
One-Pot Syntheses and Characterization of Group VI Carbonyl NHC Coordination Compounds


Abstract

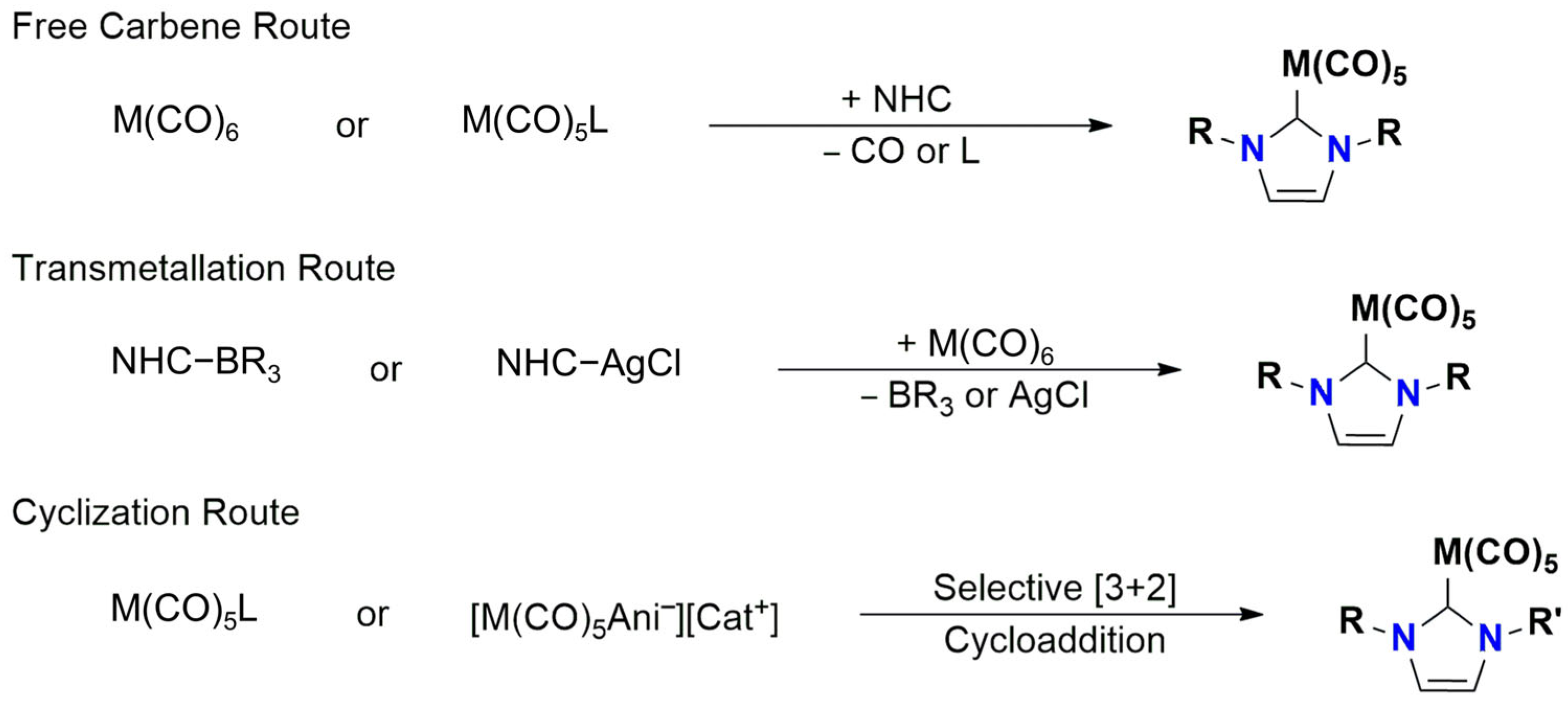
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization
Coordination Compounds with Metal Hexacarbonyls (M(CO)6; M = Cr, Mo, W)
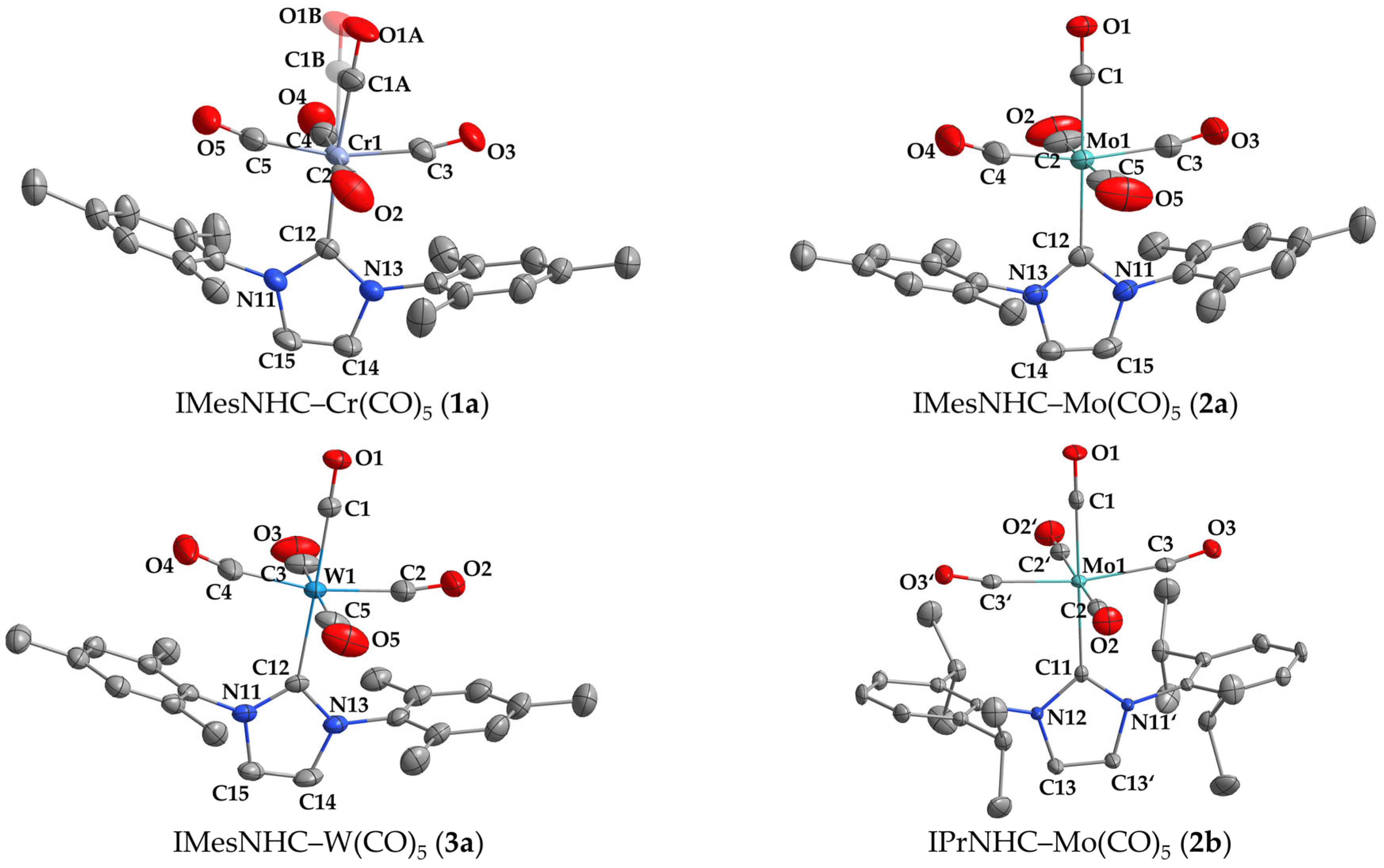
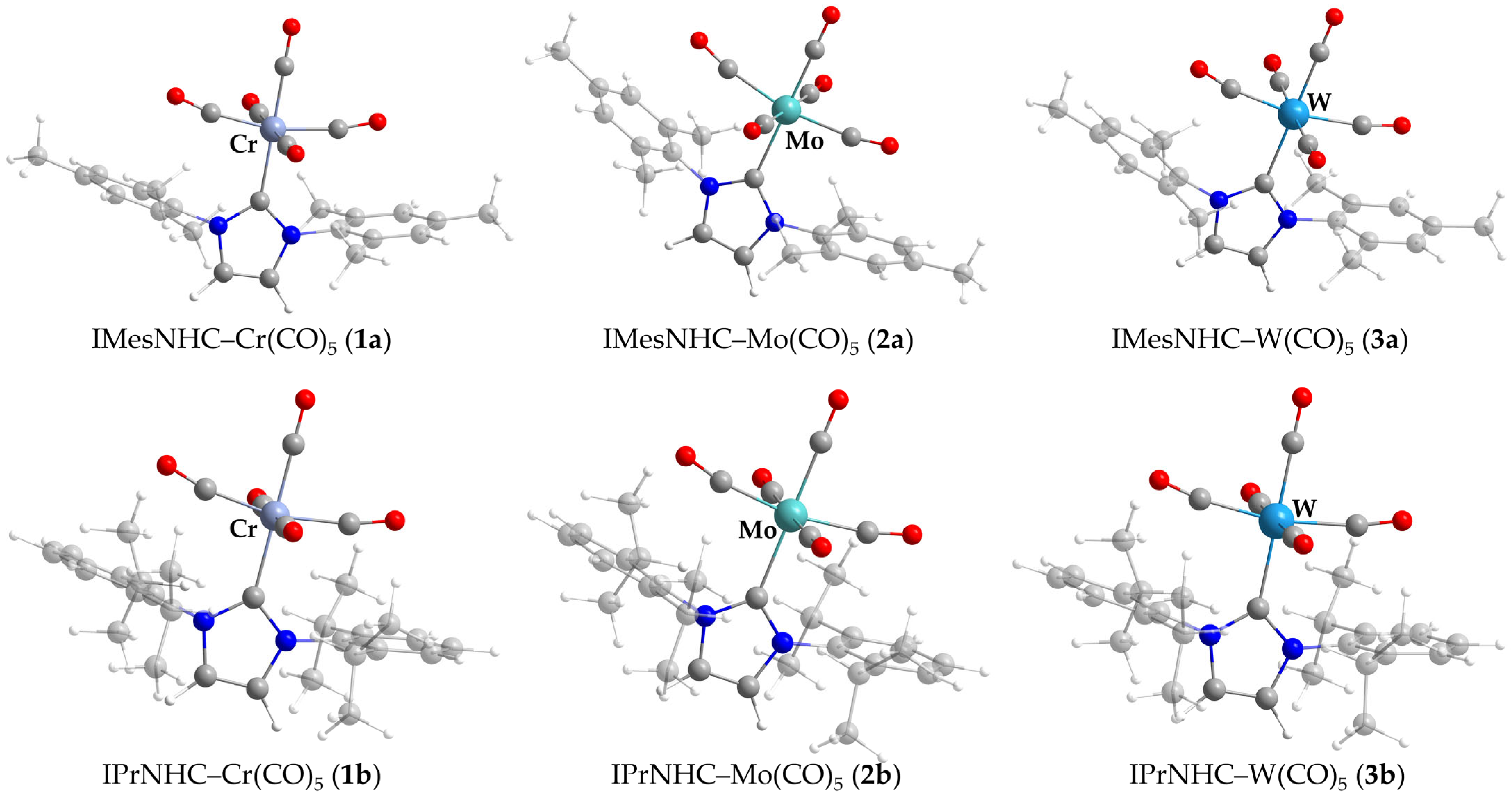
2.2. Crystal Structure Analysis
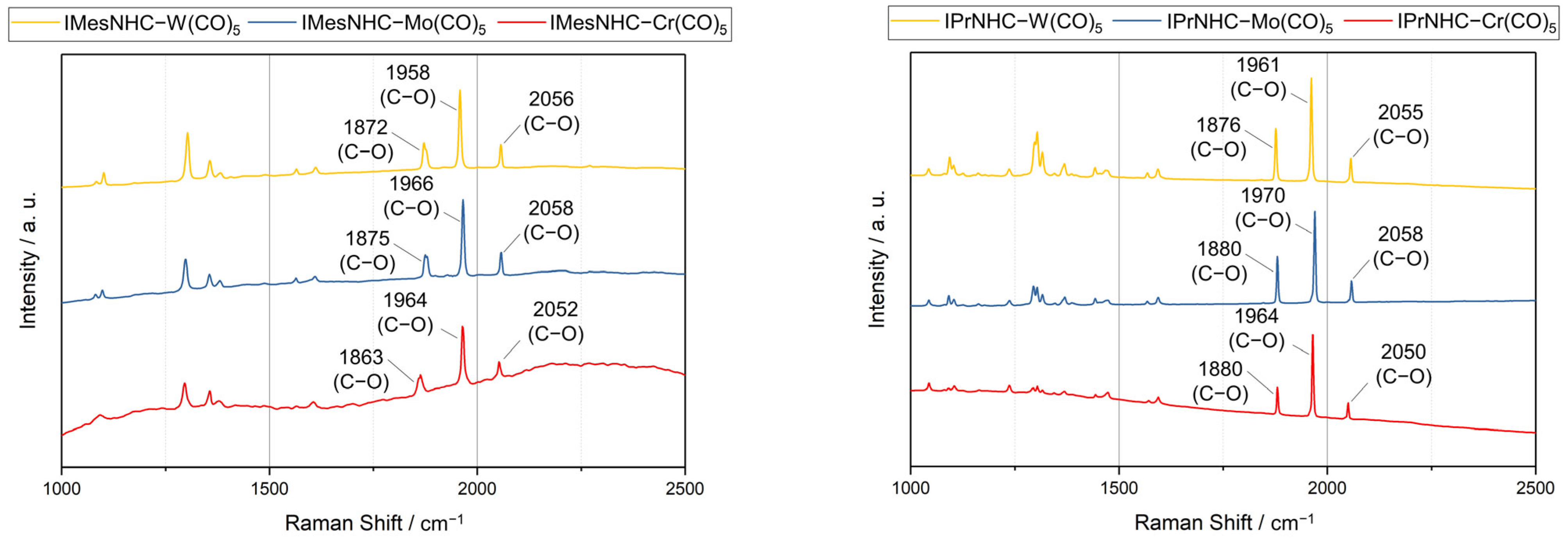
2.3. Raman Spectroscopy Results
2.4. Computational Results
3. Materials and Methods
3.1. Reagents
3.2. General
3.3. Syntheses
3.3.1. Modified Synthesis of IMesNHC Carbene
3.3.2. IMesNHC–Cr(CO)5 (1a)
3.3.3. IMesNHC–Mo(CO)5 (2a)
3.3.4. IMesNHC–W(CO)5 (3a)
3.3.5. IPrNHC–Cr(CO)5 (1b)
3.3.6. IPrNHC–Mo(CO)5 (2b)
3.3.7. IPrNHC–W(CO)5 (3b)
3.4. NMR Spectroscopy
3.5. Crystal Structure Determination
3.6. Raman Spectroscopy
3.7. Molecular Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, W.; Gust, R. Metal N-Heterocyclic Carbene Complexes as Potential Antitumor Metallodrugs. Chem. Soc. Rev. 2013, 42, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Cerquera-Montealegre, L.; Gallego, D.; Baquero, E.A. Chapter Two—Recent Advances in the Catalytic Applications of NHC-Early Abundant Metals (Mn, Co, Fe) Complexes. In Advances in Organometallic Chemistry; Pérez, P.J., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2024; Volume 81, pp. 181–270. [Google Scholar]
