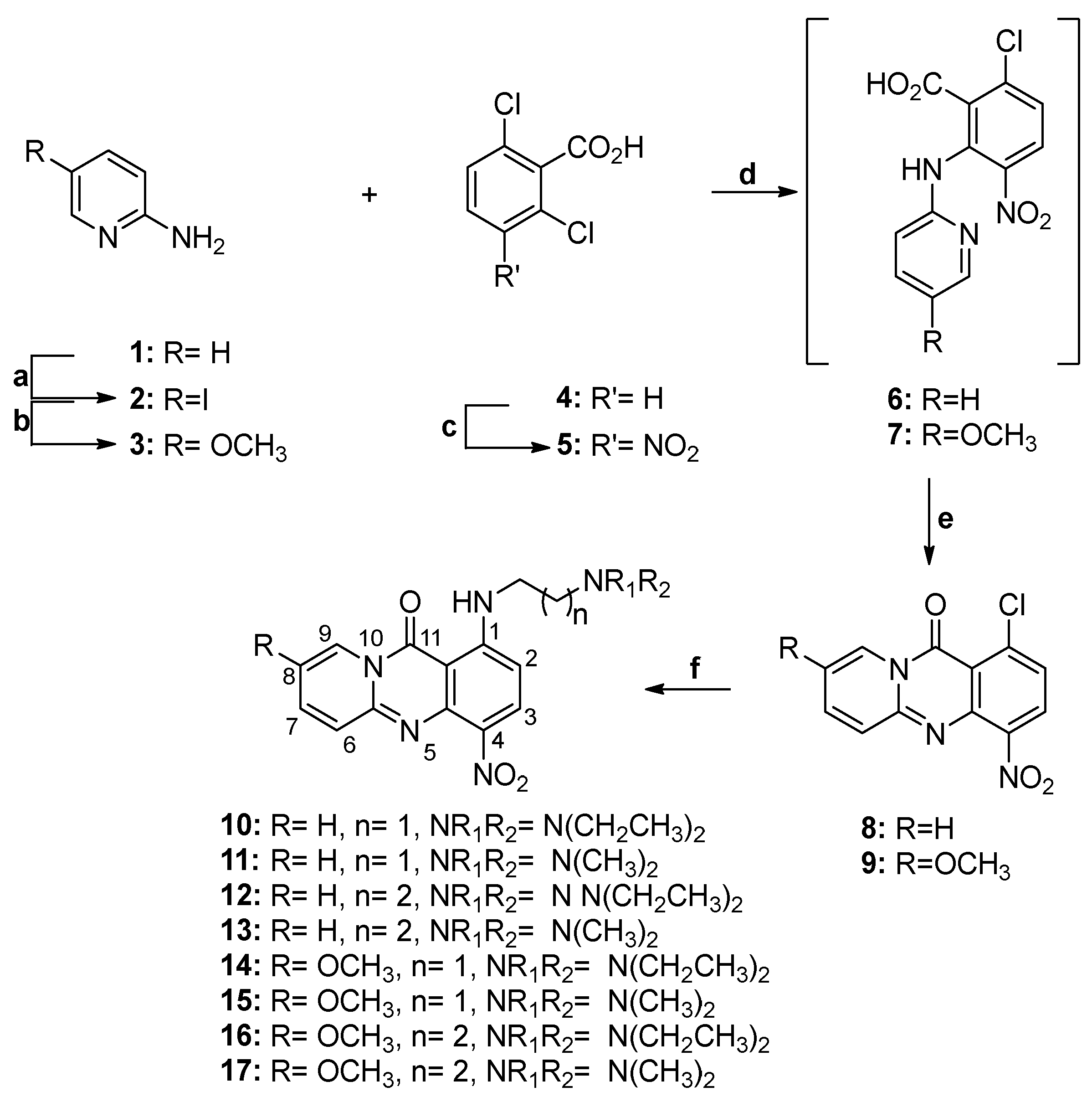
3.2. Synthesis of Compounds 2–57
2-Amino-5-iodopyridine (2)
To a suspension of 2-aminopyridine (4 g, 42.55 mmol,
1), ΚΙO
4 (1.43 g, 62.17 mmol), and Ι
2 (4.48 g, 84.56 mmol) in a mixture of acetic acid (99.6 mL) and water (5 mL) at 0 °C, sulfuric acid 98% (1.16 mL) was added dropwise, and the resulting mixture was stirred at 80 °C for 5 h. After completion of the reaction, the mixture was poured into ice-water and saponified by the addition of 20% ΝaOH solution (pH~10). The resulting precipitate was filtered off, washed with aqueous Νa
2S
2O
3 solution, and dried under vacuum over P
2O
5 to afford 8 g (67%) of the title compound
2, which was practically pure and used for the next step without further purification. Mp 126–130 °C (EtOAc) [
29].
1HNMR (400 MHz, DMSO-
d6) δ (ppm) 8.03 (d,
J = 2.1 Hz, 1H, H-6), 7.58 (dd,
J = 8.7, 2.2 Hz, 1H, H-4), 6.34 (d,
J = 8.7 Hz, 1H, H-3), 6.11 (brs, 2H, D
2Oexch., N
H2).
13C NMR (151 MHz, DMSO-
d6) δ (ppm) 157.4, 153.8, 145.3, 110.9, 77.8.
2-Amino-5-methoxy-pyridine (3)
A mixture of 2-amino-5-iodopyridine (6 g, 27.3 mmol,
2), Cs
2CO
3 (17.8 g, 54.6 mmol), CuI (520 mg, 2.73 mmol), and 1,10-phenanthroline (0.98 g, 5.46 mmol) in anhydrous MeOH (50 mL) was stirred at 120 °C in an autoclave apparatus for 12 h. The reaction mixture was then filtered, the filter cake was washed with EtOAc, the filtrate was washed with water, dried (anhydrous Na
2SO
4), and evaporated to dryness. Flash chromatography on silica gel using a mixture of cyclohexane/EtOAc 3:1 as the eluent afforded the title compound (1.5 g, 44%) as a brownish oil [
29].
1HNMR (400 MHz, CDCl
3) δ (ppm) 7.78 (d,
J = 3.2 Hz, 1H, H-6), 7.09 (dd,
J = 8.8, 3.2 Hz, 1H, H-4), 6.48 (d,
J = 9.2 Hz, 1H, H-3), 5.43 (brs, 2H, D
2O exch., N
H2), 3.68 (s, 3H, OC
H3).
13C NMR (101 MHz, CDCl
3) δ (ppm) 152.9, 149.6, 133.2, 125.7, 109.4, 56.3 (O
CH
3).
2,6-Dichloro-3-nitrobenzoic acid (5)
To a suspension of 2,6-dichlorobenzoic acid (2.00 g, 10.53 mmol) in c. H
2SO
4 (30 mL) was added dropwise to fuming HNO3 (0.6 mL, 14.28 mmol) at 0 °C, and the mixture was stirred at room temperature for 45 min. After completion of the reaction, the mixture was poured into ice, and the resulting solid was filtered, washed with cold water, and dried over P
2O
5 to afford 2.4 g (97%) of the title compound
5, which was used in the next step without any further purification. M.p.: 140–142 °C (H
2O) [
29].
1HNMR (400 MHz, CDCl
3) δ (ppm) 7.92 (d,
J = 8.8 Hz, 1H, H-4), 7.55 (d,
J = 8.8, 1H, H-5), 6.84 (brs, 1H, O
H).
13C NMR (101 MHz, CDCl
3) δ (ppm) 166.6, 146.6, 135.9, 135.4, 128.9, 126.9, 125.5.
