

Synthesis of Adenine Nucleosides with a Reactive (β-Iodovinyl)sulfone or (β-Keto)sulfone Group at the C2 Position and Their Polymerase-Catalyzed Incorporation into DNA

 , ,
, , 
Abstract

1. Introduction
2. Results and Discussions
3. Experimental Section
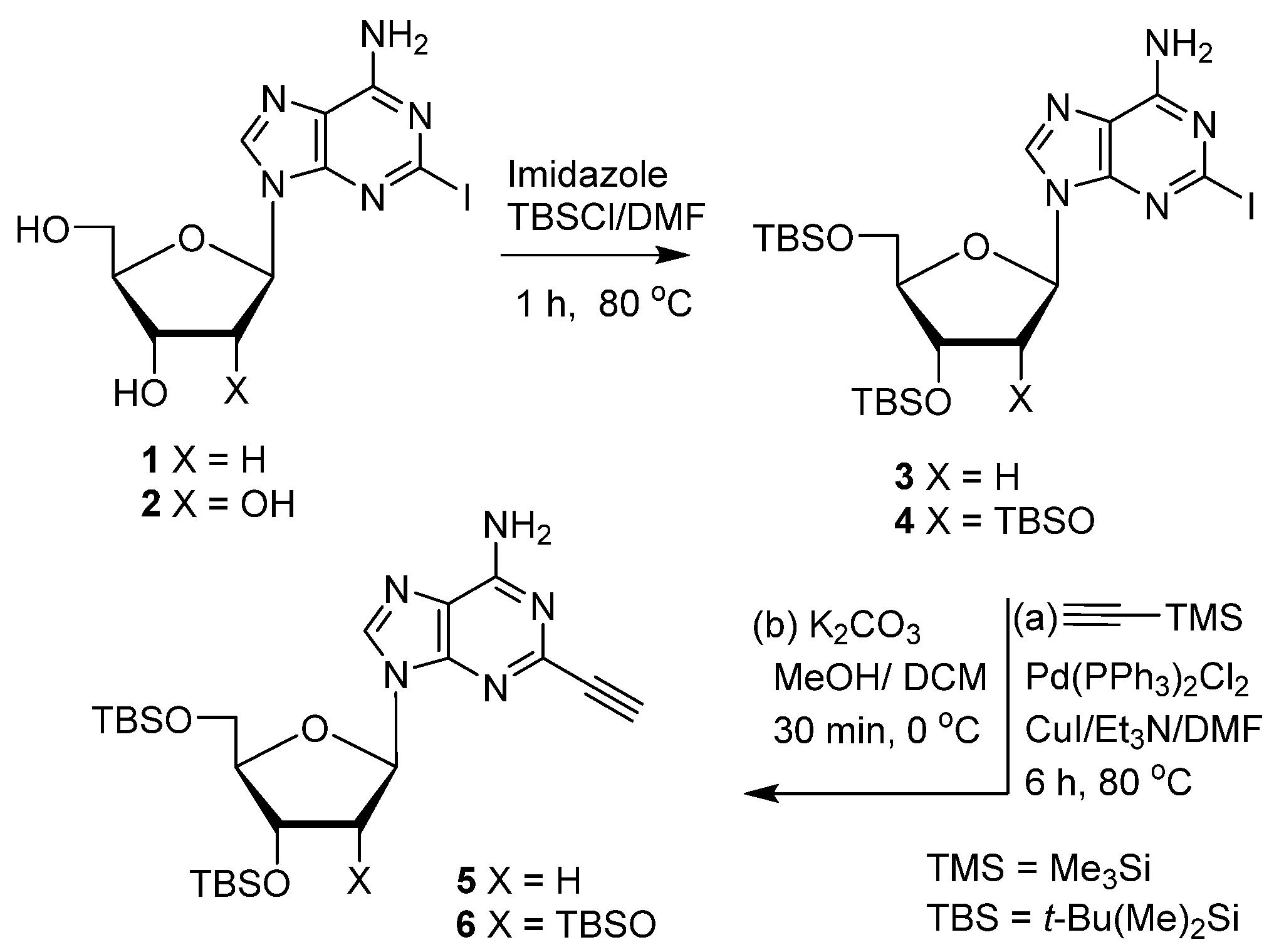
- 3′,5′-Di-O-(tert-butyldimethylsilyl)-2-ethynyl-2′-deoxyadenosine (5). Step a. Pd(PPh3)2Cl2 (70.0 mg, 0.10 mmol) and Cu(I)I (38.1 mg, 0.2 mmol) were added to dry DMF (50 mL) and Et3N (5.0 mL, 3.63 g, 35.9 mmol) in a flame-dried flask equipped with a stirring bar under N2 at rt. Then, 3 [34] (3.0 g, 4.95 mmol) was added followed by TMS–acetylene (1.41 mL, 973 mg, 9.90 mmol). The resulting mixture was stirred at 80 °C for 6 h. Volatiles were evaporated, and the residue was purified by column chromatography (20 → 50% EtOAc/hexane) to give TMS-protected alkyne 5 as a brown solid. Step b. The solid was dissolved in anhydrous MeOH (50 mL) and stirred at 0 °C. Anhydrous K2CO3 (3.2 g, 23.1 mmol) was added portion-wise at room temperature, and the mixture was stirred at rt. After 30 min, the reaction was concentrated under reduced pressure, and the residue was suspended in a mixture of H2O (30 mL) and EtOAc (50 mL). The organic layer was washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated to dryness under vacuum to give a light-yellow crude solid. The solid was recrystallized with hexane to give 5 (2.25 g, 90%, overall) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H), 6.47 (t, J = 6.3 Hz, 1H), 6.07 (s, 2H), 4.61 (dt, J = 5.9, 3.9 Hz, 1H), 3.99 (q, J = 3.5 Hz, 1H), 3.90 (dd, J = 11.2, 3.9 Hz, 1H), 3.77 (dd, J = 11.2, 3.0 Hz, 1H), 3.00 (s, 1H), 2.58 (dt, J = 12.5, 6.1 Hz, 1H), 2.45 (ddd, J = 9.1, 6.3, 3.2 Hz, 1H), 0.92 (s, 10H), 0.91 (s, 9H), 0.10 (s, 6H), 0.09 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 155.49, 149.43, 145.25, 140.19, 119.86, 88.02, 84.54, 82.68, 73.22, 71.84, 62.81, 41.69, 26.09, 25.90, 18.55, 18.14, −4.52, −4.70, −4.98, −5.26, −5.37; HRMS (TOF, ESI) m/z calcd for C24H42N5O3Si2 504.2821 [M + H]+, found 504.2832.
- 2′,3′,5′-Tri-O-(tert-butyldimethylsilyl)-2-ethynyladenosine (6). Step a. Treatment of 4 [5] (5.0 g, 6.79 mmol) with TMS–acetylene (1.93 mL, 1.33 g, 13.6 mmol) in the presence of Pd(PPh3)2Cl2 (71.5 mg, 0.102 mmol), Cu(I)I (38.8 mg, 0.204 mmol) and Et3N (5.0 mL, 3.63 g, 35.9 mmol) followed by treatment with K2CO3 (3.2 g, 23.1 mmol; Step b), as described for 5, gave 6 (3.79 g, 88%, overall) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 6.00 (d, J = 5.2 Hz, 1H), 4.70 (t, J = 4.8 Hz, 1H), 4.31 (t, J = 3.8 Hz, 1H), 4.10–4.14 (m, 1H), 4.06 (dd, J = 11.2, 4.8 Hz, 1H), 3.78 (dd, J = 11.2, 2.8 Hz, 1H), 2.93 (s, 1H), 0.95 (s, 9H), 0.93 (s, 9H), 0.81 (s, 9H), 0.14 (s, 3H), 0.13 (s, 3H), 0.103 (s, 3H), 0.097 (s, 3H), −0.04 (s, 3H), −0.21 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 155.39, 149.79, 145.27, 140.84, 120.07, 88.84, 85.69, 82.66, 75.79, 72.93, 72.08, 62.60, 26.22, 25.99, 25.84, 18.64, 18.24, 18.02, −4.24, −4.58, −4.62, −4.98, −5.241, −5.238; HRMS (TOF, ESI) m/z calcd for C30H56N5O4Si3 634.3635 [M + H]+, found 634.3641.
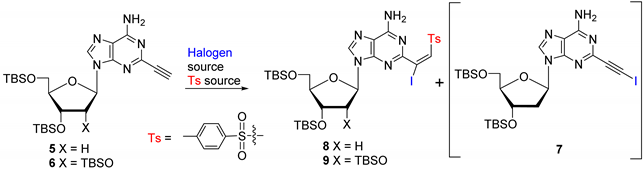
- 3′,5′-Di-O-(tert-butyldimethylsilyl)-2-(2-iodoethynyl)-2′-deoxyadenosine (7). Sodium acetate (7.4 mg, 0.09 mmol), sodium p-toluenesulfinate (32.1 mg, 0.18 mmol), and I2 (22.8 mg, 0.09 mmol) were added to a stirring solution of 5 (31 mg, 0.06 mmol) in MeCN at rt. The resulting mixture was then heated at 80 °C for 1.5 h. The reaction mixture was quenched by saturated aqueous Na2S2O3. The volatiles were removed under reduced pressure, and the residue was suspended in a mixture of H2O (30 mL) and EtOAc (50 mL). The organic layer was washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated to dryness under vacuum to give a light-yellow crude solid. The residue was purified by column chromatography (30 → 50% EtOAc/hexane) to give 7 (30 mg, 80%) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H), 6.45 (t, J = 6.4 Hz, 1H), 5.92 (s, 2H), 4.61 (dt, J = 6.0, 3.6 Hz, 1H), 3.99 (q, J = 3.4 Hz, 1H), 3.90 (dd, J = 11.2, 4.0 Hz, 1H), 3.77 (dd, J = 11.6, 3.2 Hz, 1H), 2.65–2.56 (m, 1H), 2.43 (ddd, J = 13.2, 6.0, 4.4 Hz, 1H), 0.92 (s, 9H), 0.91 (s, 9H), 0.10 (s, 12H); 13C NMR (101 MHz, CDCl3) δ 154.92, 149.31, 145.40, 140.46, 119.76, 93.73, 88.09, 84.69, 71.85, 62.78, 41.62, 26.14, 26.09, 25.96, 25.90, 24.84, 18.58, 18.18, −4.47, −4.54, −4.65, −4.71, −5.20, −5.26, −5.31, −5.37; HRMS (TOF, ESI) m/z calcd for C24H41IN5O3Si2 630.1787 [M + H]+, found 630.1778.
