
Does Metal Matter: Comparing Photophysical Properties of Bis-Cyclometalated Alkynylphosphonium Au(III) and Pt(II) Complexes



Abstract

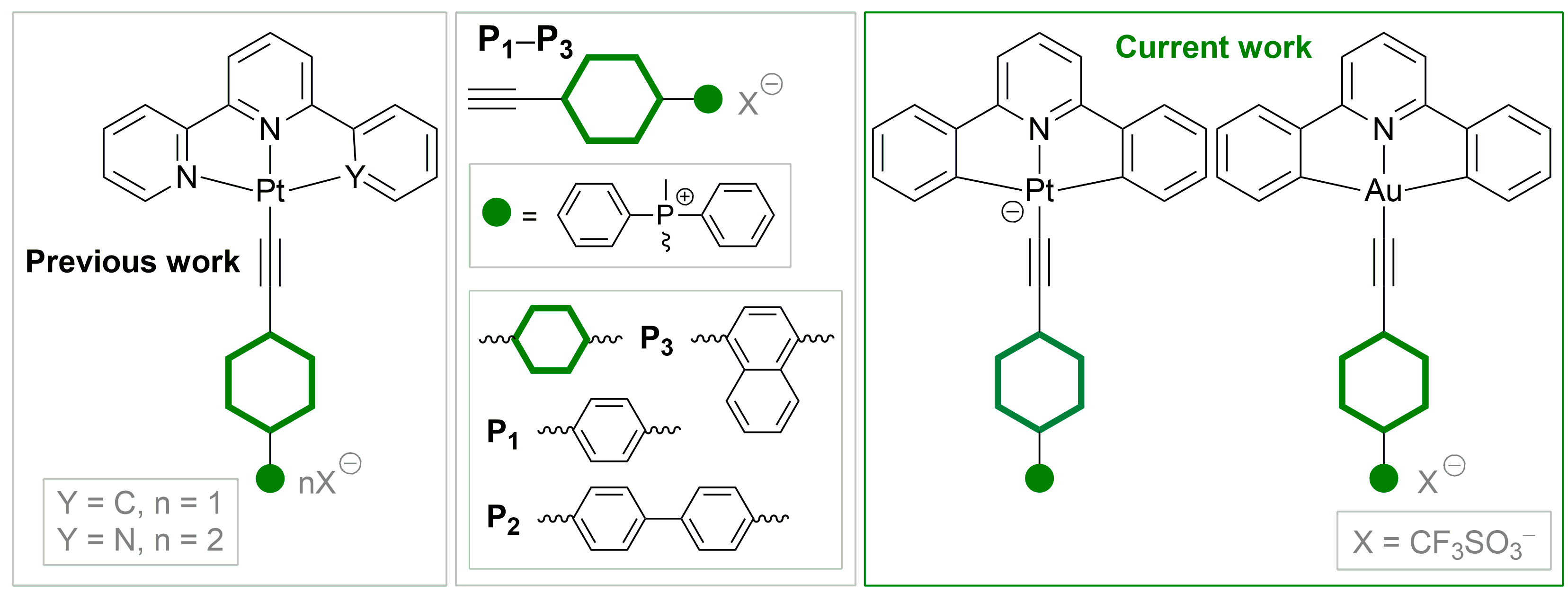
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of Pt(II) and Au(III) Complexes
2.2. Optical and Photophysical Properties
2.3. DFT Calculations
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Wang, S. Phosphorescent Pt(II) Emitters for OLEDs: From Triarylboron-Functionalized Bidentate Complexes to Compounds with Macrocyclic Chelating Ligands. Chem. Rec. 2019, 19, 1693–1709. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Ozawa, H. Homogeneous catalysis of platinum(II) complexes in photochemical hydrogen production from water. Coord. Chem. Rev. 2007, 251, 2753–2766. [Google Scholar] [CrossRef]
- Guerchais, V.; Fillaut, J.-L. Sensory luminescent iridium(III) and platinum(II) complexes for cation recognition. Coord. Chem. Rev. 2011, 255, 2448–2457. [Google Scholar] [CrossRef]
- Mauro, M.; Aliprandi, A.; Septiadi, D.; Kehr, N.S.; De Cola, L. When self-assembly meets biology: Luminescent platinum complexes for imaging applications. Chem. Soc. Rev. 2014, 43, 4144–4166. [Google Scholar] [CrossRef]
- Zhang, Y.; Ni, J.; Zhu, Y.; Zeng, Q.; Ai, Y.; Li, Y. Multi-stimuli responsive Pt(II) complexes for information storage and anti-counterfeiting. Chem. Eng. J. 2024, 498, 155049. [Google Scholar] [CrossRef]
- Wong, K.M.C.; Zhu, X.; Hung, L.L.; Zhu, N.; Yam, V.W.W.; Kwok, H.S. A novel class of phosphorescent gold(III) alkynyl-based organic light-emitting devices with tunable colour. Chem. Commun. 2005, 23, 2906–2908. [Google Scholar] [CrossRef]
- Currie, L.; Fernandez-Cestau, J.; Rocchigiani, L.; Bertrand, B.; Lancaster, S.J.; Hughes, D.L.; Duckworth, H.; Jones, S.T.E.; Credgington, D.; Penfold, T.J.; et al. Luminescent Gold(III) Thiolates: Supramolecular Interactions Trigger and Control Switchable Photoemissions from Bimolecular Excited States. Chem.—A Eur. J. 2017, 23, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Vogler, A. Photoreactivity of gold complexes. Coord. Chem. Rev. 2001, 219–221, 489–507. [Google Scholar] [CrossRef]
- Leung, M.-Y.; Tang, M.-C.; Cheung, W.-L.; Lai, S.-L.; Ng, M.; Chan, M.-Y.; Wing-Wah Yam, V. Thermally Stimulated Delayed Phosphorescence (TSDP)-Based Gold(III) Complexes of Tridentate Pyrazine-Containing Pincer Ligand with Wide Emission Color Tunability and Their Application in Organic Light-Emitting Devices. J. Am. Chem. Soc. 2020, 142, 2448–2459. [Google Scholar] [CrossRef]
