


Solid-Phase Oligosaccharide Synthesis with Highly Complexed Peptidoglycan Fragments

 ,
,
Abstract

1. Introduction
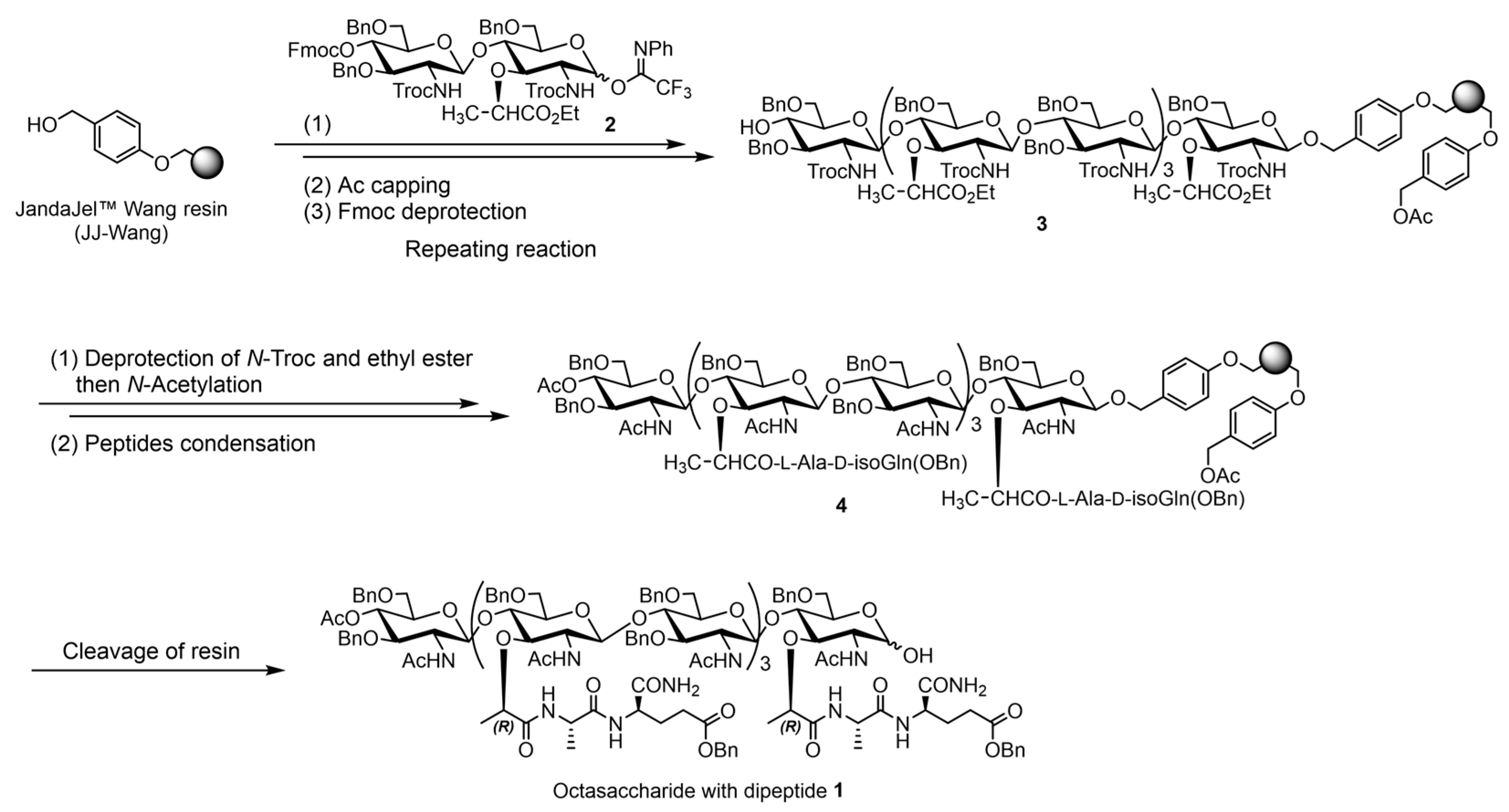
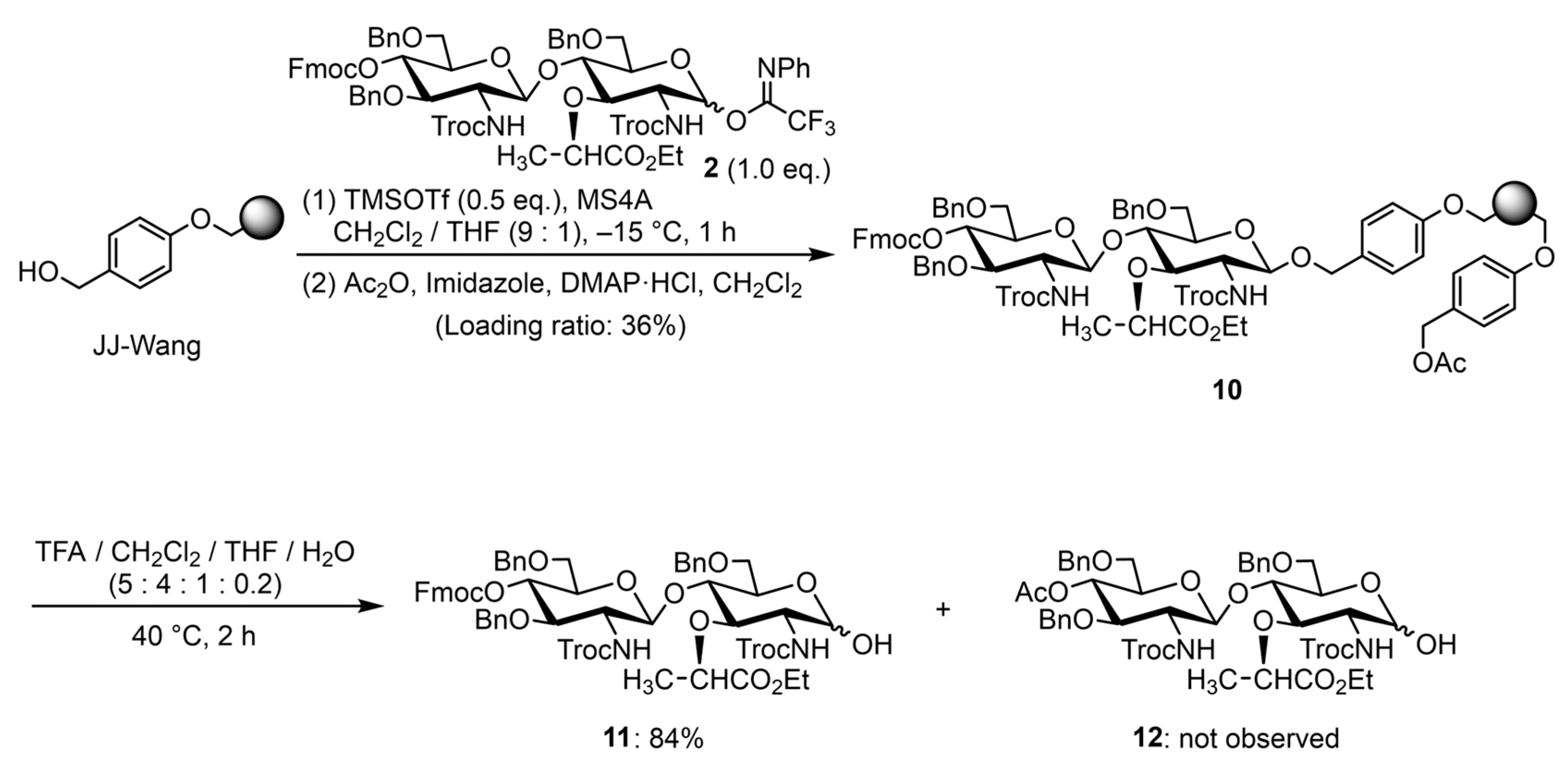
2. Results and Discussion
3. Materials and Methods
3.1. Preparation of the Glycosyl Donors
3.2. SPOS of PGN Fragments
3.3. Purification of the PGN Fragments
3.4. Analysis of the PGN Fragments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicolaou, K.C.; Watanabe, N.; Li, J.; Pastor, J.; Winssinger, N. Solid-phase synthesis of oligosaccharides: Construction of a dodecasaccharide. Angew. Chem. Int. Ed. Engl. 1998, 37, 1559–1561. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Winssinger, N.; Pastor, J.; DeRoose, F. A general and highly efficient solid phase synthesis of oligosaccharides. Total synthesis of a heptasaccharide phytoalexin elicitor (HPE). J. Am. Chem. Soc. 1997, 119, 449–450. [Google Scholar] [CrossRef]
- Plante, O.J.; Palmacci, E.R.; Seeberger, P.H. Automated solid-phase synthesis of oligosaccharides. Science 2001, 291, 1523–1527. [Google Scholar] [CrossRef]
- Joseph, A.A.; Pardo-Vargas, A.; Seeberger, P.H. Total Synthesis of Polysaccharides by Automated Glycan Assembly. J. Am. Chem. Soc. 2020, 142, 8561–8564. [Google Scholar] [CrossRef]
- Niggemeyer, G.B.; Danglad-Flores, J.A.; Seeberger, P.H. Automated synthesis of C1-functionalized oligosaccharides. J. Am. Chem. Soc. 2025, 147, 1649–1655. [Google Scholar] [CrossRef] [PubMed]
- Crawford, C.J.; Schultz-Johansen, M.; Luong, P.; Vidal-Melgosa, S.; Hehemann, J.-H.; Seeberger, P.H. Automated synthesis of algal fucoidan oligosaccharides. J. Am. Chem. Soc. 2024, 146, 18320–18330. [Google Scholar] [CrossRef]
- Zhu, Y.; Delbianco, M.; Seeberger, P.H. Automated assembly of starch and glycogen polysaccharides. J. Am. Chem. Soc. 2021, 143, 9758–9768. [Google Scholar] [CrossRef]
- Danglad-Flores, J.; Leichnitz, S.; Sletten, E.T.; Abragam Joseph, A.; Bienert, K.; Le Mai Hoang, K.; Seeberger, P.H. Microwave-assisted automated glycan assembly. J. Am. Chem. Soc. 2021, 143, 8893–8901. [Google Scholar] [CrossRef] [PubMed]
