

Functionalisation of Lignin-Derived Diols for the Synthesis of Thermoplastic Polyurethanes and Polyester Resins

 ,
,
Abstract

1. Introduction
2. Results and Discussion
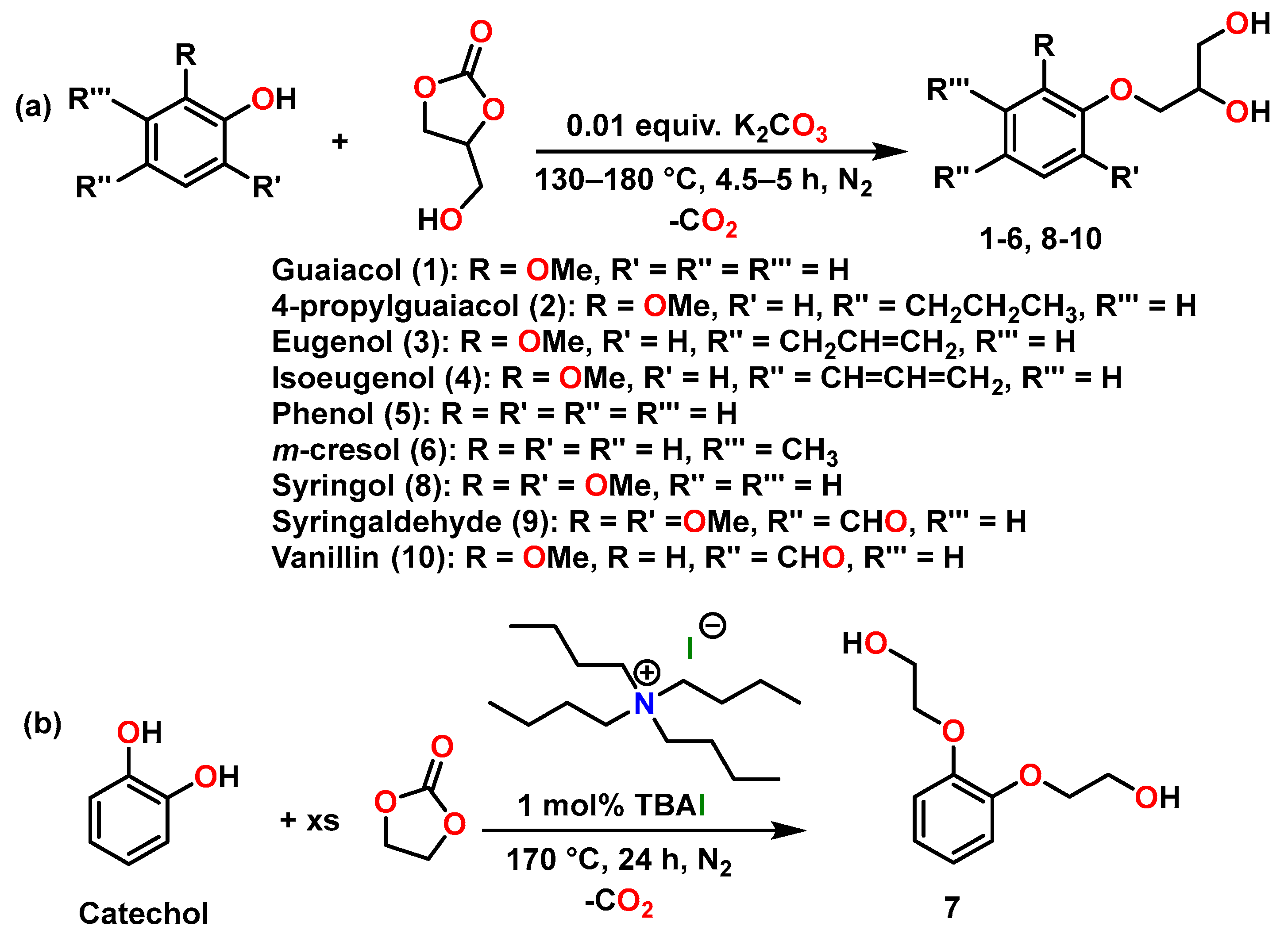
2.1. Lignin-Based Diol Synthesis
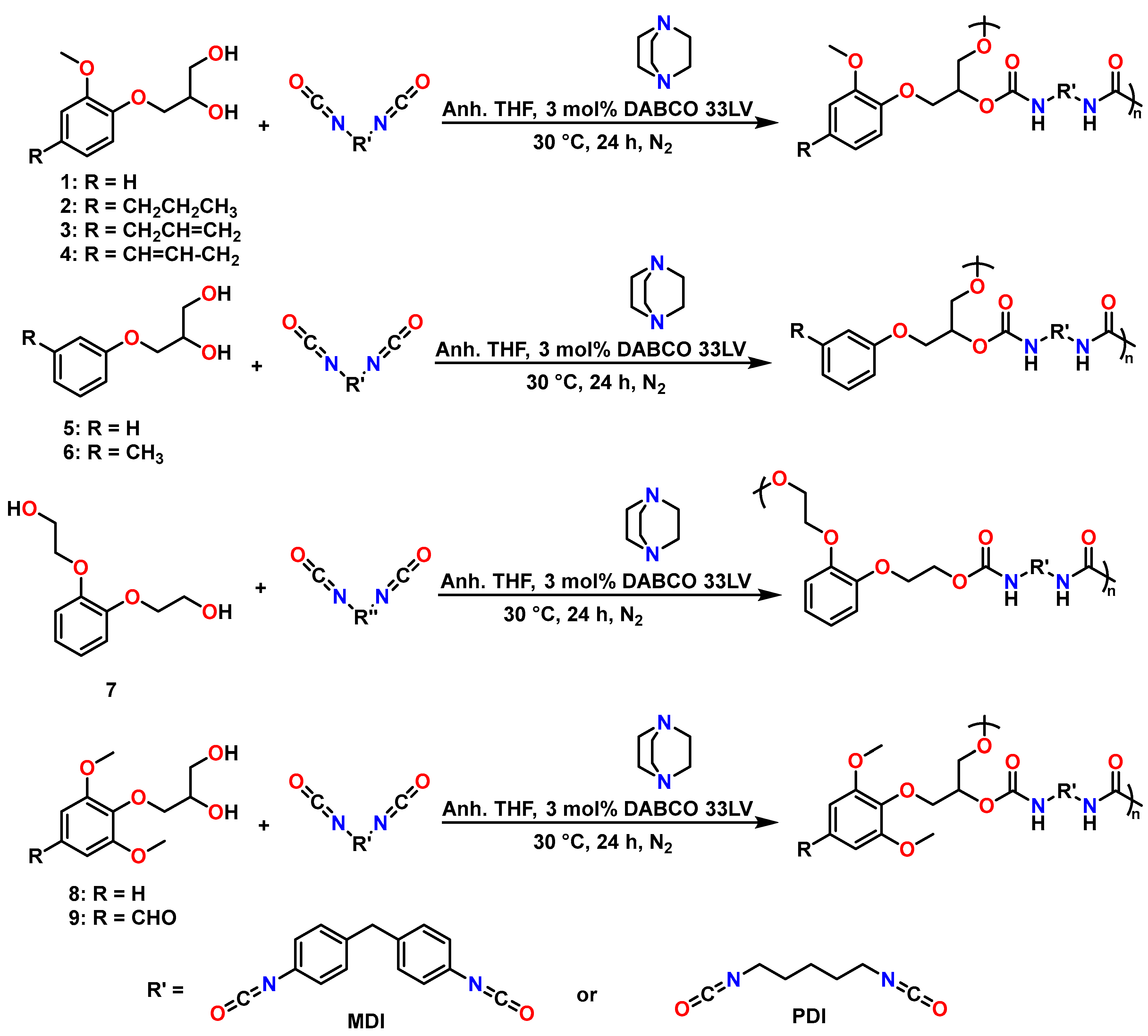
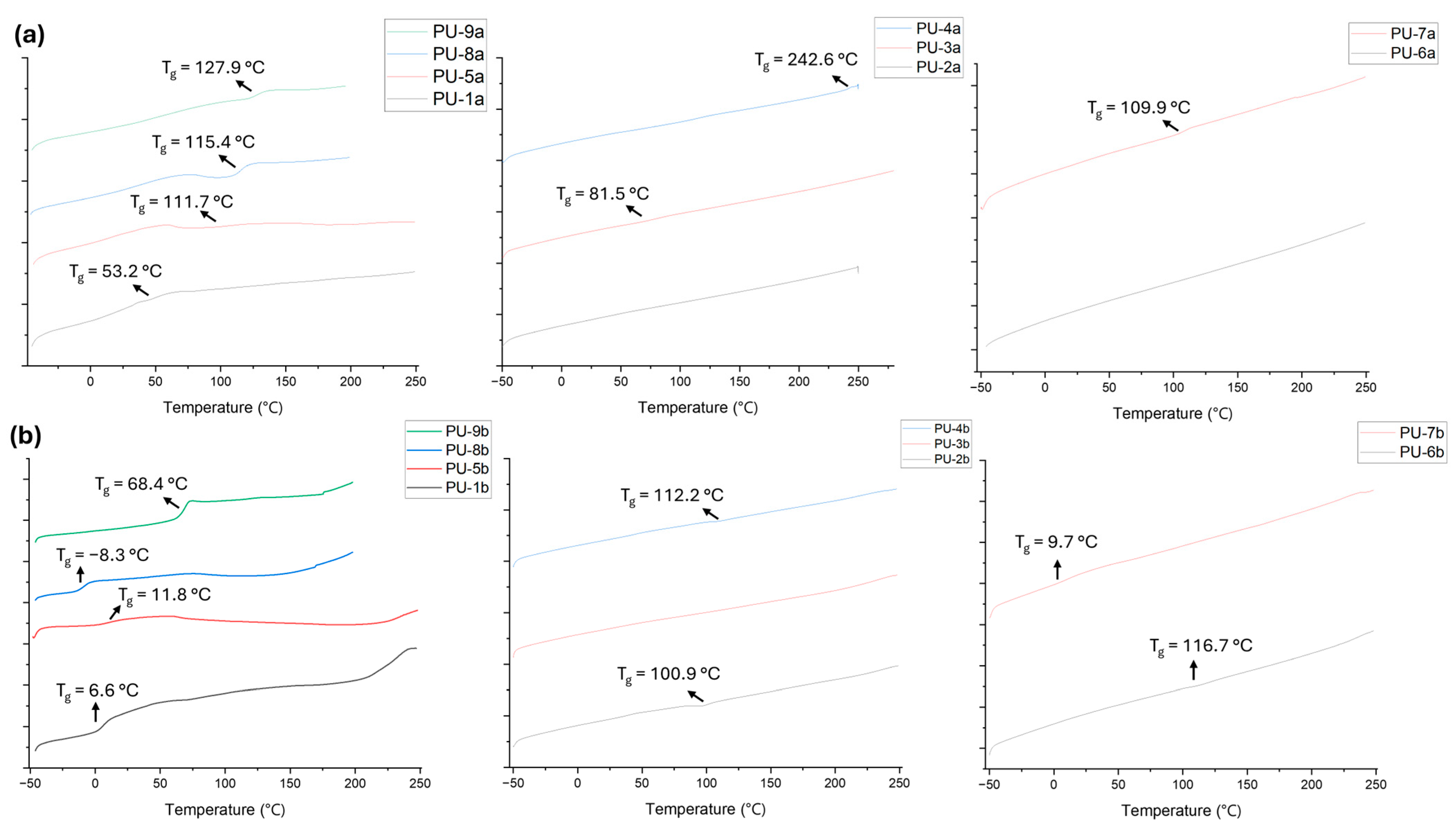
2.2. PU Synthesis and Characterisation
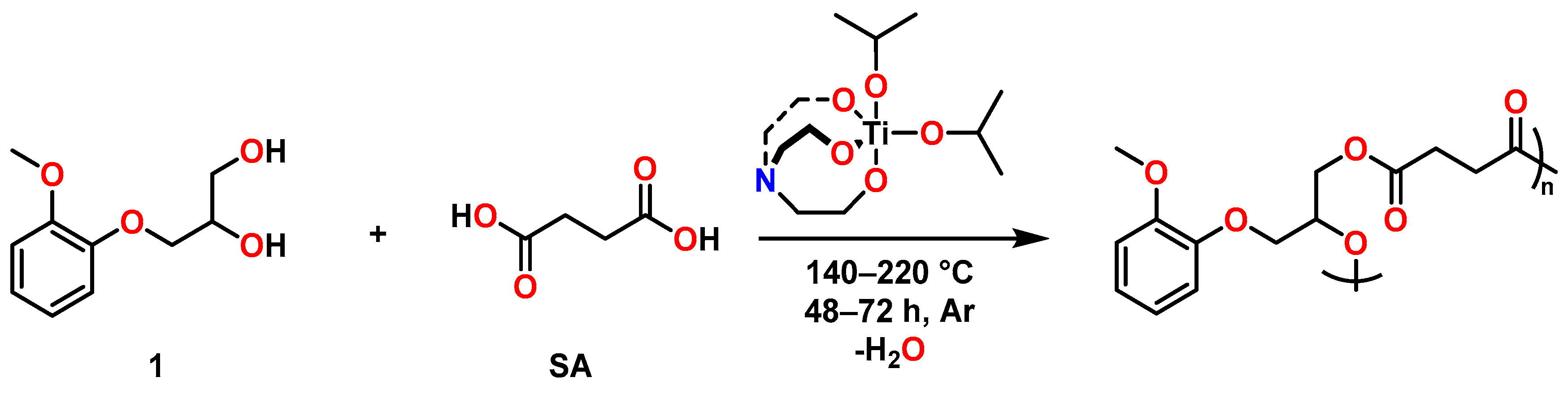
2.3. PE Synthesis and Characterisation
3. Materials and Methods
3.1. Materials
3.1.1. General Synthesis of Diols 1–6 and 8–10
3.1.2. Synthesis of Diol 7
3.2. General Preparation of PUs
3.3. General Preparation of PEs
3.3.1. Preparation of PE-1a, PE-1b and PE-1c
3.3.2. Evaluation of the Catalyst for PE-1a
3.3.3. Preparation of PE-2
3.4. Characterisation Techniques
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ar | Argon |
| CDCl3 | Deuterated chloroform |
| COSY | Correlation spectroscopy |
| Ð | Dispersity |
| DABCO | 1,4-diazabicyclo[2.2.2]octane |
| DART | Direct analysis in real time |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DMSO-d6 | Deuterated dimethyl sulfoxide |
| DSC | Differential scanning calorimetry |
| Et2O | Diethyl ether |
| EtOAc | Ethyl acetate |
| FTIR | Fourier transform infrared spectroscopy |
| GPC | Gel permeation chromatography |
| HRMS | High resolution mass spectrometry |
| IA | Isophthalic acid |
| K2CO3 | Potassium carbonate |
| MeOH | Methanol |
