
Umbrella Refinement of Ensembles—An Alternative View of Ensemble Optimization


Abstract

1. Introduction
1.1. The Conformational Ensemble
1.2. Reweighting of Ensembles
2. Umbrella Refinement of Ensembles
2.1. Optimizing the k-Vector
Umbrella Refinement and the Maximum Entropy Principle
- 1.
- Per-Observable ConstraintsThe classical solution of the maximum entropy method, leveraging Lagrange multipliers, minimizes the KL divergence under the condition of multiple per-observable constraints. If the reversed KL divergence is used, this approach offers a fast solution to the minimization problem, incentivizing the use of this direction if per-observable constraints are used.
- 2.
- The Global ConstraintAlternatively, a constraint could be set not on the individual observables but on the value, a metric that measures an average-like deviation between the experiment and simulation for all tracked observables. This metric allows for errors to compensate each other and tolerates some observables deviating as long as the majority are compliant.
2.2. Estimation of the Hyper-Parameter Theta
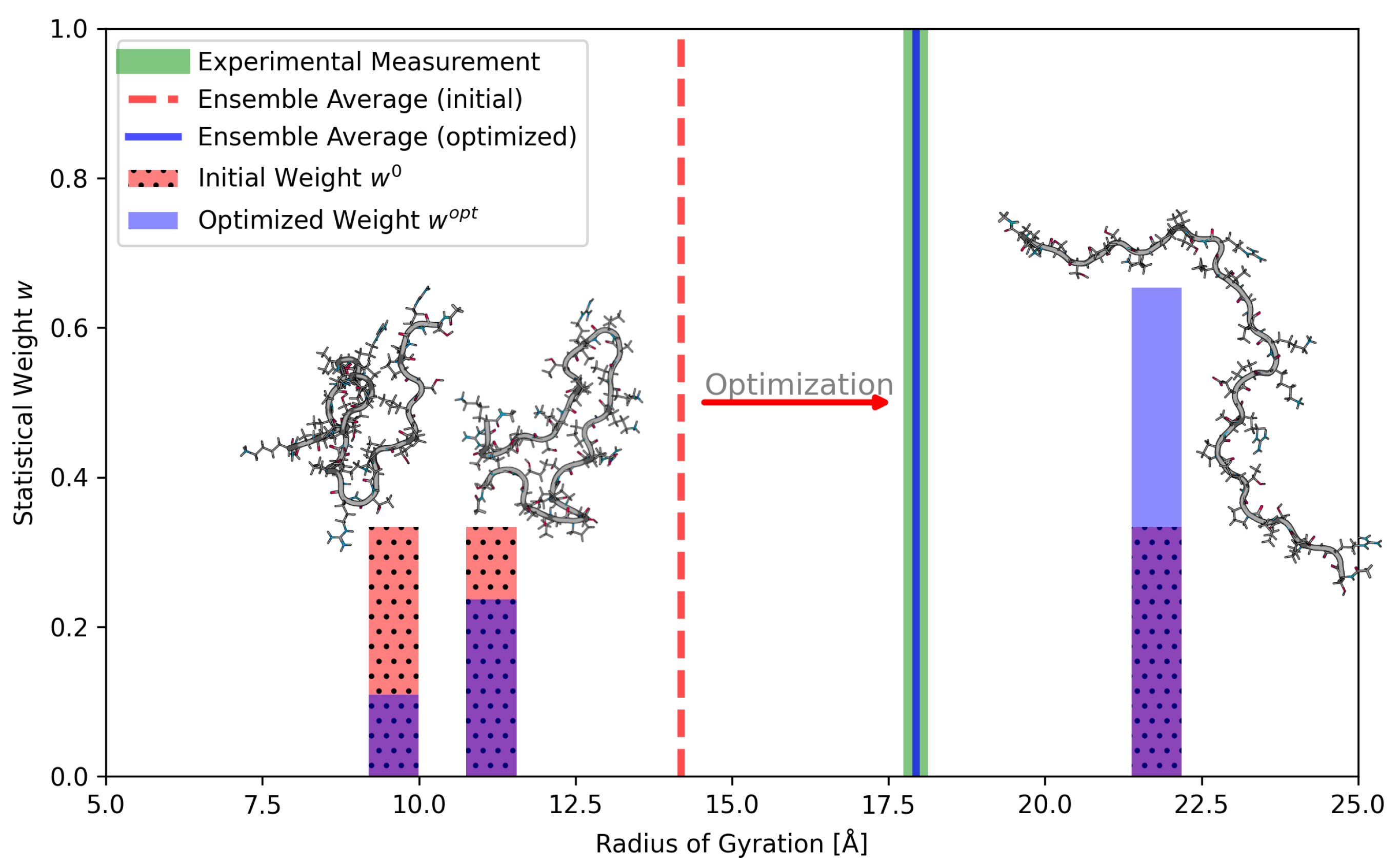
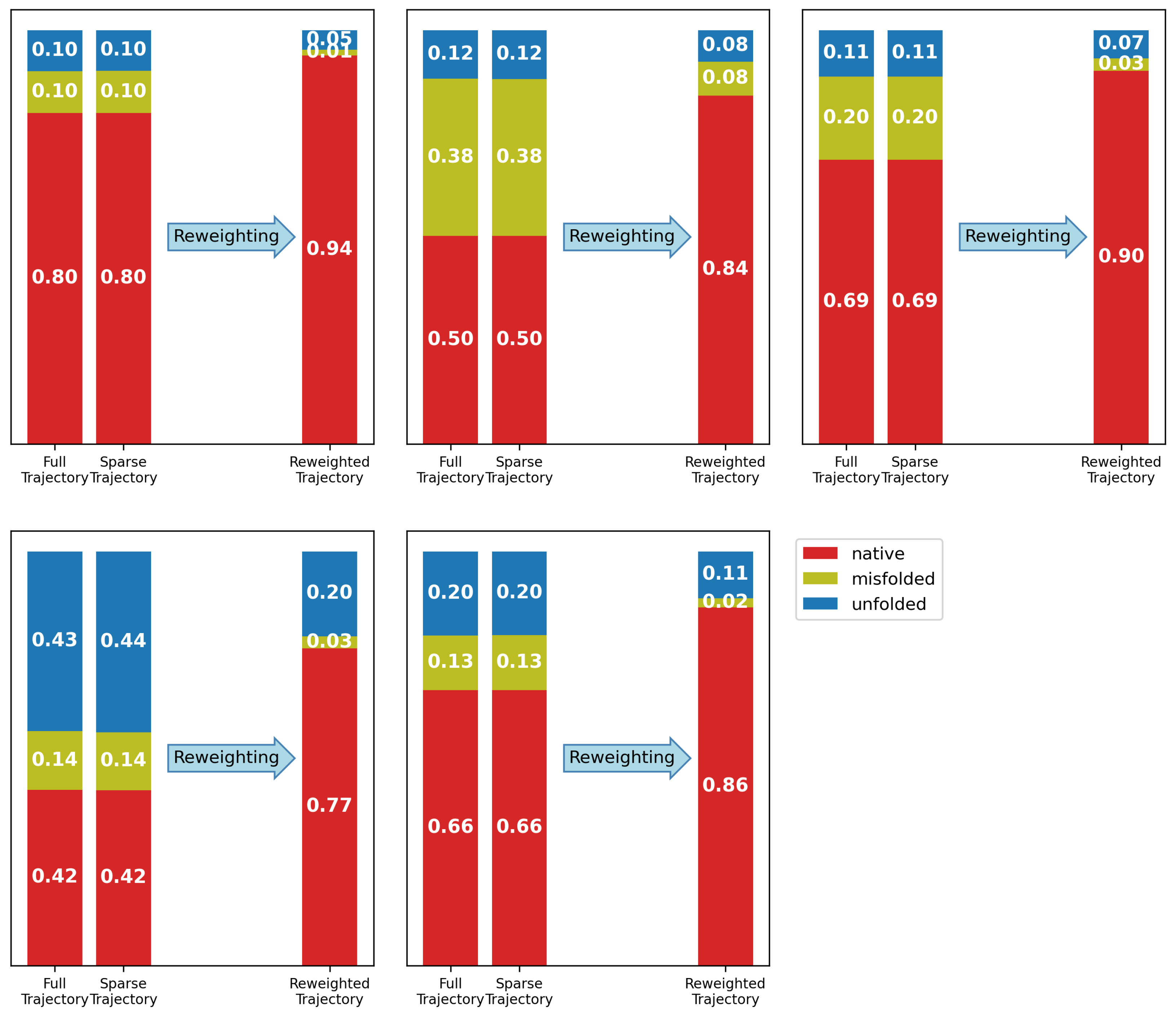
3. Validation
3.1. Methods
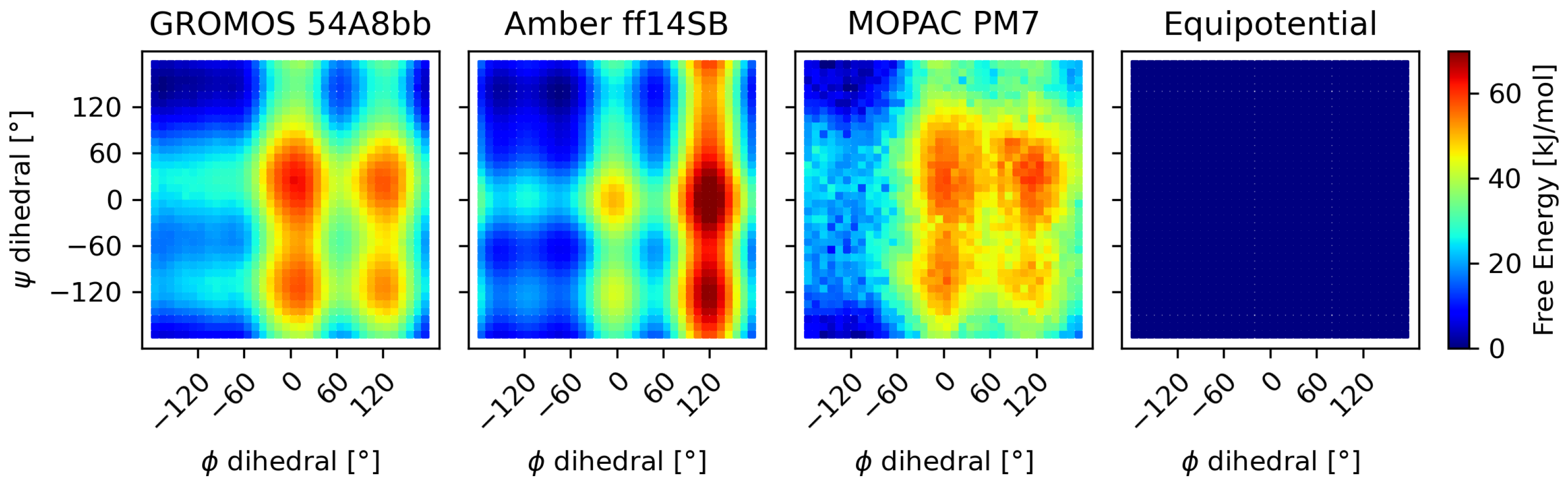
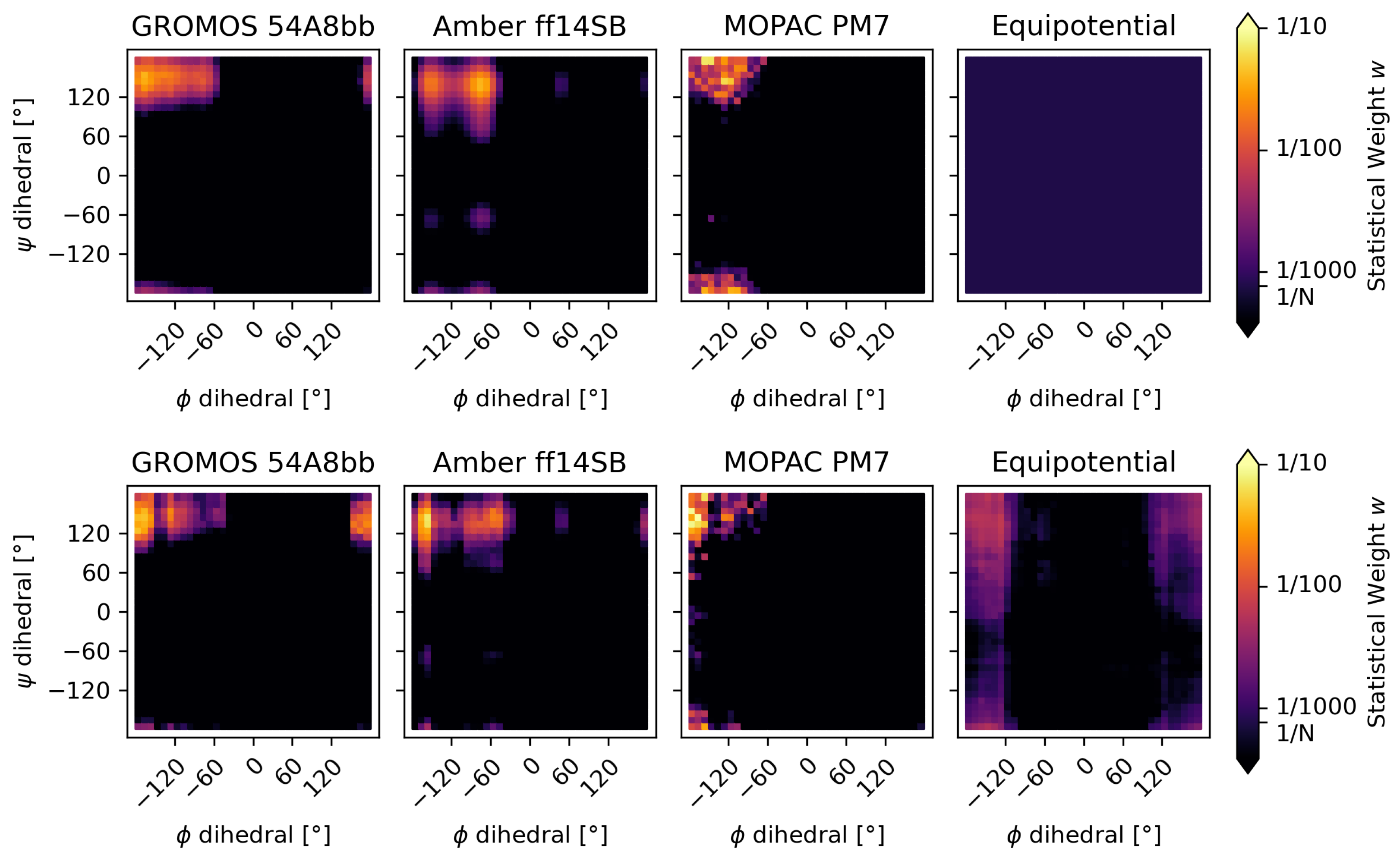
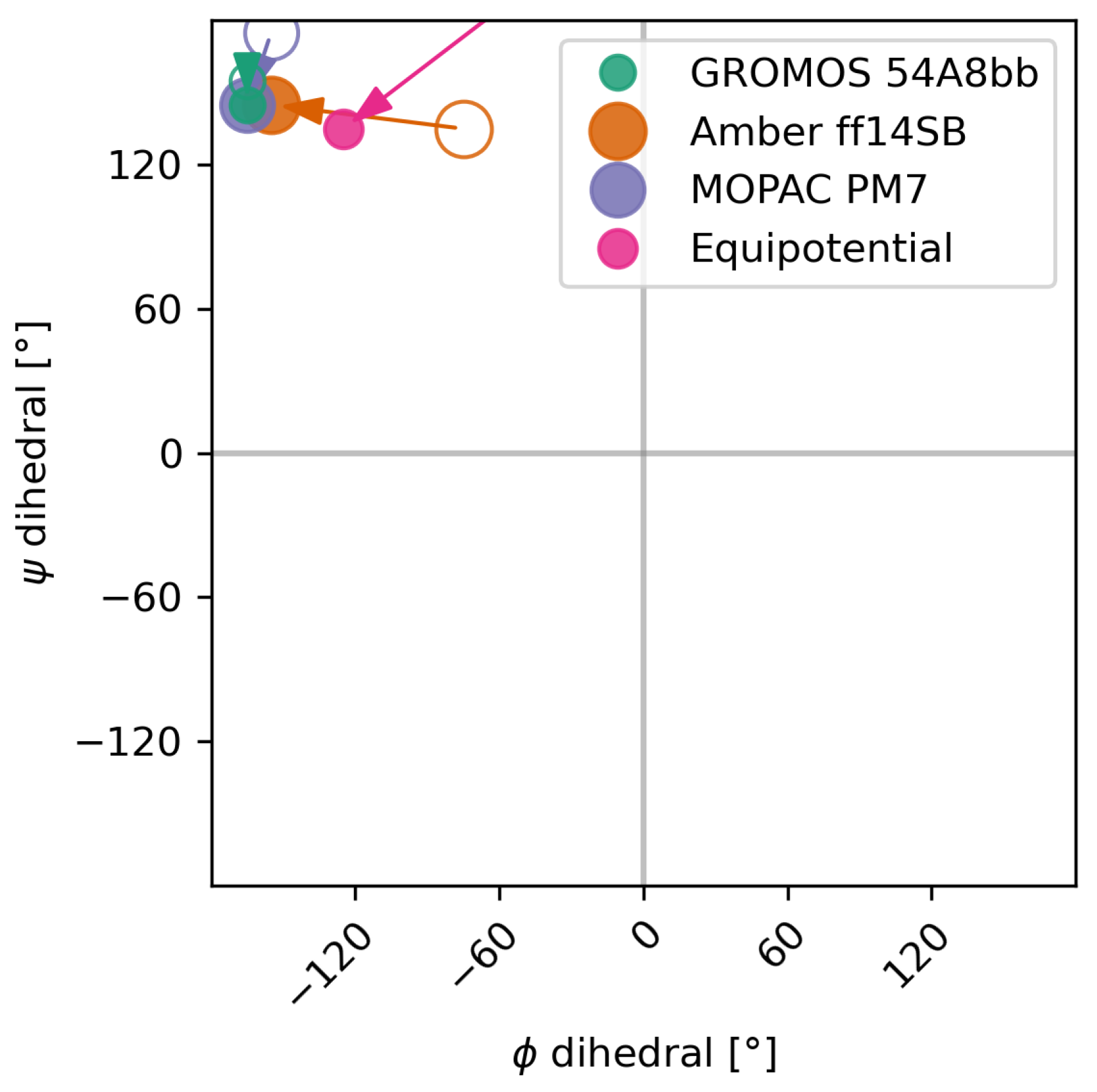
3.1.1. The Alanine–Alanine Zwitterion
3.1.2. Comparison to Bottaro et al. [52]
3.1.3. Chignolin
3.2. Results
3.2.1. The Alanine–Alanine Zwitterion
3.2.2. Comparison to Bottaro et al. [52]
3.2.3. Chignolin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme. Berichte Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.; Sodhi, J.; McGuffin, L.; Buxton, B.; Jones, D. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Kulkarni, P. Intrinsically disordered proteins: Chronology of a discovery. Biophys. Chem. 2021, 279, 106694. [Google Scholar] [CrossRef]
- Kulkarni, P.; Leite, V.B.P.; Roy, S.; Bhattacharyya, S.; Mohanty, A.; Achuthan, S.; Singh, D.; Appadurai, R.; Rangarajan, G.; Weninger, K.; et al. Intrinsically disordered proteins: Ensembles at the limits of Anfinsen’s dogma. Biophys. Rev. 2022, 3, 011306. [Google Scholar] [CrossRef]
- Goedert, M.; Klug, A.; Crowther, R.A. Tau protein, the paired helical filament and Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 195–207. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Adlard, P.A.; Masters, C.L.; Bush, A.I. Tau protein: Relevance to Parkinson’s disease. Int. J. Biochem. Cell Biol. 2010, 42, 1775–1778. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, F.; Wang, D.; Li, C.; Fu, Y.; He, W.; Zhang, J. Tau Pathology in Parkinson’s Disease. Front. Neurol. 2018, 9, 809. [Google Scholar] [CrossRef]