- Lee, C.; Tang, M.; Leung, M.; Cheng, S.; Wong, G.Y.; Cheung, W.; Lai, S.; Ko, C.; Chan, M.; Yam, V.W. Phosphine Oxide-Containing Gold(III) Complexes with Tunable Emission Color and Thermally Enhanced Luminescence Behavior. Adv. Opt. Mater. 2024, 12, 2401841. [Google Scholar] [CrossRef]
- To, W.; Zhou, D.; Tong, G.S.M.; Cheng, G.; Yang, C.; Che, C. Highly Luminescent Pincer Gold(III) Aryl Emitters: Thermally Activated Delayed Fluorescence and Solution-Processed OLEDs. Angew. Chem. Int. Ed. 2017, 56, 14036–14041. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.C.; Chan, A.K.W.; Chan, M.Y.; Yam, V.W.W. Platinum and Gold Complexes for OLEDs. Top. Curr. Chem. 2016, 374. [Google Scholar] [CrossRef] [PubMed]
- Paderina, A.; Slavova, S.; Tupikina, E.; Snetkov, D.; Grachova, E. Aggregation Game: Changing Solid-State Emission Using Different Counterions in Monoalkynylphosphonium Pt(II) Complexes. Inorg. Chem. 2024, 63, 17548–17560. [Google Scholar] [CrossRef]
- Berenguer, J.R.; Lalinde, E.; Torroba, J. Synthesis, Characterization and Photophysics of a New Series of Anionic C,N,C Cyclometalated Platinum Complexes. Inorg. Chem. 2007, 46, 9919–9930. [Google Scholar] [CrossRef] [PubMed]
- Petrovskii, S.; Paderina, A.; Sizova, A.; Grachova, E. Homoleptic Alkynylphosphonium Au(I) Complexes as Push–Pull Phosphorescent Emitters. Inorg. Chem. 2023, 62, 5123–5133. [Google Scholar] [CrossRef]
- Paderina, A.; Slavova, S.; Petrovskii, S.; Grachova, E. Alkynylphosphonium Pt(II) Complexes: Synthesis, Characterization, and Features of Photophysical Properties in Solution and in the Solid State. Inorg. Chem. 2023, 62, 18056–18068. [Google Scholar] [CrossRef]
- Grachova, E.; Paderina, A.; Sizova, A. Cationic or Neutral: Dependence of Photophysical Properties of Bis-alkynylphosphonium Pt(II) Complexes on Ancillary Ligand. Chem.—A Eur. J. 2024, 30, e202402242. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists. Coord. Chem. Rev. 2017, 345, 209–228. [Google Scholar] [CrossRef]
- Rizzato, S.; Bergès, J.; Mason, S.A.; Albinati, A.; Kozelka, J. Dispersion-driven hydrogen bonding: Predicted hydrogen bond between water and platinum(II) identified by neutron diffraction. Angew. Chemie—Int. Ed. 2010, 49, 7440–7443. [Google Scholar] [CrossRef]
- Glosz, D.; Jędrzejowska, K.; Niedzielski, G.; Kobylarczyk, J.; Zakrzewski, J.J.; Hooper, J.G.M.; Gryl, M.; Koshevoy, I.O.; Podgajny, R. Influence of O−H⋅t interactions on photoluminescent response in the (Et4N)2{[Pt(bph)(CN)2][phenylene-1,4-diresorcinol]} framework. Chem.—A Eur. J. 2024, 30, e202400797. [Google Scholar] [CrossRef]
