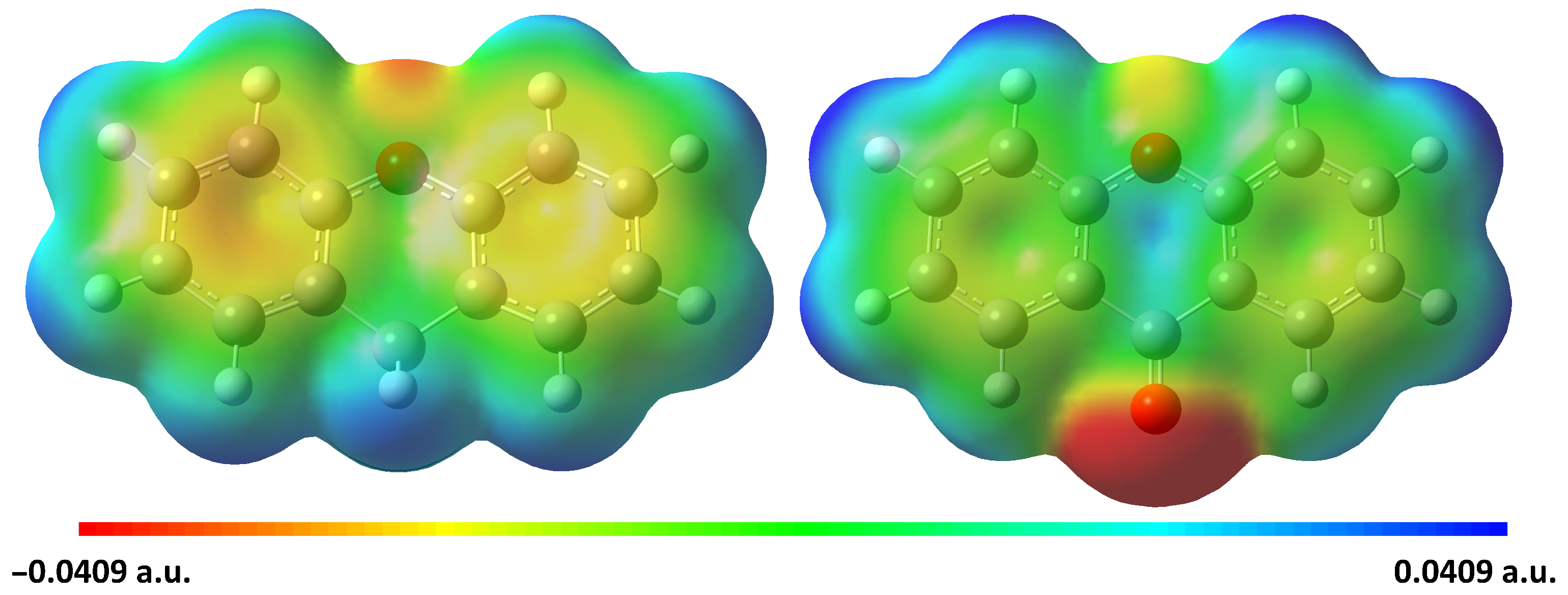
2.4. Structural Investigation
3. Materials and Methods
3.1. Theoretical Calculations
3.2. Experimental
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PAH | polycyclic aromatic hydrocarbons |
| UIB | unidentified infrared emission bands |
| ISM | interstellar medium |
| CP-FTMW | chirped pulsed Fourier transform microwave |
| FP-FTMW | Fabry–Perot Fourier transform microwave |
| DFT | density functional theory |
| RMS | root mean square |
| NCI | non-covalent interaction |
| MEP | molecular electrostatic potential |
References
- Hites, R. Polycyclic Aromatic Hydrocarbons in the Atmosphere near the Great Lakes: Why Do Their Concentrations Vary? Environ. Sci. Technol. 2021, 55, 9444–9449. [Google Scholar] [CrossRef] [PubMed]
- Wietzoreck, M.; Kyprianou, M.; Musa Bandowe, B.; Celik, S.; Crowley, J.; Drewnick, F.; Eger, P.; Friedrich, N.; Iakovides, M.; Kukucka, P.; et al. Polycyclic Aromatic Hydrocarbons (PAHs) and Their Alkylated, Nitrated and Oxygenated Derivatives in the Atmosphere over the Mediterranean and Middle East Seas. Atmos. Chem. Phys. 2022, 22, 8739–8766. [Google Scholar] [CrossRef]
- Tielens, A. Interstellar Polycyclic Aromatic Hydrocarbon Molecules. Annu. Rev. Astron. Astrophys. 2008, 46, 289–337. [Google Scholar] [CrossRef]
- Cernicharo, J.; Heras, A.; Tielens, A.; Pardo, J.; Herpin, F.; Guélin, M.; Waters, L. Infrared Space Observatory’s Discovery of C4H2, C6H2, and Benzene in CRL 618. Astrophys. J. 2001, 546, L123. [Google Scholar] [CrossRef]
- Pérez, C.; Steber, A.L.; Rijs, A.M.; Temelso, B.; Shields, G.C.; Lopez, J.C.; Kisiel, Z.; Schnell, M. Corannulene and its complex with water: A tiny cup of water. Phys. Chem. Chem. Phys. 2017, 19, 14214–14223. [Google Scholar] [CrossRef]
- Neeman, E.M.; Lesarri, A.; Bermúdez, C. Gas-Phase Conformational Landscape and Ring-Puckered Structure of 1-Aminoindane. ChemPhysChem 2025, 26, e202401012. [Google Scholar] [CrossRef]
- Maris, A.; Calabrese, C.; Melandri, S.; Blanco, S. Accurate spectroscopy of polycyclic aromatic compounds: From the rotational spectrum of fluoren-9-one in the millimeter wave region to its infrared spectrum. J. Chem. Phys. 2015, 142, 024317. [Google Scholar] [CrossRef]
- Burkhardt, A.; Lee, K.; Changala, P.; Shingledecker, C.; Cooke, I.; Loomis, R.; Wei, H.; Charnley, S.; Herbst, E.; McCarthy, M.; et al. Discovery of the Pure Polycyclic Aromatic Hydrocarbon Indene (c-C9H8) with GOTHAM Observations of TMC-1. Astrophys. J. Lett. 2021, 913, L18. [Google Scholar] [CrossRef]
- Cernicharo, J.; Agúndez, M.; Cabezas, C.; Tercero, B.; Marcelino, N.; Pardo, J.; De Vicente, P. Pure Hydrocarbon Cycles in TMC-1: Discovery of Ethynyl Cyclopropenylidene, Cyclopentadiene, and Indene. Astron. Astrophys. 2021, 649, L15. [Google Scholar] [CrossRef]