- Mészáros, B.; Hajdu-Soltész, B.; Zeke, A.; Dosztányi, Z. Mutations of Intrinsically Disordered Protein Regions Can Drive Cancer but Lack Therapeutic Strategies. Biomolecules 2021, 11, 381. [Google Scholar] [CrossRef]
- Jia, Z.C.; Yang, X.; Wu, Y.K.; Li, M.; Das, D.; Chen, M.X.; Wu, J. The Art of Finding the Right Drug Target: Emerging Methods and Strategies. Pharmacol. Rev. 2024, 76, 896–914. [Google Scholar] [CrossRef]
- Meissner, F.; Geddes-McAlister, J.; Mann, M.; Bantscheff, M. The emerging role of mass spectrometry-based proteomics in drug discovery. Nat. Rev. Drug Discov. 2022, 21, 637–654. [Google Scholar] [CrossRef] [PubMed]
- Rezaei-Ghaleh, N.; Blackledge, M.; Zweckstetter, M. Intrinsically Disordered Proteins: From Sequence and Conformational Properties toward Drug Discovery. ChemBioChem 2012, 13, 930–950. [Google Scholar] [CrossRef] [PubMed]
- Ambadipudi, S.; Zweckstetter, M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov. 2016, 11, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xiong, R.; Lai, L. Rational drug design targeting intrinsically disordered proteins. WIREs Comput. Mol. Sci. 2023, 13, e1685. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Berendsen, H.J.C. Computer Simulation of Molecular Dynamics: Methodology, Applications, and Perspectives in Chemistry. Angew. Chem. Int. Ed. Engl. 1990, 29, 992–1023. [Google Scholar] [CrossRef]
- van Gunsteren, W.F. Molecular dynamics studies of proteins. Curr. Opin. Struct. Biol. 1993, 3, 277–281. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Oostenbrink, C. Methods for Classical-Mechanical Molecular Simulation in Chemistry: Achievements, Limitations, Perspectives. J. Chem. Inf. Model. 2024, 64, 6281–6303. [Google Scholar] [CrossRef]
- Scheek, R.M.; Torda, A.E.; Kemmink, J.; van Gunsteren, W.F. Structure Determination by NMR: The Modeling of NMR Parameters as Ensemble Averages. In Computational Aspects of the Study of Biological Macromolecules by Nuclear Magnetic Resonance Spectroscopy; Hoch, J.C., Poulsen, F.M., Redfield, C., Eds.; Springer: Boston, MA, USA, 1991; pp. 209–217. [Google Scholar] [CrossRef]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The Energy Landscapes and Motions of Proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Best, R.B.; DePristo, M.A.; Dobson, C.M.; Vendruscolo, M. Simultaneous determination of protein structure and dynamics. Nature 2005, 433, 128–132. [Google Scholar] [CrossRef]