- Oehninger, L.; Rubbiani, R.; Ott, I. N-Heterocyclic Carbene Metal Complexes in Medicinal Chemistry. Dalton Trans. 2013, 42, 3269–3284. [Google Scholar] [CrossRef] [PubMed]
- Öfele, K. 1,3-Dimethyl-4-Imidazolinyliden-(2)-Pentacarbonylchrom Ein Neuer Übergangsmetall-Carben-Komplex. J. Organomet. Chem. 1968, 12, P42–P43. [Google Scholar] [CrossRef]
- Romain, C.; Bellemin-Laponnaz, S.; Dagorne, S. Recent Progress on NHC-Stabilized Early Transition Metal (Group 3–7) Complexes: Synthesis and Applications. Coord. Chem. Rev. 2020, 422, 213411. [Google Scholar] [CrossRef]
- Bolm, C.; Kesselgruber, M.; Raabe, G. The First Planar-Chiral Stable Carbene and Its Metal Complexes. Organometallics 2002, 21, 707–710. [Google Scholar] [CrossRef]
- Kim, S.; Choi, S.Y.; Lee, Y.T.; Park, K.H.; Sitzmann, H.; Chung, Y.K. Synthesis of Chromium N-Heterocyclic Carbene Complexes Using Chromium Fischer Carbenes as a Source of Chromium Carbonyls. J. Organomet. Chem. 2007, 692, 5390–5394. [Google Scholar] [CrossRef]