- Wenzel, G.; Cooke, I.; Changala, P.; Bergin, E.; Zhang, S.; Burkhardt, A.; Byrne, A.; Charnley, S.; Cordiner, M.; Duffy, M.; et al. Discovery of Interstellar 1-Cyanopyrene: A Four-Ring Polycyclic Aromatic Hydrocarbon in TMC-1. arXiv 2024, arXiv:2410.00657. [Google Scholar]
- Wenzel, G.; Speak, T.; Changala, P.; Willis, R.; Burkhardt, A.; Zhang, S.; Bergin, E.; Byrne, A.; Charnley, S.; Fried, Z.; et al. Detections of Interstellar Aromatic Nitriles 2-Cyanopyrene and 4-Cyanopyrene in TMC-1. Nat. Astron. 2024, 9, 262–270. [Google Scholar] [CrossRef]
- Wenzel, G.; Gong, S.; Xue, C.; Changala, P.; Holdren, M.; Speak, T.; Stewart, D.; Fried, Z.; Willis, R.; Bergin, E.; et al. Discovery of the Seven-Ring Polycyclic Aromatic Hydrocarbon Cyanocoronene (C24H11CN) in GOTHAM Observations of TMC-1. Astrophys. J. Lett. 2025, 984, L36. [Google Scholar] [CrossRef]
- McCarthy, M.; Lee, K.; Loomis, R.; Burkhardt, A.; Shingledecker, C.; Charnley, S.; Cordiner, M.; Herbst, E.; Kalenskii, S.; Willis, E.; et al. Interstellar Detection of the Highly Polar Five-Membered Ring Cyanocyclopentadiene. Nat. Astron. 2020, 5, 176–180. [Google Scholar] [CrossRef]
- Long, K.; Lee, K.; Changala, B.; Loomis, R.; Burkhardt, A.; Xue, C.; Cordiner, M.; Charnley, S.; McCarthy, M.; McGuire, B. Interstellar Detection of 2-Cyanocyclopentadiene, C5H5CN, a Second Five-Membered Ring toward TMC-1. Astrophys. J. Lett. 2021, 910, L2. [Google Scholar]
- Sita, M.; Changala, B.; Xue, C.; Burkhardt, A.; Shingledecker, C.; Long, K.; Lee, K.; Loomis, R.; Momjian, E.; Siebert, M.; et al. Discovery of Interstellar 2-Cyanoindene (2-C9H7CN) in GOTHAM Observations of TMC-1. Astrophys. J. Lett. 2022, 938, L12. [Google Scholar] [CrossRef]
- McGuire, B.; Loomis, R.; Burkhardt, A.; Lee, K.; Shingledecker, C.; Charnley, S.; Cooke, I.; Cordiner, M.; Herbst, E.; Kalenskii, S.; et al. Detection of Two Interstellar Polycyclic Aromatic Hydrocarbons via Spectral Matched Filtering. Science 2021, 371, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- McGuire, B.; Burkhardt, A.; Kalenskii, S.; Shingledecker, C.; Remijan, A.; Herbst, E.; McCarthy, M. Detection of the Aromatic Molecule Benzonitrile (c-C6H5CN) in the Interstellar Medium. Science 2018, 359, 202–205. [Google Scholar] [CrossRef]
- Cernicharo, J.; Cabezas, C.; Fuentetaja, R.; Agúndez, M.; Tercero, B.; Janeiro, J.; Juanes, M.; Kaiser, R.; Endo, Y.; Steber, A.; et al. Discovery of Two Cyano Derivatives of Acenaphthylene (C12H8) in TMC-1 with the QUIJOTE Line Survey. Astron. Astrophys. 2024, 690, L13. [Google Scholar] [CrossRef]
- Ghahsare, A.; Nazifi, Z.; Nazifi, S. Structure-Bioactivity Relationship Study of Xanthene Derivatives: A Brief Review. Curr. Org. Synth. 2019, 16, 1071–1077. [Google Scholar] [CrossRef]