- Fisher, C.K.; Stultz, C.M. Constructing ensembles for intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2011, 21, 426–431. [Google Scholar] [CrossRef]
- Daura, X.; van Gunsteren, W.F.; Mark, A.E. Folding-unfolding thermodynamics of a beta-heptapeptide from equilibrium simulations. Proteins 1999, 34, 269–280. [Google Scholar] [CrossRef]
- Tiberti, M.; Papaleo, E.; Bengtsen, T.; Boomsma, W.; Lindorff-Larsen, K. ENCORE: Software for Quantitative Ensemble Comparison. PLoS Comput. Biol. 2015, 11, e1004415. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, S.; Gapsys, V.; Gajda, M.J.; Zweckstetter, M.; de Groot, B.L.; Grubmüller, H. Structural Ensembles of Intrinsically Disordered Proteins Depend Strongly on Force Field: A Comparison to Experiment. J. Chem. Theory Comput. 2015, 11, 5513–5524. [Google Scholar] [CrossRef] [PubMed]
- Henriques, J.; Cragnell, C.; Skepö, M. Molecular Dynamics Simulations of Intrinsically Disordered Proteins: Force Field Evaluation and Comparison with Experiment. J. Chem. Theory Comput. 2015, 11, 3420–3431. [Google Scholar] [CrossRef]
- Palazzesi, F.; Prakash, M.K.; Bonomi, M.; Barducci, A. Accuracy of Current All-Atom Force-Fields in Modeling Protein Disordered States. J. Chem. Theory Comput. 2015, 11, 2–7. [Google Scholar] [CrossRef]
- Chebaro, Y.; Ballard, A.J.; Chakraborty, D.; Wales, D.J. Intrinsically Disordered Energy Landscapes. Sci. Rep. 2015, 5, 10386. [Google Scholar] [CrossRef]
- Viegas, R.G.; Martins, I.B.S.; Leite, V.B.P. Understanding the Energy Landscape of Intrinsically Disordered Protein Ensembles. J. Chem. Inf. Model. 2024, 64, 4149–4157. [Google Scholar] [CrossRef]
- Jensen, M.R.; Zweckstetter, M.; Huang, J.R.; Blackledge, M. Exploring Free-Energy Landscapes of Intrinsically Disordered Proteins at Atomic Resolution Using NMR Spectroscopy. Chem. Rev. 2014, 114, 6632–6660. [Google Scholar] [CrossRef]
- Cesari, A.; Reißer, S.; Bussi, G. Using the Maximum Entropy Principle to Combine Simulations and Solution Experiments. Computation 2018, 6, 15. [Google Scholar] [CrossRef]
- Bonomi, M.; Heller, G.T.; Camilloni, C.; Vendruscolo, M. Principles of protein structural ensemble determination. Curr. Opin. Struct. Biol. 2017, 42, 106–116. [Google Scholar] [CrossRef]
