
Synthesis of Ethynyl Trifluoromethyl Sulfide and Its Application to the Synthesis of CF3S-Containing Triazoles


Abstract

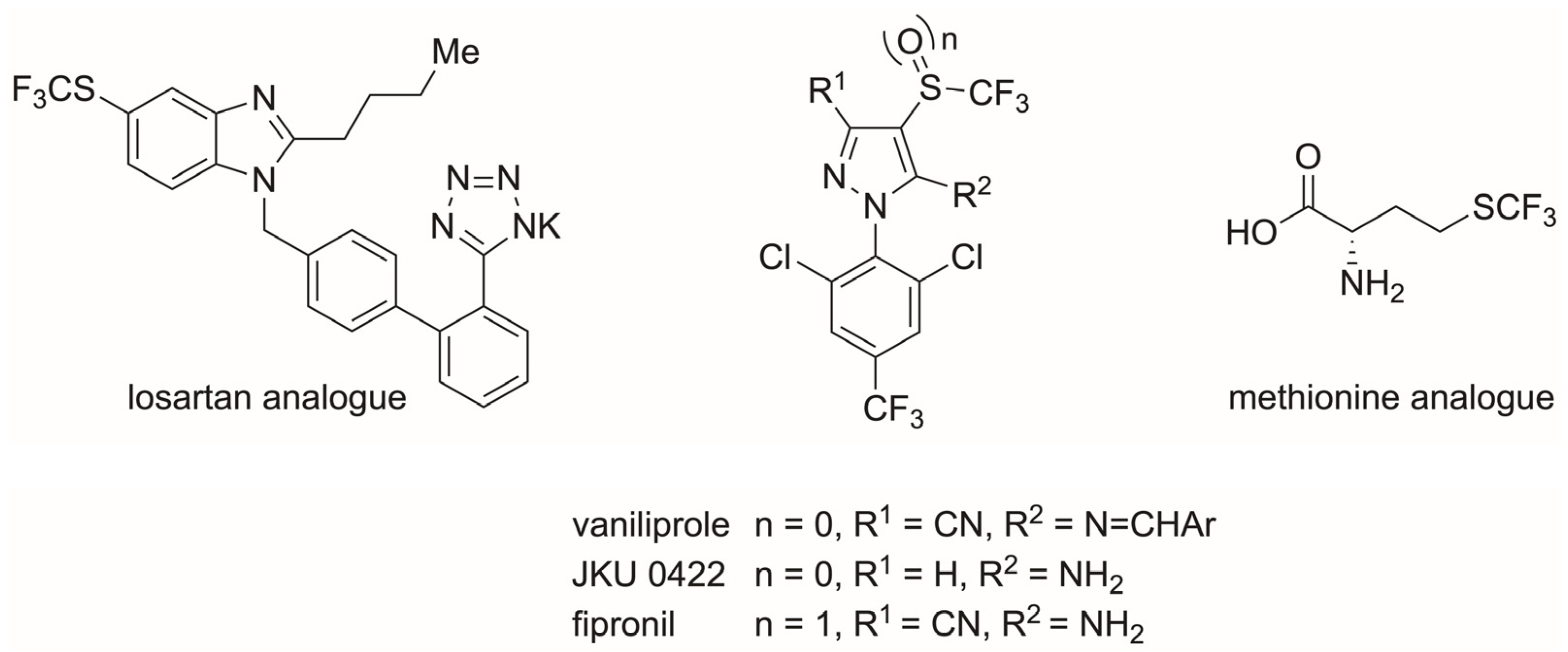
1. Introduction
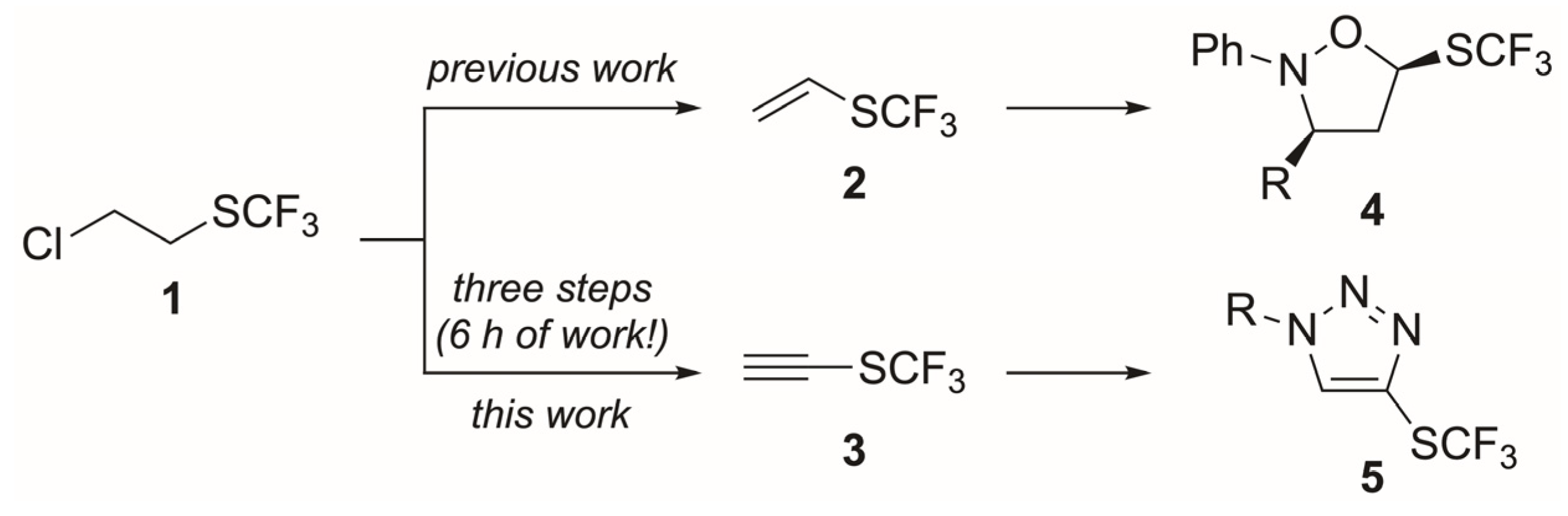
2. Results and Discussion
3. Materials and Methods
3.1. Reagents and Equipment
3.2. General Procedure for the Synthesis of Aromatic Azides 8h–k
3.2.1. 1-Azido-3-methoxybenzene (8h) [39]
3.2.2. 3-Azidobenzonitrile (8i) [40]
3.2.3. 1-Azido-2-methoxybenzene (8j) [39]
3.2.4. 2-Azidobenzonitrile (8k) [41]
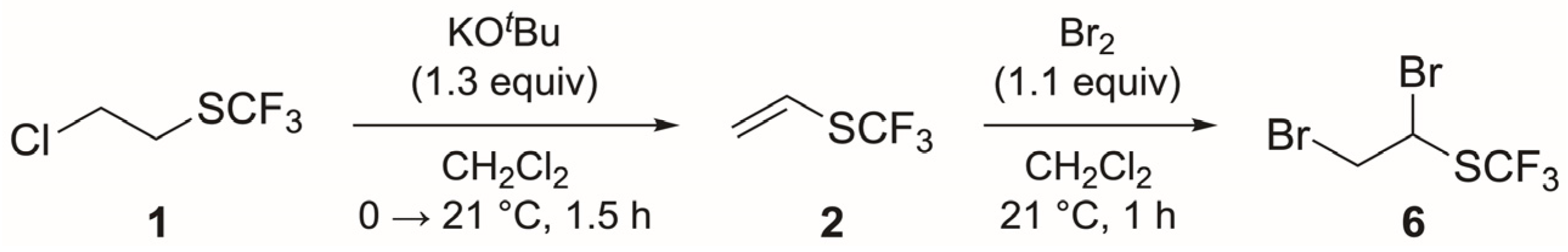
3.3. General Procedure for the Synthesis of Ethynyl Trifluoromethyl Sulfide (3)
3.3.1. Trifluoromethyl Vinyl Sulfide (2) [12]
3.3.2. 1,2-Dibromoethyl Trifluoromethyl Sulfide (6)
3.3.3. 1-Bromovinyl Trifluoromethyl Sulfide (7)
3.3.4. Ethynyl Trifluoromethyl Sulfide (3)
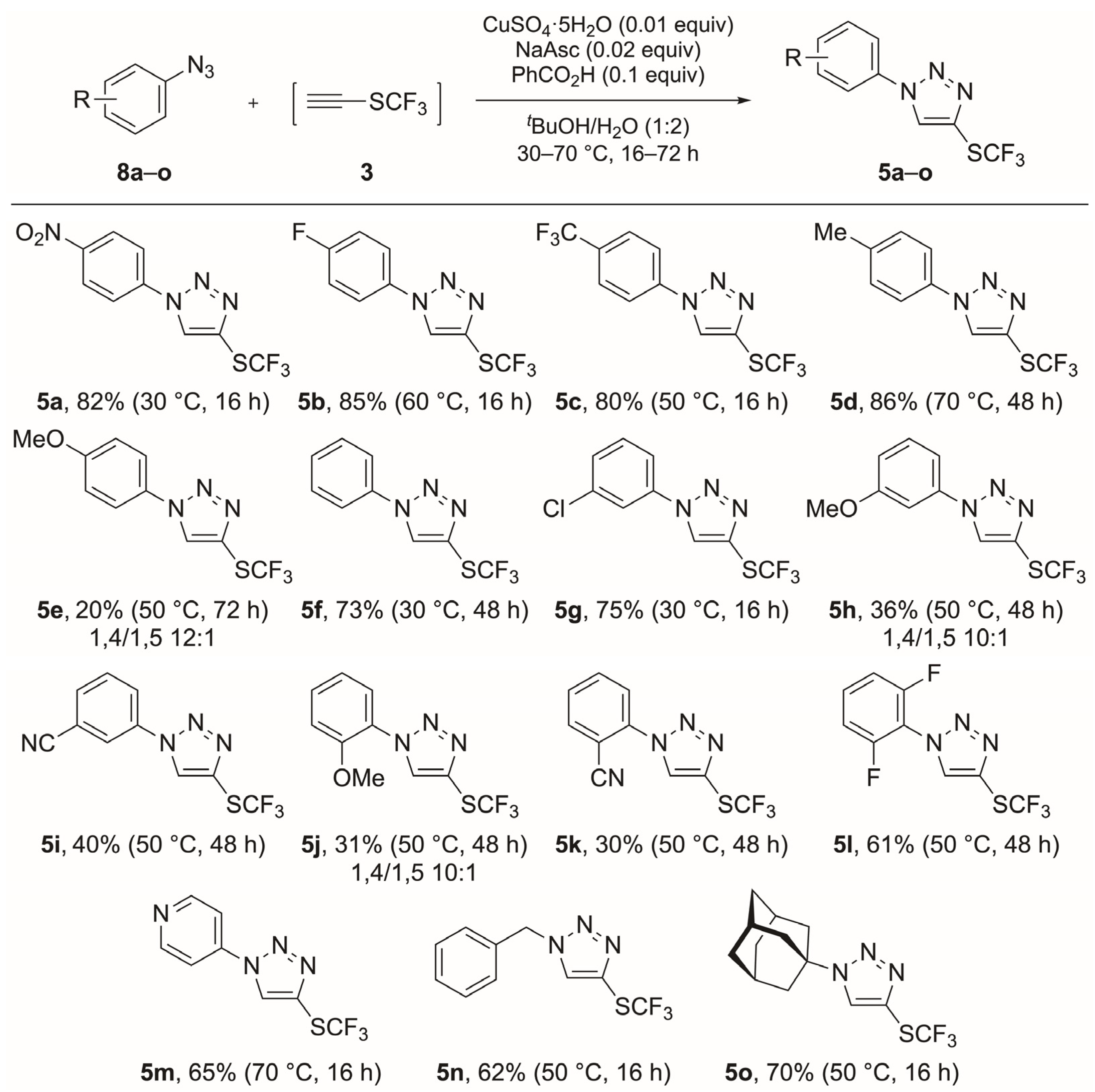
3.4. General Procedure for the Synthesis of 1,4-Disubstituted-1H-1,2,3-Triazoles 5a–o
3.4.1. 1-(4-Nitrophenyl)-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5a)
3.4.2. 1-(4-Fluorophenyl)-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5b)
3.4.3. 1-[4-(Trifluoromethyl)phenyl]-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5c)
3.4.4. 1-(4-Methylphenyl)-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5d)
3.4.5. 1-(4-Methoxyphenyl)-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5e)
3.4.6. 1-Phenyl-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5f)
3.4.7. 1-(3-Chlorophenyl)-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5g)
3.4.8. 1-(3-Methoxyphenyl)-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5h)
3.4.9. 3-{4-[(Trifluoromethyl)sulfanyl]-1H-1,2,3-triazol-1-yl}benzonitrile (5i)
3.4.10. 1-(2-Methoxyphenyl)-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5j)
3.4.11. 2-{4-[(Trifluoromethyl)sulfanyl]-1H-1,2,3-triazol-1-yl}benzonitrile (5k)
3.4.12. 1-(2,6-Difluorophenyl)-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5l)
3.4.13. 4-{4-[(Trifluoromethyl)sulfanyl]-1H-1,2,3-triazol-1-yl}pyridine (5m)
