
Surface-Confined Self-Assembly of Star-Shaped Tetratopic Molecules with Vicinal Interaction Centers

Abstract

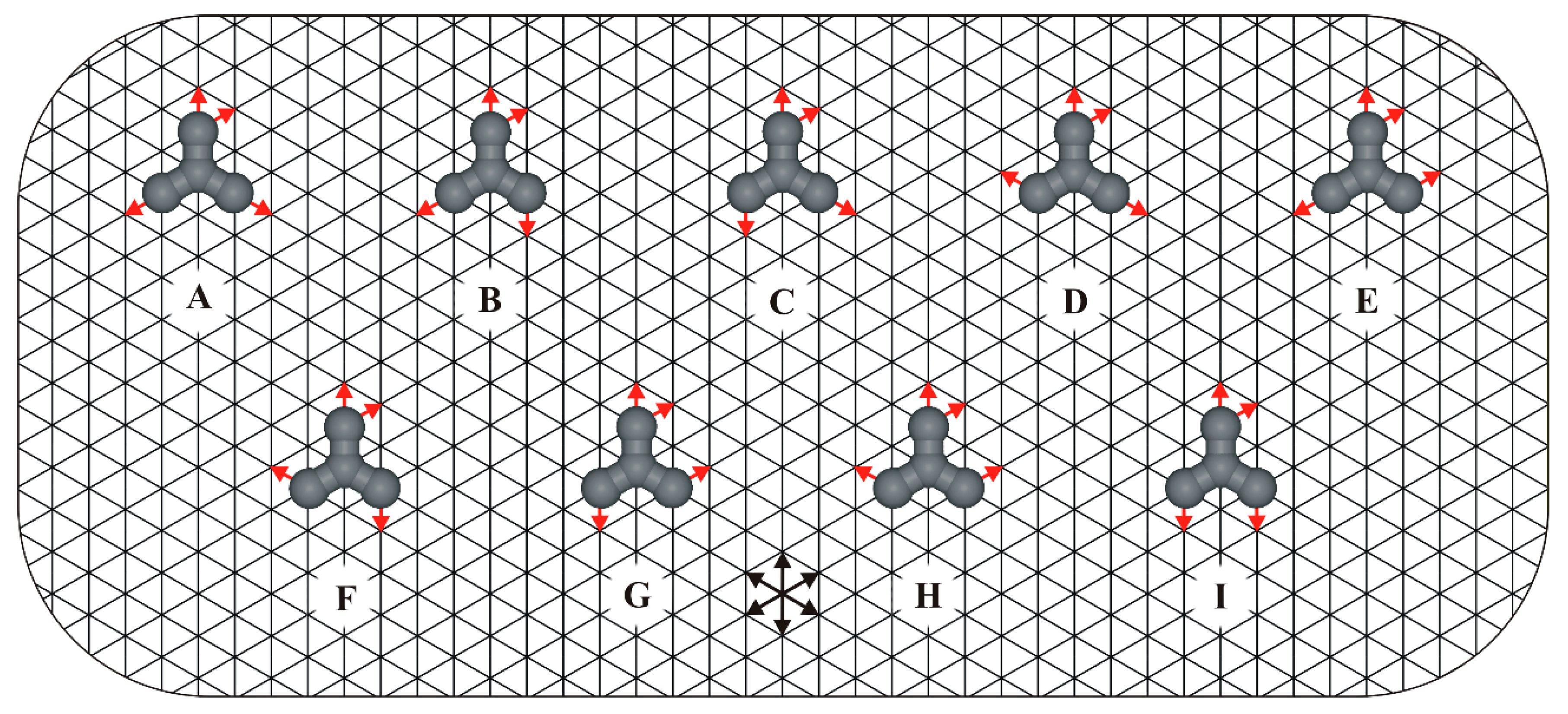
1. Introduction
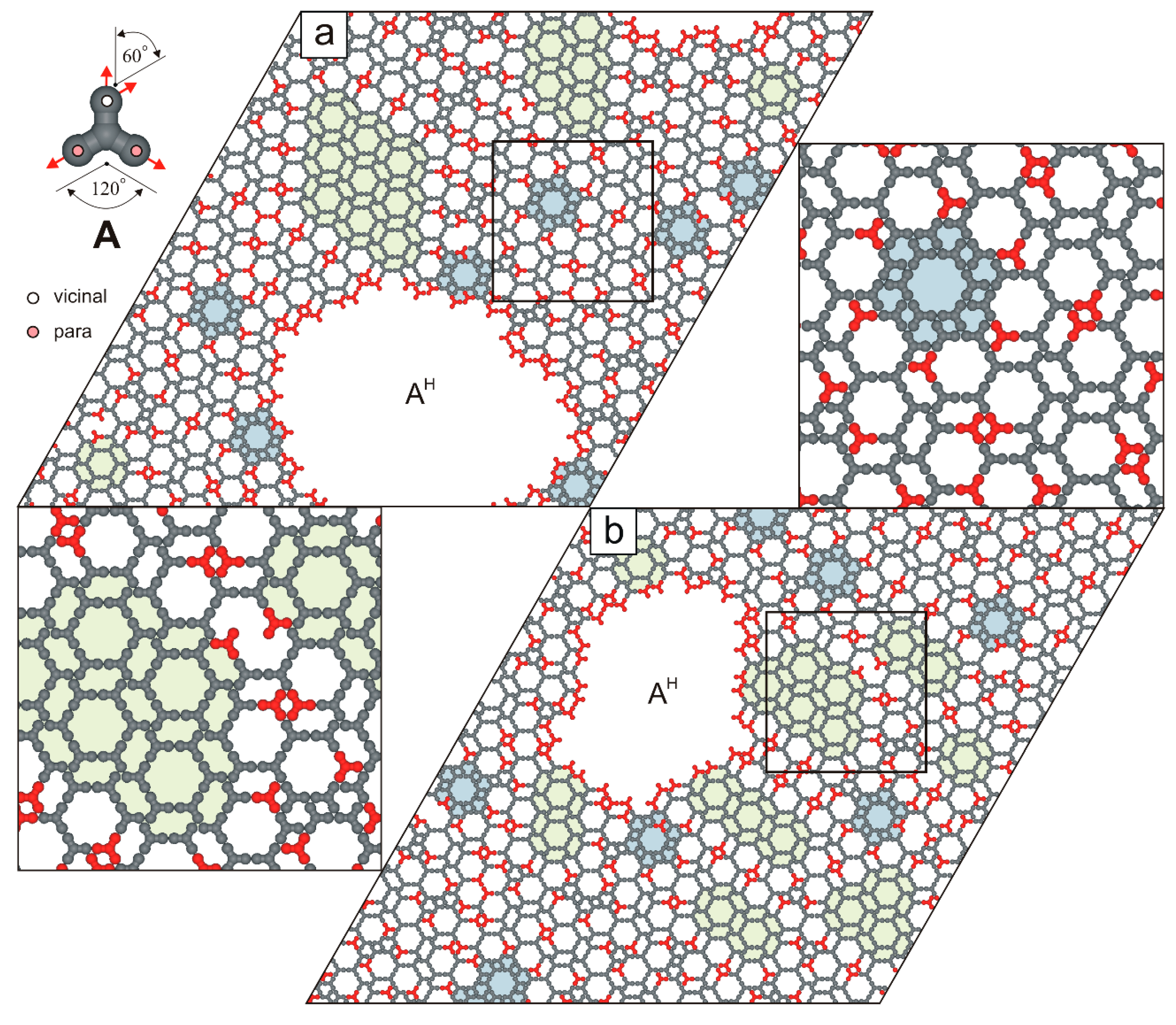
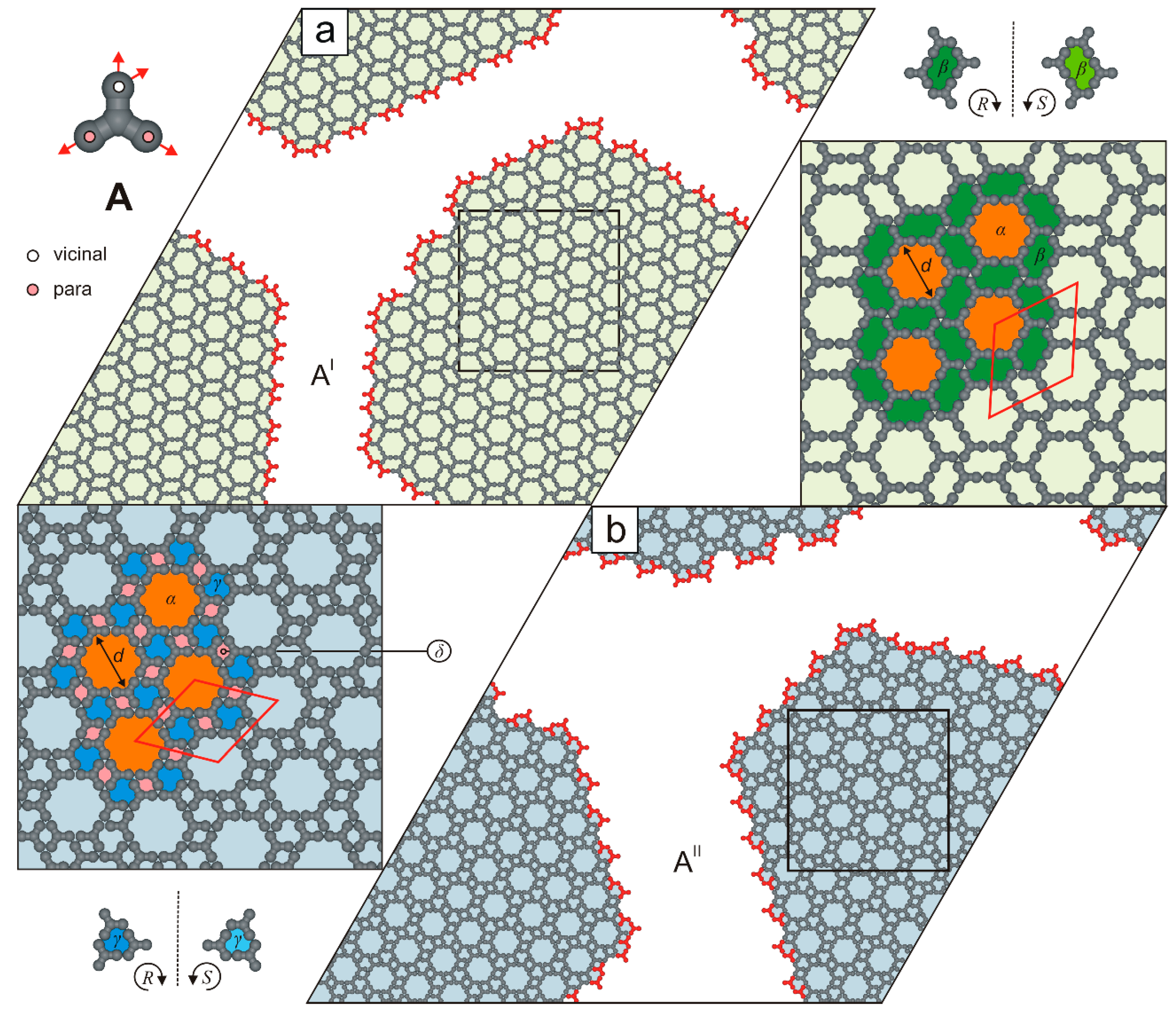
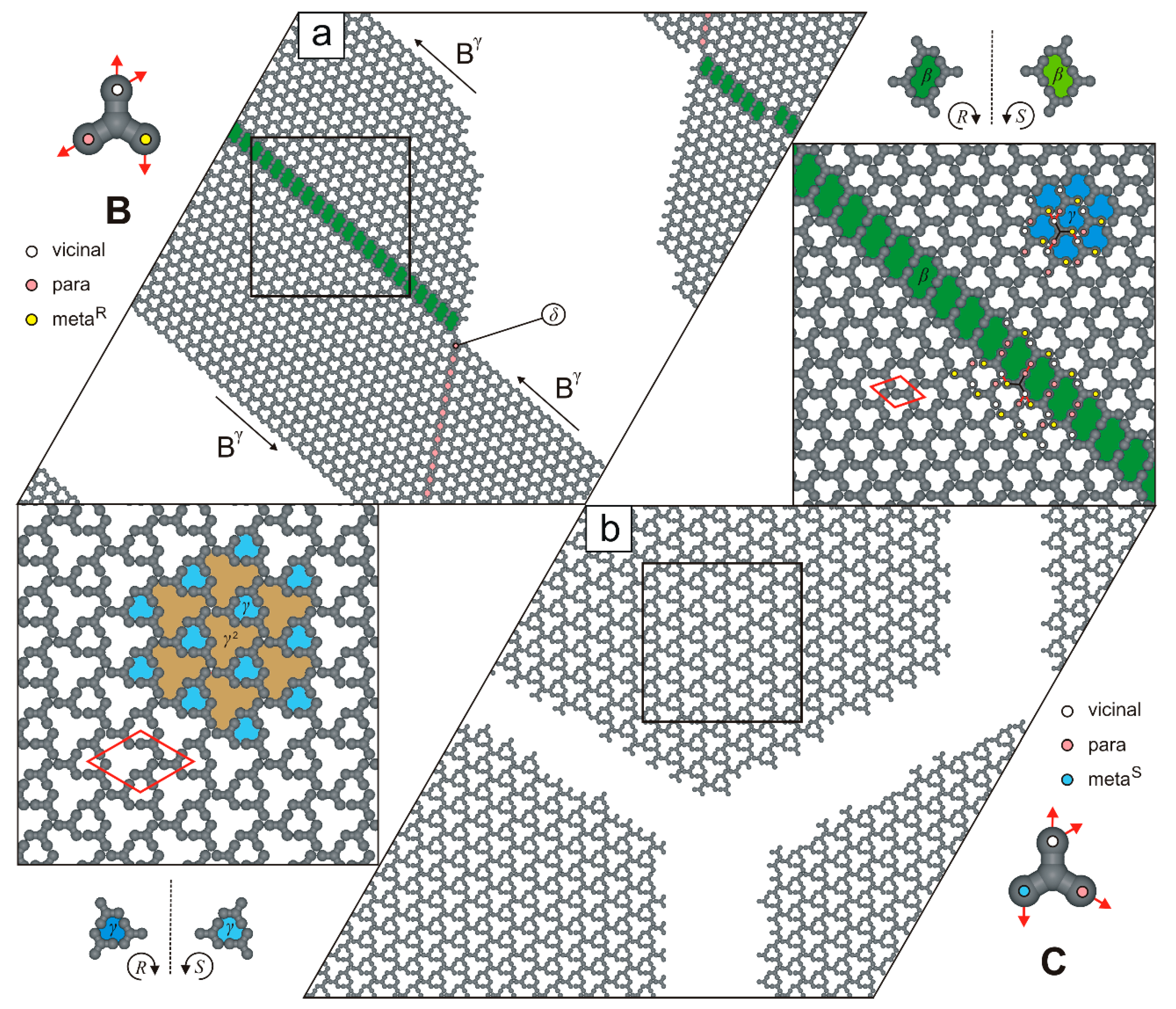
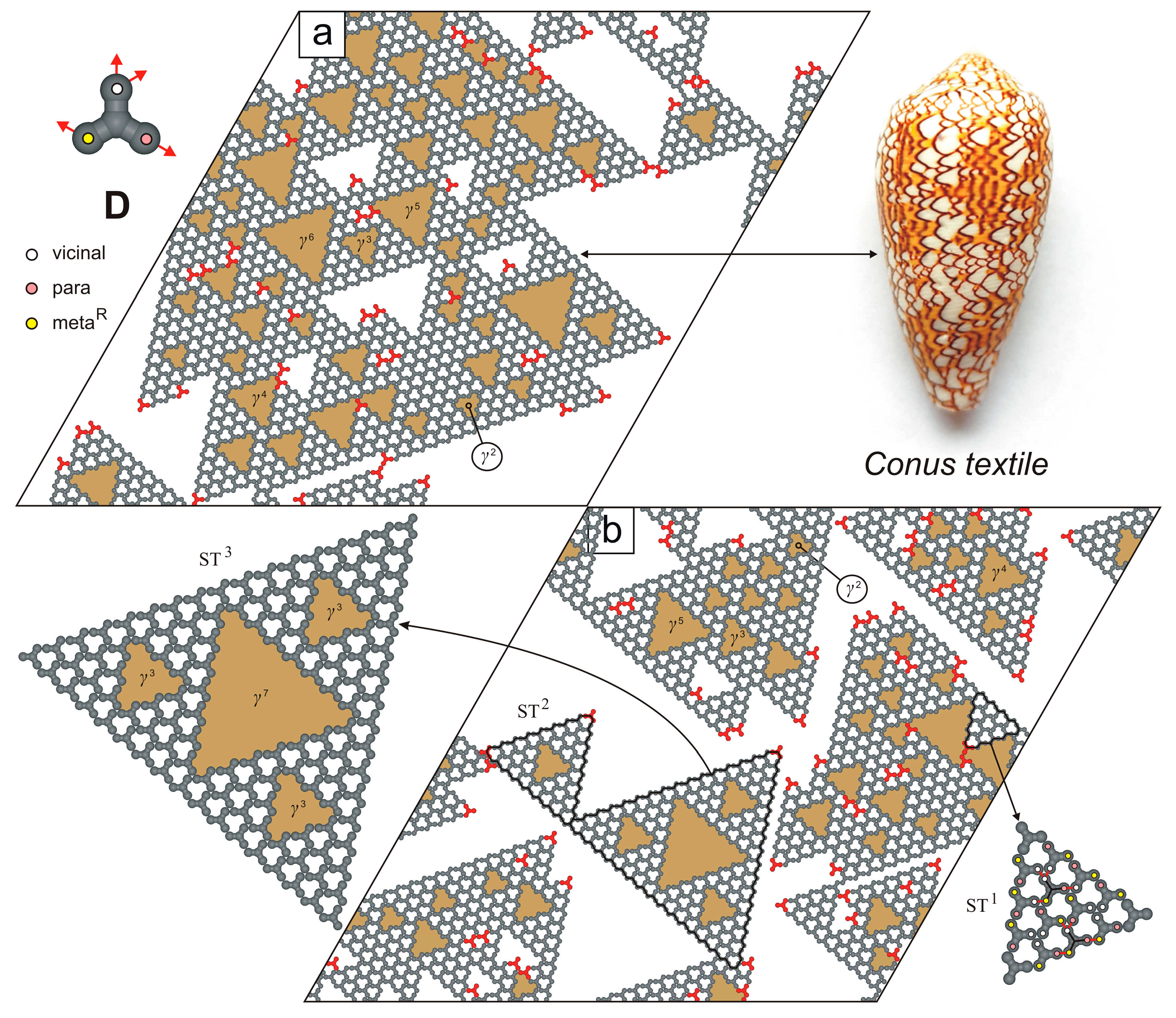
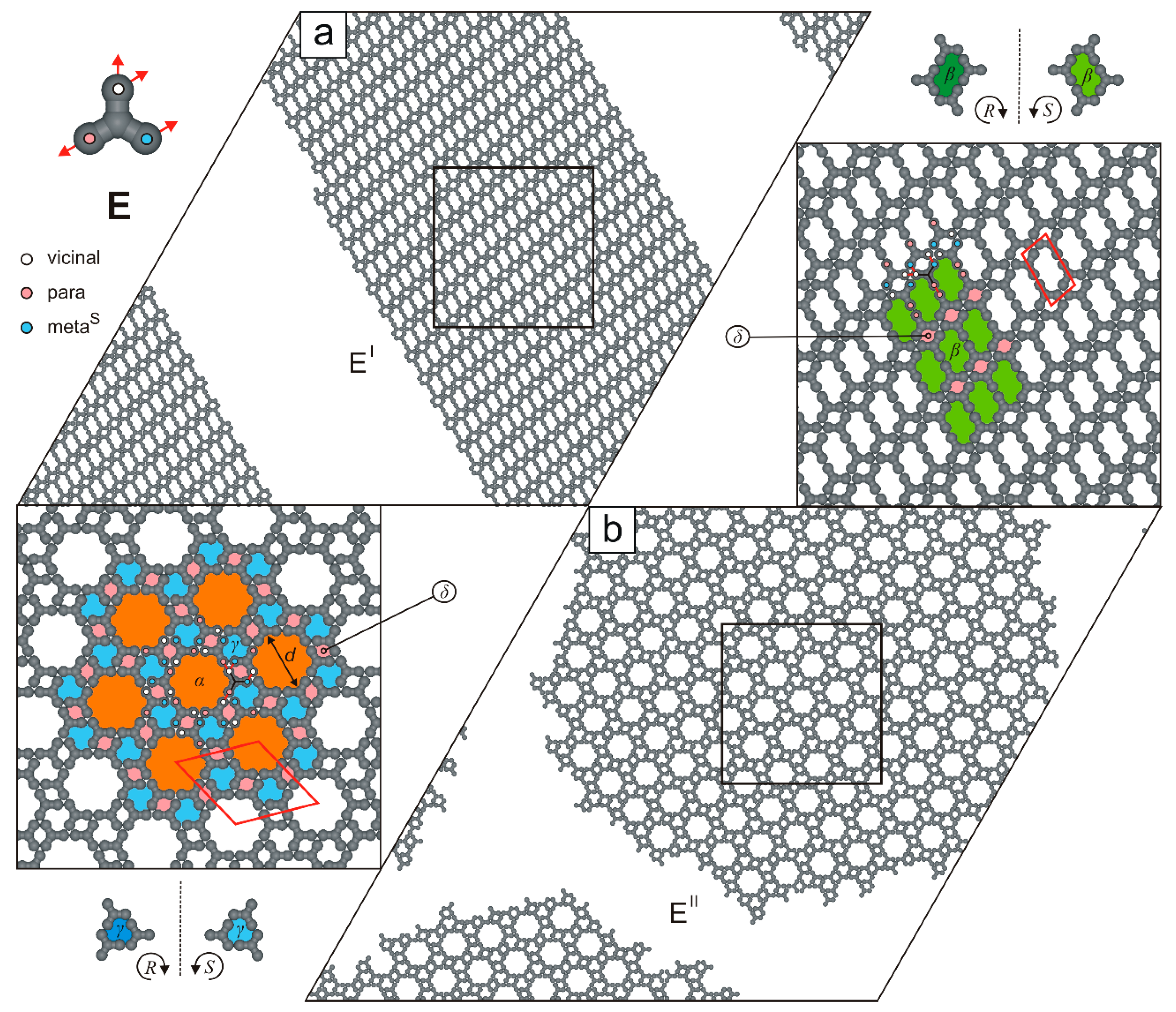
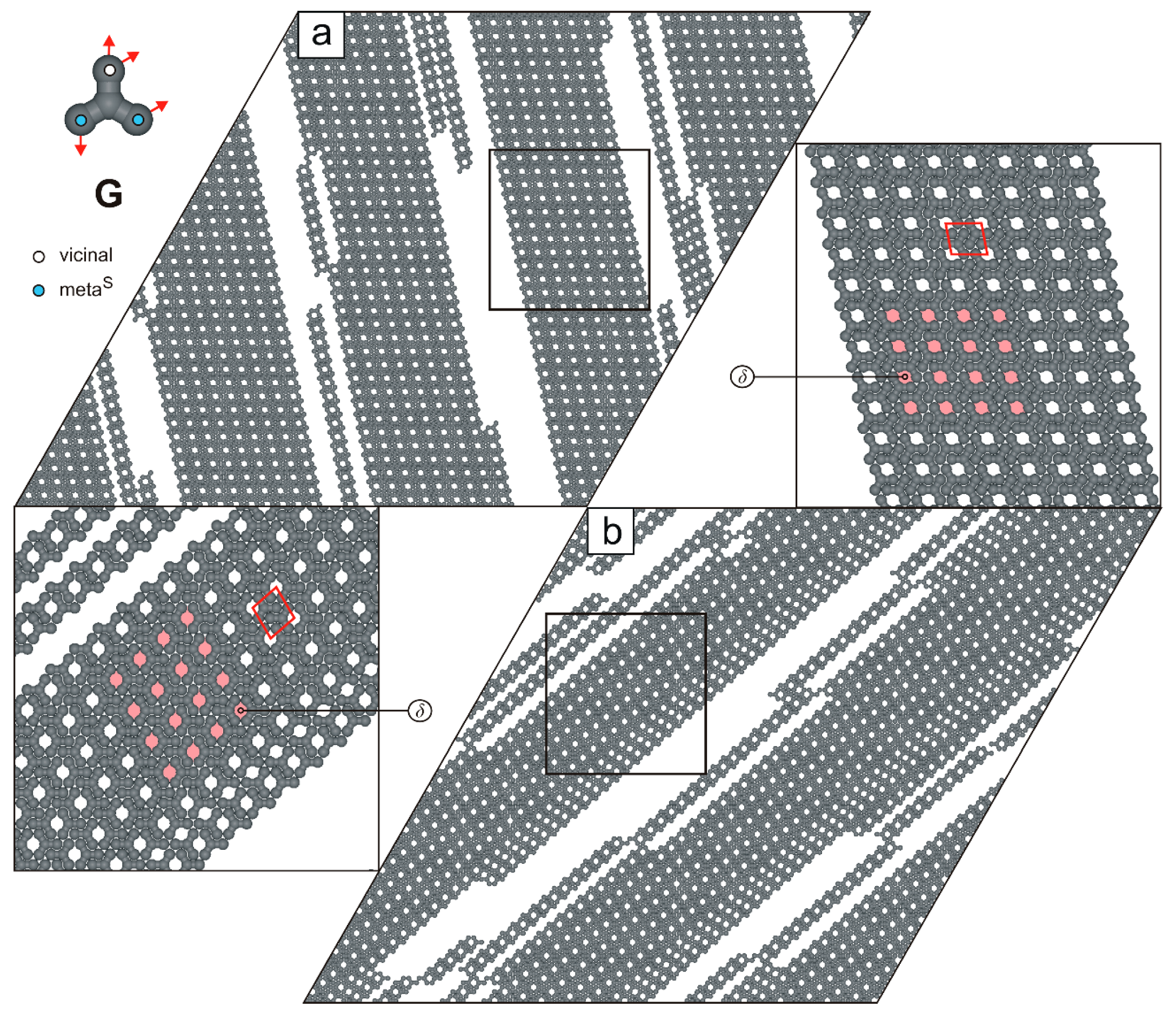
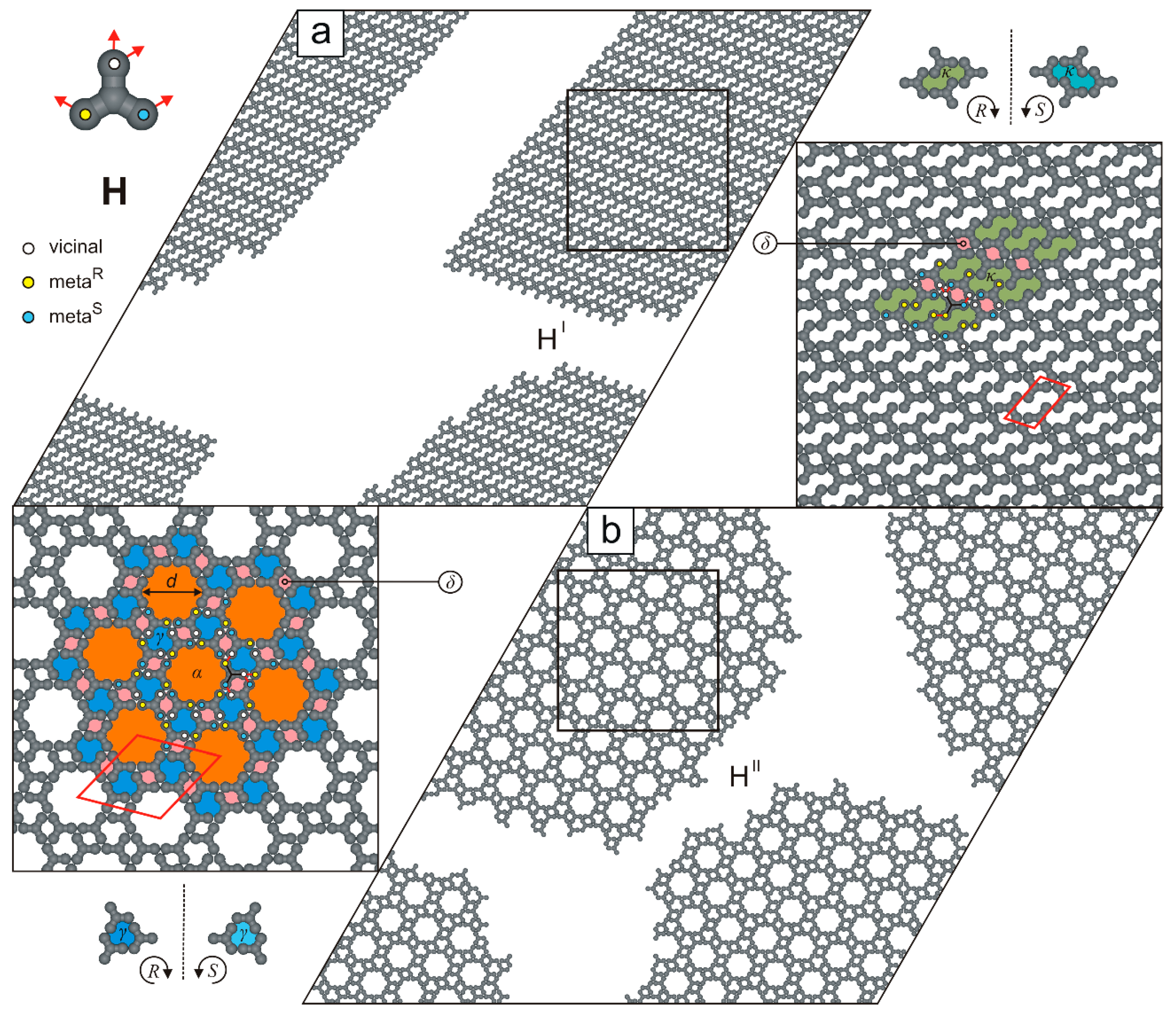
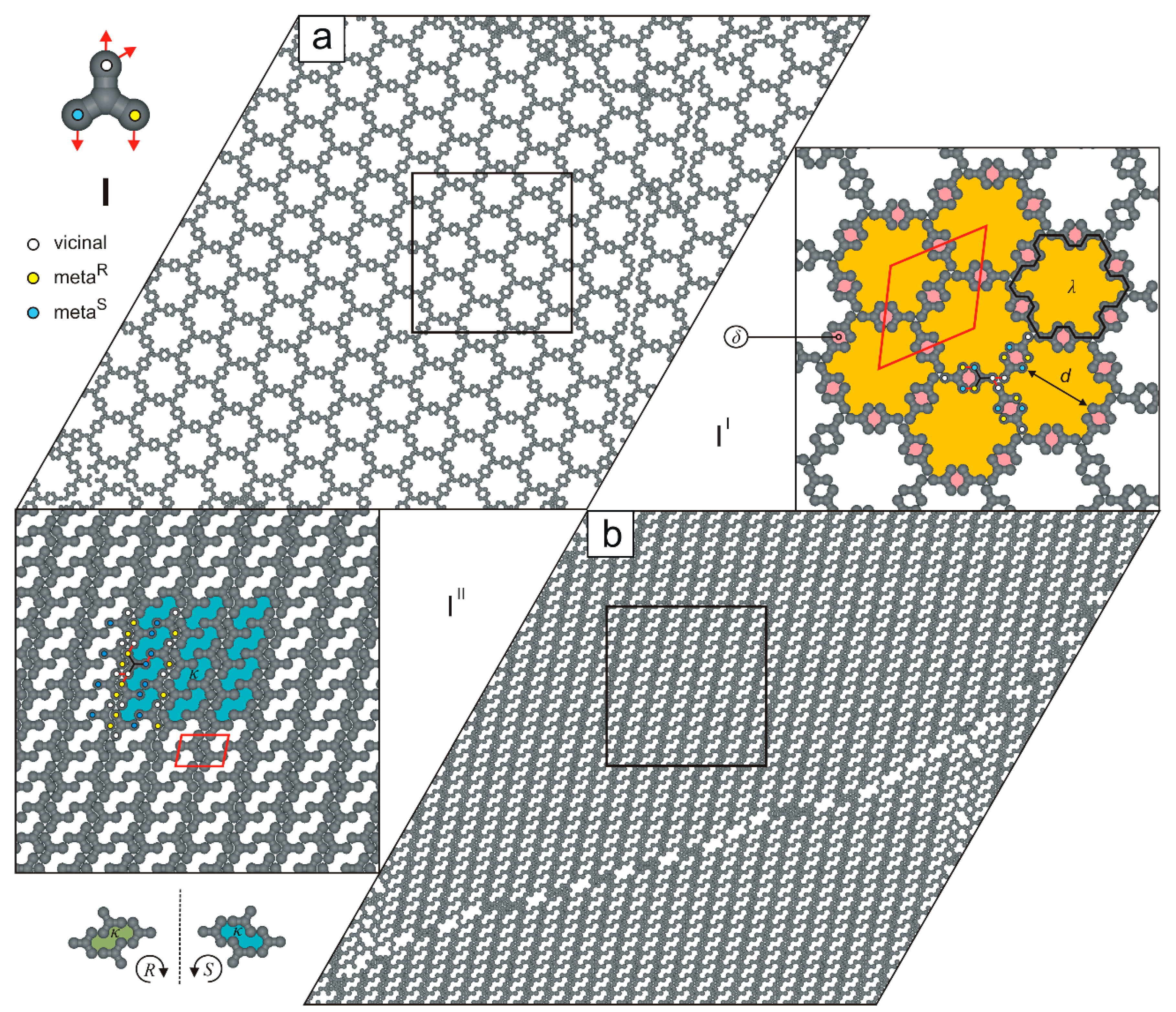
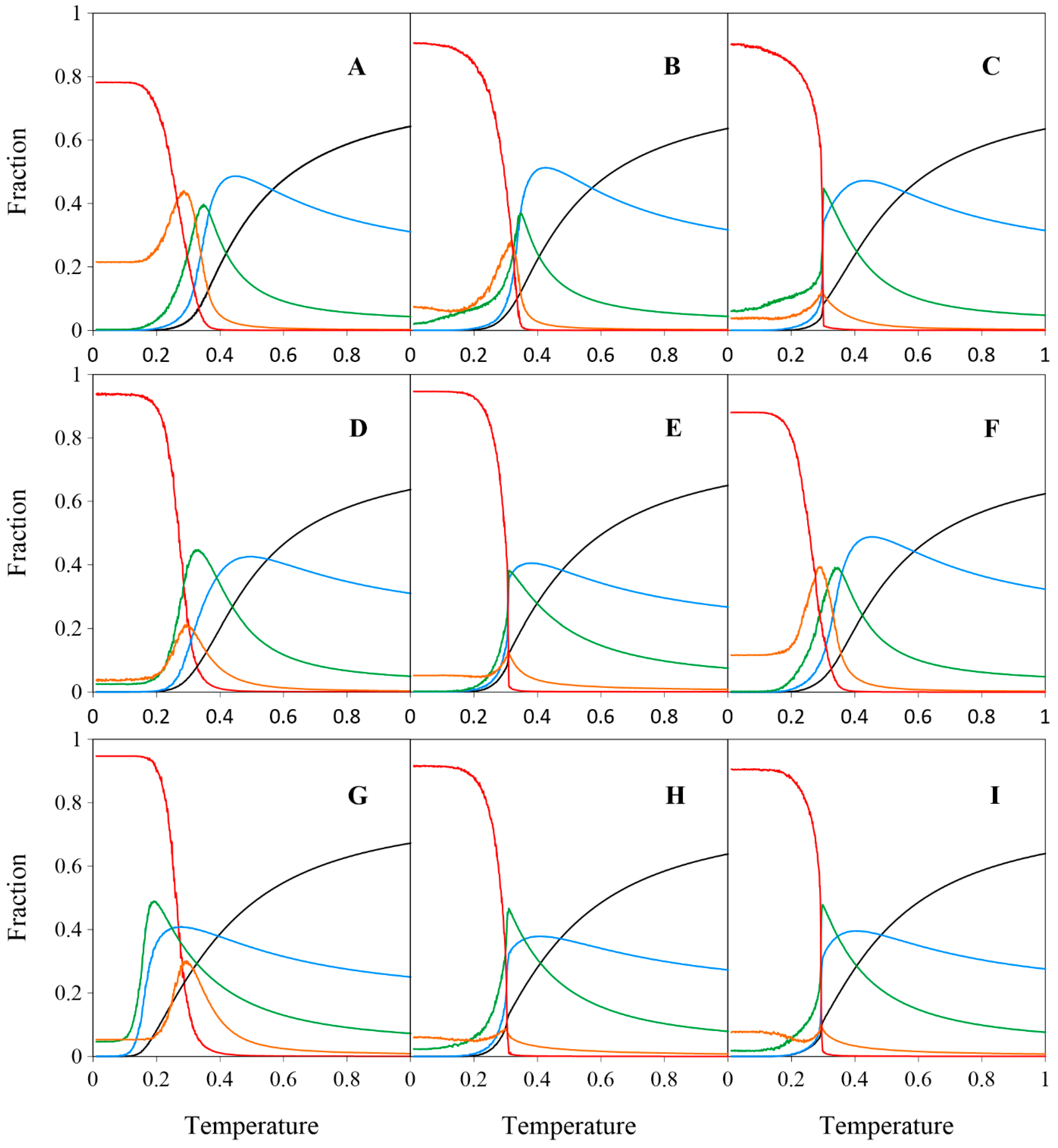
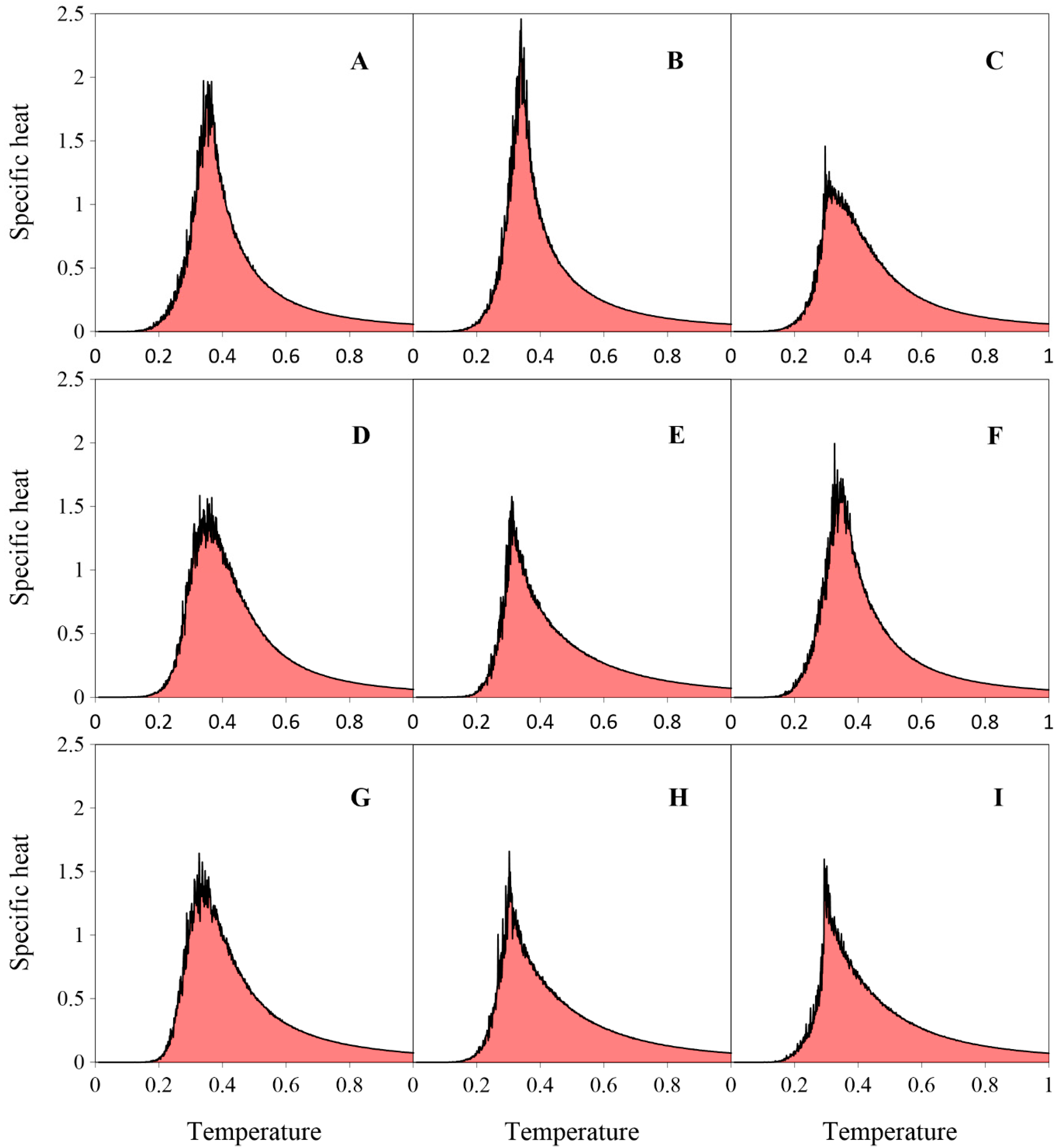
2. Results and Discussion
3. The Model and Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ye, Y.; Sun, W.; Wang, Y.; Shao, X.; Xu, X.; Cheng, F.; Li, J.; Wu, K. A Unified Model: Self-Assembly of Trimesic Acid on Gold. J. Phys. Chem. C 2007, 111, 10138–10141. [Google Scholar] [CrossRef]
- Wang, T.; Zhu, J. Confined On-Surface Organic Synthesis: Strategies and Mechanisms. Surf. Sci. Rep. 2019, 74, 97–140. [Google Scholar] [CrossRef]