| MDI | Methylene diphenyl diisocyanate |
| Mn | Number average molecular weight |
| MW | Weight average molecular weight |
| N2 | Nitrogen |
| NaBH4 | Sodium borohydride |
| Na2SO4 | Sodium sulfate |
| NMR | Nuclear magnetic resonance |
| PDI | Pentamethylene diisocyanate |
| PE | Polyester |
| PU | Polyurethane |
| SA | Succinic acid |
| TA | Terephthalic acid |
| TBAI | Tetrabutylammonium iodide |
| TBAF•3H2O | Tetrabutylammonium fluoride |
| TGA | Thermogravimetric analysis |
| Tg | Glass transition temperature |
| THF | Tetrahydrofuran |
| TLC | Thin layer chromatography |
| Ts | Softening point |
References
- Haile, A.; Gelebo, G.G.; Tesfaye, T.; Mengie, W.; Mebrate, M.A.; Abuhay, A.; Limeneh, D.Y. Pulp and Paper Mill Wastes: Utilizations and Prospects for High Value-Added Biomaterials. Bioresour. Bioprocess. 2021, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Bertella, S.; Luterbacher, J.S. Lignin Functionalization for the Production of Novel Materials. Trends Chem. 2020, 2, 440–453. [Google Scholar] [CrossRef]
- Zhao, S.; Abu-Omar, M.M. Materials Based on Technical Bulk Lignin. ACS Sustain. Chem. Eng. 2021, 9, 1477–1493. [Google Scholar] [CrossRef]
- Carafa, R.N.; Foucher, D.A.; Sacripante, G.G. Biobased Polymers from Lignocellulosic Sources. Green Chem. Lett. Rev. 2023, 16, 2153087. [Google Scholar] [CrossRef]