3.4.14. 1-Benzyl-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5n)
3.4.15. 1-(Adamantan-1-yl)-4-[(trifluoromethyl)sulfanyl]-1H-1,2,3-triazole (5o)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| THF | Tetrahydrofuran |
| HMDS | 1,1,1,3,3,3-Hexamethyldisilazanide |
| Asc | Ascorbate |
| NMR | Nuclear magnetic resonance |
| HSQC | Heteronuclear single quantum correlation |
| HMBC | Heteronuclear multiple bond correlation |
| NOESY | Nuclear Overhauser effect spectroscopy |
| HOESY | Heteronuclear Overhauser effect spectroscopy |
References
- Zhou, J.; Zhang, Q.F.; Zhao, W.H.; Jiang, G.F. Chiral phosphoric acid-catalyzed asymmetric transfer hydrogenation of 3-trifluoromethylthioquinolines. Org. Biomol. Chem. 2016, 14, 6937–6941. [Google Scholar] [CrossRef] [PubMed]
- Bootwicha, T.; Liu, X.; Pluta, R.; Atodiresei, I.; Rueping, M. N-Trifluoromethylthiophthalimide: A stable electrophilic SCF3-reagent and its application in the catalytic asymmetrictrifluoromethylsulfenylation. Angew. Chem. Int. Ed. 2013, 52, 12856–12859. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Wang, X.; Yang, T.; Lu, L.; Shen, Q. An electrophilic hypervalent iodine reagent for trifluoromethylthiolation. Angew. Chem. Int. Ed. 2013, 52, 3457–3460. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Kawai, K.; Tanaka, R.; Yoshino, T.; Matsunaga, S. Cp*CoIII-catalyzed directed C–H trifluoromethylthiolation of 2-phenylpyridines and 6-arylpurines. Chem. Commun. 2017, 53, 5974–5977. [Google Scholar] [CrossRef]
- Yagupolskii, L.M.; Maletina, I.I.; Petko, K.I.; Fedyuk, D.V.; Handrock, R.; Shavaran, S.S.; Klebanov, B.M.; Herzig, S. New fluorine-containing hypotensive preparations. J. Fluor. Chem. 2001, 109, 87–94. [Google Scholar] [CrossRef]
- Leroux, F.; Jeschke, P.; Schlosser, M. α-Fluorinated ethers, thioethers, and amines: Anomerically biased species. Chem. Rev. 2005, 105, 827–856. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Ueki, H.; Aceña, J.L.; Ellis, T.K.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Chemical deracemization and (S) to (R) interconversion of some fluorine-containing α-amino acids. J. Fluor. Chem. 2013, 152, 114–118. [Google Scholar] [CrossRef]