- Thomasen, F.E.; Lindorff-Larsen, K. Conformational ensembles of intrinsically disordered proteins and flexible multidomain proteins. arXiv 2021, arXiv:2112.05527. [Google Scholar] [CrossRef] [PubMed]
- Gama Lima Costa, R.; Fushman, D. Reweighting methods for elucidation of conformation ensembles of proteins. Curr. Opin. Struct. Biol. 2022, 77, 102470. [Google Scholar] [CrossRef] [PubMed]
- Stöckelmaier, J.; Oostenbrink, C. Conformational dependence of chemical shifts in the proline rich region of TAU protein. Phys. Chem. Chem. Phys. 2024, 26, 23856–23870. [Google Scholar] [CrossRef] [PubMed]
- Ozenne, V.; Schneider, R.; Yao, M.; Huang, J.R.; Salmon, L.; Zweckstetter, M.; Jensen, M.R.; Blackledge, M. Mapping the Potential Energy Landscape of Intrinsically Disordered Proteins at Amino Acid Resolution. J. Am. Chem. Soc. 2012, 134, 15138–15148. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Allison, J.R.; Daura, X.; Dolenc, J.; Hansen, N.; Mark, A.E.; Oostenbrink, C.; Rusu, V.H.; Smith, L.J. Deriving Structural Information from Experimentally Measured Data on Biomolecules. Angew. Chem. Int. Ed. 2016, 55, 15990–16010. [Google Scholar] [CrossRef]
- Grutsch, S.; Brüschweiler, S.; Tollinger, M. NMR Methods to Study Dynamic Allostery. PLoS Comput. Biol. 2016, 12, e1004620. [Google Scholar] [CrossRef]
- Camacho-Zarco, A.R.; Schnapka, V.; Guseva, S.; Abyzov, A.; Adamski, W.; Milles, S.; Jensen, M.R.; Zidek, L.; Salvi, N.; Blackledge, M. NMR Provides Unique Insight into the Functional Dynamics and Interactions of Intrinsically Disordered Proteins. Chem. Rev. 2022, 122, 9331–9356. [Google Scholar] [CrossRef]
- Tropp, J. Dipolar relaxation and nuclear Overhauser effects in nonrigid molecules: The effect of fluctuating internuclear distances. J. Chem. Phys. 1980, 72, 6035–6043. [Google Scholar] [CrossRef]
- Torrie, G.; Valleau, J. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Stöckelmaier, J.; Oostenbrink, C. Combining Simulations and Experiments—A Perspective on Maximum Entropy Methods. 2025, Manuscript submitted for publication.
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Jaynes, E.T. Information Theory and Statistical Mechanics. Phys. Rev. 1957, 106, 620–630. [Google Scholar] [CrossRef]
- Jaynes, E.T. Information Theory and Statistical Mechanics. II. Phys. Rev. 1957, 108, 171–190. [Google Scholar] [CrossRef]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory; John Wiley and Sons, Ltd.: New York, NY, USA, 1991; pp. 1–11. [Google Scholar]