- Abualhasan, M.; Hawash, M.; Aqel, S.; Al-Masri, M.; Mousa, A.; Issa, L. Biological Evaluation of Xanthene and Thioxanthene Derivatives as Antioxidant, Anticancer, and COX Inhibitors. ACS Omega 2023, 8, 38597–38606. [Google Scholar] [CrossRef]
- Saikia, R.; Pathak, K.; Pramanik, P.; Islam, M.; Karmakar, S.; Gogoi, S.; Pathak, M.; Das, D.; Sahariah, J.; Ahmad, M.; et al. Exploring the Therapeutic Potential of Xanthones in Diabetes Management: Current Insights and Future Directions. Eur. J. Med. Chem. Rep. 2024, 12, 100189. [Google Scholar] [CrossRef]
- Sajjadifar, S.; Pornuroz, M. Preparation of a New, Green and Recyclable Catalyst, Silica-Supported 1-(2-Sulfooxy)Ethyl)1H-Pyridine-1-Ium-Chloride, and a Solvent- and Metal-Free Protocol for the Synthesis of 14-Aryl-14H-Dibenzo[a,j]Xanthene Derivatives. Appl. Petrochem. Res. 2018, 8, 97–105. [Google Scholar] [CrossRef]
- El-Seedi, H.; El-Ghorab, D.; El-Barbary, M.; Zayed, M.; Goransson, U.; Larsson, S.; Verpoorte, R. Naturally Occurring Xanthones: Isolation, Structure Elucidation and Chemosystematic Significance. Curr. Med. Chem. 2009, 16, 2581–2626. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, D.; Soares, J.; Costa, J.; Magalhães, A.; Azevedo, C.; Pinto, M.; Afonso, C. Structures, Activities and Drug-Likeness of Anti-Infective Xanthone Derivatives Isolated from the Marine Environment: A Review. Molecules 2019, 24, 243. [Google Scholar] [CrossRef]
- Gomes, A.; Brandão, P.; Fernandes, C.; Silva, M.d.; Sousa, M.d.S.d.; Pinto, M.d.M. Drug-like Properties and ADME of Xanthone Derivatives: The Antechamber of Clinical Trials. Curr. Med. Chem. 2016, 23, 3654–3686. [Google Scholar] [CrossRef]
- Feng, Z.; Lu, X.; Gan, L.; Zhang, Q.; Lin, L. Xanthones, A Promising Anti-Inflammatory Scaffold: Structure, Activity, and Drug Likeness Analysis. Molecules 2020, 25, 598. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kang, W.; Zheng, Y.; He, Y.; Zhong, R.; Fang, S.; Wen, W.; Liu, S.; Lin, S. Development of Xanthone Derivatives as Effective Broad-Spectrum Antimicrobials: Disrupting Cell Wall and Inhibiting DNA Synthesis. Sci. Adv. 2025, 11, 4723. [Google Scholar] [CrossRef]
- Oussaid, A.; Loupy, A. Selective Oxidation of Arenes in Dry Media under FocusedMicrowaves. J. Chem. Res. Synop. 1997, 342–343. [Google Scholar] [CrossRef]
- Fujita, S.I.; Yoshida, H.; Arai, M. Nitrogen-Doped Activated Carbon as Metal-Free Catalysts Having Various Functions. C 2017, 3, 31. [Google Scholar] [CrossRef]
- Lauridsen, J.; Kragh, R.; Lee, J. 7.08-Pyrans and Their Benzo Derivatives: Synthesis. In Comprehensive Heterocyclic Chemistry IV; Black, D., Cossy, J., Stevens, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 7, pp. 329–490. ISBN 9780128186565. [Google Scholar]