- Gokul, G.; Thirumaran, S.; Vijaya, P. Synthesis and anticancer activity of Pd(II) and Pt(II) complexes of dodecyl based dithiocarbamate ligands: Insight into the C-H∙∙∙Pt interaction using X-ray structure, DFT, Hirshfeld surface and AIM analysis. J. Mol. Struct. 2025, 1321, 139998. [Google Scholar] [CrossRef]
- Sillen, A.; Engelborghs, Y. The Correct Use of “Average” Fluorescence Parameters. Photochem. Photobiol. 1998, 67, 475–486. [Google Scholar] [CrossRef]
- Lu, W.; Chan, M.C.W.; Cheung, K.K.; Che, C.M. π-π interactions in organometallic systems. Crystal structures and spectroscopic properties of luminescent mono-, bi-, and trinuclear trans-cyclometalated platinum(II) complexes derived from 2,6-diphenylpyridine. Organometallics 2001, 20, 2477–2486. [Google Scholar] [CrossRef]
- Yam, V.W.W.; Tang, R.P.L.; Wong, K.M.C.; Lu, X.X.; Cheung, K.K.; Zhu, N. Syntheses, electronic absorption, emission, and ion-binding studies of platinum(II) C^N^C and terpyridyl complexes containing crown ether pendants. Chem.—A Eur. J. 2002, 8, 4066–4076. [Google Scholar] [CrossRef]
- Leung, S.Y.; Lam, E.S.; Lam, W.H.; Wong, K.M.; Wong, W.; Yam, V.W. Luminescent Cyclometalated Alkynylplatinum(II) Complexes with a Tridentate Pyridine-Based N-Heterocyclic Carbene Ligand: Synthesis, Characterization, Electrochemistry, Photophysics, and Computational Studies. Chem.—A Eur. J. 2013, 19, 10360–10369. [Google Scholar] [CrossRef]
- Prokhorov, A.M.; Hofbeck, T.; Czerwieniec, R.; Suleymanova, A.F.; Kozhevnikov, D.N.; Yersin, H. Brightly luminescent Pt(II) pincer complexes with a sterically demanding carboranyl-phenylpyridine ligand: A new material class for diverse optoelectronic applications. J. Am. Chem. Soc. 2014, 136, 9637–9642. [Google Scholar] [CrossRef]
- Fang, B.; Zhu, Y.; Hu, L.; Shen, Y.; Jiang, G.; Zhang, Q.; Tian, X.; Li, S.; Zhou, H.; Wu, J.; et al. Series of C^N^C Cyclometalated Pt(II) Complexes: Synthesis, Crystal Structures, and Nonlinear Optical Properties in the Near-Infrared Region. Inorg. Chem. 2018, 57, 14134–14143. [Google Scholar] [CrossRef]
- Ogawa, T.; Sameera, W.M.C.; Yoshida, M.; Kobayashi, A.; Kato, M. Phosphorescence properties of anionic cyclometalated platinum(II) complexes with fluorine-substituted tridentate diphenylpyridine in the solid state. Chem. Phys. Lett. 2020, 739, 137024. [Google Scholar] [CrossRef]
- Yam, V.W.W.; Wong, K.M.C.; Hung, L.L.; Zhu, N. Luminescent gold(III) alkynyl complexes: Synthesis, structural characterization, and luminescence properties. Angew. Chem. Int. Ed. 2005, 44, 3107–3110. [Google Scholar] [CrossRef]