- De Martino, A.; De Martino, D. An introduction to the maximum entropy approach and its application to inference problems in biology. Heliyon 2018, 4, e00596. [Google Scholar] [CrossRef] [PubMed]
- Różycki, B.; Kim, Y.C.; Hummer, G. SAXS Ensemble Refinement of ESCRT-III CHMP3 Conformational Transitions. Structure 2011, 19, 109–116. [Google Scholar] [CrossRef]
- Pitera, J.W.; Chodera, J.D. On the Use of Experimental Observations to Bias Simulated Ensembles. J. Chem. Theory Comput. 2012, 8, 3445–3451. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, W.; Ferkinghoff-Borg, J.; Lindorff-Larsen, K. Combining Experiments and Simulations Using the Maximum Entropy Principle. PLoS Comput. Biol. 2014, 10, e1003406. [Google Scholar] [CrossRef]
- Leung, H.T.A.; Bignucolo, O.; Aregger, R.; Dames, S.A.; Mazur, A.; Bernèche, S.; Grzesiek, S. A Rigorous and Efficient Method to Reweight very Large Conformational Ensembles Using Average Experimental Data and to Determine Their Relative Information Content. J. Chem. Theory Comput. 2016, 12, 383–394. [Google Scholar] [CrossRef]
- Hermann, M.; Hub, J.S. SAXS-Restrained Ensemble Simulations of Intrinsically Disordered Proteins with Commitment to the Principle of Maximum Entropy. J. Chem. Theory Comput. 2019, 15, 5103–5115. [Google Scholar] [CrossRef]
- Bottaro, S.; Bengtsen, T.; Lindorff-Larsen, K. Integrating Molecular Simulation and Experimental Data: A Bayesian/Maximum Entropy Reweighting Approach. In Structural Bioinformatics: Methods and Protocols; Gáspári, Z., Ed.; Springer: New York, NY, USA, 2020; pp. 219–240. [Google Scholar] [CrossRef]
- Yamamori, Y.; Tomii, K. An ensemble reweighting method for combining the information of experiments and simulations. Chem. Phys. Lett. 2021, 779, 138821. [Google Scholar] [CrossRef]
- Gilardoni, I.; Piomponi, V.; Fröhlking, T.; Bussi, G. MDRefine: A Python package for refining molecular dynamics trajectories with experimental data. J. Chem. Phys. 2025, 162, 192501. [Google Scholar] [CrossRef]
- Wittenberg, M. An Introduction to Maximum Entropy and Minimum Cross-entropy Estimation Using Stata. Stata J. 2010, 10, 315–330. [Google Scholar] [CrossRef]
- Bottaro, S.; Bengtsen, T.; Lindorff-Larsen, K. Integrating Molecular Simulation and Experimental Data: A Bayesian/Maximum Entropy reweighting approach. bioRxiv 457952 2018. [Google Scholar] [CrossRef]