- Xu, C.; Ma, B.; Shen, Q. N-Trifluoromethylthiosaccharin: An easily accessible, shelf-stable, broadly applicable trifluoromethylthiolating reagent. Angew. Chem. Int. Ed. 2014, 53, 9316–9320. [Google Scholar] [CrossRef]
- Pluta, R.; Nikolaienko, P.; Rueping, M. Direct catalytic trifluoromethylthiolation of boronic acids and alkynes employing electrophilic shelf-stable N-(trifluoromethylthio)phthalimide. Angew. Chem. Int. Ed. 2014, 53, 1650–1653. [Google Scholar] [CrossRef]
- Alazet, S.; Zimmer, L.; Billard, T. Base-catalyzed electrophilic trifluoromethylthiolation of terminal alkynes. Angew. Chem. Int. Ed. 2013, 52, 10814–10817. [Google Scholar] [CrossRef]
- Cheng, Y.-X.; Yang, X.-G.; Du, F.-H.; Zhang, C. Direct trifluoromethylthiolation of terminal alkynes mediated by a hypervalent trifluoromethylthio-iodine(III) reagent; boosting effect of fluorinated alcohol. Green Chem. 2024, 26, 5914–5920. [Google Scholar] [CrossRef]
- Riesco-Domínguez, A.; van de Wiel, J.; Hamlin, T.A.; van Beek, B.; Lindell, S.D.; Blanco-Ania, D.; Bickelhaupt, F.M.; Rutjes, F.P.J.T. Trifluoromethyl vinyl sulfide: A building block for the synthesis of CF3S-containing isoxazolidines. J. Org. Chem. 2018, 83, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Sokolenko, L.V.; Maletina, I.I.; Yagupolskii, L.M.; Yagupolskii, Y.L. A convenient and efficient synthesis of trifluoromethyl vinyl sulfoxide and its reactivity in addition reactions. Synlett 2010, 2010, 2075–2078. [Google Scholar] [CrossRef]
- Harris, J.F., Jr.; Joyce, R.M., Jr. Ethynyl Trifluoromethyl Sulfide 3 Was Detected as a Side Product of the Synthesis of bis(trifluoromethylsulfanyl)acetylene. Fluoroalkylthioacetylenes. U.S. Patent 3062893, 6 November 1962. [Google Scholar]