- Boltje, T.J.; Kim, J.-H.; Park, J.; Boons, G.-J. Chiral-auxiliary-mediated 1,2-cis-glycosylations for the solid-supported synthesis of a biologically important branched α-glucan. Nat. Chem. 2010, 2, 552–557. [Google Scholar] [CrossRef]
- Walvoort, M.T.C.; van den Elst, H.; Plante, O.J.; Krcck, L.; Seeberger, P.H.; Overkleeft, H.S.; van der Marel, G.A.; Codee, J.D.C. Automated solid-phase synthesis of β-mannuronic acid alginates. Angew. Chem. Int. Ed. 2012, 51, 4393–4396. [Google Scholar] [CrossRef]
- Geert Volbeda, A.G.; van Mechelen, J.; Meeuwenoord, N.; Overkleeft, H.S.; van der Marel, G.A.; Codée, J.D.C. Cyanopivaloyl ester in the automated solid-phase synthesis of oligorhamnans. J. Org. Chem. 2017, 82, 12992–13002. [Google Scholar] [CrossRef] [PubMed]
- Pistorio, S.G.; Geringer, S.A.; Stine, K.J.; Demchenko, A.V. Manual and automated syntheses of the N-linked glycoprotein Core Glycans. J. Org. Chem. 2019, 84, 6576–6588. [Google Scholar] [CrossRef] [PubMed]
- Panza, M.; Stine, K.J.; Demchenko, A.V. HPLC-assisted automated oligosaccharide synthesis: The implementation of the two-way split valve as a mode of complete automation. Chem. Commun. 2020, 56, 1333–1336. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, N.V.; Fujikawa, K.; Tan, Y.H.; Stine, K.J.; Demchenko, A.V. HPLC-assisted automated oligosaccharide synthesis. Org. Lett. 2012, 14, 3036–3039. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Fujii, Y.; Tokimoto, H.; Mori, Y.; Tanaka, S.; Bao, G.M.; Siwu, E.R.O.; Nakayabu, A.; Fukase, K. Synthesis of a sialic acid containing complex-type N-glycan on a solid support. Chem. Asian J. 2009, 4, 574–580. [Google Scholar] [CrossRef]
- Ricardo, M.G.; Seeberger, P.H. Merging solid-phase peptide synthesis and automated glycan assembly to prepare lipid-peptide-glycan chimeras. Chemistry 2023, 29, e202301678. [Google Scholar] [CrossRef]
- Hurevich, M.; Seeberger, P.H. Automated glycopeptide assembly by combined solid-phase peptide and oligosaccharide synthesis. Chem. Commun. 2014, 50, 1851–1853. [Google Scholar] [CrossRef]