1-Chloro-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (8)
To a solution of 2-aminopyridine (600 mg, 6.38 mmol, 1) in anhydrous DMF (40 mL) at 0 °C under an argon atmosphere, ΝaH (60% in mineral oil) (2.5 g, 62.5 mmol) was added in portions, and the resulting mixture was stirred for 30 min at room temperature. 2-Aminopyridine (1.5 g, 6.38 mmol) was added portion-wise at 0 °C, and the resulting suspension was stirred at 50 °C for 24 h. After completion of the reaction, the mixture was allowed to cool to room temperature, poured into ice-water, and acidified by the addition of 18% HCl solution (pH~3). The resulting precipitate was filtered off and dried under vacuum over P2O5 to afford 1.59 g (85%) of the title compound 8, which was practically pure and used for the next step without further purification. M.P.: 242–244 °C (EtOH). 1H NMR (600 MHz, DMSO-d6) δ (ppm): 8.84 (d, J = 7.1 Hz, 1H, H-9), 8.26 (d, J = 8.3 Hz, 1H, H-3), 7.88 (td, J = 8.4, 1.6 Hz, 1H, H-7), 7.58 (d, J = 8.4 Hz, 1H, H-2), 7.51 (d, J = 9.0 Hz, 1H, H-6), 7.20 (td, J = 8.4, 1.6 Hz, 1H, H-8). 13C NMR (151 MHz, DMSO-d6) δ (ppm): 155.5 (CO), 149.2 (C-5a), 145.0 (C-4a), 142.2 (C-1), 138.2 (C-7), 136.6 (C-3), 127.6 (C-3), 127.2 (C-9), 125.6 (C-6), 125.0 (C-2), 114.6 (C-11a), 114.0 (C-9).
8-Methoxy-4-nitro-1-chloro-11H-pyrido[2,1-b]quinazolin-11-one (9)
This compound was synthesized using a procedure analogous to that described for the preparation of compound 8, using compound 3. Yield: 91%. M.p.: 248–250 °C (EtOAc). 1H NMR (600 MHz, DMSO-d6) δ (ppm) 8.34 (d, J = 2.5 Hz, 1H, H-9), 8.27 (d, J = 8.3 Hz, 1H, H-3), 7.77 (dd, J = 7.07, 2.65 Hz, 1H, H-7), 7.61 (d, J = 8.3 Hz, 1H, H-2), 7.56 (d, J = 9.7 Hz, 1H, H-6), 3.93 (s, 3H, OCH3). 13C NMR (151 MHz, DMSO-d6) δ (ppm) 155.0 (CO), 149.3 (C-8), 146.6 (C-5a), 145.0 (C-4a), 141.6 (C-1), 136.4 (C-4), 133.6 (C-7), 127.3 (C-3), 126.9 (C-6), 125.2 (C-2), 113.2 (C-11a), 106.2 (C-9), 56.4 (OCH3).
1-((2-(Diethylamino)ethyl)amino)-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (10)
A solution of 8 (1 g, 3.64 mmol) and N,N-diethylethylenediamine (1 mL, 7.13 mmol) in THF (40 mL) was refluxed for 24 h. Upon cooling, the mixture was vacuum-evaporated and extracted with CH2Cl2 -water; the organic layer was dried (using Na2SO4) and evaporated to dryness. The residue was purified by column chromatography (silica gel, CH2Cl2-MeOH 10:0–9:1) to afford the title compound as a yellow solid (870 mg, 67% yield). M.p.: 123–125 °C (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 9.87 ((brs, 1H, D2O exch., NH), 8.82 (d, J = 7.2 Hz, 1H, H-9), 8.32 (d, J = 9.3 Hz, 1H, H-3), 7.62 (t, J = 6.61 Hz, 1H, H-7), 7.55 (d, J = 9.0 Hz, 1H, H-6), 6.94 (t, J = 6.27 Hz, 1H, H-8), 6.34 (d, J = 9.3 Hz, 1H, H-2), 3.41 (q, J = 6.3 Hz, 2H, NHCH2CH2), 2.83 (t, J = 6.3 Hz, 2H, NHCH2CH2), 2.66 (q, J = 7.1 Hz, 4H, N(CH2CH3)2), 1.09 (t, J = 7.1 Hz, 6H, N(CH2CH3)2). 13C NMR (151 MHz, CDCl3) δ (ppm): 159.4 (CO), 154.9 (C-1), 148.7 (C-5a), 145.5 (C-4a), 136.2 (C-7), 134.6 (C-3), 131.1 (C-4), 126.7 (C-6), 126.4 (C-9), 113.6 (C-8), 101.8 (C-2), 100.7 (C-11a), 50.7 (NHCH2CH2), 46.9 (NH(CH2CH3)2), 41.3 (NHCH2CH2), 11.8 (NH(CH2CH3)2). m/z: calcd. for C18H22N5O3+: [M1 + H]+ = 356.1717, found 356.1724.
1-((2-(Dimethylamino)ethyl)amino)-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (11)
This compound was synthesized using a procedure analogous to that described for the preparation of 10, using compound 8 and N,N-dimethylethylenediamine. Yield: 76%. M.p.: 160–162 °C (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 9.85 (brs, 1H, D2O exch., NH), 8.83 (d, J = 7.3 Hz, 1H, H-9), 8.33 (d, J = 9.3 Hz, 1H, H-3), 7.58–7.63 (m, 2H, H-6, H-7), 6.91 (t, J = 7.4 Hz, 1H, H-8), 6.34 (d, J = 9.3 Hz, 1H, H-2), 3.4 (q, J = 6.1 Hz, 2H, NHCH2CH2), 2.67 (t, J = 6.2 Hz, 2H, NHCH2CH2), 2.34 (s, 6H, N(CH3)2). 13C NMR (151 MHz, CDCl3) δ (ppm): 159.8 (CO), 155.1 (C-1), 148.9 (C-5a), 145.7 (C-4a), 136.2 (C-7), 134.9 (C-3), 131.6 (C-4), 127.0 (C-6), 126.6 (C-9), 113.7 (C-8), 101.8 (C-2), 101.0 (C-11a), 57.5 (NHCH2CH2), 45.6 (N(CH3)2), 41.2 (NHCH2CH2). m/z: calcd. for C16H18N5O3+: [M1 + H]+ = 328.1404, found 328.1411.
1-((3-(Diethylamino)propyl)amino)-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (12)
This compound was synthesized using a procedure analogous to that described for the preparation of 10, using compound 8 and N,N-diethyl-1,3-propanediamine. Yield: 56%. M.p.: 217–219 °C (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 9.76 (brs, 1H, D2O exch., NH), 8.80 (d, J = 7.2 Hz, 1H, H-9), 8.34 (d, J = 9.3 Hz, 1H, H-3), 7.58–7.63 (m, 2H, H-6, H-7), 6.95 (t, J = 6.7 Hz, 1H, H-8), 6.40 (d, J = 9.3 Hz, 1H, H-2), 3.43 (q, J = 6.2 Hz, 2H, NHCH2CH2CH2), 2.61–2.63 (m, 6H, NHCH2CH2CH2, N(CH2CH3)2), 1.94 (quintet, J = 6.9 Hz, 2H, NHCH2CH2CH2), 1.06 (t, J = 7.2 Hz, 6H, N(CH2CH3)2). 13C NMR (151 MHz, CDCl3) δ (ppm): 159.9 (CO), 155.3 (C-1), 148.8 (C-5a), 145.6 (C-4a), 136.3 (C-7), 134.9 (C-3), 131.4 (C-4), 127.0 (C-6), 126.4 (C-9), 113.9 (C-8), 101.8 (C-2), 100.8 (C-11a), 50.3 (NHCH2CH2CH2 ), 47.1 (N(CH2CH3)2), 41.7 (NHCH2CH2CH2), 26.4 (NHCH2CH2CH2), 11.6 (N(CH2CH3)2). m/z: calcd. for C19H24N5O3+: [M1 + H]+ = 370.1874, found 370.1867.
1-((3-(Dimethylamino)propyl)amino)-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (13)
This compound was synthesized using a procedure analogous to that described for the preparation of 10, using compound 8 and N,N-dimethyl-1,3-propanediamine. Yield 94%. M.p.: 134–136 °C (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 9.78 (brs, 1H, D2O exch., NH), 8.82 (d, J = 7.3 Hz, 1H, H-9), 8.35 (d, J = 9.4 Hz, 1H, H-3), 7.59–7.64 (m, 2H, H-6, H-7), 6.95 (t, J = 8.2 Hz, 1H, H-8), 6.42 (d, J = 9.4 Hz, 1H, H-2), 3.44 (q, J = 6.2 Hz, 2H, NHCH2CH2CH2), 2.45 (t, J = 6.9 Hz, 2H, NHCH2CH2CH2), 2.27 (s, 6H, N(CH3)2) 1.93 (quintet, J = 6.9 Hz, 2H, NHCH2CH2CH2). 13C NMR (151 MHz, CDCl3) δ (ppm): 159.9 (CO), 155.3 (C-1), 148.8 (C-5a), 145.7 (C-4a), 136.2 (C-7), 134.9 (C-3), 131.5 (C-4), 127.1 (C-6), 126.4 (C-9), 113.8 (C-8), 101.8 (C-2), 100.9 (C-11a), 57.1 (NHCH2CH2CH2), 45.6 (N(CH3)2), 41.56 (NHCH2CH2CH2), 26.9 (NHCH2CH2CH2). m/z: calcd. for C17H20N5O3+: [M1 + H]+ = 342.1561, found 342.1569.