- Du, L.; Wang, Z.; Li, S.; Song, W.; Lin, W. A Comparison of Monomeric Phenols Produced from Lignin by Fast Pyrolysis and Hydrothermal Conversions. Int. J. Chem. React. Eng. 2013, 11, 135–145. [Google Scholar] [CrossRef]
- Gandini, A.; Lacerda, T.M. From Monomers to Polymers from Renewable Resources: Recent Advances. Prog. Polym. Sci. 2015, 48, 1–39. [Google Scholar] [CrossRef]
- Tabanelli, T.; Giliberti, C.; Mazzoni, R.; Cucciniello, R.; Cavani, F. An Innovative Synthesis Pathway to Benzodioxanes: The Peculiar Reactivity of Glycerol Carbonate and Catechol. Green Chem. 2019, 21, 329–338. [Google Scholar] [CrossRef]
- Sternberg, J.; Pilla, S. Materials for the Biorefinery: High Bio-Content, Shape Memory Kraft Lignin-Derived Non-Isocyanate Polyurethane Foams Using a Non-Toxic Protocol. Green Chem. 2020, 22, 6922–6935. [Google Scholar] [CrossRef]
- Kao, S.-C.; Lin, Y.-C.; Ryu, I.; Wu, Y.-K. Revisiting Hydroxyalkylation of Phenols with Cyclic Carbonates. Adv. Synth. Catal. 2019, 361, 3639–3644. [Google Scholar] [CrossRef]
- de Caro, P.; Bandres, M.; Urrutigoïty, M.; Cecutti, C.; Thiebaud-Roux, S. Recent Progress in Synthesis of Glycerol Carbonate and Evaluation of Its Plasticizing Properties. Front. Chem. 2019, 7, 308. [Google Scholar] [CrossRef]
- Truscello, A.M.; Gambarotti, C.; Lauria, M.; Auricchio, S.; Leonardi, G.; Shisodia, S.U.; Citterio, A. One-Pot Synthesis of Aryloxypropanediols from Glycerol: Towards Valuable Chemicals from Renewable Sources. Green Chem. 2013, 15, 625–628. [Google Scholar] [CrossRef]