- Wang, Z.; Li, S.; Teo, W.J.; Poh, Y.T.; Zhao, J.; Hor, T.S.A. Molybdenum (0) and Tungsten (0) Carbonyl N-Heterocyclic Carbene Complexes as Catalyst for Olefin Epoxidation. J. Organomet. Chem. 2015, 775, 188–194. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kashiwabara, T.; Ogata, K.; Miura, Y.; Nakamura, Y.; Kobayashi, K.; Ito, T. Synthesis and Reactivity of Triethylborane Adduct of N-Heterocyclic Carbene: Versatile Synthons for Synthesis of N-Heterocyclic Carbene Complexes. Chem. Commun. 2004, 2004, 2160–2161. [Google Scholar] [CrossRef]
- Fehlhammer, W.P.; Völkl, A.; Plaia, U.; Beck, G. Metallkomplexe Funktioneller Isocyanide, XVI. 1,3-Dipolare Cycloadditionen von Heteroallenen an Die Metallorganischen Nitrilylide [(OC)5M–C≡N–CHR]− (M = Cr, W; R = CO2Et). Chem. Berichte 1987, 120, 2031–2040. [Google Scholar] [CrossRef]
- Rieger, D.; Lotz, S.D.; Kernbach, U.; Schröder, S.; André, C.; Fehlhammer, W.P. A Novel Reaction of the Cyano Ligand Opening an Organometallic Route to 4-Amino Imidazoles: Four Component Condensation (4CC) with Isocyanide, Aldehyde and Amine. Inorg. Chim. Acta 1994, 222, 275–290. [Google Scholar] [CrossRef]
- Kernbach, U.; Mühl, M.; Polborn, K.; Fehlhammer, W.P.; Jaouen, G. Attachment of Amino Acid Derivatives to Tungsten Carbonyl Complexes via Four Component Condensations. Inorg. Chim. Acta 2002, 334, 45–53. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, L.; Mohamed, D.K.B.; Zhao, J.; Hor, T.S.A. N-Heterocyclic Carbene Complexes of Group 6 Metals. Coord. Chem. Rev. 2015, 293–294, 292–326. [Google Scholar] [CrossRef]
- Cole, M.L.; Gyton, M.R.; Harper, J.B. Metal Complexes of an Ionic Liquid-Derived Carbene. Aust. J. Chem. 2011, 64, 1133–1140. [Google Scholar] [CrossRef]
- Rosen, E.L.; Varnado, C.D., Jr.; Tennyson, A.G.; Khramov, D.M.; Kamplain, J.W.; Sung, D.H.; Cresswell, P.T.; Lynch, V.M.; Bielawski, C.W. Redox-Active N-Heterocyclic Carbenes: Design, Synthesis, and Evaluation of Their Electronic Properties. Organometallics 2009, 28, 6695–6706. [Google Scholar] [CrossRef]
- Nonnenmacher, M.; Kunz, D.; Rominger, F.; Oeser, T. First Examples of Dipyrido[1,2-c:2′,1′-e]Imidazolin-7-Ylidenes Serving as NHC-Ligands: Synthesis, Properties and Structural Features of Their Chromium and Tungsten Pentacarbonyl Complexes. J. Organomet. Chem. 2005, 690, 5647–5653. [Google Scholar] [CrossRef]
- Ghadwal, R.S.; Rottschäfer, D.; Andrada, D.M.; Frenking, G.; Schürmann, C.J.; Stammler, H.-G. Normal-to-Abnormal Rearrangement of an N-Heterocyclic Carbene with a Silylene Transition Metal Complex. Dalton Trans. 2017, 46, 7791–7799. [Google Scholar] [CrossRef] [PubMed]