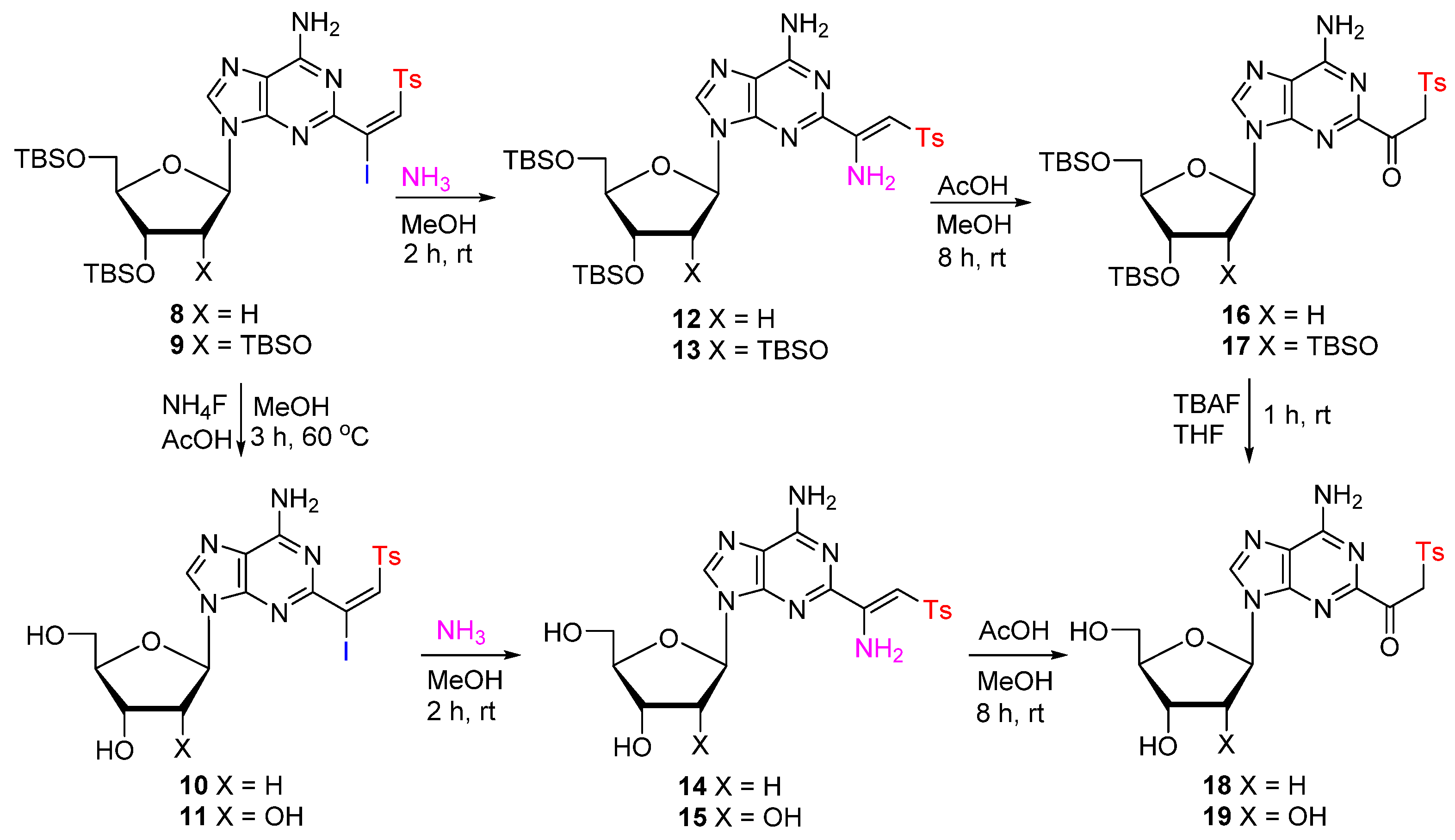
- (E)-3′,5′-Di-O-(tert-butyldimethylsilyl)-2-(1-iodo-2-tosylvinyl)-2′-deoxyadenosine (8). NaOAc (342 mg, 4.17 mmol) and freshly prepared TsI (784 mg, 2.78 mmol) were added to a stirring solution of 5 (700 mg, 1.39 mmol) in THF (15 mL) at rt. After stirring for 40 h, the reaction mixture was quenched with saturated aqueous Na2S2O3. The volatiles were removed under reduced pressure, and the residue was suspended in a mixture of H2O (30 mL) and EtOAc (50 mL). The organic layer was washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated to dryness under vacuum. The residue was purified by column chromatography (30 → 50% EtOAc/hexane) to give 8 (885 mg, 81%) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.19 (s, 1H), 7.75, (d, J = 8.4 Hz, 2H), 7.22 (d, J = 7.8 Hz, 2H), 7.23 (s, 1H), 6.41 (t, J = 6.4 Hz, 1H), 6.22 (s, 2H), 4.63 (dt, J = 6.0, 3.6 Hz, 1H), 4.03 (q, J = 3.6 Hz, 1H), 3.92 (dd, J = 11.2, 4.4 Hz, 1H), 3.79 (dd, J = 10.8, 3.2 Hz, 1H), 2.76–2.68 (m, 1H), 2.44 (ddd, J = 13.2, 6.0, 4.0 Hz, 1H), 2.31 (s, 3H), 0.93 (s, 9H), 0.92 (s, 9H), 0.12–0.11 (m, 12H); 13C NMR (101 MHz, CDCl3) δ 158.29, 155.16, 149.08, 144.94, 140.63, 140.54, 137.22, 129.86, 128.56, 119.33, 110.84, 88.29, 84.89, 72.12, 63.12, 41.46, 26.24, 26.04, 21.18, 18.68, 18.27, −4.36, −4.53, −5.08, −5.17; HRMS (TOF, ESI) m/z calcd for C31H49IN5O5SSi2 786.2032 [M + H]+, found 786.2021.
- (E)-2′,3′,5′-Tri-O-(tert-butyldimethylsilyl)-2-(1-iodo-2-tosylvinyl)adenosine (9). Treatment of 6 (1.5 g, 2.36 mmol) with NaOAc (582 mg, 7.09 mmol) and freshly prepared TsI (1.33 g, 4.72 mmol), as described for 8, gave 9 (1.99 g, 92%) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H); 7.73 (d, J = 8.4 Hz, 2H), 7.23–7.18 (m, 3H), 6.28 (s, 2H), 5.93 (d, J = 4.0 Hz, 1H), 4.60 (t, J = 4.0 Hz, 1H), 4.34–4.32 (m, 1H), 4.16 (td, J = 4.8, 2.8 Hz, 1H), 4.10 (dd, J = 11.2, 4.4 Hz, 1H), 3.83 (dd, J = 11.2, 2.6 Hz, 1H), 2.30 (s, 3H), 0.97 (s, 9H), 0.93 (s, 9H), 0.84 (s, 9H), 0.17 (s, 3H), 0.16 (s, 3H), 0.11 (s, 3H), 0.09 (s, 3H), 0.02 (s, 3H), −0.07 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 158.34, 155.04, 149.13, 144.83, 140.68, 140.40, 137.02, 129.74, 128.53, 119.24, 110.06, 89.29, 84.93, 75.83, 71.31, 62.25, 26.29, 26.01, 25.97, 21.70, 18.72, 18.23, 18.07, −4.14, −4.51, −4.60, −4.67, −5.09, −5.17; HRMS (TOF, ESI) m/z calcd for C37H63IN5O6SSi3 916.2846 [M + H]+, found 916.2867.
- (E)-2-(1-Iodo-2-tosylvinyl)-2′-deoxyadenosine (10). CH3CO2H (1.7 mL, 1.78 g, 29.6 mmol) and NH4F (944 mg, 25.5 mmol) were added to a stirring solution of 8 (400 mg, 0.51 mmol) in MeOH (15 mL) at rt. The resulting mixture was stirred at 60 °C for 3.0 h. The volatiles were evaporated at reduced pressure and co-evaporated with acetonitrile (3 × 5 mL) yielding an off-white solid, which was suspended in 20% MeOH in CH2Cl2. The white precipitate was removed by vacuum filtration, and the mother liquor evaporated at reduced pressure. The residue was purified by column chromatography (5 → 10% MeOH/CH2Cl2) to give 10 (222 mg, 78%) as a white solid. 1H NMR (400 MHz, MeOD-d4) δ 8.38 (s, 1H), 7.71 (d, J = 8.0 Hz, 2H), 7.45 (s, 1H), 7.32 (d, J = 8.0 Hz, 2H), 6.42 (t, J = 6.8 Hz, 1H), 4.60 (quint, J = 2.8 Hz, 1H), 4.09 (q, J = 3.2 Hz, 1H), 3.87 (dd, J = 12.4, 3.2 Hz, 1H), 3.75 (dd, J = 12.0, 3.6 Hz, 1H), 2.86–2.77 (m, 1H), 2.43 (ddd, J = 13.6, 6.4, 3.2 Hz, 1H), 2.37 (s, 3H); 13C NMR (101 MHz, MeOD-d4) δ 160.11, 156.90, 149.69, 146.53, 142.38, 141.36, 138.06, 130.82, 129.43, 119.80, 110.85, 89.85, 86.78, 73.04, 63.64, 41.57, 21.64; HRMS (TOF, ESI) m/z calcd for C19H21IN5O5S 558.0303 [M + H]+, found 558.0330.