- Galletti, G.; Prete, P.; Vanzini, S.; Cucciniello, R.; Fasolini, A.; De Maron, J.; Cavani, F.; Tabanelli, T. Glycerol Carbonate as a Versatile Alkylating Agent for the Synthesis of β-Aryloxy Alcohols. ACS Sustain. Chem. Eng. 2022, 10, 10922–10933. [Google Scholar] [CrossRef]
- Kosalka, J.J.S.; Sacripante, G.G.; Lough, A.J.; Foucher, D.A.; Gossage, R.A. Biomass Utilisation Strategies for Applications in Novel Polymer and Polymer Resin Production. Polym. From Renew. Resour. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Kinsyo, T.; Nakanishi, H.; Hirai, K.; Noda, H.; Takikawa, T.; Yahiro, S. Development of Polyester Resin Particles for Toner with a Controlled Particle Size Distribution and Shape. Polym. J. 2017, 49, 593–600. [Google Scholar] [CrossRef]
- Alinejad, M.; Henry, C.; Nikafshar, S.; Gondaliya, A.; Bagheri, S.; Chen, N.; Singh, S.K.; Hodge, D.B.; Nejad, M. Lignin-Based Polyurethanes: Opportunities for Bio-Based Foams, Elastomers, Coatings and Adhesives. Polymers 2019, 11, 1202. [Google Scholar] [CrossRef]
- Wendels, S.; Avérous, L. Biobased Polyurethanes for Biomedical Applications. Bioact. Mater. 2021, 6, 1083–1106. [Google Scholar] [CrossRef]
- Xu, T.; Shen, W.; Lin, X.; Xie, Y.M. Mechanical Properties of Additively Manufactured Thermoplastic Polyurethane (TPU) Material Affected by Various Processing Parameters. Polymers 2020, 12, 3010. [Google Scholar] [CrossRef]