- Shang, J.; Wang, Y.; Chen, M.; Dai, J.; Zhou, X.; Kuttner, J.; Hilt, G.; Shao, X.; Gottfried, J.M.; Wu, K. Assembling Molecular Sierpiński Triangle Fractals. Nat. Chem. 2015, 7, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Chinaud-Chaix, C.; Marchenko, N.; Fernique, T.; Tricard, S. Do Chemists Control Plane Packing, i.e., Two-Dimensional Self-Assembly, At All Scales? New J. Chem. 2023, 47, 7014–7025. [Google Scholar] [CrossRef]
- Urgel, J.I.; Écija, D.; Lyu, G.; Zhang, R.; Palma, C.A.; Auwärter, W.; Lin, N.; Barth, J.V. Quasicrystallinity Expressed in Two-Dimensional Coordination Networks. Nat. Chem. 2016, 8, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Vargas, L.; Kim, E.; Attias, A.J. Beyond “Decorative” 2D Supramolecular Self-Assembly: Strategies Towards Functional Surfaces for Nanotechnology. Mater. Horiz. 2017, 4, 570–583. [Google Scholar] [CrossRef]
- Cai, L.; Gao, T.; Wee, A.T.S. Topology Selectivity of a Conformationally Flexible Precursor Through Selenium Doping. Nat. Commun. 2024, 15, 3235. [Google Scholar] [CrossRef]