- (E)-2-(1-Iodo-2-tosylvinyl)adenosine (11). Treatment of 9 (800 mg, 0.87 mmol) with CH3CO2H (3.1 mL, 3.25 g, 54.2 mmol) and NH4F (1.61 g, 43.5 mmol) in MeOH (20 mL), as described for 10, gave 11 (499 mg, 80%) as a white solid. 1H NMR (400 MHz, MeOD-d4) δ 8.38 (s, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.46 (s, 1H), 7.31 (d, J = 8.0 Hz, 2H), 5.98 (d, J = 6.4 Hz, 1H), 4.75 (dd, J = 6.2, 5.0 Hz, 1H), 4.36 (dd, J = 5.0, 3.0 Hz, 1H), 4.20 (q, J = 2.8 Hz, 1H), 3.92 (dd, J = 12.6, 2.6 Hz, 1H), 3.75 (dd, J = 12.6, 3.0 Hz, 1H), 2.35 (s, 3H); 13C NMR (101 MHz, MeOD-d4) δ 160.13, 156.96, 149.72, 146.56, 142.74, 141.48, 137.81, 130.83, 129.49, 119.98, 110.37, 90.97, 87.97, 75.66, 72.59, 63.38, 21.64; HRMS (TOF, ESI) m/z calcd for C19H21IN5O6S 574.0252 [M + H]+, found 574.0253.
- (Z)-2′,3′,5′-Tri-O-(tert-butyldimethylsilyl)-2-(1-amino-2-tosylvinyl)adenosine (13). Iodovinylsulfone 9 (303 mg, 0.33 mmol) was dissolved in methanolic ammonia (1 M, 12 mL), and the resulting mixture was stirred at rt for 2 h. The volatiles were removed under reduced pressure, and the residue was suspended in a mixture of H2O (30 mL) and EtOAc (50 mL). The organic layer was washed with brine (50 mL), dried over Na2SO4, filtered, and evaporated to give 13 (256 mg, 96%) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 7.6 Hz, 2H), 6.9 (s, 2H), 6.27 (s, 1H), 6.04 (d, J = 3.6 Hz, 1H), 5.62 (s, 2H), 4.32 (t, J = 4.0 Hz, 1H), 4.29 (t, J = 4.6 Hz, 1H), 4.13–4.11 (m, 1H), 4.01 (dd, J = 11.6, 2.8 Hz, 1H), 3.81 (dd, J = 11.6, 2.0 Hz, 1H), 2.39 (s, 3H), 0.96 (s, 9H), 0.92 (s, 9H), 0.79 (s, 9H), 0.16 (s, 3H), 0.14 (s, 3H), 0.09 (s, 3H), 0.07 (s, 3H), −0.05 (s, 3H), −0.18 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 154.64, 153.41, 149.98, 149.84, 143.04, 141.87, 140.74, 129.62, 126.37, 120.19, 92.90, 88.32, 84.73, 77.02, 71.20, 62.16, 26.27, 25.98, 25.75, 21.64, 18.72, 18.24, 17.96, −4.14, −4.65, −4.67, −4.93, −5.18, −5.23; HRMS (TOF, ESI) m/z calcd for C37H65N6O6SSi3 805.3989 [M + H]+, found 805.3971.
- (Z)-2-(1-Amino-2-tosylvinyl)-2′-deoxyadenosine (14). Iodovinylsulfone 10 (100 mg, 0.18 mmol) was dissolved in methanolic ammonia (1 M, 5 mL), and the resulting mixture was stirred at rt for 2h. The volatiles were evaporated at reduced pressure and co-evaporated with acetonitrile (3 × 5 mL), yielding an off-white solid, which was suspended in 10% MeOH in CH2Cl2. The off-white precipitate was removed by vacuum filtration, and the mother liquor was evaporated at reduced pressure to give 14 (56 mg, 70%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 1H), 7.80 (d, J = 8.3 Hz, 2H), 7.55 (s, 2H), 7.39 (d, J = 7.6 Hz, 2H), 6.98 (s, 2H), 6.40 (t, J = 6.8 Hz, 1H), 6.00 (s, 1H), 5.34 (s, 1H), 4.93 (s, 1H), 4.40 (s, 1H), 3.87–2.82 (m, 1H), 3.59–3.49 (m, 2H), 2.70–2.63 (m, 1H), 2.37 (s, 3H), 2.30 (ddd, J = 13.6, 6.4, 3.6 Hz, 1H); 13C NMR (101 MHz, MeOD-d4) δ 156.75, 154.60, 152.29, 150.71, 144.65, 143.07, 142.23, 130.66, 127.00, 120.50, 91.72, 89.22, 85.62, 72.47, 63.12, 41.34, 21.46; HRMS (TOF, ESI) m/z calcd for C19H23N6O5S 447.1445 [M + H]+, found 447.1445.
- (Z)-2-(1-Amino-2-tosylvinyl)adenosine (15). Treatment of 11 (150 mg, 0.26 mmol) with methanolic ammonia (1 M, 10 mL), as described for 14, gave 15 (85 mg, 70%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.47 (s, 1H), 7.86–7.74 (m, 2H), 7.57 (s, 2H), 7.43–7.34 (m, 2H), 7.06 (s, 1H), 6.98 (s, 2H), 6.00 (s, 1H), 5.94 (d, J = 5.8 Hz, 1H), 5.48 (d, J = 6.2 Hz, 1H), 5.21 (d, J = 5.0 Hz, 1H), 5.02 (t, J = 5.5 Hz, 1H), 4.52 (q, J = 5.8 Hz, 1H), 4.14 (td, J = 5.0, 3.5 Hz, 1H), 3.93 (q, J = 3.9 Hz, 1H), 3.64 (dt, J = 11.9, 4.9 Hz, 1H), 3.55 (dt, J = 11.9, 4.8 Hz, 1H), 2.37 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 13C NMR (101 MHz, DMSO-d6) δ 155.39, 152.37, 150.23, 149.45, 142.81, 141.78, 140.90, 129.70, 125.63, 119.32, 90.73, 87.05, 85.52, 73.95, 70.28, 61.29, 54.93, 20.99; HRMS (TOF, ESI) m/z calcd for C19H23N6O6S 463.1394 [M + H]+, found 463.1383.
- 3′,5′-Di-O-(tert-butyldimethylsilyl)-2-(2-tosylacetyl)-2′-deoxyadenosine (16). Step a. Treatment of iodovinylsulfone 8 (200 mg, 0.25 mmol) with methanolic ammonia (1 M, 10 mL), as described for 13, gave (Z)-3′,5′-Di-O-(tert-butyldimethylsilyl)-2-(1-amino-2-tosylvinyl)-2′-deoxyadenosine 12 (160 mg, 95%) as an off-white solid of sufficient purity to be used directly in the next step. Step b. CH3CO2H/H2O (1:1, 0.5 mL) was added to a stirring solution of 12 (150 mg, 0.22 mmol) in MeOH (5 mL) at rt. The resulting solution was stirred at rt for 8.0 h. The volatiles were removed under reduced pressure, and the residue was suspended in mixture of H2O (20 mL) and EtOAc (30 mL). The organic layer was washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated to dryness under vacuum. The residue was purified by column chromatography (40 → 50% EtOAc/hexane) to give 16 (129 mg, 86%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H), 6.69 (s, 2H),6.42 (t, J = 6.2 Hz, 1H), 5.10 (s, 2H), 4.62–4.57 (m, 1H), 4.03 (q, J = 3.2 Hz, 1H), 3.89 (dd, J = 11.2, 3.6 Hz, 1H), 3.80 (dd, J = 11.2, 2.8 Hz, 1H), 2.54–2.46 (m, 2H), 2.36 (s, 3H), 0.93 (s, 9H), 0.92 (s, 9H), 0.12 (s, 3H), 0.12 (s, 3H), 0.11 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 187.75, 155.82, 153.54, 148.75, 145.09, 141.63, 136.42, 129.75, 128.77, 121.08, 88.12, 84.44, 71.70, 62.80, 62.69, 42.16, 26.09, 25.89, 21.72, 18.55, 18.14, −4.46, −4.66, −5.25, −5.35; HRMS (TOF, ESI) m/z calcd for C31H50N5O6SSi2 676.3015 [M + H]+, found 676.3001.
- 2′,3′,5′-Tri-O-(tert-butyldimethylsilyl)-2-(2-tosylacetyl)adenosine (17). Treatment of 13 (250 mg, 0.31 mmol) with CH3CO2H/H2O (1:1, 1.0 mL), as described for 16 (Step b), gave 17 (213 mg, 85%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 8.0 Hz, 2H), 6.78 (s,2H), 5.98 (d, J = 4.0 Hz, 1H), 5.38 (d, J = 14.0 Hz, 1H), 4.76 (d, J = 14.0 Hz, 1H), 4.37 (t, J = 4.4 Hz, 1H), 4.32 (t, J = 4.4 Hz, 1H), 4.18–4.15 (m, 1H), 4.05 (dd, J = 11.6, 3.0 Hz, 1H), 3.84 (dd, J = 11.6, 2.4 Hz, 1H), 2.35 (s, 3H), 0.98 (s, 9H), 0.94 (s, 9H), 0.81 (s, 9H), 0.18 (s, 3H), 0.16 (s, 3H), 0.12 (s, 3H), 0.10 (s, 3H), −0.10 (s, 3H), −0.19 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 187.62, 155.98, 153.54, 148.95, 145.14, 141.93, 136.40, 129.75, 128.78, 121.22, 88.25, 85.18, 77.06, 71.54, 62.64, 62.37, 26.26, 25.98, 25.75, 21.70, 18.70, 18.24, 17.96, −4.15, −4.58, −4.65, −4.72, −5.19, −5.23; HRMS (TOF, ESI) m/z calcd for C37H64N5O7SSi3 806.3829 [M + H]+, found 806.3833.