1-((2-(Diethylamino)ethyl)amino)-8-methoxy-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (14)
This compound was synthesized using a procedure analogous to that described for the preparation of 10, using compound 9 and N,N-diethylethylenediamine. Yield: 63%. M.p.: 140–142 °C (EtoAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 9.94 (brs, 1H, D2O exch., NH), 8.38 (d, J = 9.3 Hz, 1H, H-3), 8.32 (s, J = 2.67 Hz, 1H, H-9), 7.58 (d, J = 9.7 Hz,1H, H-6), 7.45 (dd, J = 9.73, 2.74 Hz 1H, H-7), 6.37 (d, J = 9.4 Hz, 1H, H-2), 3.93 (s, 3H, OCH3), 3.41 (q, J = 6.4 Hz, 2H, NHCH2CH2), 2.83 (t, J = 6.3 Hz, 2H, NHCH2CH2), 2.65 (q, J = 7.1 Hz, 4H, N(CH2CH3)2), 1.10 (t, J = 7.12 Hz, 6H, N(CH2CH3)2). 13C NMR (151 MHz, CDCl3) δ (ppm): 159.4 (CO), 155.1 (C-1), 149.7 (C-8), 146.5 (C-5a), 145.3 (C-4a), 134.7 (C-3), 132.6 (C-7), 131.3 (C-4), 127.9 (C-6), 105.5 (C-9), 101.7 (C-2), 100.8 (C-11a), 56.5 (OCH3), 51.0 (NHCH2CH2), 47.1 (N(CH2CH3)2), 41.6 (NHCH2CH2), 12.0 (N(CH2CH3)2). m/z: calcd. for C19H24N5O4+: [M1 + H]+ = 386.1823, found 386.1830.
1-((2-(Dimethylamino)ethyl)amino)-8-methoxy-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (15)
This compound was synthesized using a procedure analogous to that described for the preparation of 10, using compound 9 and N,N-dimethylethylenediamine. Yield: 50%. M.p.: 193–195 °C (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 9.90 (brs, 1H, D2O exch., NHCO), 8.37 (d, J = 9.3 Hz, 1H, H-3), 8.34 (s, 1H, H-9), 7.58 (d, J = 9.7 Hz, 1H, H-6), 7.44 (dd, J = 9.7, 2.26Hz, 1H, H-7), 6.36 (d, J = 9.4, Hz, 1H, H-2), 3.92 (s, 3H, OCH3), 3.42 (q, J = 11.0, 6.0 Hz, 2H, NHCH2CH2), 2.70 (t, J= 9.7 Hz, 2H, NHCH2CH2), 2.36 (s, 6H, N(CH3)2). 13C NMR (151 MHz, CDCl3) δ (ppm): 159.5 (CO), 155.0 (C-1), 149.7 (C-8), 146.5 (C-5a), 145.2 (C-4a), 134.7 (C-3), 132.7 (C-7), 131.4 (C-4), 127.9 (C-6), 105.5 (C-9), 101.6 (C-2), 100.8 (C-11a), 57.6 (NHCH2CH2), 56.5 (OCH3), 45.6 (N(CH3)2), 41.2 (NHCH2CH2). m/z: calcd. for C17H20N5O4+: [M1 + H]+ = 358.1510, found 358.1517.
1-((3-(Diethylamino)propyl)amino)-8-methoxy-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (16)
This compound was synthesized using a procedure analogous to that described for the preparation of 10, using compound 9 and N,N-diethyl-1,3-propanediamine. Yield: 53%. M.p.: 230 °C (dec). 1H NMR (600 MHz, CDCl3) δ (ppm) 9.94 (t, 1H, J = 5.0 Hz, D2Oexch., NH), 8.36 (d, J = 9.2 Hz, 1H, H-3), 8.27 (d, J = 2.6 Hz, 1H, H-9), 7.59 (d, J = 9.8 Hz, 1H, H-6), 7.48 (dd, J = 9.8, 2.7 Hz, 1H, H-7), 6.39 (d, J = 9.3, Hz, 1H, H-2), 3.94 (s, 3H, OCH3), 3.56 (q, J = 6.2 Hz, 2H, NHCH2CH2CH2), 2.30 (quintet, J = 6.8 Hz, 2H, NHCH2CH2CH2), 3.07–3.09 (m, 6H, N(CH2CH3)2, NHCH2CH2CH2), 1.73 (t, J = 7.2 Hz, 6H, N(CH2CH3)2). 13C NMR (50 MHz, CDCl3) δ (ppm) 159.5 (CO), 155.1 (C-1), 149.8 (C-8), 146.4 (C-5a), 145.1 (C-4a), 134.7 (C-3), 132.7 (C-7), 131.2 (C-4), 127.9 (C-6), 105.2 (C-9), 101.6 (C-2), 100.6 (C-11a), 56.4 (OCH3), 51.0 (NHCH2CH2CH2), 46.9 (N(CH2CH3)2), 41.5 (NHCH2CH2CH2), 26.2 (NHCH2CH2CH2), 11.4 (N(CH2CH3)2). m/z: calcd. for C20H26N5O4+: [M1 + H]+ = 400.1979, found 400.1972.
1-((3-(Dimethylamino)propyl)amino)-8-methoxy-4-nitro-11H-pyrido[2,1-b]quinazolin-11-one (17)
This compound was synthesized using a procedure analogous to that described for the preparation of 10, using compound 9 and N,N-dimethyl-1,3-propanediamine. Yield: 61%. M.p.: 164–167 °C (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 9.80 (brs, 1H, D2O exch., NH), 8.34 (d, J = 9.2 Hz, 1H, H-3), 8.27 (d, J = 2.66 Hz, 1H, H-9), 7.55 (d, J = 9.7 Hz,1H, H-6), 7.44 (dd, J = 9.7, 2.7 Hz, 1H, H-7), 6.39 (d,J = 8.9 Hz, 1H, H-2), 3.92 (s, 3H, OCH3), 3.42 (q, J = 6.8 Hz, 2H, NHCH2CH2CH2), 2.45 (t, J = 6.9 Hz, 2H, NHCH2CH2CH2), 2.27 (s, 1H, 6H, N(CH3)2), 1.94 (quintet, J = 7.0 Hz, 2H, NHCH2CH2CH2). 13C NMR (50 MHz, CDCl3) δ (ppm): 159.5 (CO), 155.2 (C-1), 149.8 (C-8), 146.4 (C-5a), 145.1 (C-4a), 134.7 (C-3), 132.6 (C-7), 131.2 (C-4), 127.9 (C-6), 105.2 (C-9), 101.6 (C-2), 100.7 (C-11a), 57.0 (NHCH2CH2CH2), 56.4 (OCH3), 45.6 (N(CH3)2), 41.5 (NHCH2CH2CH2), 26.9 (NHCH2CH2CH2). m/z: calcd. for C18H22N5O4+: [M1 + H]+ = 372.1666, found 372.1673.