- Mesnage, S.; Dellarole, M.; Baxter, N.J.; Rouget, J.-B.; Dimitrov, J.D.; Wang, N.; Fujimoto, Y.; Hounslow, A.M.; Lacroix-Desmazes, S.; Fukase, K.; et al. Molecular basis for bacterial peptidoglycan recognition by LysM domains. Nat. Commun. 2014, 5, 4269. [Google Scholar] [CrossRef]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; et al. Host Recognition of Bacterial Muramyl Dipeptide Mediated through NOD2. J. Biol. Chem. 2003, 278, 5509–5512. [Google Scholar] [CrossRef]
- Wang, Z.-M.; Li, X.; Cocklin, R.R.; Wang, M.; Wang, M.; Fukase, K.; Inamura, S.; Kusumoto, S.; Gupta, D.; Dziarski, R. Human peptidoglycan recognition protein-L Is an N-acetylmuramoyl-L-alanine amidase. J. Biol. Chem. 2003, 278, 49044–49052. [Google Scholar] [CrossRef]
- Resko, A.J.; Anderson, C.M.; Federle, M.J.; Alonzo, F., III. A staphylococcal glucosaminidase drives inflammatory responses by processing peptidoglycan chains to physiological lengths. Infect. Immun. 2023, 91, e00500-22. [Google Scholar] [CrossRef] [PubMed]
- Chamaillard, M.; Hashimoto, M.; Horie, Y.; Masumoto, J.; Qiu, S.; Saab, L.; Ogura, Y.; Kawasaki, A.; Fukase, K.; Kusumoto, S.; et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 2003, 4, 702–707. [Google Scholar] [CrossRef]
- Liang, Y.; Yang, L.; Wang, Y.; Tang, T.; Liu, F.; Zhang, F. Peptidoglycan recognition protein SC (PGRP-SC) shapes gut microbiota richness, diversity and composition by modulating immunity in the house fly Musca domestica. Insect Mol. Biol. 2023, 32, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Bersch, K.L.; DeMeester, K.E.; Zagani, R.; Chen, S.; Wodzanowski, K.A.; Liu, S.; Mashayekh, S.; Reinecker, H.-C.; Grimes, C.L. Bacterial peptidoglycan fragments differentially regulate innate immune signaling. ACS Cent. Sci. 2021, 7, 688–696. [Google Scholar] [CrossRef]
- Wolf, A.J.; Underhill, D.M. Peptidoglycan recognition by the innate immune system. Nat. Rev. Immunol. 2018, 18, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Lien, E.; Ingalls, R.R.; Tuomanen, E.; Dziarski, R.; Golenbock, D. Cutting Edge: Recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via toll-like receptor 2. J. Immunol. 1999, 163, 1–5. [Google Scholar] [CrossRef]