- 2-(2-Tosylacetyl)-2′-deoxyadenosine (18). Method A. TBAF [330 µL, 0.33 mmol (1.0 M in THF)] was added to a stirring solution of 16 (101 mg, 0.15 mmol) in THF (5 mL) at 0 °C, and the resulting mixture was stirred at rt for 1 h. The volatiles were evaporated, and the residue was column-chromatographed (5 → 10% MeOH/CH2Cl2) to give 18 (58 mg, 87%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.56 (s, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.68 (s, 2H), 7.38 (d, J = 8.0 Hz, 2H), 6.33 (t, J = 6.8 Hz, 1H), 5.37 (d, J = 4.0 Hz, 1H), 5.30 (d, J = 2.4 Hz, 2H), 4.94 (t, J = 5.6 Hz, 1H), 4.44–3.38 (m, 1H), 3.88 (q, J = 4.4 Hz, 1H), 3.63–3.56 (m 1H), 3.55–3.49 (m 1H), 2.73–2.66 (m, 1H), 2.35 (s, 3H), 2.32–2.27 (m, 1H); 13C NMR (101 MHz, MeOD-d4) δ 189.15, 157.05, 154.60, 150.04, 146.60, 143.82, 137.78, 130.72, 129.58, 121.66, 89.53, 86.28, 72.62, 63.26, 54.81, 41.64, 21.50; HRMS (TOF, ESI) m/z calcd for C19H22N5O6S 448.1285 [M + H]+, found 448.1284.
- 2-(2-Tosylacetyl)adenosine (19). Method A. Treatment of 17 (202 mg, 0.25 mmol) in THF (10 mL) with TBAF [830 µL, 0.83 mmol (1.0 M in THF)], as described for 18, gave 19 (102 mg, 88%) as a white solid. 1H NMR (400 MHz, MeOD-d4) δ 8.52 (s, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 6.00 (d, J = 5.6 Hz, 1H), 4.63 (t, J = 5.6 Hz, 1H), 4.35 (t, J = 4.4 Hz, 1H), 4.16 (q, J = 3.2 Hz, 1H), 3.91 (dd, J = 12.4, 2.8 Hz, 1H), 3.79 (dd, J = 12.6, 3.4 Hz, 1H), 2.31 (s, 3H); 13C NMR (101 MHz, MeOD-d4) δ 189.07, 157.11, 154.66, 150.28, 146.65, 143.96, 137.70, 130.73, 129.62, 121.79, 90.59, 87.38, 76.07, 71.93, 62.86, 21.49; HRMS (TOF, ESI) m/z calcd for C19H22N5O7S 464.1234 [M + H]+, found 464.1233.
- (E)-2-(1-Propanethio-2-tosylvinyl)adenosine (20). Et3N (9.0 μL, 6.6 mg, 0.065 mmol) and PrSH (4.8 μL, 4.1 mg, 0.054 mmol) were sequentially added to a stirred solution of 11 (31 mg, 0.054 mmol) in MeOH (2 mL) at ambient temperature. After 24 h, the volatiles were evaporated, and the residue was purified by silica gel column chromatography [MeOH/DCM (dichloromethane); 2 → 5%] to give 20 (17 mg, 60%) as a white solid. 1H NMR (400 MHz, MeOD-d4) δ 8.41 (s, 1H), 8.05 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 7.7 Hz, 2H), 6.01 (d, J = 5.9 Hz, 1H), 4.73 (t, J = 5.5 Hz, 1H), 4.33 (dd, J = 5.1, 3.4 Hz, 1H), 4.13 (q, J = 3.2 Hz, 1H), 3.86 (dd, J = 12.4, 2.9 Hz, 1H), 3.77–3.73 (m, 1H), 2.73–2.68 (m, 2H), 2.46 (s, 3H), 1.26–1.20 (m, 2H), 0.67 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, MeOD-d4) δ 157.14, 157.02, 150.64, 146.44, 145.63, 142.94, 141.93, 136.90, 130.76, 130.26, 119.80, 90.81, 87.65, 75.62, 72.33, 63.21, 39.16, 23.17, 21.59, 13.32; HRMS (TOF, ESI) m/z calcd for C22H28N5O6S2 522.1476 [M + H]+, found 522.1477.
- (E/Z)-2-(1-Diisopropylamino-2-tosylvinyl)adenosine (21). (Me2CH)2NH (17 μL, 12.3 mg, 0.12 mmol) was added to a stirred solution of 11 (30 mg, 0.052 mmol) in MeOH (3 mL) at ambient temperature. After 30 min, the volatiles were evaporated, and the residue was purified by silica gel column chromatography (MeOH/DCM; 2 → 5%) to give 21 (E/Z, ~46:54; 21 mg, 74%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (s, 0.46H), 8.41 (s, 0.54H), 7.59 (d, J = 8.4 Hz, 2H), 7.50 (s, 0.92H), 7.48 (s, 1.08H) 7.35 (d, J = 8.0 Hz, 0.92H), 7.28 (d, J = 8.0 Hz, 1.08H), 5.89 (d, J = 6.4 Hz, 0.46H), 5.83 (d, J = 6.4 Hz, 0.54H), 5.53 (dd, J = 6.0, 2.8 Hz, 0.46H), 5.40 (d, J = 6.4 Hz, 0.54H) 5.25 (dt, J = 7.9, 3.5 Hz, 2H), 4.97 (s, 0.54H), 4.94 (s, 0.46H), 4.59–4.51 (m, 1H), 4.14 (qd, J = 5.0, 3.3, 2.2 Hz, 1H), 4.02 (q, J = 3.2 Hz, 0.54H), 3.95 (q, J = 3.2 Hz, 0.46H) 3.71–3.47 (m, 3.46H), 3.25 (d, J = 6.4 Hz, 0.54H), 2.36 (s, 3H), 1.16 (br s, 12H). 13C NMR (101 MHz, DMSO) δ 155.81, 155.72, 154.11, 153.76, 149.31, 149.22, 143.70, 143.63, 141.01, 140.99, 139.96, 139.87, 129.30, 129.00, 126.05, 118.46, 118.45, 93.58, 93.40, 87.42, 87.09, 86.11, 85.97, 73.96, 73.80, 70.90, 70.76, 61.72, 53.21, 21.01, 20.96, 20.20, 19.58; HRMS (TOF, ESI) m/z calcd for C25H35N6O6S 547.2333 [M + H]+, found 547.2330.
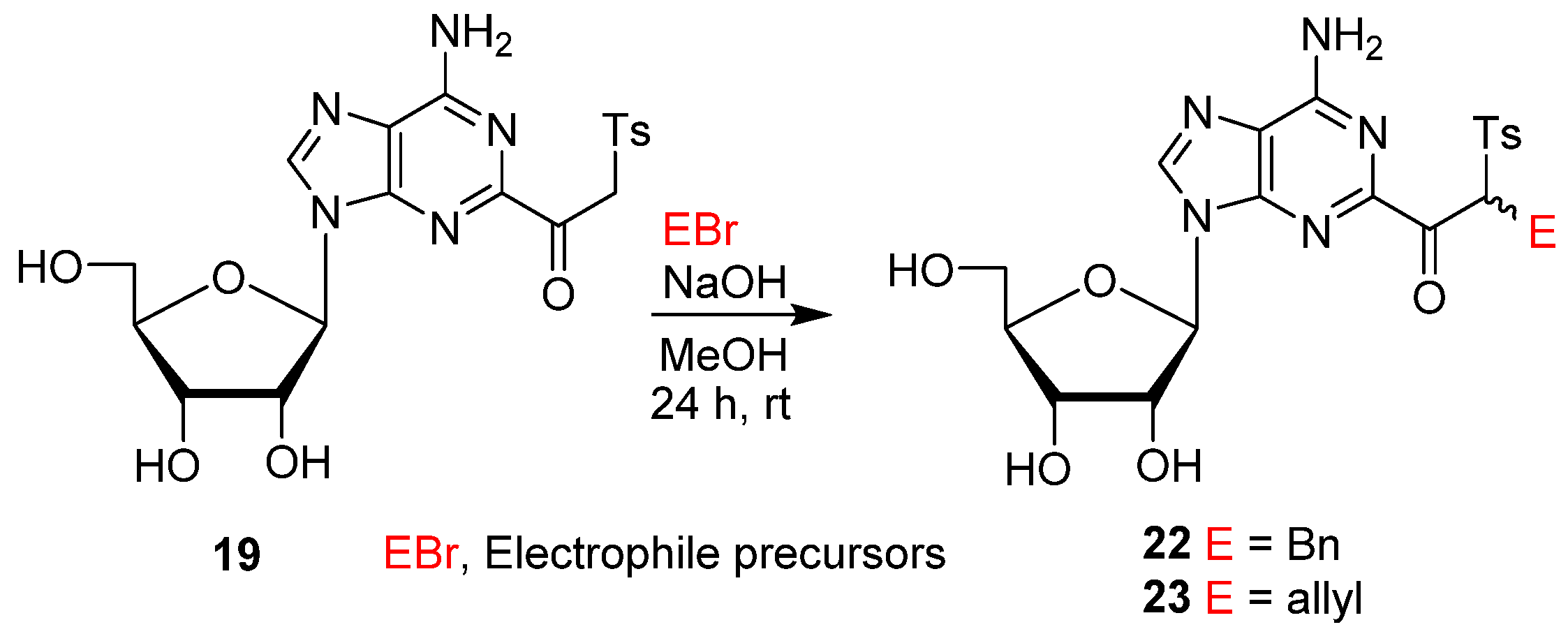
- 2-(2-Benzyl-2-tosylacetyl)adenosine (22). Aqueous NaOH solution (1 M, 130 µL, 0.13 mmol) was added to a stirred solution of 19 (30 mg, 0.065 mmol) in MeOH (2 mL) at rt. After 30 min, BnBr (15.4 µL, 22.3 mg, 0.13 mmol) was added, and the resulting mixture was stirred for 24 h. The reaction mixture was then neutralized with dil. HCl to pH ~7, and the volatiles were evaporated. The residue was column-chromatographed (5 → 10% MeOH/DCM) to give 22 (27 mg, 75%) as a 50:50 mixture of diastereomers. 1H NMR (400 MHz, MeOD-d4) δ 8.48 (s, 0.5H), 8.46 (s, 0.5H), 7.67 (d, J = 8.0 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.29–7.06 (m, 7.5H), 6.52–6.45 (m, 0.5H), 5.98 (t, J = 5.2 Hz, 1H), 4.61 (t, J = 5.2 Hz, 1H), 4.36–4.32 (m, 1H), 4.19 (“q”, J = 3.2 Hz, 0.5H), 4.16 (“q”, J = 3.2 Hz, 0.5H), 3.94–3.89 (m, 1H), 3.80–3.76 (m, 1H), 3.52–3.42 (m, 2H), 2.25 (s, 1.5H), 2.22 (s, 1.5H); 13C NMR (101 MHz, MeOD-d4) δ 192.47, 192.26, 156.89, 155.13, 154.99, 150.12, 150.06, 146.92, 144.05, 144.02, 137.65, 137.62, 137.60, 137.57, 136.16, 136.09, 130.69, 130.48, 130.36, 130.05, 130.03, 129.63, 127.93, 127.90, 121.66, 90.71, 90.65, 87.50, 87.46, 75.98, 75.90, 72.11, 72.02, 70.39, 63.07, 62.97, 32.72, 32.44, 21.46, 21.44; HRMS (TOF, ESI) m/z calcd for C26H28N5O7S 554.1704 [M + H]+, found 554.1704.