- Harris, J.F., Jr. Trifluoromethylthioalkanes, -olefins, and -acetylenes. J. Org. Chem. 1967, 32, 2063–2074. [Google Scholar] [CrossRef]
- Lo, J.M.H.; Marriott, R.A.; Giri, B.R.; Roscoe, J.M.; Klobukowski, M. A theoretical analysis of the kinetics of the reaction of atomic bromine with tetrahydrofuran. Can. J. Chem. 2010, 88, 1136–1145. [Google Scholar] [CrossRef]
- Giri, B.R.; Roscoe, J.M. Temperature dependence of the rate coefficients for the reactions of atomic bromine with toluene, tetrahydrofuran, and tetrahydropyran. J. Phys. Chem. A 2009, 113, 8001–8010. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Ania, D.; Aben, R.W.M.; van Berkom, L.W.A.; Scheeren, H.W.; Rutjes, F.P.J.T. Synthesis of steroidal D-ring-fused pyrrolidines of dehydroepiandrosterone. Eur. J. Org. Chem. 2017, 2017, 3729–3737. [Google Scholar] [CrossRef]
- Blanco-Ania, D.; Aben, R.W.M.; van Berkom, L.W.A.; Scheeren, H.W.; Rutjes, F.P.J.T. High-pressure-mediated extension of the privileged steroid scaffold. Eur. J. Org. Chem. 2014, 2014, 1438–1444. [Google Scholar] [CrossRef]
- Jenner, G. Correlation between pressure and steric interactions in organic reactions. Tetrahedron 2005, 61, 3621–3635. [Google Scholar] [CrossRef]
- Singh, M.S.; Chowdhury, S.; Koley, S. Advances of azide-alkyne cycloaddition-click chemistry over the recent decade. Tetrahedron 2016, 72, 5257–5283. [Google Scholar] [CrossRef]
- Thirumurugan, P.; Matosiuk, D.; Jozwiak, K. Click chemistry for drug development and diverse chemical–biology applications. Chem. Rev. 2013, 113, 4905–4979. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide–alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Haldon, E.; Nicasio, M.C.; Perez, P. Copper-catalysed azide–alkyne cycloadditions (CuAAC): An update. J. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Vale, D.P.; Vala, R.M.; Patel, H.M. Versatile synthetic platform for 1,2,3-triazole chemistry. ACS Omega 2022, 7, 36945–36987. [Google Scholar] [CrossRef]