- Carafa, R.N.; Fernandes, B.V.; Repiquet, C.; Rana, S.; Foucher, D.A.; Sacripante, G.G. Functionalization of Phenolic Aldehydes for the Preparation of Sustainable Polyesters and Polyurethanes. Polymers 2025, 17, 643. [Google Scholar] [CrossRef]
- Sheldon, R.A. Metrics of Green Chemistry and Sustainability: Past, Present, and Future. ACS Sustain. Chem. Eng. 2018, 6, 32–48. [Google Scholar] [CrossRef]
- Kosalka, J.J.S. Green Resins from Renewable Resources. Master’s Thesis, Ryerson University, Toronto, ON, Canada, 2014. [Google Scholar]
- Sayyed, I.A.; Thakur, V.V.; Nikalje, M.D.; Dewkar, G.K.; Kotkar, S.P.; Sudalai, A. Asymmetric Synthesis of Aryloxypropanolamines via OsO4-Catalyzed Asymmetric Dihydroxylation. Tetrahedron 2005, 61, 2831–2838. [Google Scholar] [CrossRef]
- Bredikhin, A.A.; Bredikhina, Z.A.; Antonovich, O.A.; Zakharychev, D.V.; Krivolapov, D.B. Crystallization Features and Spontaneous Resolution of 3-(2,6-Dimethoxyphenoxy)Propane-1,2-Diol: The Case of Stable Conglomerate and Metastable Solid Solution. J. Mol. Struct. 2017, 1144, 443–450. [Google Scholar] [CrossRef]
- Zhao, C.; Huang, C.; Chen, Q.; Ingram, I.D.V.; Zeng, X.; Ren, T.; Xie, H. Sustainable Aromatic Aliphatic Polyesters and Polyurethanes Prepared from Vanillin-Derived Diols via Green Catalysis. Polymers 2020, 12, 586. [Google Scholar] [CrossRef] [PubMed]