- Au, V.K.-M.; Wong, K.M.-C.; Tsang, D.P.-K.; Chan, M.-Y.; Zhu, N.; Yam, V.W.-W. High-Efficiency Green Organic Light-Emitting Devices Utilizing Phosphorescent Bis-cyclometalated Alkynylgold(III) Complexes. J. Am. Chem. Soc. 2010, 132, 14273–14278. [Google Scholar] [CrossRef]
- Au, V.K.-M.; Tsang, D.P.-K.; Wong, K.M.-C.; Chan, M.-Y.; Zhu, N.; Yam, V.W.-W. Functionalized Bis-Cyclometalated Alkynylgold(III) Complexes: Synthesis, Characterization, Electrochemistry, Photophysics, Photochemistry, and Electroluminescence Studies. Inorg. Chem. 2013, 52, 12713–12725. [Google Scholar] [CrossRef] [PubMed]
- Au, V.K.-M.; Tsang, D.P.-K.; Wong, Y.-C.; Chan, M.-Y.; Yam, V.W.-W. Synthesis of alkynylgold(III) complexes with bis-cyclometalating ligand derived from ethyl 2,6-diphenylisonicotinate and their structural, electrochemical, photo- and electroluminescence studies. J. Organomet. Chem. 2015, 792, 109–116. [Google Scholar] [CrossRef]
- Tong, G.S.; Che, C. Emissive or Nonemissive? A Theoretical Analysis of the Phosphorescence Efficiencies of Cyclometalated Platinum(II) Complexes. Chem.—A Eur. J. 2009, 15, 7225–7237. [Google Scholar] [CrossRef]
- Zehnder, T.N.; Blacque, O.; Venkatesan, K. Luminescent monocyclometalated cationic gold(iii) complexes: Synthesis, photophysical characterization and catalytic investigations. Dalton Trans. 2014, 43, 11959. [Google Scholar] [CrossRef]
- Hua, F.; Kinayyigit, S.; Rachford, A.A.; Shikhova, E.A.; Goeb, S.; Cable, J.R.; Adams, C.J.; Kirschbaum, K.; Pinkerton, A.A.; Castellano, F.N. Luminescent charge-transfer platinum(II) metallacycle. Inorg. Chem. 2007, 46, 8771–8783. [Google Scholar] [CrossRef]
- Leung, M.Y.; Leung, S.Y.L.; Yim, K.C.; Chan, A.K.W.; Ng, M.; Yam, V.W.W. Multiresponsive Luminescent Cationic Cyclometalated Gold(III) Amphiphiles and Their Supramolecular Assembly. J. Am. Chem. Soc. 2019, 141, 19466–19478. [Google Scholar] [CrossRef] [PubMed]
- Yim, K.C.; Au, V.K.M.; Wong, K.M.C.; Yam, V.W.W. Luminescent Bis-Cyclometalated Gold(III) Complexes with Alkynyl Ligands of Hexaphenylbenzene and Hexabenzocoronene Derivatives and Their Supramolecular Assembly. Chem.—A Eur. J. 2017, 23, 5772–5786. [Google Scholar] [CrossRef]
- Wong, K.M.-C.; Hung, L.-L.; Lam, W.H.; Zhu, N.; Yam, V.W.-W. A Class of Luminescent Cyclometalated Alkynylgold(III) Complexes: Synthesis, Characterization, and Electrochemical, Photophysical, and Computational Studies of [Au(C∧N∧C)(C⋮CR)] (C∧N∧C = κ3C,N,C Bis-cyclometalated 2,6-Diphenylpyridyl). J. Am. Chem. Soc. 2007, 129, 4350–4365. [Google Scholar] [CrossRef]
- The IUPAC Compendium of Chemical Terminology; International Union of Pure and Applied Chemistry (IUPAC): Research Triangle Park, NC, USA, 2014.