- Al-Rafia, S.M.I.; Malcolm, A.C.; Liew, S.K.; Ferguson, M.J.; Rivard, E. Stabilization of the Heavy Methylene Analogues, GeH2 and SnH2, within the Coordination Sphere of a Transition Metal. J. Am. Chem. Soc. 2011, 133, 777–779. [Google Scholar] [CrossRef]
- Al-Rafia, S.M.I.; Shynkaruk, O.; McDonald, S.M.; Liew, S.K.; Ferguson, M.J.; McDonald, R.; Herber, R.H.; Rivard, E. Synthesis and Mössbauer Spectroscopy of Formal Tin(II) Dichloride and Dihydride Species Supported by Lewis Acids and Bases. Inorg. Chem. 2013, 52, 5581–5589. [Google Scholar] [CrossRef]
- Singh, S.K.; Das, A. The n → π* Interaction: A Rapidly Emerging Non-Covalent Interaction. Phys. Chem. Chem. Phys. 2015, 17, 9596–9612. [Google Scholar] [CrossRef]
- Kia, R.; Shojaei, H.; Boyarskiy, V.P.; Mikherdov, A.S. Noncovalent n → π*, C–H⋯π, and C–H⋯O Interaction Mediated Supramolecular Assembly in a Re(CO)3(Trifluoroacetate) Complex Bearing a Bulky Tetraazaphenanthrene Ligand: A Combined CSD Study and Theoretical Calculations. CrystEngComm 2023, 25, 1803–1816. [Google Scholar] [CrossRef]
- Krahfuß, M.J.; Nitsch, J.; Bickelhaupt, F.M.; Marder, T.B.; Radius, U. N-Heterocyclic Silylenes as Ligands in Transition Metal Carbonyl Chemistry: Nature of Their Bonding and Supposed Innocence. Chem. Eur. J. 2020, 26, 11276–11292. [Google Scholar] [CrossRef]