- Liu, Q.; Gao, Y.; Zhang, C. On-Surface Ullmann-Type Coupling Reactions of Aryl Halide Precursors with Multiple Substituted Sites. Nanomaterials 2025, 15, 646. [Google Scholar] [CrossRef]
- Ammon, M.; Sander, T.; Maier, S. On-Surface Synthesis of Porous Carbon Nanoribbons from Polymer Chains. J. Am. Chem. Soc. 2017, 139, 12976–12984. [Google Scholar] [CrossRef]
- Peyrot, D.; Silly, F. Toward Two-Dimensional Tessellation through Halogen Bonding between Molecules and On-Surface-Synthesized Covalent Multimers. J. Mol. Sci. 2023, 24, 11291. [Google Scholar] [CrossRef]
- Song, Y.; Wang, Y.; Jin, Q.; Zhou, K.; Shi, Z.; Liu, P.N.; Ma, Y.Q. Self-Assembly and Local Manipulation of Au-Pyridyl Coordination Networks on Metal Surfaces. ChemPhysChem 2017, 18, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Kuang, G.; Zhang, Q.; Shang, X.; Liu, P.N.; Lin, N. Self-Assembly of a Binodal Metal-Organic Framework Exhibiting a Demi-Regular Lattice. Faraday Discuss. 