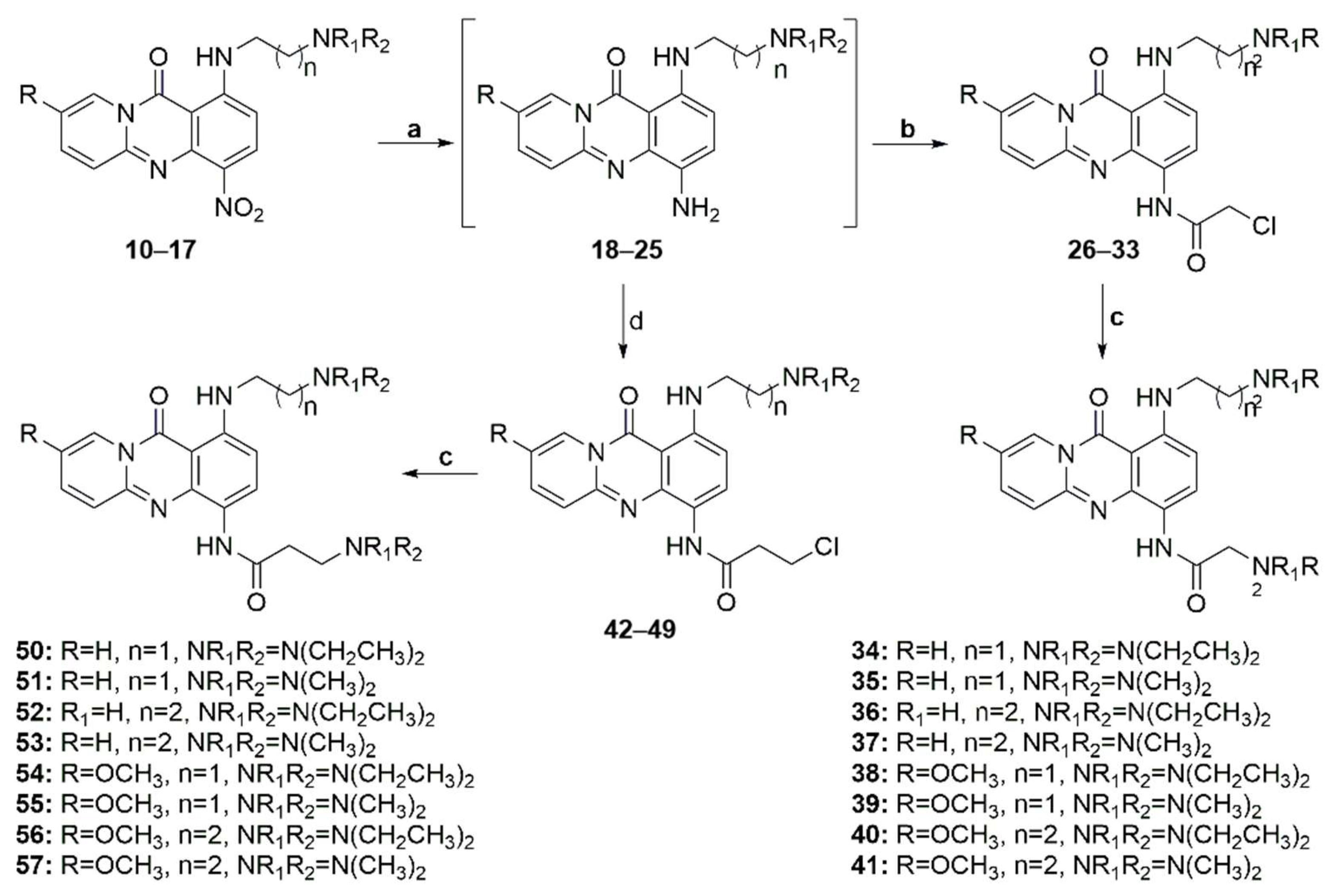
2-(Diethylamino)-N-(1-((2-(diethylamino)ethyl)amino)-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)acetamide (34)
A mixture of compound 10 (150 mg, 0.42 mmol), ammonium formate (400 mg, 6.30 mmol), and a catalytic amount of 10% palladium charcoal (45 mg) in anhydrous methanol (20 mL) was placed in a quartz tube, and the resulting suspension was heated under microwaves at 70 °C (150 W) for 2 min. The catalyst was filtered through celite, and the solvent was evaporated under a vacuum. The obtained crude compound 18 was immediately dissolved in THF (25 mL) and CH2Cl2 (5 mL) without any further purification, and Na2CO3 (1.6 g, 15.12 mmol) and chloroacetyl chloride (0.6 mL, 7.56 mmol) were added. The resulting suspension was stirred at room temperature for 12 h, and the organic solvent was evaporated under vacuum. The residue was dissolved in CH2Cl2, washed with a 10% Na2CO3 solution and water, dried over Na2SO4, and evaporated. Without further purification, the residue was dissolved in a mixture of abs. ethanol (20 mL) and CH2Cl2 (2 mL), diethylamine (3.5 mL, 33.8 mmol) was added. The resulting solution was then refluxed for 72 h. Upon cooling, the mixture was vacuum-evaporated and extracted with CH2Cl2-water. The organic layer was dried (using Na2SO4) and evaporated to dryness. The residue was purified by column chromatography (silica gel, CH2Cl2-MeOH 97:3–90:10) to afford the title compound as an orange solid (80 mg, 74%). M.p.: 90–92 °C (EtOAc). 1H NMR (400 MHz, CDCl3) δ (ppm): 10.81 (brs, 1H, D2O exch., NHCO), 8.80 (d, J = 8.9 Hz, 1H, H-3), 8.76 (d, J = 7.4 Hz, 1H, H-9), 8.49 (brs, 1H, D2O exch., NHCH2CH2,) 7.40 (t, J = 7.8 Hz, 1H, H-7), 7.34 (d, J = 9.0 Hz, 1H, H-6), 6.77 (t, J = 6.7 Hz, 1H, H-8), 6.47 (d, J = 8.9 Hz, 1H, H-2), 3.50 (brs, 2H, NHCH2CH2), 3.24 (s, 2H, COCH2), 2.98 (t, J = 6.7 Hz, 2H, NHCH2CH2), 2.85 (q, J = 7.1 Hz, 4H, CH2CH2N(CH2CH3)2), 2.70 (q, J = 7.1 Hz, 4H, COCH2N(CH2CH3)2), 1.20–1.15 (m, 12H, CH2CH2N(CH2CH3)2, COCH2N(CH2CH3)2). 13C NMR (50 MHz, CDCl3) δ (ppm): 169.7 (NHCO), 160.0 (CO), 146.7 (C-4a, C-5a), 139.6 (C-1), 134.2 (C-7), 126.5 (C-9), 126.1 (C-3), 126.0 (C-6), 121.0 (C-4), 112.0 (C-8), 103.0 (C-2), 101.7 (C-11a), 59.2 (COCH2), 51.3 (CH2CH2N(CH2CH3)2), 48.6 (COCH2N(CH2CH3)2), 47.2 (CH2CH2N(CH2CH3)2), 41.2 (CH2CH2N(CH2CH3)2), 12.9 (COCH2N(CH2CH3)2), 11.7 (CH2CH2N(CH2CH3)2). m/z: calcd. for C24H35N6O2+: [M1 + H]+ = 439.2816, found 439.2811.
2-(Dimethylamino)-N-(1-((2-(dimethylamino)ethyl)amino)-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)acetamide (35)
This compound was synthesized using a procedure analogous to that described for the preparation of 34, using compound 11 and dimethylamine. Yield: 47%. M.p.: 230 °C (dec) (EtOAc). 1H NMR (400 MHz, CDCl3) δ (ppm): 10.40 (brs, 1H, D2O exch., NHCO), 8.73 (d, J = 7.2 Hz, 1H, H-3), 8.68 (d, J = 8.8 Hz, 1H, H-9), 8.45 (t,J = 4.5 Hz, 1H, D2O exch., NHCH2CH2,), 7.46 (t, J = 9.0 Hz, 1H, H-7), 7.35 (d, J = 9.0 Hz, 1H, H-6), 6.81 (t, J = 10.04 Hz, 1H, H-8), 6.47 (d, J = 8.9 Hz, 1H, H-2), 3.64 (q, J = 6.3 Hz, 2H, NHCH2CH2), 3.17 (s, 2H, COCH2), 3.02 (t, J = 6.3 Hz, 2H, NHCH2CH2), 2.63 (s, 6H, CH2CH2N(CH3)2), 2.45 (s, 6H, COCH2N(CH3)2). 13C NMR (50 MHz, CDCl3+1drop of MeOD) δ (ppm): 168.5 (NHCO), 160.1 (CO), 146.9 (C-5a), 145.6 (C-4a), 140.2 (C-1), 134.7 (C-7), 126.3 (C-9, C-3, C-6), 121.2 (C-4), 112.7 (C-8), 102.6 (C-2), 101.7 (C-11a), 63.6 (COCH2), 56.4 (CH2CH2N(CH3)2), 46.0 (COCH2N(CH3)2), 44.0 (CH2CH2N(CH3)2), 39.0 CH2CH2N(CH3)2). m/z: calcd. for C20H27N6O2+: [M1 + H]+ = 383.2190, found 383.2199.