- Zhang, L.L.; Li, M.T.; Shen, L.L.; Wu, Q.P. Efficient synthesis of 5-trifluoromethylthio-1,2,3-triazoles: One-pot multicomponent reaction from elemental sulfur and TMSCF3. Synthesis 2020, 52, 304–310. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Lázaro-Milla, C. Metal-free [3+2] cycloaddition of azides with Tf2C=CH2 for the regioselective preparation of elusive 4-(trifluoromethylsulfonyl)-1,2,3-triazoles. Chem. Commun. 2015, 51, 6992–6995. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Fülöp, F. Flow chemistry as a versatile tool for the synthesis of triazoles. Catal. Sci. Technol. 2015, 5, 4926–4941. [Google Scholar] [CrossRef]
- Baumann, M.; Baxendale, I.R.; Ley, S.V.; Nikbin, N. An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals. Beilstein J. Org. Chem. 2011, 7, 442–495. [Google Scholar] [CrossRef]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click chemistry: 1,2,3-Triazoles as pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F.; Steck, P.L. High Pressure in Organic Synthesis: Influence on Selectivity. In High Pressure Chemistry; van Eldik, R., Klärner, F.G., Eds.; Wiley-VCH: Weinheim, Germany, 2002; pp. 239–283. [Google Scholar]
- Shao, C.; Wang, X.; Xu, J.; Zhao, J.; Zhang, Q.; Hu, Y. Carboxylic acid-promoted copper(I)-catalyzed azide–alkyne cycloaddition. J. Org. Chem. 2010, 75, 7002–7005. [Google Scholar] [CrossRef] [PubMed]
- Kutonova, K.V.; Trusova, M.E.; Postnikov, P.S.; Filimonov, V.D.; Parello, J. A simple and effective synthesis of aryl azides via arenediazonium tosylates. Synthesis 2013, 45, 2706–2710. [Google Scholar]
- Blastik, Z.E.; Voltrová, S.; Matoušek, V.; Jurásek, B.; Manley, D.W.; Klepetářová, B.; Beier, P. Azidoperfluoroalkanes: Synthesis and application in copper(I)-catalyzed azide–alkyne cycloaddition. Angew. Chem. Int. Ed. 2017, 56, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, M.K.; Singh, M.K.; Parrish, D.; Balachandran, R.; Day, B.W. Azide–tetrazole equilibrium of C-6 azidopurine nucleosides and their ligation reactions with alkynes. J. Org. Chem. 2010, 75, 2461–2473. [Google Scholar] [CrossRef]