- Armstrong, R.S.; Aroney, M.J.; Barnes, C.M.; Nugent, K.W. Infrared and Raman Spectra of (η6-mesitylene)M(CO)3 Complexes (M = Cr, Mo or W): An Insight into Metal–Arene Bonding. Appl. Organomet. Chem. 1990, 4, 569–580. [Google Scholar] [CrossRef]
- Jafarpour, L.; Stevens, E.D.; Nolan, S.P. A Sterically Demanding Nucleophilic Carbene: 1,3-Bis(2,6-Diisopropylphenyl)Imidazol-2-Ylidene). Thermochemistry and Catalytic Application in Olefin Metathesis. J. Organomet. Chem. 2000, 606, 49–54. [Google Scholar] [CrossRef]
- Hintermann, L. Expedient Syntheses of the N-Heterocyclic Carbene Precursor Imidazolium Salts IPr·HCl, IMes·HCl and IXy·HCl. Beilstein J. Org. Chem. 2007, 3, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Becker, E.D.; De Menezes, S.M.C.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further Conventions for NMR Shielding and Chemical Shifts (IUPAC Recommendations 2008). Magn. Reson. Chem. 2008, 46, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Rigaku Oxford Diffraction, CrysAlisPro, Software System, version 1.171.41.120a; (Release Date 26-10-2021); Rigaku Corporation: Wroclaw, Poland, 2021.