- Shinji, K.; Funato, R.; Tajiri, N.; Ito, H.; Iwasaki, H. Polyester Resin for Toner and Process for Its Production. US4866158, 12 September 1989. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Diisocyanate | Appearance | Yield (%) | Tg (°C) | Td (5%) (°C) | Td (50%) (°C) |
|---|---|---|---|---|---|---|
| PU-1a | MDI | Gummy | 94 | 53.2 | 185 | 348 |
| PU-1b | PDI | Gel | 75 | 6.6 | 209 | 279 |
| PU-2a | MDI | Powder | 33 | - | 215 | 331 |
| PU-2b | PDI | Gel | 27 | 100.9 | 140 | 319 |
| PU-3a | MDI | Gummy | 52 | 81.5 | 224 | 335 |
| PU-3b | PDI | Powder | 44 | - | 212 | 350 |
| PU-4a | MDI | Powder | 94 | 242.6 | 155 | 338 |
| PU-4b | PDI | Gummy | 61 | 112.2 | 145 | 316 |
| PU-5a | MDI | Gummy | 58 | 111.7 | 222 | 339 |
| PU-5b | PDI | Gummy | 66 | 11.8 | 206 | 285 |
| PU-6a | MDI | Gummy | 12 | - | 142 | 336 |
| PU-6b | PDI | Gel | 17 | 116.7 | 124 | 317 |
| PU-7a | MDI | Powder | 98 | 109.9 | 284 | 335 |
| PU-7b | PDI | Gel | 33 | 9.7 | 193 | 320 |
| PU-8a | MDI | Gummy | 83 | 115.4 | 239 | 337 |
| PU-8b | PDI | Gel | 30 | −8.3 | 175 | 253 |
| PU-9a | MDI | Gummy | 92 | 127.9 | 143 | 362 |
| PU-9b | PDI | Powder | 57 | 68.4 | 176 | 335 |
| Sample | Diacid | Yield (%) | Tg (°C) | Ts (°C) | Mw (g/mol) | Mn (g/mol) | Ð (Mw/Mn) |
|---|---|---|---|---|---|---|---|
| PE-1a | SA | 91 | 17.7 | 100.1 | 26,440 | 3730 | 7.1 |
| PE-1b | TA | 94 | 41.8 | 102.9 | 3900 | 2790 | 1.4 |