- Wang, Q.; Marchetti, R.; Prisic, S.; Ishii, K.; Arai, Y.; Ohta, I.; Inuki, S.; Uchiyama, S.; Silipo, A.; Molinaro, A.; et al. A comprehensive study of the interaction between peptidoglycan fragments and the extracellular domain of Mycobacterium tuberculosis Ser/Thr kinase PknB. ChemBioChem 2017, 18, 2094–2098. [Google Scholar] [CrossRef]
- Wang, N.; Hasegawa, H.; Huang, C.-Y.; Fukase, K.; Fujimoto, Y. Synthesis of peptidoglycan fragments from Enterococcus faecalis with Fmoc-strategy for glycan elongation. Chem. Asian J. 2017, 12, 27–30. [Google Scholar] [CrossRef]
- Wang, N.; Hirata, A.; Nokihara, K.; Fukase, K.; Fujimoto, Y. Peptidoglycan microarray as a novel tool to explore protein–ligand recognition. Biopolymers 2016, 106, 422–429. [Google Scholar] [CrossRef]
- Wang, Q.; Matsuo, Y.; Pradipta, A.R.; Inohara, N.; Fujimoto, Y.; Fukase, K. Synthesis of characteristic Mycobacterium peptidoglycan (PGN) fragments utilizing with chemoenzymatic preparation of meso-diaminopimelic acid (DAP), and their modulation of innate immune responses. Org. Biomol. Chem. 2016, 14, 1013–1023. [Google Scholar] [CrossRef]
- Wang, N.; Huang, C.-Y.; Hasegawa, M.; Inohara, N.; Fujimoto, Y.; Fukase, K. Glycan sequence-dependent Nod2 activation investigated by using a chemically synthesized bacterial peptidoglycan fragment library. ChemBioChem 2013, 14, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Fukase, K. Structures, synthesis, and human Nod1 stimulation of immunostimulatory bacterial peptidoglycan fragments in the environment. J. Nat. Prod. 2011, 74, 518–525. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Konishi, Y.; Kubo, O.; Hasegawa, M.; Inohara, N.; Fukase, K. Synthesis of crosslinked peptidoglycan fragments for investigation of their immunobiological functions. Tetrahedron Lett. 2009, 50, 3631–3634. [Google Scholar] [CrossRef]
- Kawasaki, A.; Inohara, N.; Fujimoto, Y.; Fukase, K. Synthesis of diaminopimelic acid containing peptidoglycan fragments and tracheal cytotoxin (TCT) and investigation of their biological functions. Chem. Eur. J. 2008, 14, 10318–10330. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Inamura, S.; Kawasaki, A.; Shiokawa, Z.; Shimoyama, A.; Hashimoto, T.; Kusumoto, S.; Fukase, K. IEIIS Meeting minireview: Chemical synthesis of peptidoglycan fragments for elucidation of the immunostimulating mechanism. J. Endotoxin Res. 2007, 13, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Inamura, S.; Fujimoto, Y.; Kawasaki, A.; Shiokawa, Z.; Woelk, E.; Heine, H.; Lindner, B.; Inohara, N.; Kusumoto, S.; Fukase, K. Synthesis of peptidoglycan fragments and evaluation of their biological activity. Org. Biomol. Chem. 2006, 4, 232–242. [Google Scholar] [CrossRef]
- Inamura, S.; Fukase, K.; Kusumoto, S. Synthetic study of peptidoglycan partial structures. Synthesis of tetrasaccharide and octasaccharide fragments. Tetrahedron Lett. 2001, 42, 7613–7616. [Google Scholar] [CrossRef]
- Kadonaga, Y.; Wang, N.; Fujimoto, Y.; Fukase, K. Solid-phase synthesis of bacterial cell wall peptidoglycan fragments. Chem. Lett. 2014, 43, 1461–1463. [Google Scholar] [CrossRef]
- Vaino, A.R.; Goodin, D.B.; Janda, K.D. Investigating resins for solid phase organic synthesis: The relationship between swelling and microenvironment as probed by EPR and fluorescence spectroscopy. J. Comb. Chem. 2000, 2, 330–336. [Google Scholar] [CrossRef]
- Gambs, C.; Dickerson, T.J.; Mahajan, S.; Pasternack, L.B.; Janda, K.D. High-resolution diffusion-ordered spectroscopy to probe the microenvironment of JandaJel and Merrifield resins. J. Org. Chem. 2003, 68, 3673–3678. [Google Scholar] [CrossRef]
- Collot, M.; Eller, S.; Weishaupt, M.; Seeberger, P.H. Glycosylation efficiencies on different solid supports using a hydrogenolysis-labile linker. Beilstein J. Org. Chem. 2013, 9, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Lanari, D.; Ballini, R.; Bonollo, S.; Palmieri, A.; Pizzo, F.; Vaccaro, L. JandaJel as a polymeric support to improve the catalytic efficiency of immobilized-1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) under solvent-free conditions. Green Chem. 2011, 13, 3181–3186. [Google Scholar] [CrossRef]
- Yu, B.; Tao, H. Glycosyl trifuoroacetimidates. Part 1: Preparation and application as new glycosyl donors. Tetrahedron Lett. 2001, 42, 2405–2407. [Google Scholar] [CrossRef]