- Torregrosa-Chinillach, A.; Chinchilla, R. Synthesis of Xanthones, Thioxanthones and Acridones by a Metal-Free Photocatalytic Oxidation. Molecules 2021, 26, 974. [Google Scholar] [CrossRef]
- Neeman, E.M.; Huet, T. Identification of the Maze in the Conformational Landscape of Fenchol. Phys. Chem. Chem. Phys. 2018, 20, 24708–24715. [Google Scholar] [CrossRef] [PubMed]
- Neeman, E.M.; Huet, T. Conformational Changes in Hydroxyl Functional Groups upon Hydration: The Case Study of Endo Fenchol. Phys. Chem. Chem. Phys. 2021, 23, 2179–2185. [Google Scholar] [CrossRef]
- Evangelisti, L.; Gou, Q.; Feng, G.; Caminati, W.; Mead, G.; Finneran, I.; Carroll, P.; Blake, G. Conformational Equilibrium and Internal Dynamics in the Iso-Propanol-Water Dimer. Phys. Chem. Chem. Phys. 2017, 19, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Neeman, E.M.; Aviles Moreno, J.; Huet, T. Gas-Phase Hydration of Nopinone: The Interplay between Theoretical Methods and Experiments. Phys. Chem. Chem. Phys. 2021, 23, 18137–18144. [Google Scholar] [CrossRef] [PubMed]
- Neeman, E.M.; Huet, T. Unravelling the Structural Features of Monosaccharide Glyceraldehyde upon Mono-Hydration. J. Chem. Phys. 2023, 159, 194303. [Google Scholar] [CrossRef]
- Loru, D.; Sun, W.; Nootebos, H.; Steber, A.; Ferrari, P.; Schnell, M.; Schnell, M. Probing the Structure and Dynamics of the Heterocyclic PAH Xanthene and Its Water Complexes with Infrared and Microwave Spectroscopy. Phys. Chem. Chem. Phys. 2024, 26, 25341–25351. [Google Scholar] [CrossRef]
- Neeman, E.M.; Avilés-Moreno, J.R.; Huet, T. The quasi-unchanged gas-phase molecular structures of the atmospheric aerosol precursor β-pinene and its oxidation product nopinone. Phys. Chem. Chem. Phys. 2017, 19, 13819–13827. [Google Scholar] [CrossRef]
- Neeman, E.M.; Osseiran, N.; Huet, T. The gas-phase structure determination of α-pinene oxide: An endo-cyclic epoxide of atmospheric interest. J. Chem. Phys. 2023, 158, 154304. [Google Scholar] [CrossRef]
- Neeman, E.M.; Avilés Moreno, J.; Huet, T. The Gas Phase Structure of α-Pinene, a Main Biogenic Volatile Organic Compound. J. Chem. Phys. 2017, 147, 214305. [Google Scholar] [CrossRef]
- Pickett, H. The Fitting and Prediction of Vibration-Rotation Spectra with Spin Interactions. J. Mol. Spectrosc. 1991, 148, 371–377. [Google Scholar] [CrossRef]
- Watson, J. Aspects of Quartic and Sextic Centrifugal Effects on Rotational Energy Levels. In Vibrational Spectra and Structure; Durig, J.R., Ed.; Elsevier: Amsterdam, The Netherlands, 1977; Volume 6, pp. 1–89. ISBN 9780444415882. [Google Scholar]