| PE-1c | IA | 89 | 49.5 | 104.1 | 5070 | 2530 | 2.0 |
| PE-2 | SA | 88 | 20.1 | <100 | 94,500 | 2840 | 33.3 |
| PE-3 [13] | SA | 90 | 29.9 | <100 | 132,900 | 1850 | 71.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carafa, R.N.; Kosalka, J.J.S.; Fernandes, B.V.; Desai, U.; Foucher, D.A.; Sacripante, G.G. Functionalisation of Lignin-Derived Diols for the Synthesis of Thermoplastic Polyurethanes and Polyester Resins. Molecules 2025, 30, 2604. https://doi.org/10.3390/molecules30122604
Carafa RN, Kosalka JJS, Fernandes BV, Desai U, Foucher DA, Sacripante GG. Functionalisation of Lignin-Derived Diols for the Synthesis of Thermoplastic Polyurethanes and Polyester Resins. Molecules. 2025; 30(12):2604. https://doi.org/10.3390/molecules30122604
Chicago/Turabian StyleCarafa, Rachele N., Justin J. S. Kosalka, Brigida V. Fernandes, Unnati Desai, Daniel A. Foucher, and Guerino G. Sacripante. 2025. "Functionalisation of Lignin-Derived Diols for the Synthesis of Thermoplastic Polyurethanes and Polyester Resins" Molecules 30, no. 12: 2604. https://doi.org/10.3390/molecules30122604
APA StyleCarafa, R. N., Kosalka, J. J. S., Fernandes, B. V., Desai, U., Foucher, D. A., & Sacripante, G. G. (2025). Functionalisation of Lignin-Derived Diols for the Synthesis of Thermoplastic Polyurethanes and Polyester Resins. Molecules, 30(12), 2604. https://doi.org/10.3390/molecules30122604