2-(Diethylamino)-N-(1-((3-(diethylamino)propyl)amino)-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)acetamide (36)
This compound was synthesized using a procedure analogous to that described for the preparation of 34, using compound 12 and diethylamine. Yield: 35%. M.p.: 86–88 °C (EtOAc). 1H NMR (400 MHz, CDCl3) δ (ppm): 10.70 (brs, 1H, D2O exch., NHCO), 8.67 (d, J = 8.9 Hz, 1H, H-3), 8.63 (d, J = 7.4 Hz, 1H, H-9), 8.26 (t, J = 4.7 Hz, 1H, D2O exch., NHCH2CH2CH2), 7.39 (t, J = 7.8 Hz, 1H, H-7), 7.23 (d, J = 7.7 Hz, 1H, H-6), 6.71 (t, J = 7.0 Hz, 1H, H-8), 6.34 (d, J = 10.8 Hz, 1H, H-2), 3.27 (q, J = 6.8 Hz, 2H, NHCH2CH2CH2), 3.16 (s, 2H, COCH2), 2.96 (t, 2H, J = 7.1 Hz, NHCH2CH2CH2), 2.84 (q, J = 7.1 Hz, 4H, CH2CH2CH2N(CH2CH3)2), 2.62 (q, J = 7.2 Hz, 4H, COCH2N(CH2CH3)2), 2.12 (quintet, J = 7.59, 2H, NHCH2CH2CH2), 1.27 (t, J = 7.12 Hz, 6H, CH2CH2CH2N(CH2CH3)2), 1.09 (t, J = 7.1 Hz, 6H, COCH2N(CH2CH3)2,). 13C NMR (50 MHz, CDCl3) δ (ppm): 170.0 (HNCO), 160.0 (CO), 146.6 (C-5a), 146.1 (C-4a), 139.7 (C-1), 134.5 (C-7), 126.1 (C-9), 125.9 (C-3), 125.7 (C-6), 121.2 (C-4), 112.4 (C-8), 102.6 (C-2), 101.6 (C-11a), 58.2 (COCH2), 48.8 (NHCH2CH2CH2), 47.9 (COCH2N(CH2CH3)2), 46.2 (NHCH2CH2CH2N(CH2CH3)2), 39.7 (NHCH2CH2CH2), 22.6 (NHCH2CH2CH2), 12.1 (COCH2CH2N(CH2CH3), 8.3 (CH2CH2CH2N(CH2CH3)2). m/z: calcd. for C25H37N6O2+: [M1 + H]+ = 453.2973, found 453.2965.
2-Dimethylamino-N-(1-((3-(dimethylamino)propyl)amino)-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)acetamide (37)
This compound was synthesized using a procedure analogous to that described for the preparation of 34, using compound 13 and dimethylamine. Yield: 27%. M.p.: 98–100 °C (EtOAc). 1H NMR (400 MHz, CDCl3) δ (ppm): 10.40 (brs, 1H, D2O exch., NHCO), 8.71–8.74 (m, 2H, H-3, H-9), 8.36 (brs, 1H, D2O exch., NHCH2CH2CH2, D2Oexchange), 7.43 (t, J = 6.3 Hz, 1H, H-7), 7.36 (d, J = 9.0 Hz, 1H, H-6), 6.77 (t, J = 7.5 Hz, 1H, H-8), 6.46 (d, J = 9.0 Hz, 1H, H-2), 3.30 (q, J = 12.1, 6.7 Hz, 2H, NHCH2CH2CH2), 3.16 (s, 2H, COCH2) 2.43–2.47 (m, 8H, NHCH2CH2CH2, CH2CH2CH2N(CH3)2), 2.29 (s, 6H, COCH2N(CH3)2), 1.92 (quintet, J = 7.12 Hz, 2H, NHCH2CH2CH2). 13C NMR (50 MHz, CDCl3) δ (ppm): 168.4 (HNCO), 160.3 (CO), 147.2 (C-5a), 146.8 (C-4a), 139.9(C-1), 134.2 (C-7), 126.7 (C-9), 126.5 (C-3, C-6), 120.7 (C-4), 112.2 (C-8), 103.1 (C-2), 101.7 (C-11a), 64.4 (COCH2), 57.3 (NHCH2CH2CH2), 46.4 (COCH2N(CH3)2), 45.7 (NHCH2CH2CH2N(CH3)2), 41.5 (NHCH2CH2CH2), 27.3 (NHCH2CH2CH2). m/z: calcd. for C21H29N6O2+: [M1 + H]+ = 397.2347, found 397.2354.
2-Diethylamino-N-(1-((2-(diethylamino)ethyl)amino)-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)acetamide (38)
A suspension of 14 (200 mg, 0.52 mmol), ammonium formate (50 mg, 0.78 mmol), and activated zinc (136 mg, 2.08 mmol) in anhydrous methanol (10 mL) was stirred at room temperature for 1 h. After completion of the reaction, the mixture was filtered through a Celite pad, and the filtrate was evaporated to dryness. Without further purification, the obtained crude 22 was immediately dissolved in a mixture of THF (25 mL) and CH2Cl2 (5 mL), and Na2CO3 (170 mg, 1.56 mmol) and chloroacetyl chloride (0.06 mL, 0.78 mmol) were added to this solution. The resulting suspension was stirred for 15 min at room temperature. The solvent was then evaporated under vacuum, and the residue was dissolved in CH2Cl2, washed with water, dried over Na2SO4, and evaporated to dryness to obtain the oil corresponding to compound 30. Without further purification, the residue was dissolved in a mixture of absolute. ethanol (20 mL) and) and2 (2 mL), and diethylamine (3.5 mL, 33.8 mmol) was added to this solution. The resulting mixture was then refluxed for 72 h. Upon cooling, the mixture was vacuum-evaporated and extracted with CH2Cl2 -water; the organic layer was dried (anhydrous Na2SO4) and evaporated to dryness. The residue was purified by column chromatography (silica gel, CH2Cl2-MeOH 100:0–75:25) to afford the title compound as an orange solid (20 mg, 18% yield). M.p.: 144–146 °C (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 10.76 (brs, 1H, D2O exch., NHCO), 8.77 (d, J = 8.9 Hz, 1H, H-3), 8.44 (t, J = 4.6 Hz, 1H, D2O exch., NHCH2CH2), 8.22 (d, J = 2.6 Hz, 1H, H-9), 7.29 (d, J = 9.6 Hz, 1H, H-6), 7.26 (dd, J = 9.75, 2.57 Hz, 1H, H-7), 6.44 (d, J = 8.9 Hz, 1H, H-2), 3.87 (s, 3H, OCH3), 3.405 (q, J = 6.2 Hz, 3H, NHCH2CH2), 3.22 (s, 2H, COCH2), 3.01 (t, J = 6.5 Hz, 2H, NHCH2CH2), 2.62–2.68 (m, 4H, NHCH2CH2N(CH2CH3)2, COCH2N(CH2CH3)2), 1.21–1.07 (m, 12H, HNCH2CH2N(CH2CH3)2, COCH2N(CH2CH3)2). 13C NMR (151 MHz, CDCl3) δ (ppm): 169.7 (NHCO), 159.8 (CO), 148.4 (C-8), 146.3 (C-5a), 144.3 (C-4a), 139.5 (C-1), 131.0 (C-7), 127.0 (C-6), 125.3 (C-3), 121.1 (C-4), 105.0 (C-9), 102.9 (C-2), 101.6 (C-11a), 59.2 (COCH2), 56.3 (OCH3), 51.4 (NHCH2CH2), 48.7 (COCH2N(CH2CH3)2), 47.3 (NHCH2CH2N(CH2CH3)2), 41.3 (NHCH2CH2), 12.9 (COCH2N(CH2CH3)2), 11.7 (NHCH2CH2N(CH2CH3)2). m/z: calcd. for C25H37N6O3+: [M1 + H]+ = 469.2922, found 469.2915.