2017, 204, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lin, T.; Shi, Z.; Xia, F.; Dong, L.; Liu, P.N.; Lin, N. Structural Transformation of Two-Dimensional Metal–Organic Coordination Networks Driven by Intrinsic In-Plane Compression. J. Am. Chem. Soc. 2011, 133, 18760–18766. [Google Scholar] [CrossRef]
- Szabelski, P. Theoretical Modeling of the Structure Formation in Adsorbed Overlayers Comprising Molecular Building Blocks with Different Symmetries. Molecules 2025, 30, 866. [Google Scholar] [CrossRef]
- Nieckarz, D.; Nieckarz, K. Steering the Surface-Confined Self-Assembly of Multifunctional Star-Shaped Molecules. J. Phys. Chem. C 2023, 127, 12035–12054. [Google Scholar] [CrossRef]
- Nieckarz, D. Computer Simulations of Surface-Confined Cocrystals Cemented by 3-Fold Halogen Bonds. J. Phys. Chem. C 2025, 129, 9368–9382. [Google Scholar] [CrossRef]
- Szabelski, P.; Nieckarz, D.; Rżysko, W. Structure Formation in 2D Assemblies Comprising Functional Tripod Molecules with Reduced Symmetry. J. Phys. Chem. C 2017, 121, 25104–25117. [Google Scholar] [CrossRef]
- Baran, Ł.; Rżysko, W.; Szajnar, S. Archimedean Tessellation Found by the Variation of Building Blocks’ and Linkers’ Geometry: In Silico Investigations. J. Phys. Chem. C 2020, 124, 20101–20108. [Google Scholar] [CrossRef]
- Gorbunov, V.A.; Uliankina, A.I.; Myshlyavtsev, A.V. Off-Lattice Coarse-Grained Model of Surface-Confined Metal–Organic Architectures. J. Phys. Chem. C 2023, 127, 8281–8293. [Google Scholar] [CrossRef]
- Szabelski, P.; Rżysko, W.; Nieckarz, D. Directing the Self-Assembly of Tripod Molecules on Solid Surfaces: A Monte Carlo Simulation Approach. J. Phys. Chem. C 2016, 120, 13139–13147. [Google Scholar] [CrossRef]
- Fadeeva, A.I.; Gorbunov, V.A.; Solovyeva, O.S.; Stishenko, P.V.; Myshlyavtsev, A.V. Homologous Series of Flower Phases in Metal–Organic Networks on Au(111) Surface. J. Phys. Chem. C 2020, 124, 11506–11515. [Google Scholar] [CrossRef]