- Schmidt, R.R.; Michel, J. Facile synthesis of α- and β-O-glycosyl imidates; preparation of glycosides and disaccharides. Angew. Chem. Int. Ed. Engl. 1980, 19, 731–732. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, Q.; Liu, Y.; Wang, Q. 4-(N,N-dimethylamino)pyridine hydrochloride as a recyclable catalyst for acylation of inert alcohols: Substrate scope and reaction mechanism. Org. Lett. 2014, 16, 236–239. [Google Scholar] [CrossRef]
- Huang, C.-y.; Wang, N.; Fujiki, K.; Otsuka, Y.; Akamatsu, M.; Fujimoto, Y.; Fukase, K. Widely applicable deprotection method of 2,2,2-trichloroethoxycarbonyl (troc) group using tetrabutylammonium fluoride. J. Carbohydr. Chem. 2010, 29, 289–298. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Donor 5 (e.q.) | Solvent (5.0 w/v %) | Time (h) | Loading Ratio (%) *1 |
| 1 *2 | 1.0 | CH2Cl2 | 1 | 22 |
| 2 | 2.0 | CH2Cl2 | 1 | 35 |
| 3 | 3.0 | CH2Cl2 | 1 | 45 |
| 4 *2 | 1.0 | THF | 1 | 34 |
| 5 *2 | 1.0 | CH2Cl2/THF (9:1) | 1 | 44 |
| 6 | 1.0 | CH2Cl2/THF (9:1) | 3 | 44 |
| Entry | Solvent | Glycosylation Yield (%) |
|---|---|---|
| 1 | CH2Cl2/THF (9:1) | 56 |
| 2 | CH2Cl2/C4F9OEt/THF (5:4:1) | 83 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadonaga, Y.; Wang, N.; Shimoyama, A.; Fujimoto, Y.; Fukase, K. Solid-Phase Oligosaccharide Synthesis with Highly Complexed Peptidoglycan Fragments. Molecules 2025, 30, 2787. https://doi.org/10.3390/molecules30132787
Kadonaga Y, Wang N, Shimoyama A, Fujimoto Y, Fukase K. Solid-Phase Oligosaccharide Synthesis with Highly Complexed Peptidoglycan Fragments. Molecules. 2025; 30(13):2787. https://doi.org/10.3390/molecules30132787
Chicago/Turabian StyleKadonaga, Yuichiro, Ning Wang, Atsushi Shimoyama, Yukari Fujimoto, and Koichi Fukase. 2025. "Solid-Phase Oligosaccharide Synthesis with Highly Complexed Peptidoglycan Fragments" Molecules 30, no. 13: 2787. https://doi.org/10.3390/molecules30132787
APA StyleKadonaga, Y., Wang, N., Shimoyama, A., Fujimoto, Y., & Fukase, K. (2025). Solid-Phase Oligosaccharide Synthesis with Highly Complexed Peptidoglycan Fragments. Molecules, 30(13), 2787. https://doi.org/10.3390/molecules30132787