- Clark, R.C.; Reid, J.S. The Analytical Calculation of Absorption in Multifaceted Crystals. Acta Crystallogr. Sect. A Found. Crystallogr. 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Diamond—Crystal and Molecular Structure Visualization (v4.6.8), Crystal Impact—Dr. H. Putz & Dr. K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany. Available online: https://www.crystalimpact.com/diamond/ (accessed on 6 May 2025).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental | Literature | ||
|---|---|---|---|
| Compound | Raman (cm−1) | IR (cm−1) | Reference |
| IMesNHC–Cr(CO)5 (1a) | 1863, 1964, 2052 | 1917, 2044 | [7] |
| IMesNHC–Mo(CO)5 (2a) | 1875, 1966, 2058 | 1879, 1924, 2059 | [8] |
| IMesNHC–W(CO)5 (3a) | 1872, 1958, 2056 | 1876, 1911, 2057 | [8] |
| IPrNHC–Cr(CO)5 (1b) | 1880, 1964, 2050 | 1880, 1918, 1981, 2056 | [19] |
| IPrNHC–Mo(CO)5 (2b) | 1880, 1970, 2058 | 1924, 2060 | [7] |
| IPrNHC–W(CO)5 (3b) | 1876, 1961, 2055 | 1880, 1918, 1981, 2056 | [18] |
| M–C(carbene) (Å) | trans-CO–M (Å) | |||
|---|---|---|---|---|
| Compound | Experimental | Calculated | Experimental | Calculated |
| IMesNHC–Cr(CO)5 (1a) | 2.142(2) * | 2.131 | 1.847(2) * | 1.853 |
| IMesNHC–Mo(CO)5 (2a) | 2.278(2) | 2.273 | 1.987(2) | 2.000 |
| IMesNHC–W(CO)5 (3a) | 2.264(3) | 2.271 | 1.990(4) | 2.018 |
| IPrNHC–Cr(CO)5 (1b) | 2.142(4) [7] | 2.119 | 1.843(5) [7] | 1.854 |
| IPrNHC–Mo(CO)5 (2b) | 2.264(3) | 2.259 | 1.985(3) | 2.003 |
| IPrNHC–W(CO)5 (3b) | 2.260(2) [17] | 2.259 | 1.997(2) [17] | 2.020 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stopar, Z.; Gruden, E.; Tramšek, M.; Tavčar, G. One-Pot Syntheses and Characterization of Group VI Carbonyl NHC Coordination Compounds. Molecules 2025, 30, 2433. https://doi.org/10.3390/molecules30112433
Stopar Z, Gruden E, Tramšek M, Tavčar G. One-Pot Syntheses and Characterization of Group VI Carbonyl NHC Coordination Compounds. Molecules. 2025; 30(11):2433. https://doi.org/10.3390/molecules30112433
Chicago/Turabian StyleStopar, Zala, Evelin Gruden, Melita Tramšek, and Gašper Tavčar. 2025. "One-Pot Syntheses and Characterization of Group VI Carbonyl NHC Coordination Compounds" Molecules 30, no. 11: 2433. https://doi.org/10.3390/molecules30112433
APA StyleStopar, Z., Gruden, E., Tramšek, M., & Tavčar, G. (2025). One-Pot Syntheses and Characterization of Group VI Carbonyl NHC Coordination Compounds. Molecules, 30(11), 2433. https://doi.org/10.3390/molecules30112433