- 2-(2-Allyl-2-tosylacetyl)adenosine (23). Aqueous NaOH solution (1 M, 130 µL, 0.13 mmol) was added to a stirred solution of 19 (30 mg, 0.065 mmol) in MeOH (2 mL) at rt. After 30 min, allyl bromide (11.2 µL 15.7 mg, 0.13 mmol) was added, and the resulting mixture was stirred for 24 h. The reaction mixture was then neutralized with dil. HCl to pH~7, and the volatiles were evaporated. The residue was column-chromatographed (5 → 10% MeOH/DCM) to give 23 (22.3 mg, 68%) as a 50:50 mixture of diastereomers in the form of a white solid. 1H NMR (400 MHz, MeOD-d4) δ 1H NMR (400 MHz, MeOD-d4) δ 8.52 (s, 0.5H), 8.51 (s, 0.5H), 7.69–7.58 (m, 2H), 7.28–7.17 (m, 2H), 6.23 (ddd, J = 14.4, 10.4, 4.4 Hz, 1H), 6.04 (d, J = 5.6 Hz, 0.5H), 6.00 (d, J = 5.6 Hz, 0.5H), 5.82–5.70 (m, 1H), 5.12 (q, J = 1.6 Hz, 0.5H), 5.10 (q, J = 1.6 Hz, 0.5H), 5.09 (t, J = 1.2 Hz, 1H), 4.98 (t, J = 1.2 Hz, 1H), 4.65 (dt, J = 6.4, 5.2 Hz, 1H), 4.35 (ddd, J = 5.2, 3.6, 1.6 Hz, 1H), 4.19 (“q”, J = 3.2 Hz, 0.5H), 4.17 (“q”, J = 3.2 Hz, 0.5H), 3.92 (“ddd”, J = 12.4, 7.6, 2.8 Hz, 1H), 3.82–3.77 (m, 1H), 2.99–2.85 (m, 2H), 2.25 (s, 1.5H), 2.22 (s, 1.5H); 13C NMR (101 MHz, MeOD-d4) δ 192.40, 192.22, 157.04, 155.29, 155.14, 150.30, 150.19, 146.91, 144.07, 143.97, 136.16, 136.08, 133.98, 133.95, 133.94, 133.91, 130.65, 130.46, 130.33, 121.72, 118.65, 118.63, 90.64, 90.59, 87.55, 87.52, 76.07, 76.03, 72.11, 72.06, 68.70, 68.61, 63.03, 62.95, 31.14, 31.05, 30.95, 30.86, 21.44, 21.43; HRMS (TOF, ESI) m/z calcd for C22H26N5O7S 504.1547 [M + H]+, found 504.1545.
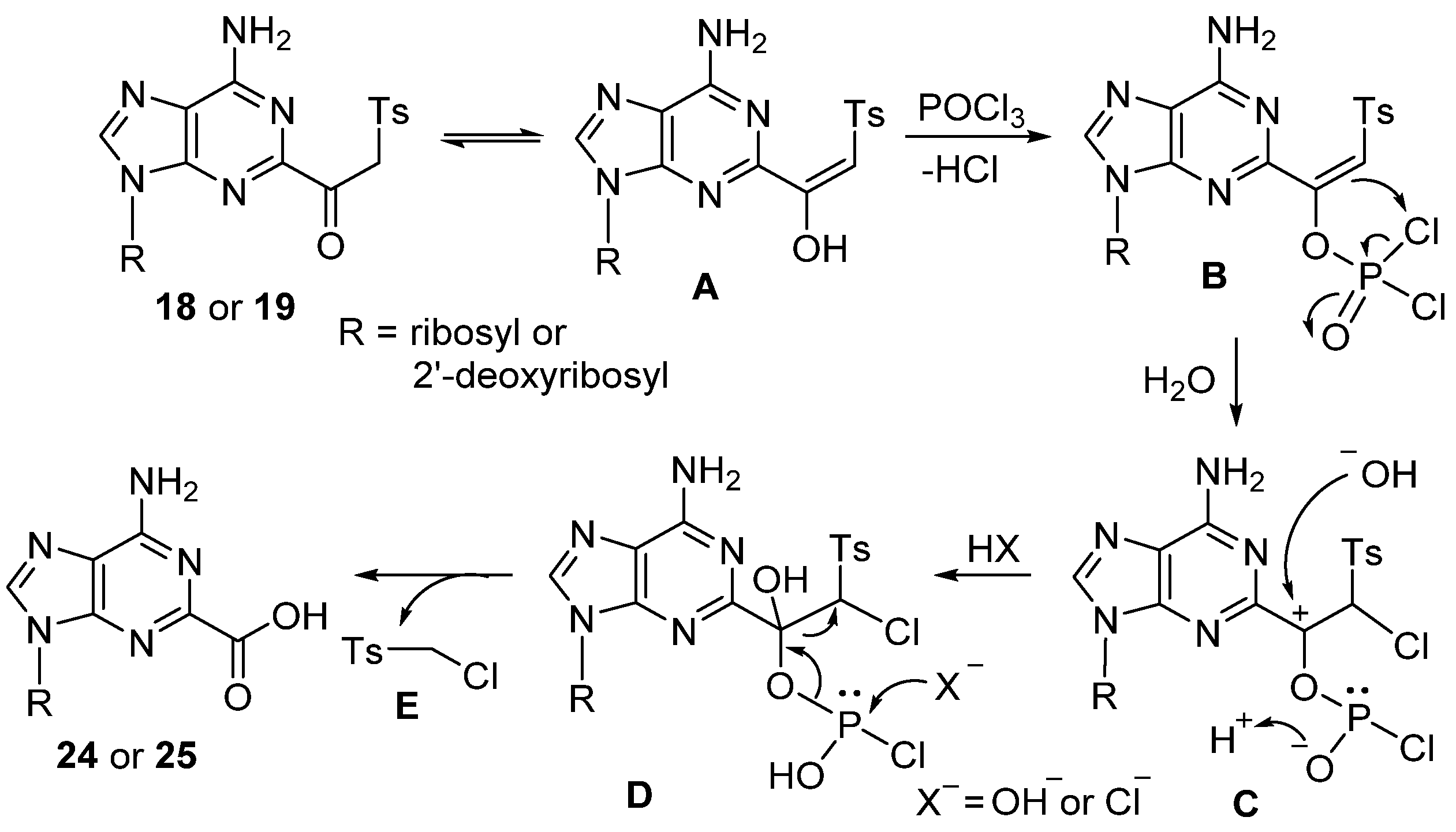
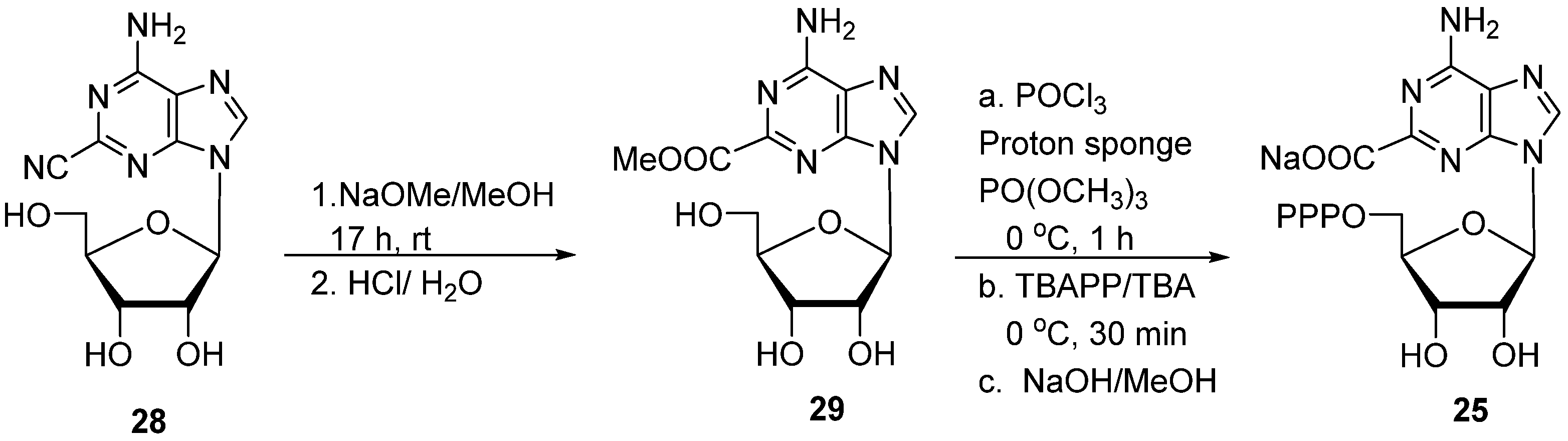
- 2′-Deoxyadenosine-2-carboxylic acid 5′-O-triphosphate (24). (MeO)3PO (1.0 mL; dried over 3A molecular sieves) was added to the flame-dried flask containing 8-(β-keto)sulfone 18 [30 mg, 0.067 mmol; dried in vacuum (40 °C, over P2O5)] and proton sponge (21.5 mg, 0.10 mmol), and the resulting solution was stirred at 0 °C for 5 min under an Ar atmosphere. Freshly distilled POCl3 (8.1 μL, 13.4 mg, 0.087 mmol) was then added, and stirring was continued for 1.0 h at 0 °C. The mixture of tributylammonium pyrophosphate (TBAPP; 0.5 M/dimethylformamide (DMF); 670 μL, 0.34 mmol) and Bu3N (49.7 mg, 0.27 mmol) was added and stirred for another 30 min at 0 °C. The reaction mixture was quenched by adjusting pH to 7.5–7.8 with triethylammonium bicarbonate (TEAB) buffer (2 M, several drops). The residue was dissolved in water (5 mL) and was extracted with EtOAc (3 × 5 mL). The water layer was evaporated and co-evaporated (three times) with a mixture of EtOH/H2O (1:1, 5 mL). The residue was chromatographed on a DEAE-Sephadex A-25 column with TEAB (0.1 → 0.6 M), and the appropriate fractions (TLC, Rf 0.28; i-PrOH/H2O/NH4OH, 5:2:3) were evaporated in vacuum and co-evaporated three times with a mixture of EtOH/H2O (1:1, 10 mL) to remove excess of TEAB salt to give 24 as a triethylammonium salt, which was then converted to sodium salt 24 (11.0 mg, 25%) with Dowex-Na+. 1H NMR (400 MHz, D2O) δ 8.56 (s, 1H), 6.61 (s, 1H), 4.89 (s, 1H), 4.32–4.13 (m, 3H), 2.85–2.74 (m, 1H), 2.69–2.58 (m, 1H); 31P NMR (162 MHz, D2O) δ −10.89 (d, J = 19.8 Hz), −11.44 (d, J = 20.1 Hz), −23.24 (t, J = 19.8 Hz); HRMS (TOF, ESI) m/z calcd for C11H15N5O14P3 533.9834 [M − H]−, found 533.9830.