- Song, W.; Zheng, N.; Li, M.; He, J.; Li, J.; Dong, K.; Ullah, K.; Zheng, Y. Rhodium(I)-catalyzed regioselective azi-internal alkynyl trifluoromethyl sulfide cycloaddition and azide-internal thioalkyne cycloaddition under mild conditions. Adv. Synth. Catal. 2019, 361, 469–475. [Google Scholar] [CrossRef]
- Kwok, S.W.; Fotsing, J.R.; Fraser, R.J.; Rodionov, V.O.; Fokin, V.V. Transition-metal-free catalytic synthesis of 1,5-diaryl-1,2,3-triazoles. Org. Lett. 2010, 12, 4217–4219. [Google Scholar] [CrossRef]
- Akula, H.K.; Lakshman, M.K. Synthesis of deuterated 1,2,3-triazoles. J. Org. Chem. 2012, 77, 8896–8904. [Google Scholar] [CrossRef]
- Budruev, A.V.; Dzhons, D.Y.; Faerman, V.I.; Fukin, G.K.; Shavyrin, A.S. Photochemical synthesis of 6-substituted 12-oxo-6,12-dihydroazepino [2,1-c]quinazolines. Chem. Heterocycl. Compd. 2016, 52, 694–699. [Google Scholar] [CrossRef]
- Jin, L.M.; Xu, X.; Lu, H.; Cui, X.; Wojtas, L.; Zhang, X.P. Effective synthesis of chiral N-fluoroaryl aziridines through enantioselective aziridination of alkenes with fluoroaryl azides. Angew. Chem. Int. Ed. 2013, 52, 5309–5313. [Google Scholar] [CrossRef]
- Colombano, G.; Travelli, C.; Galli, U.; Caldarelli, A.; Chini, M.G.; Canonico, P.L.; Sorba, G.; Bifulco, G.; Tron, G.C.; Genazzani, A.A. A novel potent nicotinamide phosphoribosyltransferase inhibitor synthesized via click chemistry. J. Med. Chem. 2010, 53, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, H.C.; Schaap, M.; Baird, L.; Georgakopoulos, N.D.; Fowkes, A.; Thiollier, C.; Kachi, H.; Dinkova-Kostova, A.T.; Wells, G. Design, Synthesis, and Evaluation of Triazole Derivatives That Induce Nrf2 Dependent Gene Products and Inhibit the Keap1-Nrf2 Protein-Protein Interaction. J. Med. Chem. 2015, 58, 7186. [Google Scholar] [CrossRef] [PubMed]
- Lanke, S.R.; Bhanage, B.M. Copper bis(2,2,6,6-Tetramethyl-3,5-heptanedionate)–Catalyzed Coupling of Sodium Azide with Aryl Iodides/Boronic Acids to Aryl Azides or Aryl Amines. Synth. Commun. 2014, 44, 399. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Base | Equiv | Time (h) | Ratio 7/3 1 |