2-Dimethylamino-N-(1-((2-(dimethylamino)ethyl)amino)-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)acetamide (39)
This compound was synthesized using a procedure analogous to that described for the preparation of 38, using compound 15 and dimethylamine. Yield: 35%. M.p.: 108–110 °C (dec) (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 10.38 (s, br,1H,NHCO, D2Oexchange), 8.69 (d, J = 8.8 Hz, 1H, H-3), 8.52 (s, br, 1H, NHCH2CH2, D2Oexchange), 8.24 (d, J= 2.5 Hz, 1H, H-9), 7.35 (d, J = 9.8 Hz, 1H, H-6), 7.29 (dd, J = 9.7, 2.6 Hz, 1H, H-7), 6.45 (d, J = 8.9 Hz, 1H, H-2), 3.89 (s, 3H, OCH3), 3.51 (q, J = 4.8 Hz, 2H, NHCH2CH2), 3.17 (s, 2H, COCH2), 2.89 (t, 2H, J = 6.0 Hz, NHCH2CH2), 2.71 (s, 6H, COCH2N(CH3)2), 2.45 (s, 6H, NHCH2CH2N(CH3)2). 13C NMR (151 MHz, MeOD-d4) δ (ppm): (NHCO), 161.0 (CO), 150.7 (C-8), 147.8 (C-5a), 146.6 (C-4a), 142.4 (C-1), 133.3 (C-7), 128.8 (C-6), 127.7 (C-3), 121.6 (C-4), 106.2 (C-9), 103.5 (C-2), 101.2 (C-11a), 60.3 (COCH2), 57.2 (NHCH2CH2), 56.8 (OCH3), 44.6 (NHCH2CH2N(CH3)2), 44.1 (COCH2N(CH3)2), 39.2 (NHCH2CH2). m/z: calcd. for C21H29N6O3+: [M1 + H]+ = 413.2296, found 413.2290.
2-Diethylamino-N-(1-((3-(diethylamino)propyl)amino)-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)acetamide (40)
This compound was synthesized using a procedure analogous to that described for the preparation of 38, using compound 16 and diethylamine. Yield: 29%. M.p.: 148–150 °C (dec) (EtOAc). 1H NMR (400 MHz, CDCl3) δ (ppm): 10.77 (brs, 1H, D2O exch., NHCO), 8.75 (d, J = 9.3 Hz, 1H, H-3), 8.39 (brs, 1H, D2O exch., NHCH2CH2CH2, D2Oexchange), 8.19 (s, 1H, H-9), 7.37 (d, J = 9.7 Hz, 1H, H-6), 7.30 (dd, J = 9.8, 2.6 Hz, 1H, H-7), 6.40 (d,J = 11.5 Hz, 1H, H-2), 6.43 (d, J = 10 Hz, 1H, H-2), 3.91 (s, 3H, OCH3), 3.41 (q, J = 2.6 Hz, 2H, NHCH2CH2CH2), 3.12 (s, 2H, COCH2), 3.09 (m, 6H, NHCH2CH2CH2, CH2CH2CH2N(CH2CH3)2), 2.70 (q, J = 7.1 Hz, 4H, COCH2N(CH2CH3)2), 2.20 (quintet, J = 8.02 Hz, 2H, NHCH2CH2CH2), 1.36 (t, J = 7.3 Hz, 6H, CH2CH2CH2N(CH2CH3)2), 1.16 (t, J = 7.11 Hz, 6H, COCH2N(CH2CH3)2). 13C NMR (50 MHz, CDCl3) δ (ppm): 169.5 (NHCO), 160.050 (CO), 148.6 (C-8), 146.1 (C-5a), 144.5 (C-4a), 139.5 (C-1), 131.3 (C-7), 126.8 (C-6), 126.2 (C-3), 121.5 (C-4), 104.9 (C-9), 102.6 (C-2), 101.6 (C-11a), 56.3 (COCH2), 49.5 (OCH3), 48.5 (NHCH2CH2CH2), 46.4 (COCH2CH2N(CH2CH3)2), 46.4 (CH2CH2CH2N(CH2CH3)2), 41.1 (NHCH2CH2CH2), 24.4 (NHCH2CH2CH2), 10.5 (COCH2CH2N(CH2CH3)2), 9.9 (CH2CH2CH2N(CH2CH3)2). m/z: calcd. for C26H39N6O3+: [M1 + H]+ = 483.3078, found 483.3084.
2-Dimethylamino-N-(1-((3-(dimethylamino)propyl)amino)-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)acetamide (41)
This compound was synthesized using a procedure analogous to that described for the preparation of 38, using compound 17 and dimethylamine. Yield: 35%. M.p.: 210 °C (dec) (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 10.37 (brs, 1H, D2O exch., NHCO), 8.69 (d, J = 8.8 Hz, 1H, H-3), 8.21 (s, J = 2.5 Hz, 1H, H-9), 7.38 (d, J = 9.8 Hz, 1H, H-6), 7.31 (dd, J = 9.8, 2.6 Hz, 1H, H-7), 6.40 (d, J = 8.9Hz, 1H,H-2), 3.91 (s, 3H, OCH3), 3.42 (q, J = 6.4 Hz, 2H, NHCH2CH2CH2), 3.20 (s, 2H, COCH2), 3.09 (t, J = 4.06 Hz, 2H, NHCH2CH2CH2), 2.76 (s, 6H, NHCH2CH2CH2N(CH3)2), 2.49 (s, 6H, COCH2N(CH3)2), 2.24 (quintet, J = 7.9 Hz, 2H, NHCH2CH2CH2). 13C NMR (50 MHz, CDCl3+1drop Acetone-d6) δ (ppm): 168.4 (NHCO), 160.0 (CO), 148.8 (C-8), 145.9 (C-5a), 144.6 (C-4a), 139.8 (C-1), 131.2 (C-7), 127.3 (C-6), 125.68 (C-3), 121.4 (C-4), 104.8 (C-9), 102.8 (C-2), 101.9 (C-11a), 64.2 (COCH2), 56.3 (NHCH2CH2CH2), 56.1 (OCH3), 46.1 (COCH2N(CH3)2), 43.0 (NHCH2CH2CH2N(CH3)2), 40.4 (NHCH2CH2CH2), 24.2 (NHCH2CH2CH2). m/z: calcd. for C22H31N6O3+: [M1 + H]+ = 427.2452, found 427.2457.
3-(Diethylamino)-N-(1-((2-(diethylamino)ethyl)amino)-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)propanamide (50)
This compound was synthesized using a procedure analogous to that described for the preparation of 34, using compound 10, 3-cloropropionyl chloride, and diethylamine. Yield: 32%. 1H NMR (400 MHz, CDCl3) δ (ppm): 9.87 (brs, 1H, D2O exch., NHCO), 8.69 (d, J = 7.3 Hz, 1H, H-3), 8.61 (d, J = 8.8 Hz, 1H, H-9), 8.43 (t, J = 4.6 Hz, 1H, D2O exch., NHCH2CH2), 7.45 (t, J = 5.1 Hz, 1H, H-7), 7.35 (d, J = 9.1 Hz, 1H, H-6), 6.76 (t, J = 6.4 Hz, 1H, H-8), 6.41 (d, J = 8.8 Hz, 2H, H-2), 3.19 (q, J = 6.5 Hz, 2H, NHCH2CH2), 2.89–3.07 (m, 10H, NHCH2CH2, COCH2CH2, NHCH2CH2N(CH2CH3)2, 2.77 (q, J = 7. 1Hz, 4H, COCH2CH2N(CH2CH3)2), 1.27 (t, J = 7.1 Hz, 6H, NHCH2CH2N(CH2CH3)2, 1.14 (t, J = 6.9 Hz, 6H, COCH2CH2N(CH2CH3)2). 13C NMR (50 MHz, CDCl3) δ (ppm): 169.7 (HNCO), 160.0 (CO), 146.7 (C-5a), 146.6 (C-4a), 139.5 (C-1), 134.3 (C-7), 126.9 (C-3), 126.5 (C-9), 125.7 (C-6), 121.4 (C-4), 111.9 (C-8), 102.9 (C-2), 101.6 (C-11a), 51.3 (NHCH2CH2,) 48.6 (COCH2CH2), 47.2 (COCH2CH2N(CH2CH3)2), 46.5 (NHCH2CH2N(CH2CH3)2), 41.1 (NHCH2CH2), 34.3(COCH2CH2), 11.7 (COCH2CH2N(CH2CH3)2), 10.8 (NHCH2CH2N(CH2CH3)2). m/z: calcd. for C25H37N6O2+: [M1 + H]+ = 453.2973, found 453.2978.