- Bradbury, J.; Frostig, R.; Hawkins, P.; Johnson, M.J.; Leary, C.; Maclaurin, D.; Necula, G.; Paszke, A.; VanderPlas, J.; Wanderman-Milne, S.; et al. JAX: Composable Transformations of Python+NumPy Programs. 2018. Available online: http://github.com/jax-ml/jax (accessed on 2 June 2025).
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domanski, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Sebastian Benthall, Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 56–61. [Google Scholar] [CrossRef]
- The pandas development team. pandas-dev/pandas: Pandas. Zenodo, 2023. [CrossRef]
- The Matplotlib Development Team. Matplotlib: Visualization with Python. Zenodo. [CrossRef]
- Bouř, P.; Buděšínský, M.; Špirko, V.; Kapitán, J.; Šebestík, J.; Sychrovský, V. A Complete Set of NMR Chemical Shifts and Spin–Spin Coupling Constants for l-Alanyl-l-alanine Zwitterion and Analysis of Its Conformational Behavior. J. Am. Chem. Soc. 2005, 127, 17079–17089. [Google Scholar] [CrossRef]
- Diem, M.; Oostenbrink, C. Hamiltonian Reweighing To Refine Protein Backbone Dihedral Angle Parameters in the GROMOS Force Field. J. Chem. Inf. Model. 2020, 60, 279–288. [Google Scholar] [CrossRef]
- Schmid, N.; Christ, C.D.; Christen, M.; Eichenberger, A.P.; van Gunsteren, W.F. Architecture, implementation and parallelisation of the GROMOS software for biomolecular simulation. Comput. Phys. Commun. 2012, 183, 890–903. [Google Scholar] [CrossRef]
- Eichenberger, A.P.; Allison, J.R.; Dolenc, J.; Geerke, D.P.; Horta, B.A.C.; Meier, K.; Oostenbrink, C.; Schmid, N.; Steiner, D.; Wang, D.; et al. GROMOS++ Software for the Analysis of Biomolecular Simulation Trajectories. J. Chem. Theory Comput. 2011, 7, 3379–3390. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Eastman, P.; Galvelis, R.; Peláez, R.P.; Abreu, C.R.A.; Farr, S.E.; Gallicchio, E.; Gorenko, A.; Henry, M.M.; Hu, F.; Huang, J.; et al. OpenMM 8: Molecular Dynamics Simulation with Machine Learning Potentials. J. Phys. Chem. B 2024, 128, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Gieseking, R.L.M. A new release of MOPAC incorporating the INDO/S semiempirical model with CI excited states. J. Comput. Chem. 2021, 42, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces: Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry Held in Jerusalem, Israel, April 13–16, 1981; Springer: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Huber, T.; Torda, A.E.; van Gunsteren, W.F. Local elevation: A method for improving the searching properties of molecular dynamics simulation. J. Comput.-Aided Mol. Des. 1994, 8, 695–708. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. PRoceedings Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Sweet, C.R.; Pande, V.S. Multiscale dynamics of macromolecules using Normal Mode Langevin. Pac. Symp. Biocomput. 2010, 15, 240–251. [Google Scholar]