| 1 | NaHMDS | 2.0 then 1.0 | 17 2 | 1:0 disappearance of 7 |
| 2 | NaHMDS | 2.0 then 1.0 | 1.5 8 | 1:0 1:0 |
| 3 | KOtBu then NaHMDS | 1.5 0.6 | 1.5 1 | 4:1 1:0.9 |
| 4 | KOtBu then NaHMDS | 1.3 0.7 | 1.5 1 | 7:3 4:13 |
| 5 | KOtBu then NaHMDS | 1.3 1.7 | 1.5 1.5 | 4:1 0:1 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | R | t (°C) | Time (h) | Promoter | Product | Yield (%) 2 |
| 1 3 | 8a | NO2 | 21 | 72 | 15 kbar | 5a | 78 4 |
| 2 | 8a | NO2 | 21 → 30 | 72 | – | 5a | – |
| 3 | 8b | F | 21 → 50 5 | 120 | 15 kbar | 5b | 49 |
| 4 | 8c | CF3 | 21 → 50 5 | 120 | 15 kbar | 5c | 38 |
| 5 | 8d | Me | 21 → 50 6 | 120 | 15 kbar | 5d | – |
| 6 | 8e | OMe | 21 → 50 6 | 120 | 15 kbar | 5e | – |
| 7 | 8a | NO2 | 30 | 16 | Cu(I) 7 | 5a | 82 |
| 8 | 8b | F | 60 | 16 | Cu(I) 7 | 5b | 85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riesco-Domínguez, A.; Hammoudeh, H.; Blanco-Ania, D.; Rutjes, F.P.J.T. Synthesis of Ethynyl Trifluoromethyl Sulfide and Its Application to the Synthesis of CF3S-Containing Triazoles. Molecules 2025, 30, 2358. https://doi.org/10.3390/molecules30112358
Riesco-Domínguez A, Hammoudeh H, Blanco-Ania D, Rutjes FPJT. Synthesis of Ethynyl Trifluoromethyl Sulfide and Its Application to the Synthesis of CF3S-Containing Triazoles. Molecules. 2025; 30(11):2358. https://doi.org/10.3390/molecules30112358
Chicago/Turabian StyleRiesco-Domínguez, Alejandra, Hussein Hammoudeh, Daniel Blanco-Ania, and Floris P. J. T. Rutjes. 2025. "Synthesis of Ethynyl Trifluoromethyl Sulfide and Its Application to the Synthesis of CF3S-Containing Triazoles" Molecules 30, no. 11: 2358. https://doi.org/10.3390/molecules30112358
APA StyleRiesco-Domínguez, A., Hammoudeh, H., Blanco-Ania, D., & Rutjes, F. P. J. T. (2025). Synthesis of Ethynyl Trifluoromethyl Sulfide and Its Application to the Synthesis of CF3S-Containing Triazoles. Molecules, 30(11), 2358. https://doi.org/10.3390/molecules30112358