- Neeman, E.M.; Dréan, P.; Huet, T. The structure and molecular parameters of camphene determined by Fourier transform microwave spectroscopy and quantum chemical calculations. J. Mol. Spectrosc. 2016, 322, 50–54. [Google Scholar] [CrossRef]
- Chakraborty, T.; Lim, E.; Lima, E. Conformational Studies of Xanthene by Laser-induced Fluorescence in a Supersonic Free Jet: Inversion Potentials. J. Chem. Phys. 1993, 98, 836–840. [Google Scholar] [CrossRef]
- Laane, J. Eigenvalues of the Potential Function V = z4 ± Bz2 and the Effect of Sixth Power Terms. Appl. Spectrosc. 1970, 24, 73–80. [Google Scholar] [CrossRef]
- Mjöberg, P.; Almlöf, J. A Conformational Study of Bicyclo[3.1.0]Hexane and of the 3-Bicyclo[3.1.0]Hexyl Carbonium Ion: An Ab Initio SCF Investigation. Chem. Phys. 1978, 29, 201–208. [Google Scholar] [CrossRef]
- Moran, D.; Simmonett, A.C.; Leach, F.E.; Allen, W.D.; Schleyer, P.V.R.; Schaefer, H.F. Popular theoretical methods predict benzene and arenes to be nonplanar. J. Am. Chem. Soc. 2006, 128, 9342–9343. [Google Scholar] [CrossRef]
- Evangelisti, L.; Spada, L.; Li, W.; Vazart, F.; Barone, V.; Caminati, W. The Borderline between Reactivity and Pre-Reactivity of Binary Mixtures of Gaseous Carboxylic Acids and Alcohols. Angew. Chem. 2017, 129, 3930–3933. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Politzer, P.; Truhlar, D. Introduction: The Role of the Electrostatic Potential in Chemistry. In Chemical Applications of Atomic and Molecular Electrostatic Potentials; Springer: New York, NY, USA, 1981. [Google Scholar]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16, Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Møller, C.; Plesset, M. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Chai, J.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Curtiss, L.; Raghavachari, K.; Redfern, P.; Rassolov, V.; Pople, J. Gaussian-3 (G3) Theory for Molecules Containing First and Second-Row Atoms. J. Chem. Phys. 1998, 109, 7764–7776. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kassi, S.; Petitprez, D.; Wlodarczak, G. Microwave Fourier Transform Spectroscopy of T-Butylchloride and T-Butylbromide Isotopic Species. J. Mol. Struct. 2000, 517, 375–386. [Google Scholar] [CrossRef]
- Kassi, S.; Petitprez, D.; Wlodarczak, G. Microwave Spectrum of Isotopic Species of Urea (NH2)2CO. J. Mol. Spectrosc. 2004, 228, 293–297. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Par. a | 0 | 1 | B97xD | B3LYP | M06-2X | MP2 |
|---|---|---|---|---|---|---|
| A (MHz) | 2032.111253(62) | 2031.038848(69) | 2048.0 | 2056.6 | 2042.0 | 1904.9 |
| B (MHz) | 465.950920(23) | 466.056198(24) | 468.2 | 462.4 | 467.9 | 477.6 |
| C (MHz) | 385.125452(20) | 385.251448(21) | 387.3 | 381.9 | 387.1 | 399.4 |
| (kHz) | 0.014016(30) | 0.012734(33) | 0.0165 | 0.0098 | 0.0129 | 0.0131 |