- Adenosine-2-carboxylic acid 5′-O-triphosphate (25). Method A: Treatment of 19 (31 mg, 0.067 mmol) with (MeO)3PO (1.0 mL), proton sponge (21.5 mg, 0.10 mmol), and freshly distilled POCl3 (8.1 μL, 13.4 mg, 0.087 mmol), followed by a mixture of tributylammonium pyrophosphate (TBAPP; 0.5 M/dimethylformamide (DMF); 670 μL, 0.34 mmol) and Bu3N (49.7 mg, 0.27 mmol), as described for 24, gave 25 (9.6 mg, 22%) as a triethylammonium salt.
- 2-(Methoxycarbonyl)adenosine (29). A mixture of 28 [50] (100 mg, 0.34 mmol) and NaOMe (5.4 M; 95 μL, 0.51 mmol) in MeOH (10 mL) was stirred at rt for 15 h. After neutralization with Dowex 50 (H+) and filtration of resin, the volatiles were removed at reduced pressure. The residue was dissolved in a mixture of MeOH/H2O (10 mL; 1:1) and 1.0 M HCl (340 μL, 0.34 mmol) was added. The mixture was stirred at rt for 2 h. After neutralization with 1.0 M NaOH, the volatiles were removed at reduced pressure, and the residue was column-chromatographed (10 → 20% MeOH/DCM) to give 29 (89 mg, 80%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H), 7.71 (s, 2H), 5.93 (d, J = 6.4 Hz, 1H), 5.47 (d, J = 6.0 Hz, 1H), 5.22 (d, J = 4.7 Hz, 1H), 5.05 (dd, J = 6.4, 5.2 Hz, 1H), 4.62–4.56 (m, 1H), 4.15 (td, J = 4.8, 2.8 Hz, 1H), 3.96 (q, J = 3.6 Hz, 1H), 3.84 (s, 3H), 3.71–3.64 (m, 1H), 3.56 (ddd, J = 12.0, 6.4, 4.0 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 164.47, 156.15, 150.61, 149.47, 141.85, 120.12, 87.41, 86.08, 73.84, 70.73, 61.67, 52.57. HRMS (TOF, ESI) m/z calcd for C12H16N5O6 326.1095 [M + H]+, found 326.1012.
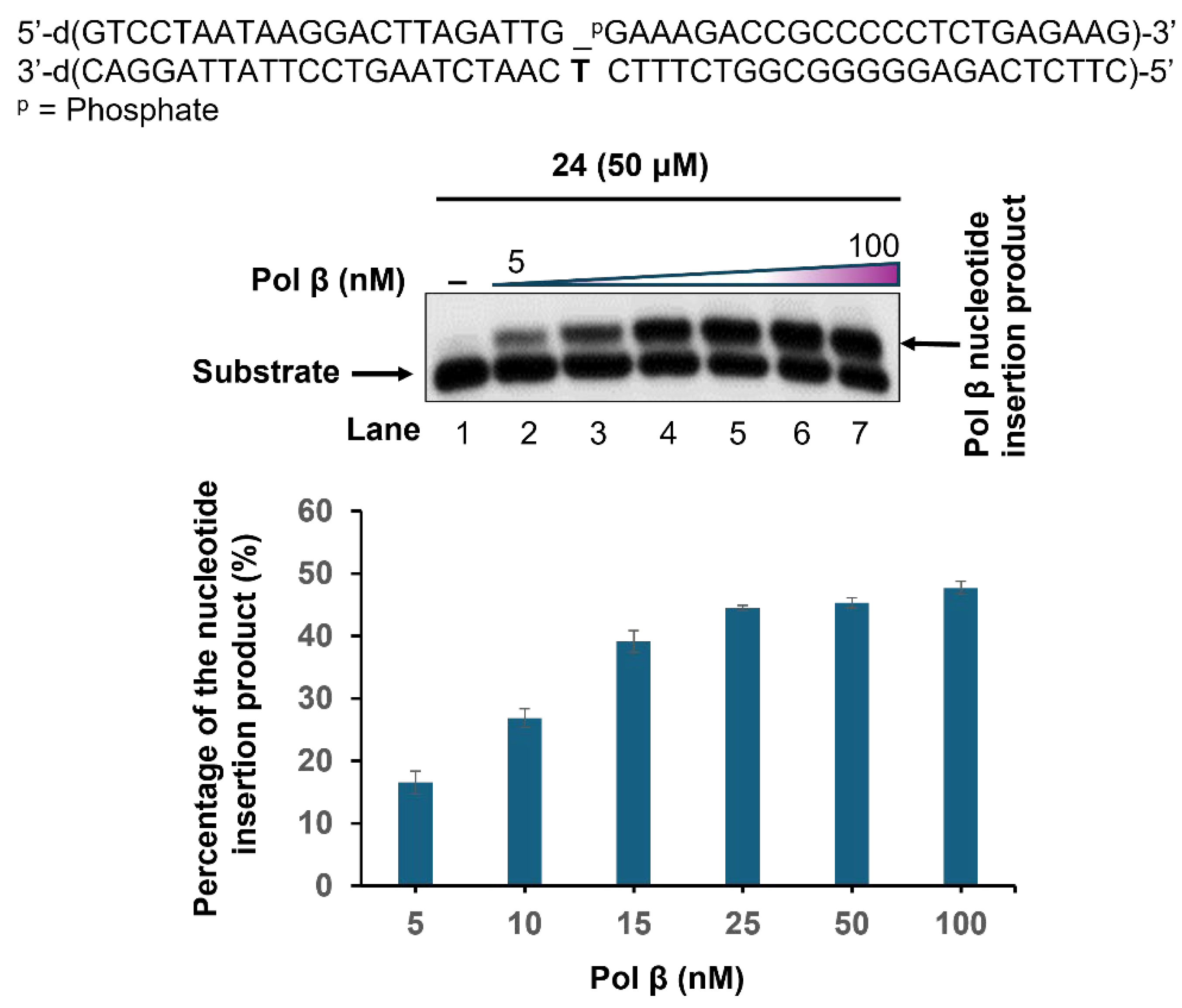
- Incorporation of the nucleotide analog 24 by Pol β
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, Y.; Wen, Z.; Cabrera, M.; Howlader, A.H.; Wnuk, S.F. Purines. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations: Knowledge Updates 2020/1; Christmann, M., Huang, Z., Jiang, X., Li, J.J., Oestreich, M., Petersson, E.J., Schaumann, E., Wang, M., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 2020; pp. 195–384. [Google Scholar]
- Li, Y.; Butt, M.; Bao, W.; Xie, R.; Chen, G. Progress in the Functionalization of Purines and Purine Nucleosides by Minisci Reactions Over the Past 50 Years. ChemCatChem 2025, 17, e202401550. [Google Scholar] [CrossRef]
- Sengupta, S.; Das, P. Application of diazonium chemistry in purine modifications: A focused review. J. Heterocycl. Chem. 2022, 59, 5–21. [Google Scholar] [CrossRef]
- Kumamoto, H.; Tanaka, H.; Tsukioka, R.; Ishida, Y.; Nakamura, A.; Kimura, S.; Hayakawa, H.; Kato, K.; Miyasaka, T. First Evident Generation of Purin-2-yllithium: Lithiation of an 8-Silyl-Protected 6-Chloropurine Riboside as a Key Step for the Synthesis of 2-Carbon-Substituted Adenosines. J. Org. Chem. 1999, 64, 7773–7780. [Google Scholar] [CrossRef]
- Nair, V.; Turner, G.A.; Buenger, G.S.; Chamberlain, S.D. New methodologies for the synthesis of C-2 functionalized hypoxanthine nucleosides. J. Org. Chem. 1988, 53, 3051–3057. [Google Scholar] [CrossRef]
- Van Aerschot, A.A.; Mamos, P.; Weyns, N.J.; Ikeda, S.; De Clercq, E.; Herdewijn, P.A. Antiviral activity of C-alkylated purine nucleosides obtained by cross-coupling with tetraalkyltin reagents. J. Med. Chem. 1993, 36, 2938–2942. [Google Scholar] [CrossRef]
- Cain, R.; Salimraj, R.; Punekar, A.S.; Bellini, D.; Fishwick, C.W.G.; Czaplewski, L.; Scott, D.J.; Harris, G.; Dowson, C.G.; Lloyd, A.J.; et al. Structure-Guided Enhancement of Selectivity of Chemical Probe Inhibitors Targeting Bacterial Seryl-tRNA Synthetase. J. Med. Chem. 2019, 62, 9703–9717. [Google Scholar] [CrossRef]
- Liang, Y.; Wnuk, S. Transition Metal-Catalyzed C–H Functionalization of Nucleoside Bases. In Transition-Metal-Catalyzed C-H Functionalization of Heterocycles; Kumar, A., Punniyamurthy, T., Eds.; Wiley: Hoboken, NJ, USA, 2023; pp. 631–655. [Google Scholar]
- Li, Y.; Zhou, Y.; Zhou, D.; Jiang, Y.; Butt, M.; Yang, H.; Que, Y.; Li, Z.; Chen, G. Regioselective Homolytic C2–H Borylation of Unprotected Adenosine and Adenine Derivatives via Minisci Reaction. J. Am. Chem. Soc. 2024, 146, 21428–21441. [Google Scholar] [CrossRef]
- Ingall, A.H.; Dixon, J.; Bailey, A.; Coombs, M.E.; Cox, D.; McInally, J.I.; Hunt, S.F.; Kindon, N.D.; Teobald, B.J.; Willis, P.A.; et al. Antagonists of the Platelet P2T Receptor: A Novel Approach to Antithrombotic Therapy. J. Med. Chem. 1999, 42, 213–220. [Google Scholar] [CrossRef]
- Springthorpe, B.; Bailey, A.; Barton, P.; Birkinshaw, T.N.; Bonnert, R.V.; Brown, R.C.; Chapman, D.; Dixon, J.; Guile, S.D.; Humphries, R.G.; et al. From ATP to AZD6140: The discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg. Med. Chem. Lett. 2007, 17, 6013–6018. [Google Scholar] [CrossRef]
- Sharif, E.U.; Kalisiak, J.; Lawson, K.V.; Miles, D.H.; Newcomb, E.; Lindsey, E.A.; Rosen, B.R.; Debien, L.P.P.; Chen, A.; Zhao, X.; et al. Discovery of Potent and Selective Methylenephosphonic Acid CD73 Inhibitors. J. Med. Chem. 2021, 64, 845–860. [Google Scholar] [CrossRef]
- Cachatra, V.; Martins, A.; Oliveira, M.C.; Oliveira, M.C.; Gano, L.; Paulo, A.; López, Ó.; Fernández-Bolaños, J.G.; Contino, M.; Colabufo, N.A.; et al. Purine nucleosides as selective inhibitors of butyrylcholinesterase—A multidisciplinary study. Org. Biomol. Chem. 2024. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Šinkevičiūtė, U.; Dvořáková, M.; Tichý, M.; Gurská, S.; Ječmeňová, K.; Poštová Slavětínská, L.; Džubák, P.; Hajdúch, M.; Hocek, M. Synthesis and Biological Activity of 2,6-Disubstituted 7-Deazapurine Ribonucleosides. Eur. J. Org. Chem. 2025, 28, e202401187. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Merighi, S.; Varani, K.; Borea, P.A.; Baraldi, S.; Aghazadeh Tabrizi, M.; Romagnoli, R.; Baraldi, P.G.; Ciancetta, A.; Tosh, D.K.; et al. A(3) Adenosine Receptors as Modulators of Inflammation: From Medicinal Chemistry to Therapy. Med. Res. Rev. 2018, 38, 1031–1072. [Google Scholar] [CrossRef] [PubMed]
- Bednarska-Szczepaniak, K.; Mieczkowski, A.; Kierozalska, A.; Pavlović Saftić, D.; Głąbała, K.; Przygodzki, T.; Stańczyk, L.; Karolczak, K.; Watała, C.; Rao, H.; et al. Synthesis and evaluation of adenosine derivatives as A(1), A(2A), A(2B) and A(3) adenosine receptor ligands containing boron clusters as phenyl isosteres and selective A(3) agonists. Eur. J. Med. Chem. 2021, 223, 113607. [Google Scholar] [CrossRef]