- Li, J.; Bennett, K.C.; Liu, Y.; Martin, M.V.; Head-Gordon, T. Accurate prediction of chemical shifts for aqueous protein structure on “Real World” data. Chem. Sci. 2020, 11, 3180–3191. [Google Scholar] [CrossRef]
- Karplus, M. Contact Electron-Spin Coupling of Nuclear Magnetic Moments. J. Chem. Phys. 1959, 30, 11–15. [Google Scholar] [CrossRef]
- Karplus, M. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar] [CrossRef]
- Wang, A.C.; Bax, A. Determination of the Backbone Dihedral Angles φ in Human Ubiquitin from Reparametrized Empirical Karplus Equations. J. Am. Chem. Soc. 1996, 118, 2483–2494. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Best, R.B.; Vendruscolo, M. Interpreting Dynamically-Averaged Scalar Couplings in Proteins. J. Biomol. NMR 2005, 32, 273–280. [Google Scholar] [CrossRef]
- Vögeli, B.; Ying, J.; Grishaev, A.; Bax, A. Limits on Variations in Protein Backbone Dynamics from Precise Measurements of Scalar Couplings. J. Am. Chem. Soc. 2007, 129, 9377–9385. [Google Scholar] [CrossRef]
- Sychrovský, V.; Buděšínský, M.; Benda, L.; Špirko, V.; Vokáčová, Z.; Šebestík, J.; Bouř, P. Dependence of the l-Alanyl-l-Alanine Conformation on Molecular Charge Determined from Ab Initio Computations and NMR Spectra. J. Phys. Chem. B 2008, 112, 1796–1805. [Google Scholar] [CrossRef]
- Honda, S.; Yamasaki, K.; Sawada, Y.; Morii, H. 10 Residue Folded Peptide Designed by Segment Statistics. Structure 2004, 12, 1507–1518. [Google Scholar] [CrossRef]
- Kührová, P.; De Simone, A.; Otyepka, M.; Best, R. Force-Field Dependence of Chignolin Folding and Misfolding: Comparison with Experiment and Redesign. Biophys. J. 2012, 102, 1897–1906. [Google Scholar] [CrossRef]
- Fischer, A.L.M.; Tichy, A.; Kokot, J.; Hoerschinger, V.J.; Wild, R.F.; Riccabona, J.R.; Loeffler, J.R.; Waibl, F.; Quoika, P.K.; Gschwandtner, P.; et al. The Role of Force Fields and Water Models in Protein Folding and Unfolding Dynamics. J. Chem. Theory Comput. 2024, 20, 2321–2333. [Google Scholar] [CrossRef]
- Marshall, T.; Raddi, R.; Voelz, V. An Evaluation of Force Field Accuracy for the Mini-Protein Chignolin using Markov State Models. ChemRxiv xmztm 2024. [Google Scholar] [CrossRef]