- Ibenskas, A.; Tornau, E.E. Modeling of Ribbon and Oblique Structures of Benzene-1,3,5-triyl-tribenzoic Acid. J. Phys. Chem. C 2020, 124, 18650–18659. [Google Scholar] [CrossRef]
- Björk, J. Reaction Mechanisms for On-Surface Synthesis of Covalent Nanostructures. J. Phys. Condens. Matter 2016, 28, 083002. [Google Scholar] [CrossRef]
- Jacquelín, D.K.; Soria, F.A.; Paredes-Olivera, P.A.; Patrito, E.M. Reactive Force Field-Based Molecular Dynamics Simulations on the Thermal Stability of Trimesic Acid on Graphene: Implications for the Design of Supramolecular Networks. ACS Appl. Nano Mater. 2021, 4, 9241–9253. [Google Scholar] [CrossRef]
- Nieckarz, D.; Szabelski, P. Simulation of the Self-Assembly of Simple Molecular Bricks into Sierpiński Triangles. Chem. Commun. 2014, 50, 6843–6845. [Google Scholar] [CrossRef]
- Rockel, D.; Korn, W.; Kohn, A.J. Manual of the Living Conidae; Mal de Mer Enterprises: Devon, UK, 1995. [Google Scholar]
- Wolfram, S. A New Kind of Science; Wolfram Media: Champaign, IL, USA, 2019. [Google Scholar]