- Abiru, T.; Miyashita, T.; Watanabe, Y.; Yamaguchi, T.; Machida, H.; Matsuda, A. Nucleosides and nucleotides. 107. 2-(Cycloalkylalkynyl)adenosines: Adenosine A2 receptor agonists with potent antihypertensive effects. J. Med. Chem. 1992, 35, 2253–2260. [Google Scholar] [CrossRef]
- Kim, G.; Jarhad, D.B.; Lee, G.; Kim, G.; Hou, X.; Yu, J.; Lee, C.S.; Warnick, E.; Gao, Z.-G.; Ahn, S.Y.; et al. Structural Modification and Biological Evaluation of 2,8-Disubstituted Adenine and Its Nucleosides as A2A Adenosine Receptor Antagonists: Exploring the Roles of Ribose at Adenosine Receptors. J. Med. Chem. 2024, 67, 10490–10507. [Google Scholar] [CrossRef]
- Krömer, M.; Brunderová, M.; Ivancová, I.; Poštová Slavětínská, L.; Hocek, M. 2-Formyl-dATP as Substrate for Polymerase Synthesis of Reactive DNA Bearing an Aldehyde Group in the Minor Groove. ChemPlusChem 2020, 85, 1164–1170. [Google Scholar] [CrossRef]
- Duan, H.-C.; Zhang, C.; Song, P.; Yang, J.; Wang, Y.; Jia, G. C2-methyladenosine in tRNA promotes protein translation by facilitating the decoding of tandem m2A-tRNA-dependent codons. Nat. Commun. 2024, 15, 1025. [Google Scholar] [CrossRef]
- Ferguson, L.; Madieh, N.S.; Vaideanu, A.; Schatzlein, A.; Festa, J.; Singh, H.; Wells, G.; Bhakta, S.; Brucoli, F. C2-linked alkynyl poly-ethylene glycol(PEG) adenosine conjugates as water-soluble adenosine receptor agonists. Chem. Biol. Drug Des. 2023, 101, 340–349. [Google Scholar] [CrossRef]
- Matyašovský, J.; Perlíková, P.; Malnuit, V.; Pohl, R.; Hocek, M. 2-Substituted dATP Derivatives as Building Blocks for Polymerase-Catalyzed Synthesis of DNA Modified in the Minor Groove. Angew. Chem. Int. Ed. 2016, 55, 15856–15859. [Google Scholar] [CrossRef]
- Parker, W.B.; Shaddix, S.C.; Chang, C.-H.; White, E.L.; Rose, L.M.; Brockman, R.W.; Shortnacy, A.T.; Montgomery, J.A.; Secrist, J.A.; Bennett, L.L. Effects of 2-Chloro-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)adenine on K562 Cellular Metabolism and the Inhibition of Human Ribonucleotide Reductase and DNA Polymerases by Its 5′-Triphosphate. Cancer Res. 1991, 51, 2386–2394. [Google Scholar] [PubMed]
- Chollet, A.; Kawashima, E. DNA containing the base analogue 2-aminoadenine: Preparation, use as hybridization probes and cleavage by restriction endonucleases. Nucleic Acids Res. 1988, 16, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Kutyavin, I.V. Use of Base-Modified Duplex-Stabilizing Deoxynucleoside 5′-Triphosphates To Enhance the Hybridization Properties of Primers and Probes in Detection Polymerase Chain Reaction. Biochemistry 2008, 47, 13666–13673. [Google Scholar] [CrossRef] [PubMed]
- Matyašovský, J.; Pohl, R.; Hocek, M. 2-Allyl- and Propargylamino-dATPs for Site-Specific Enzymatic Introduction of a Single Modification in the Minor Groove of DNA. Chem. Eur. J. 2018, 24, 14938–14941. [Google Scholar] [CrossRef]
- Matyašovský, J.; Hocek, M. 2-Substituted 2′-deoxyinosine 5′-triphosphates as substrates for polymerase synthesis of minor-groove-modified DNA and effects on restriction endonuclease cleavage. Org. Biomol. Chem. 2020, 18, 255–262. [Google Scholar] [CrossRef]
- Liang, Y.; Suzol, S.H.; Wen, Z.; Artiles, A.G.; Mathivathanan, L.; Raptis, R.G.; Wnuk, S.F. Uracil Nucleosides with Reactive Group at C5 Position: 5-(1-Halo-2-sulfonylvinyl)uridine Analogues. Org. Lett. 2016, 18, 1418–1421. [Google Scholar] [CrossRef]
- Wen, Z.; Suzol, S.H.; Peng, J.; Liang, Y.; Snoeck, R.; Andrei, G.; Liekens, S.; Wnuk, S.F. Antiviral and Cytostatic Evaluation of 5-(1-Halo-2-sulfonylvinyl)- and 5-(2-Furyl)uracil Nucleosides. Arch. Pharm. 2017, 350, 1700023. [Google Scholar] [CrossRef]
- Suzol, S.H.; Howlader, A.H.; Wen, Z.; Ren, Y.; Laverde, E.E.; Garcia, C.; Liu, Y.; Wnuk, S.F. Pyrimidine Nucleosides with a Reactive (β-Chlorovinyl)sulfone or (β-Keto)sulfone Group at the C5 Position, Their Reactions with Nucleophiles and Electrophiles, and Their Polymerase-Catalyzed Incorporation into DNA. ACS Omega 2018, 3, 4276–4288. [Google Scholar] [CrossRef]
- Howlader, H.; Suzol, S.H.; Blanco, K.; Martin-Rafa, L.; Laverde, E.E.; Liu, Y.; Wnuk, S.F. Purine Nucleosides with a Reactive (β-Iodovinyl)sulfone or a (β-Keto)sulfone Group at the C8 Position and Their Polymerase-Catalyzed Incorporation into DNA. Asian J. Org. Chem. 2022, 11, e202100764. [Google Scholar] [CrossRef]
- Tanaka, M.; Kozakai, R.; Saito, Y.; Saito, I. Stabilization of DNA duplex by 2-substituted adenine as a minor groove modifier. Bioorg. Med. Chem. Lett. 2011, 21, 7021–7024. [Google Scholar] [CrossRef]
- Grünewald, C.; Kwon, T.; Piton, N.; Förster, U.; Wachtveitl, J.; Engels, J.W. RNA as scaffold for pyrene excited complexes. Bioorg. Med. Chem. 2008, 16, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Purdy, D.F. Synthetic approaches to new doubly modified nucleosides: Congeners of cordycepin and related 2′-deoxyadenosine. Tetrahedron 1991, 47, 365–382. [Google Scholar] [CrossRef]
- Zhang, Y.; Vessally, E. Direct halosulfonylation of alkynes: An overview. RSC Adv. 2021, 11, 33447–33460. [Google Scholar] [CrossRef]
- Reddy, R.J.; Kumar, J.J.; Kumari, A.H. Recent trends in the synthesis and applications of β-iodovinyl sulfones: A decade of progress. Org. Biomol. Chem. 2024, 22, 2492–2509. [Google Scholar] [CrossRef]
- Reddy, R.J.; Kumar, J.J.; Kumari, A.H. Unprecedented Reactivity of β-Iodovinyl Sulfones: An Efficient Synthesis of β-Keto Sulfones and β-Keto Thiosulfones. Eur. J. Org. Chem. 2019, 2019, 3771–3775. [Google Scholar] [CrossRef]
- Nair, V.; Augustine, A.; Suja, T.D. CAN Mediated Reaction of Aryl Sulfinates with Alkenes and Alkynes: Synthesis of Vinyl Sulfones, β-Iodovinyl Sulfones and Acetylenic Sulfones. Synthesis 2002, 2002, 2259–2265. [Google Scholar] [CrossRef]
- Sun, Y.; Abdukader, A.; Lu, D.; Zhang, H.; Liu, C. Synthesis of (E)-β-iodo vinylsulfones via iodine-promoted iodosulfonylation of alkynes with sodium sulfinates in an aqueous medium at room temperature. Green Chem. 2017, 19, 1255–1258. [Google Scholar] [CrossRef]