3-(Dimethylamino)-N-(1-((2-(dimethylamino)ethyl)amino)-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)propanamide (51)
This compound was synthesized using a procedure analogous to that described for the preparation of 34, using compound 11, 3-cloropropionyl chloride, and dimethylamine. Yield: 38%. M.p.: 258–260 °C (EtOAc). 1H NMR (400 MHz, CDCl3) δ(ppm): 10.25 (brs, 1H, D2O exch., NHCO), 8.64 (d, J = 7.3 Hz, 1H, H-3), 8.55 (d, J = 8.9 Hz, 1H, H-9), 8.37 (brs, 1H, D2O exch., 1H, NHCH2CH2CH2), 7.39 (t, J = 8.1 Hz, 1H, H-7), 7.27 (d, J = 7.8 Hz, 1H, H-6), 6.72 (t, J = 6.9 Hz, 1H, H-8), 6.34 (d, J = 9.0 Hz 1H, H-2), 3.29 (q, J = 7.0 Hz, 2H, NHCH2CH2), 2.82 (t, J = 6.6 Hz, 2H, COCH2CH2), 2.65–2.69 (m, 4H, NHCH2CH2, COCH2CH2,), 2.41 (s, 6H, CH2CH2CH2N(CH3)2), 2.28 (s, 6H, COCH2CH2N(CH3)2). 13C NMR (151 MHz, CDCl3) δ (ppm): 168.8 (HNCO), 160.1 (CO), 146.8 (C-5a), 146.7 (C-4a), 139.7 (C-1), 134.6 (C-7), 126.8 (C-3), 126.5 (C-9), 125.9 (C-6), 121.3 (C-4), 112.9 (C-8), 102.8 (C-2), 101.7 (C-11a), 57.8 (NHCH2CH2), 54.7 (COCH2CH2), 45.4 (COCH2CH2N(CH3)2), 44.6 (NHCH2CH2CH2N(CH3)2), 40.8 (NHCH2CH2), 34.2 (COCH2CH2). m/z: calcd. for C21H29N6O2+: [M1 + H]+ = 397.2347, found 397.2341.
3-(Diethylamino)-N-(1-((3-(diethylamino)propyl)amino)-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)propanamide (52)
This compound was synthesized using a procedure analogous to that described for the preparation of 34, using compound 12, 3-cloropropionyl chloride, and diethylamine. Yield: 40%. 1H NMR (400 MHz, CDCl3) δ (ppm): 10.70 (brs, 1H, D2O exch., NHCO), 8.78 (d, J = 8.9 Hz, 1H, H-3), 8.69 (d, J = 6.9 Hz, 1H, H-9), 8.34 (brs, 1H, D2O exch., NHCH2CH2CH2), 7.38 (t, J = 8.4, 7.3 Hz, 1H, H-7), 7.27 (d, J = 7.7 Hz, 1H, H-6), 6.71 (t, J = 6.3 Hz, 1H, H-8), 6.42 (d, J= 8.7 Hz, 1H, H-2), 3.25 (q, J = 5.9 Hz, 2H, NHCH2CH2CH2), 2.82 (t, J = 6.3 Hz, 2H, COCH2CH2), 2.67 (q, J = 7.0 Hz, 4H, CH2CH2CH2N(CH2CH3)2) 2.50–2.58 (m, 8H, NHCH2CH2CH2, COCH2CH2, COCH2CH2N(CH2CH3)2), 1.86 (quintet, J = 6.9 Hz, 2H, NHCH2CH2CH2), 1.10 (t, J = 7.1 Hz, 6H, CH2CH2CH2N(CH2CH3)2), 1.02 (t, J = 7.1 Hz, 6H, COCH2CH2N(CH2CH3)2). 13C NMR (50 MHz, CDCl3) δ (ppm): 170.6 (HNCO), 160.2 (CO), 146.9 (C-5a), 146.4 (C-4a), 139.5 (C-1), 134.1 (C-7), 127.2 (C-3), 126.4 (C-9), 125.8 (C-6), 121.7 (C-4), 111.9 (C-8), 103.1 (C-2), 101.5 (C-11a), 50.6 (NHCH2CH2CH2), 48.8 (COCH2CH2), 47.0 (COCH2CH2N(CH2CH3)2), 46.6 (CH2CH2CH2N(CH2CH3)2), 41.6 (NHCH2CH2CH2), 34.9 (COCH2CH2), 26.6 (NHCH2CH2CH2), 11.8 (COCH2CH2N(CH2CH3)2), 11.5 (NHCH2CH2CH2N(CH2CH3)2). m/z: calcd. for C26H39N6O2+: [M1 + H]+ = 467.3129, found 467.3135.
3-Dimethylamino-N-(1-((3-(dimethylamino)propyl)amino)-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)propanamide (53)
This compound was synthesized using a procedure analogous to that described for the preparation of 34, using compound 13, 3-cloropropionyl chloride, and dimethylamine. Yield: 24%. 1H NMR (400 MHz, CDCl3) δ (ppm): 10.68 (brs, 1H, D2O exch., NHCO), 8.73–8.78 (m, 2H, H-3, H-9), 8.36 (brs, 1H, D2O exch., NHCH2CH2CH2), 7.44 (t, J = 7.7 Hz, 1H, H-7), 7.34 (d, J = 9.0 Hz, 1H, H-6), 6.77 (t, J = 6.9 Hz, 1H, H-8), 6.46 (d, J = 9.0 Hz, 1H, H-2), 3.31 (q, J = 6.2 Hz, 2H, NHCH2CH2CH2), 2.78 (t, J = 6.2 Hz, 2H, COCH2CH2), 2.65 (t, J = 6.2 Hz, 2H, COCH2CH2), 2.54 (t, J = 7.3 Hz, 2H, NHCH2CH2CH2), 2.44 (s, 6H, NHCH2CH2CH2N(CH3)2), 2.35 (s, 6H, COCH2CH2N(CH3)2), 1.95 (quintet, J = 14.2, 7.0 Hz, 2H, NHCH2CH2CH2). 13C NMR (151 MHz, CDCl3) δ (ppm): 169.8 (HNCO), 160.3 (CO), 146.8 (C-5a), 146.6 (C-4a), 139.6 (C-1), 134.3 (C-7), 127.1 (C-3), 126.5 (C-9), 126.1 (C-6), 121.7 (C-4), 112.1 (C-8), 103.2 (C-2), 101.6 (C-11a), 57.3 (NHCH2CH2CH2), 55.2 (COCH2CH2), 45.1 (COCH2CH2N(CH3)2), 45.1 (NHCH2CH2CH2N(CH3)2), 41.3 (NHCH2CH2CH2), 34.9 (COCH2CH2), 26.6 (NHCH2CH2CH2). m/z: calcd. for C22H31N6O2+: [M1 + H]+ = 411.2503, found 411.2496.
