
Electronic Modulation of Cu Catalytic Interfaces by Functionalized Ionic Liquids for Enhanced CO2 Reduction

Abstract
1. Introduction
2. Simulation Details
3. Results and Discussion
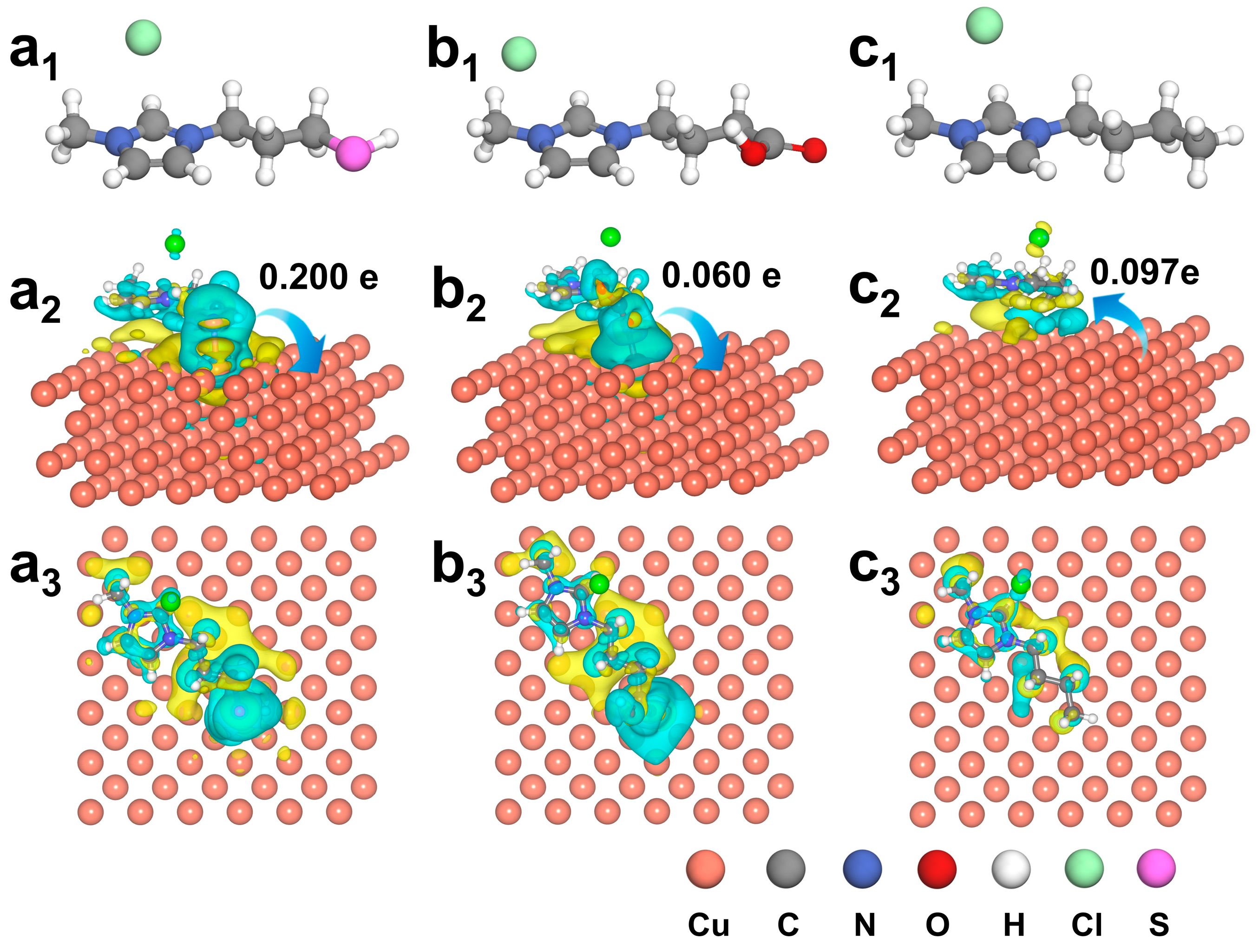
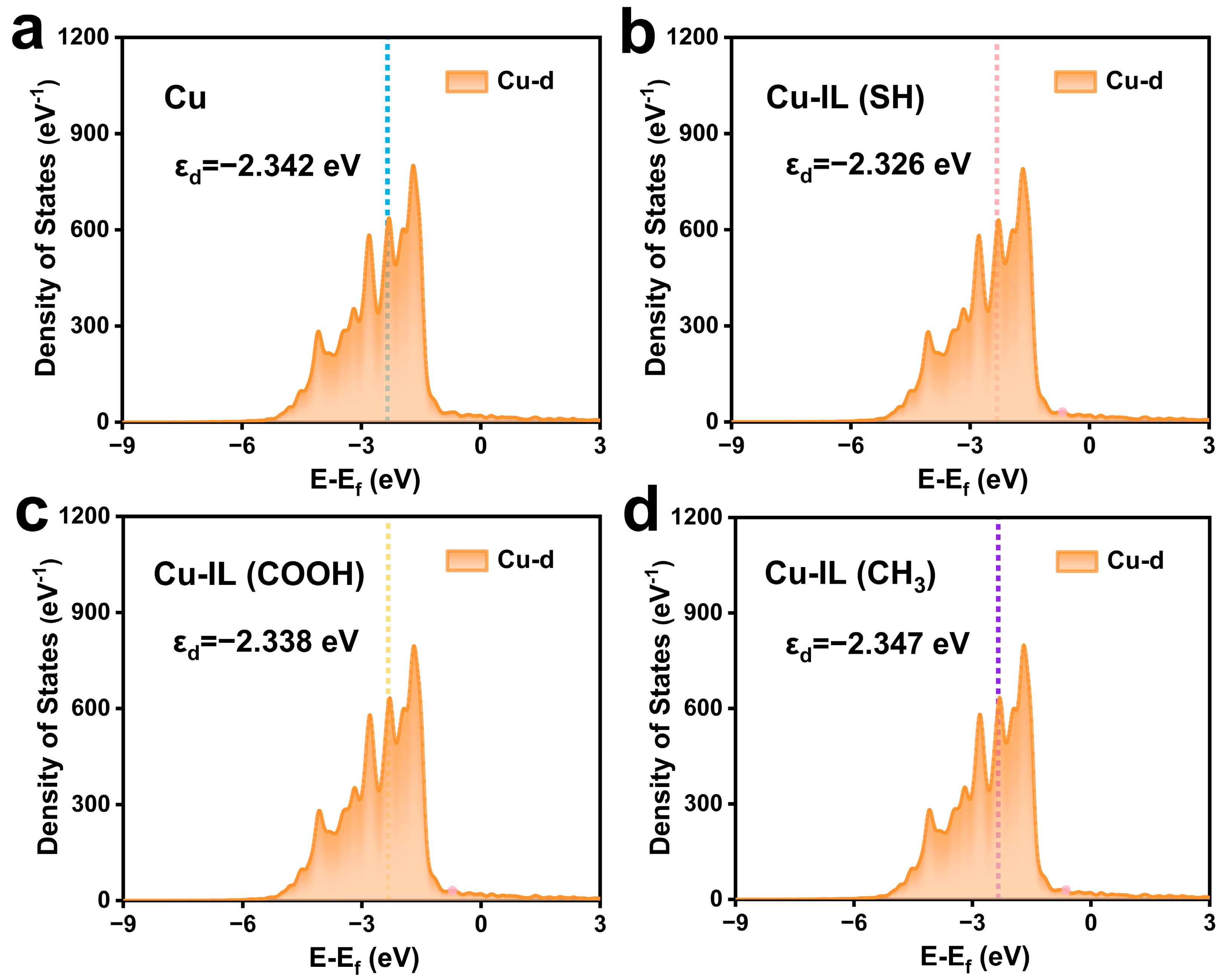
3.1. Functional Group-Governed Electronic Reconstruction in IL-Modified Cu Catalysts