- Li, C.; Zhang, X.; Li, N.; Wang, Y.; Jang, J.; Gu, G.; Zhang, Y.; Hou, S.; Peng, L.; Wu, K. Construction of Sierpiński Triangles up to the Fifth Order. J. Am. Chem. Soc. 2017, 139, 13749–13753. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Resenbluth, M.N.; Teller, A.H. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Replica No. | |||
|---|---|---|---|
| 1 | 1888 | 1952 | 1952 |
| 2 | 1891 | 1952 | 1953 |
| 3 | 1893 | 1953 | 1952 |
| 4 | 1892 | 1953 | 1952 |
| 5 | 1890 | 1952 | 1953 |
| 6 | 1890 | 1952 | 1952 |
| 7 | 1887 | 1953 | 1952 |
| 8 | 1888 | 1953 | 1952 |
| 9 | 1889 | 1952 | 1952 |
| 10 | 1892 | 1952 | 1952 |
| Average | 1890.00 | 1952.40 | 1952.20 |
| Standard deviation | 1.90 | 0.49 | 0.40 |
| Range | 6.00 | 1.00 | 1.00 |
| Replica No. | Under-Coordinated Molecules D | % |
|---|---|---|
| 1 | 59 | 5.90 |
| 2 | 64 | 6.40 |
| 3 | 66 | 6.60 |
| 4 | 59 | 5.90 |
| 5 | 58 | 5.80 |
| 6 | 60 | 6.00 |
| 7 | 67 | 6.70 |
| 8 | 64 | 6.40 |
| 9 | 61 | 6.10 |
| 10 | 54 | 5.40 |
| Average | 61.20 | 6.12 |
| Standard deviation | 3.82 | - |
| Range | 13 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisiecki, J.; Nieckarz, D. Surface-Confined Self-Assembly of Star-Shaped Tetratopic Molecules with Vicinal Interaction Centers. Molecules 2025, 30, 2656. https://doi.org/10.3390/molecules30122656
Lisiecki J, Nieckarz D. Surface-Confined Self-Assembly of Star-Shaped Tetratopic Molecules with Vicinal Interaction Centers. Molecules. 2025; 30(12):2656. https://doi.org/10.3390/molecules30122656
Chicago/Turabian StyleLisiecki, Jakub, and Damian Nieckarz. 2025. "Surface-Confined Self-Assembly of Star-Shaped Tetratopic Molecules with Vicinal Interaction Centers" Molecules 30, no. 12: 2656. https://doi.org/10.3390/molecules30122656
APA StyleLisiecki, J., & Nieckarz, D. (2025). Surface-Confined Self-Assembly of Star-Shaped Tetratopic Molecules with Vicinal Interaction Centers. Molecules, 30(12), 2656. https://doi.org/10.3390/molecules30122656