3-(Diethylamino)-N-(1-((2-(diethylamino)ethyl)amino)-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)propanamide (54)
This compound was synthesized using a procedure analogous to that described for the preparation of 38, using compound 14, 3-cloropropionyl chloride, and diethylamine. Yield: 28%. M.p.: 128–130 °C (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 9.66 (brs, 1H, D2O exch., NHCO), 8.51 (d, J = 8.9 Hz, 2H, H-3), 8.37 (brs, 1H, D2O exch., NHCH2CH2), 8.08 (d, J = 2.4 Hz, 1H, H-9), 7.27 (d, J = 9.7 Hz, 1H, H-6), 7.18 (dd, J = 9.7, 2.5 Hz, 1H, H-7), 6.30 (d, J = 11.9 Hz, 1H, H-2), 3.78 (s, 3H, OCH3), 3.40 (t, J = 4.1 Hz, 3H, NHCH2CH2), 3.12 (t, J = 6.8 Hz, 2H, COCH2CH2), 2.82–2.91 (m, 8H, NHCH2CH2N(CH2CH3)2), NHCH2CH2CH2, COCH2CH2), 2.73 (q, J = 7.1 Hz, 4H, COCH2CH2N(CH2CH3)2), 1.17 (t, J = 7.2 Hz, 6H, HNCH2CH2N(CH2CH3)2), 1.07 (t, J = 7.2 Hz, 6H, COCH2CH2N(CH2CH3)2). 13C NMR (151 MHz, CDCl3) δ (ppm): 168.4 (NHCO), 159.9 (CO), 148.9 (C-8), 146.2 (C-5a), 144.8 (C-4a), 139.9 (C-1), 131.6 (C-7), 126.9 (C-3), 126.0 (C-6), 121.5 (C-4), 105.2 (C-9), 102.7 (C-2), 101.8 (C-11a), 56.5 (OCH3), 51.0 (NHCH2CH2), 48.5 (COCH2CH2), 47.3 (COCH2CH2N(CH2CH3)2), 46.7 (NHCH2CH2N(CH2CH3)2), 40.4 (NHCH2CH2), 33.5 (COCH2CH2), 10.9 (COCH2CH2N(CH2CH3)2), 9.8 (NHCH2CH2CH2N(CH2CH3)2). m/z: calcd. for C26H39N6O3+: [M1 + H]+ = 483.3078, found 483.3085.
3-Dimethylamino-N-(1-((2-(dimethylamino)ethyl)amino)-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)propanamide (55)
This compound was synthesized using a procedure analogous to that described for the preparation of 38, using compound 15, 3-cloropropionyl chloride, and dimethylamine. Yield: 20%. M.p.: 202–204 °C (EtOAc). 1H NMR (600 MHz, CDCl3) δ (ppm): 10.33 (brs, 1H, D2O exch., NHCO), 8.72 (d, J = 8.8 Hz, 1H, H-3), 8.51 (t, J = 4.2 Hz, 1H, D2O exch., NHCH2CH2e), 8.25 (d, J = 2.5 Hz, 1H, H-9), 7.35 (d, J = 9.74 Hz, 1H, H-6), 7.29 (dd, J = 9.7, 2.6 Hz, 1H, H-7), 6.42 (d, J = 8.9 Hz, 1H, H-2), 3.88 (s, 3H, OCH3), 3.37 (q, J = 5.5 Hz, 2H, NHCH2CH2), 2.94 (t, J = 6.3 Hz, 2H, COCH2CH2), 2.77 (t, J = 6.4 Hz, 2H, NHCH2CH2), 2.72 (t, J = 6.3 Hz, 2H, COCH2CH2), 2.52 (s, 6H, HNCH2CH2N(CH3)2), 2.38 (s, 6H, COCH2CH2N(CH3)2). 13C NMR (151 MHz, CDCl3) δ (ppm): 169.0 (NHCO), 159.9 (CO), 148.6 (C-8), 146.3 (C-5a), 144.6 (C-4a), 139.5 (C-1), 131.3 (C-7), 126.9 (C-6), 126.1 (C-3), 121.5 (C-4), 105.2 (C-9), 102.8 (C-2), 101.6 (C-11a), 57.8 (NHCH2CH2), 56.4 (OCH3), 54.7 (COCH2CH2) 45.3 (COCH2CH2N(CH3)2) 44.6 (HNCH2CH2N(CH3)2) 40.8 (NHCH2CH2), 34.4 (COCH2CH2). m/z: calcd. for C22H31N6O3+: [M1 + H]+ = 427.2452, found 427.2457.
3-Diethylamino-N-(1-((3-(diethylamino)propyl)amino)-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)propanamide (56)
This compound was synthesized using a procedure analogous to that described for the preparation of 38, using compound 16, 3-cloropropionyl chloride, and dimethylamine. Yield: 10%. 1H NMR (600 MHz, CDCl3) δ (ppm): 10.26 (brs, 1H, D2O exch., NHCO), 8.78 (d, J = 8.9 Hz, 1H, H-3), 8.59 (brs, 1H, D2O exch., NHCH2CH2CH2), 8.22 (d, J = 2.56 Hz 1H, H-9), 7.37 (d, J = 9.7 Hz,1H, H-6), 7.31 (dd, J = 9.8, 2.6 Hz, 1H, H-7), 6.39 (d, J = 9.3 Hz, 1H, H-2), 6.43 (d, J = 8.9 Hz, 1H, H-2), 3.90 (s, 3H, OCH3), 3.35 (t, J = 6.6 Hz, 2H, NHCH2CH2CH2), 3.03 (t, J = 6.6 Hz, 2H, COCH2CH2), 2.82–2.93 (m, 10H, NHCH2CH2CH2, NHCH2CH2CH2N(CH2CH3)2, COCH2CH2N(CH2CH3)2), 2.77 (t, J = 6.6 Hz, 2H,COCH2CH2), 2.06 (quintet, J = 7.6 Hz, 2H, NHCH2CH2CH2), 1.20–1.24 (m, 12H, NHCH2CH2CH2N(CH2CH3)2, COCH2CH2N(CH2CH3)2). 13C NMR (151 MHz, CDCl3) δ (ppm): 169.5 (NHCO), 160.0 (CO), 148.7 (C-8), 146.1 (C-5a), 144.5 (C-4a), 139.5 (C-1), 131.3 (C-7), 126.2 (C-3), 126.8 (C-6), 121.8 (C-4), 104.9 (C-9), 102.8 (C-2), 101.6 (C-11a), 56.3 (OCH3), 49.5 (NHCH2CH2CH2), 48.5 (COCH2CH2), 46.4 (COCH2CH2N(CH2CH3)2), 46.4 (NHCH2CH2CH2N(CH2CH3)2), 41.1 (NHCH2CH2CH2), 34.1 (COCH2CH2), 24.4 (NHCH2CH2CH2), 10.5 (COCH2CH2N(CH2CH3)2), 9.9 (NHCH2CH2CH2N(CH2CH3)2). m/z: calcd. for C27H41N6O3+: [M1 + H]+ = 497.3235, found 497.3228.
3-Dimethylamino-N-(1-((3-(dimethylamino)propyl)amino)-8-methoxy-11-oxo-11H-pyrido[2,1-b]quinazolin-4-yl)propanamide (57)
This compound was synthesized using a procedure analogous to that described for the preparation of 38, using compound 17, 3-cloropropionyl chloride, and dimethylamine. M.p.: 178–180 °C (dec) (EtOAc). 1H NMR (400 MHz, CDCl3) δ (ppm): 10.34 (brs, 1H, D2O exch., NHCO), 8.64 (d, J = 8.9 Hz, 1H), 8.3 (t, J = 8.4 Hz 1H, D2O exch., NHCH2CH2CH2), 8.19 (s, J = 2.3 Hz, 1H, H-9), 7.34 (d, J = 9.8 Hz, 1H, H-6), 7.29 (dd, J = 9.8, 2.5 Hz, 1H, H-7), 6.41 (d, J = 8.9 Hz, 1H, H-3), 3.89 (s, 3H, OCH3), 3.31 (q, J = 7.2 Hz, 2H, NHCH2CH2CH2), 2.87 (t, J = 6.3 Hz, 2H, COCH2CH2), 2.67–2.72(m, 4H, NHCH2CH2CH2, COCH2CH2), 2.47 (s, 6H, NHCH2CH2CH2N(CH3)2), 2.43 (s, 6H, COCH2CH2N(CH3)2), 2.02 (quintet, J = 6.9 Hz, 2H, NHCH2CH2CH2). 13C NMR (151 MHz, CDCl3) δ (ppm): 169.9 (NHCO), 160.0 (CO), 148.6 (C-8), 146.3 (C-5a), 144.4 (C-4a), 139.4 (C-1), 131.1 (C-7), 126.9 (C-6), 126.3 (C-3), 121.7 (C-4), 104.9 (C-9), 103.0 (C-2), 101.6 (C-11a), 57.3 (NHCH2CH2CH2), 56.3 (OCH3), 55.2 (COCH2CH2), 45.1 (NHCH2CH2CH2N(CH3)2, COCH2CH2N(CH3)2) 41.3 (NHCH2CH2CH2), 34.9 (COCH2CH2), 26.6 (NHCH2CH2CH2). m/z: calcd. for C23H33N6O3+: [M1 + H]+ = 441.2609, found 441.2616.




 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}