- Liu, Z.H.; Zhang, O.; Teixeira, J.M.; Li, J.; Head-Gordon, T.; Forman-Kay, J.D. SPyCi-PDB: A modular command-line interface for back-calculating experimental datatypes of protein structures. J. Open Source Softw. 2023, 8, 4861. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Silva, H.; Lim, S.; Kozuno, T.; Mahmood, A.R.; White, M. Greedification operators for policy optimization: Investigating forward and reverse KL divergences. J. Mach. Learn. Res. 2022, 23, 1–79. [Google Scholar]
- Vaitl, L.; Nicoli, K.A.; Nakajima, S.; Kessel, P. Gradients should stay on path: Better estimators of the reverse- and forward KL divergence for normalizing flows. Mach. Learn. Sci. Technol. 2022, 3, 045006. [Google Scholar] [CrossRef]
- Shen, M.; Diamant, N. On KL Divergence in Discrete Spaces, 2022. Available online: https://argmax.blog/posts/kl-discrete/ (accessed on 14 August 2024).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial Weights | Mininitial [°] | Minoptimized [°] | ||||
|---|---|---|---|---|---|---|
| 54A8bb (GROMOS) | 0.483 | 0.200 | 1.307 | 0.800 | (, 155) | (, 145) |
| Amber ff14SB (OpenMM) | – | 0.200 | 1.355 | 0.961 | (, 135) | (, 145) |
| PM7 (MOPAC) | 0.183 | 0.200 | 2.159 | 0.904 | (, 175) | (, 145) |
| Equipotential | 0.070 | 0.200 | 1.514 | 0.931 | – | (, 135) |
| URE Method | BME Method | |||||
|---|---|---|---|---|---|---|
| Reweighting Strength | e.p. | e.p. | ||||
| underfitting | 15.10 | 1.42 | 97 | 111.38 | 1.42 | 97 |
| good | 0.20 | 0.93 | 55 | 1.30 | 0.92 | 54 |
| good | 0.04 | 0.80 | 27 | 0.40 | 0.78 | 26 |
| overfitting | 0.01 | 0.72 | 3 | 0.02 | 0.71 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stöckelmaier, J.; Capraz, T.; Oostenbrink, C. Umbrella Refinement of Ensembles—An Alternative View of Ensemble Optimization. Molecules 2025, 30, 2449. https://doi.org/10.3390/molecules30112449
Stöckelmaier J, Capraz T, Oostenbrink C. Umbrella Refinement of Ensembles—An Alternative View of Ensemble Optimization. Molecules. 2025; 30(11):2449. https://doi.org/10.3390/molecules30112449
Chicago/Turabian StyleStöckelmaier, Johannes, Tümay Capraz, and Chris Oostenbrink. 2025. "Umbrella Refinement of Ensembles—An Alternative View of Ensemble Optimization" Molecules 30, no. 11: 2449. https://doi.org/10.3390/molecules30112449
APA StyleStöckelmaier, J., Capraz, T., & Oostenbrink, C. (2025). Umbrella Refinement of Ensembles—An Alternative View of Ensemble Optimization. Molecules, 30(11), 2449. https://doi.org/10.3390/molecules30112449