- Katrun, P.; Chiampanichayakul, S.; Korworapan, K.; Pohmakotr, M.; Reutrakul, V.; Jaipetch, T.; Kuhakarn, C. PhI(OAc)2/KI-Mediated Reaction of Aryl Sulfinates with Alkenes, Alkynes, and α,β-Unsaturated Carbonyl Compounds: Synthesis of Vinyl Sulfones and β-Iodovinyl Sulfones. Eur. J. Org. Chem. 2010, 2010, 5633–5641. [Google Scholar] [CrossRef]
- Wan, J.-P.; Hu, D.; Bai, F.; Wei, L.; Liu, Y. Stereoselective Z-halosulfonylation of terminal alkynes using sulfonohydrazides and CuX (X = Cl, Br, I). RSC Adv. 2016, 6, 73132–73135. [Google Scholar] [CrossRef]
- Liu, L.K.; Chi, Y.; Jen, K.-Y. Copper-catalyzed additions of sulfonyl iodides to simple and cyclic alkenes. J. Org. Chem. 1980, 45, 406–410. [Google Scholar] [CrossRef]
- Tsui, G.C.; Glenadel, Q.; Lau, C.; Lautens, M. Rhodium(I)-Catalyzed Addition of Arylboronic Acids to (Benzyl-/Arylsulfonyl)acetonitriles: Efficient Synthesis of (Z)-β-Sulfonylvinylamines and β-Keto Sulfones. Org. Lett. 2011, 13, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Huang, L.; Xu, Y.; Yang, J.; Wu, W.; Jiang, H. Copper-Catalyzed Coupling of Oxime Acetates with Sodium Sulfinates: An Efficient Synthesis of Sulfone Derivatives. Angew. Chem. Int. Ed. 2014, 53, 4205–4208. [Google Scholar] [CrossRef] [PubMed]
- Markitanov, Y.M.; Timoshenko, V.M.; Shermolovich, Y.G. β-Keto sulfones: Preparation and application in organic synthesis. J. Sulphur Chem. 2014, 35, 188–236. [Google Scholar] [CrossRef]
- Nasuhipur, F.; Ghasemi, Z.; Poupon, M.; Dušek, M. POCl(3) mediated one-pot deoxygenative aromatization and electrophilic chlorination of dihydroxy-2-methyl-4-oxo-indeno[1,2-b]pyrroles. RSC Adv. 2023, 13, 17812–17816. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Kolodiazhna, A. Nucleophilic substitution at phosphorus: Stereochemistry and mechanisms. Tetrahedron Asymmetry 2017, 28, 1651–1674. [Google Scholar] [CrossRef]
- Suryakiran, N.; Srikanth Reddy, T.; Suresh, V.; Lakshman, M.; Venkateswarlu, Y. Synthesis of α-iodo β-ketosulfones and α-iodo methylsulfones using iodine monochloride. Tetrahedron Lett. 2006, 47, 4319–4323. [Google Scholar] [CrossRef]
- Suryakiran, N.; Prabhakar, P.; Srikanth Reddy, T.; Chinni Mahesh, K.; Rajesh, K.; Venkateswarlu, Y. Chemoselective mono halogenation of β-keto-sulfones using potassium halide and hydrogen peroxide; synthesis of halomethyl sulfones and dihalomethyl sulfones. Tetrahedron Lett. 2007, 48, 877–881. [Google Scholar] [CrossRef]
- Matsuda, A.; Nomoto, Y.; Ueda, T. Synthesis of 2- and 8-Cyanoadenosines and Their Derivatives: Nucleosides and Nucleotides. XXVII. Chem. Pharm. Bull. 1979, 27, 183–192. [Google Scholar] [CrossRef]
- Ding, T.; Tang, F.; Ni, G.; Liu, J.; Zhao, H.; Chen, Q. The development of isoguanosine: From discovery, synthesis, and modification to supramolecular structures and potential applications. RSC Adv. 2020, 10, 6223–6248. [Google Scholar] [CrossRef]
- Xia, Z.; Kondhare, D.; Budow-Busse, S.; Leonard, P.; Seela, F. 7-Deaza-2’-deoxyisoguanosine, a Noncanonical Nucleoside for Nucleic Acid Code Expansion and New DNA Constructs: Nucleobase Functionalization of Inverse Watson–Crick and Purine–Purine Base Pairs. Bioconjugate Chem. 2024, 35, 1233–1250. [Google Scholar] [CrossRef]
- Hollenstein, M. Deoxynucleoside triphosphates bearing histamine, carboxylic acid, and hydroxyl residues—Synthesis and biochemical characterization. Org. Biomol. Chem. 2013, 11, 5162–5172. [Google Scholar] [CrossRef] [PubMed]
- Franzini, R.M.; Samain, F.; Abd Elrahman, M.; Mikutis, G.; Nauer, A.; Zimmermann, M.; Scheuermann, J.; Hall, J.; Neri, D. Systematic Evaluation and Optimization of Modification Reactions of Oligonucleotides with Amines and Carboxylic Acids for the Synthesis of DNA-Encoded Chemical Libraries. Bioconjugate Chem. 2014, 25, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, V.P.; Vozdvizhenskaya, O.A.; Baryshnikova, M.A.; Pershina, A.G.; Musiyak, V.V.; Matveeva, T.V.; Nevskaya, K.V.; Brikunova, O.Y.; Gruzdev, D.A.; Levit, G.L. Synthesis and Cytotoxic Activity of the Derivatives of N-(Purin-6-yl)aminopolymethylene Carboxylic Acids and Related Compounds. Molecules 2023, 28, 1853. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.M.; Lai, Y.; Xu, M.; Casin, A.H.; Laverde, E.E.; Liu, Y. AP endonuclease 1 prevents trinucleotide repeat expansion via a novel mechanism during base excision repair. Nucleic Acids Res. 2015, 43, 5948–5960. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Halogen Source | Ts Source | Solvent | Additive | Temp, Time | Product | Yields (%) |
| 1 | I2 | TsNa | MeCN | NaOAc | 80 °C, 1.5 h | 7 | 80 |
| 2 | I2 | TsNa | H2O | 80 °C, 1.5 h | 7 | 40 | |
| 3 | I2 | TsNa | H2O/MeCN | 80 °C, 1.5 h | 7 | 60 | |
| 4 | KI | TsNHNH2 | DMSO | Bz2O | rt, 15 h | 8 | 0 [a] |
| 5 | CuI | TsNHNH2 | DMSO | Bz2O | rt, 15 h | 8 | 0 |
| 6 | NaI | TsNa | MeCN | CAN [b] | 80 °C, 2 h | 8 | 0 |
| 7 | KI | TsNa | MeCN | PhI(OAc)2 | 80 °C, 2 h | 8 | 0 |
| 8 | TsI | THF | rt, 40 h | 8 | 0 [c] | ||
| 9 | TsI | THF | NaOAc | rt, 40 h | 8 | 80 | |
| 10 | TsI | THF | NaOAc | rt, 40 h | 9 | 9 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Howlader, A.H.; Fernandez, R.; Tsegay, P.S.; Liu, Y.; Wnuk, S.F. Synthesis of Adenine Nucleosides with a Reactive (β-Iodovinyl)sulfone or (β-Keto)sulfone Group at the C2 Position and Their Polymerase-Catalyzed Incorporation into DNA. Molecules 2025, 30, 1358. https://doi.org/10.3390/molecules30061358
Howlader AH, Fernandez R, Tsegay PS, Liu Y, Wnuk SF. Synthesis of Adenine Nucleosides with a Reactive (β-Iodovinyl)sulfone or (β-Keto)sulfone Group at the C2 Position and Their Polymerase-Catalyzed Incorporation into DNA. Molecules. 2025; 30(6):1358. https://doi.org/10.3390/molecules30061358
Chicago/Turabian StyleHowlader, A. Hasan, Richard Fernandez, Pawlos S. Tsegay, Yuan Liu, and Stanislaw F. Wnuk. 2025. "Synthesis of Adenine Nucleosides with a Reactive (β-Iodovinyl)sulfone or (β-Keto)sulfone Group at the C2 Position and Their Polymerase-Catalyzed Incorporation into DNA" Molecules 30, no. 6: 1358. https://doi.org/10.3390/molecules30061358
APA StyleHowlader, A. H., Fernandez, R., Tsegay, P. S., Liu, Y., & Wnuk, S. F. (2025). Synthesis of Adenine Nucleosides with a Reactive (β-Iodovinyl)sulfone or (β-Keto)sulfone Group at the C2 Position and Their Polymerase-Catalyzed Incorporation into DNA. Molecules, 30(6), 1358. https://doi.org/10.3390/molecules30061358