| (kHz) | −0.23104(17) | −0.20487(21) | −0.281 | −0.1527 | −0.2110 | −0.177 |
| (kHz) | 1.2645(17) | 1.1285(16) | 1.502 | 0.8841 | 1.1587 | 0.882 |
| (kHz) | −0.000560(15) | −0.000469(17) | 0.0008 | 0.0003 | 0.0006 | 0.0004 |
| (kHz) | 0.0001420(66) | 0.0001351(70) | −0.0001 | −0.0001 | −0.0001 | −0.0001 |
| (MHz) | 4689.7095(10) | - | - | - | - | |
| (MHz) | 0.360263(32) | - | - | - | - | |
| (D) | - | 0.0 | 0.0 | 0.0 | 0.0 | |
| (D) | - | 1.1 | 1.1 | 1.1 | 1.1 | |
| (D) | - | 0.1 | 0.1 | 0.1 | 0.2 | |
| N b | 511 | - | - | - | - | |
| c (kHz) | 2.8 | - | - | - | - | |
 | (cm−1) c | (cm−1) d | |
| v | This work model | Experimental [44] | |
| 1 | 0.158 | 0 | |
| 2 | 24.7 | 26 | |
| 3 | 29.4 | 30 | |
| 4 | 45.3 | 46 | |
| 5 | 60.2 | 60 | |
| 6 | 78.0 | 78 | |
| 7 | 97.6 | 97 | |
| 8 | 119 | 119 | |
| = 0.15643190(3) a,b | Barrier | 45 | 49 |
| Method | Barrier Height (cm−1) | Angle to Planarity (°) |
|---|---|---|
| B3LYP | 34 | 162.1 |
| M06-2X | 54.7 | 158.5 |
| B97xD | 44.4 | 158.7 |
| MP2 | 748 | 144.8 |
| Par. | Exp. a | B97xD | B3LYP | M06-2X | MP2 |
|---|---|---|---|---|---|
| A (MHz) | 1461.290022(36) | 1472.3 | 1463.0 | 1471.6 | 1451.5 |
| B (MHz) | 465.138292(19) | 467.4 | 464.1 | 466.8 | 463.4 |
| C (MHz) | 352.988498(14) | 354.7 | 352.3 | 354.4 | 351.3 |
| (kHz) | 0.001721(20) | 0.0017 | 0.0017 | 0.0017 | 0.0017 |
| (kHz) | 0.00700(12) | 0.0069 | 0.0069 | 0.0067 | 0.0070 |
| (kHz) | 0.03076(48) | 0.0289 | 0.0291 | 0.0291 | 0.0287 |
| (kHz) | −0.000493(10) | −0.0005 | −0.0005 | −0.0005 | −0.0005 |
| (kHz) | −0.0000884(48) | −0.0001 | −0.0001 | −0.0001 | −0.0001 |
| (D) | - | 0.0 | 0.0 | 0.0 | 0.0 |
| (D) | - | 3.0 | 3.1 | 2.8 | 2.8 |
| (D) | - | 0.0 | 0.0 | 0.0 | 0.0 |
| N b | 336 | - | - | - | - |
| c (kHz) | 2.3 | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bermúdez, C.; Goubet, M.; Neeman, E.M. Inversion Motion of Xanthene and Detection of Its Oxidation Product Xanthone from Gas-Phase Rotational Spectroscopy. Molecules 2025, 30, 2801. https://doi.org/10.3390/molecules30132801
Bermúdez C, Goubet M, Neeman EM. Inversion Motion of Xanthene and Detection of Its Oxidation Product Xanthone from Gas-Phase Rotational Spectroscopy. Molecules. 2025; 30(13):2801. https://doi.org/10.3390/molecules30132801
Chicago/Turabian StyleBermúdez, Celina, Manuel Goubet, and Elias M. Neeman. 2025. "Inversion Motion of Xanthene and Detection of Its Oxidation Product Xanthone from Gas-Phase Rotational Spectroscopy" Molecules 30, no. 13: 2801. https://doi.org/10.3390/molecules30132801
APA StyleBermúdez, C., Goubet, M., & Neeman, E. M. (2025). Inversion Motion of Xanthene and Detection of Its Oxidation Product Xanthone from Gas-Phase Rotational Spectroscopy. Molecules, 30(13), 2801. https://doi.org/10.3390/molecules30132801