- Brookhart, M.; Green, M.L.H.; Parkin, G. Agostic interactions in transition metal compounds. Proc. Natl. Acad. Sci. USA 2007, 104, 6908–6914. [Google Scholar] [CrossRef]
- Lein, M. Characterization of agostic interactions in theory and computation. Coord. Chem. Rev. 2009, 253, 625–634. [Google Scholar] [CrossRef]
- Luginin, M.; Snetkov, D.; Sizova, A.; Paderina, A.V.; Sizov, V.; Grachova, E.V. Cyclometalated Au(III) complexes with alkynylphosphine oxide ligands: Synthesis and photophysical properties. Dalton Trans. 2025, 54, 2950–2963. [Google Scholar] [CrossRef] [PubMed]
- Cave, G.W.V.; Fanizzi, F.P.; Deeth, R.J.; Errington, W.; Rourke, J.P. C−H Activation Induced by Water. Monocyclometalated to Dicyclometalated: C∧N∧C Tridentate Platinum Complexes. Organometallics 2000, 19, 1355–1364. [Google Scholar] [CrossRef]
- Wong, K.-H.; Cheung, K.-K.; Chan, M.C.-W.; Che, C.-M. Application of 2,6-Diphenylpyridine as a Tridentate [C^N^C] Dianionic Ligand in Organogold(III) Chemistry. Structural and Spectroscopic Properties of Mono- and Binuclear Transmetalated Gold(III) Complexes. Organometallics 1998, 17, 3505–3511. [Google Scholar] [CrossRef]
- Armareg, W.L.F.; Cha, C. Purification of Laboratory Chemicals; Elsevier: Amsterdam, The Netherlands, 2013; ISBN 9780123821614. [Google Scholar]
- CrysAlisPro, Rigaku Oxford Diffraction, Version 1.171.39.35a; Rigaku: Cedar Park, TX, USA, 2017.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 59–75. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- MacHado, S.F.; Camiletti, G.G.; Neto, A.C.; Jorge, F.E.; Jorge, R.S. Gaussian basis set of triple zeta valence quality for the atoms from K to Kr: Application in DFT and CCSD(T) calculations of molecular properties. Mol. Phys. 2009, 107, 1713–1727. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. van der Waals potential: An important complement to molecular electrostatic potential in studying intermolecular interactions. J. Mol. Model. 2020, 26, 315. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, K.; Reed, E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO, Version 7.0; University of Wisconsin/Madison: Madison, WI, USA, 2018.
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.0; Gaussian Inc.: Wallingford, CT, USA, 2009.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | λabs, nm | λem, * nm | λem, * nm | τ, ** ns | ||
|---|---|---|---|---|---|---|
| DMSO r.t. | Solid 77K | Solid r.t. | DMSO r.t. | Solid r.t. | ||
| Pt1 | 284, 332, 378, ~410, ~522 | 521 | 710 | 692 | 89 (aer) 147 (deg) | 17 |
| Pt2 | 286, 332, 381, 423, ~525 | – | 700 | 680 | – | 23 |
| Au1 | 247, 276, 312, 368, 384, 403 | – | 536 | 563 | – | 55,218 |
| Au2 | 250sh, 275sh, 283, 328, 381sh, 402, | – | 506 | 563 | – | 33,221 |
| Au3 | 247, 278sh, 283sh, 328sh, 343, 360, 385sh, 403 | – | 587 | 538 | – | 14,958 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luginin, M.; Paderina, A.; Sizova, A.; Tupikina, E.; Grachova, E. Does Metal Matter: Comparing Photophysical Properties of Bis-Cyclometalated Alkynylphosphonium Au(III) and Pt(II) Complexes. Molecules 2025, 30, 2434. https://doi.org/10.3390/molecules30112434
Luginin M, Paderina A, Sizova A, Tupikina E, Grachova E. Does Metal Matter: Comparing Photophysical Properties of Bis-Cyclometalated Alkynylphosphonium Au(III) and Pt(II) Complexes. Molecules. 2025; 30(11):2434. https://doi.org/10.3390/molecules30112434
Chicago/Turabian StyleLuginin, Maksim, Aleksandra Paderina, Anastasia Sizova, Elena Tupikina, and Elena Grachova. 2025. "Does Metal Matter: Comparing Photophysical Properties of Bis-Cyclometalated Alkynylphosphonium Au(III) and Pt(II) Complexes" Molecules 30, no. 11: 2434. https://doi.org/10.3390/molecules30112434
APA StyleLuginin, M., Paderina, A., Sizova, A., Tupikina, E., & Grachova, E. (2025). Does Metal Matter: Comparing Photophysical Properties of Bis-Cyclometalated Alkynylphosphonium Au(III) and Pt(II) Complexes. Molecules, 30(11), 2434. https://doi.org/10.3390/molecules30112434