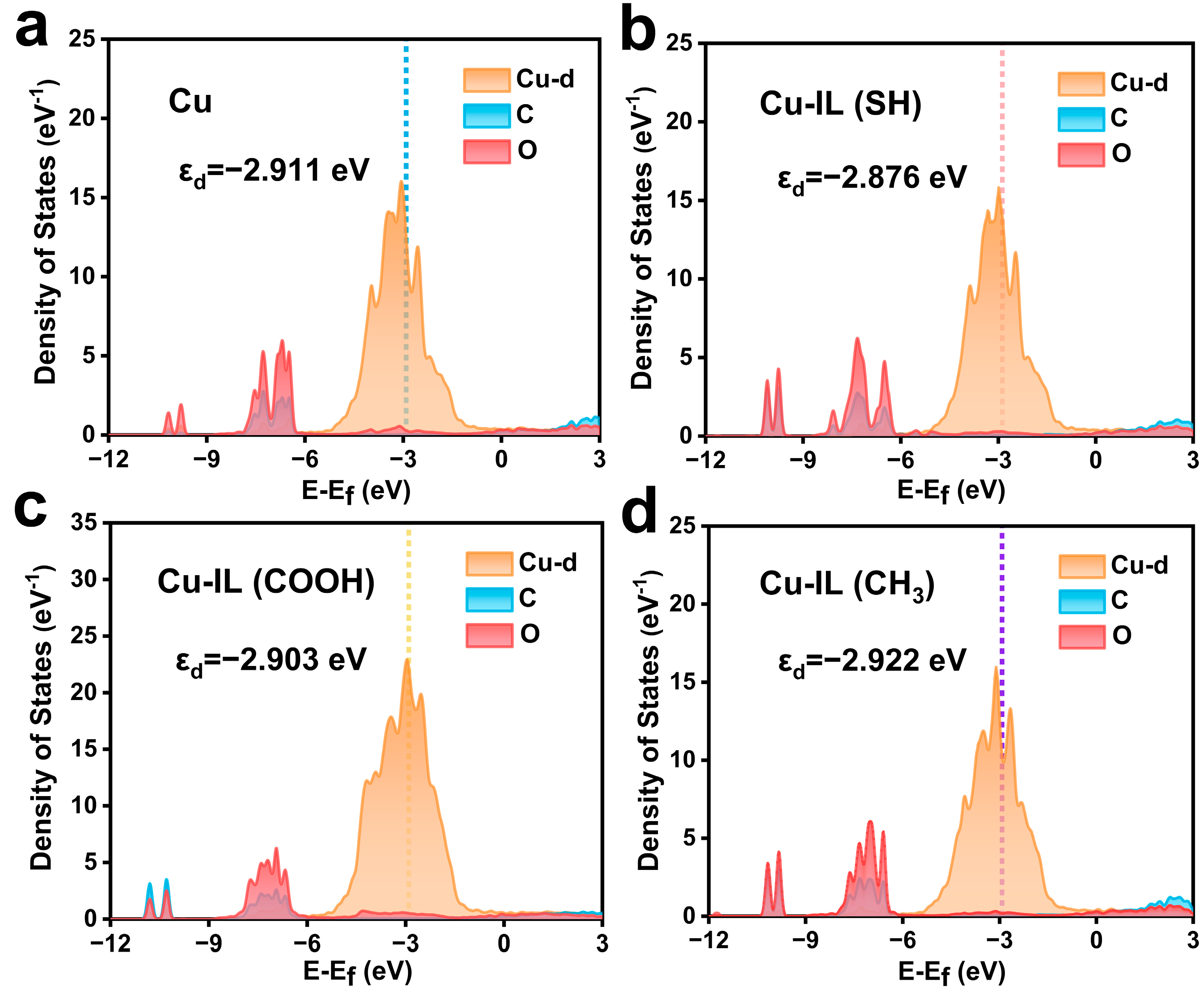
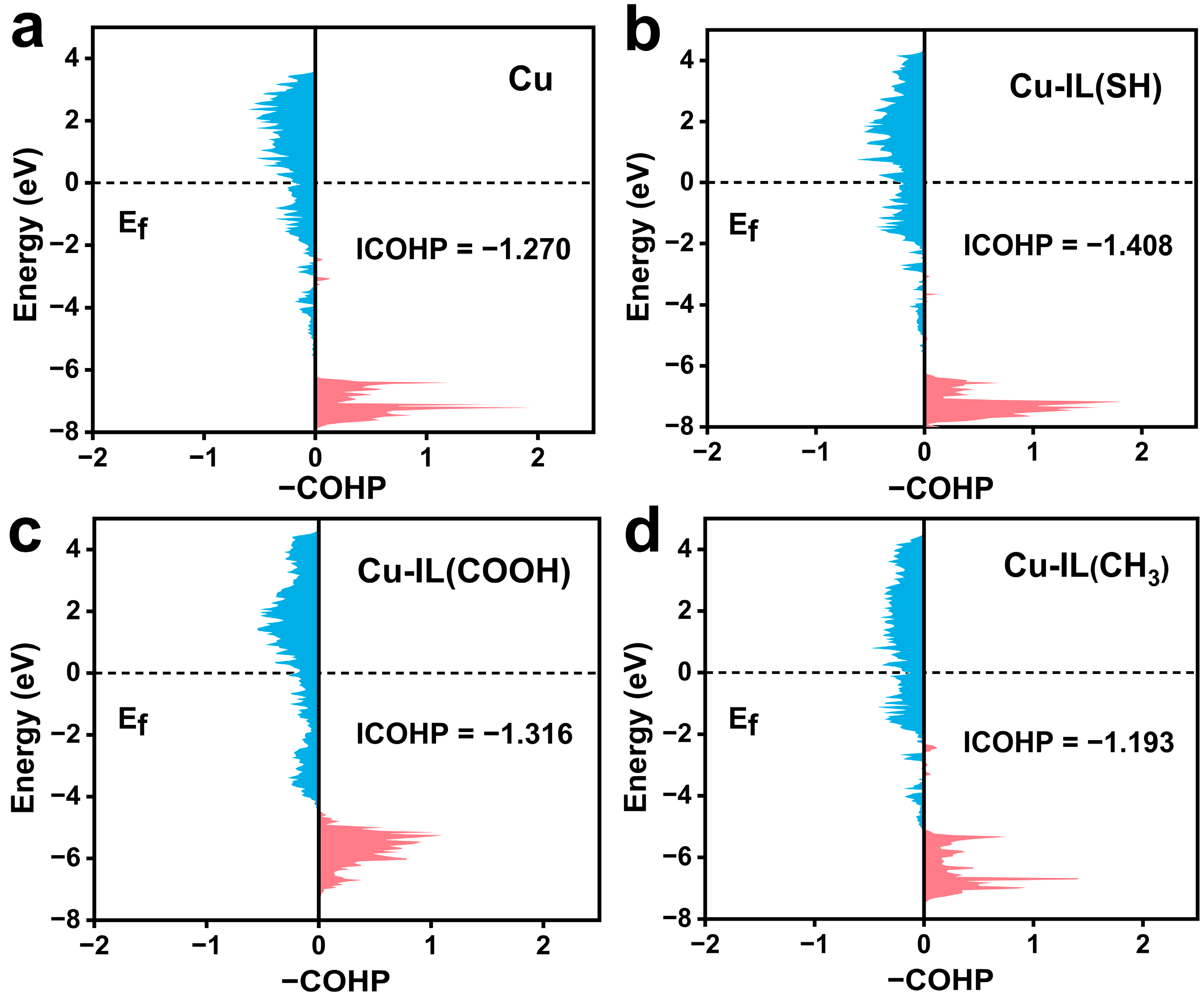
3.2. Electronic Structure Modulation and Intermediate Stabilization via IL-Modified Cu Surfaces
3.3. IL-Mediated Modulation of Coupling Barriers and Product Selectivity in CO2RR on Cu-Based Catalysts—Dual Modulation of Thermodynamics and Kinetics in C–C Coupling by ILs
3.4. IL-Guided Pathway Divergence and Suppression of Hydrogen Evolution Reaction of Cu-Based Catalysts for CO2RR Products
3.4.1. Competitive Reaction Pathways Toward C1 and C2 Products of CO2RR on IL-Modified Cu
3.4.2. Suppression of HER
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yin, Z.-Z.; Huang, T.-T.; Shao, Z.-B.; Zhao, B. Development of fire-resistant PVA films with superior toughness, self-healing, and antibacterial properties via a phosphazene molecule. Prog. Org. Coat. 2025, 198, 108881. [Google Scholar] [CrossRef]
- Li, X.-J.; Ning, K.; Huang, T.-T.; Shao, Z.-B.; Zhao, B. Arone-containing cyclomatrix-type polyphosphazene hybrid microspheres for epoxy resin: Enhancing fire safety and mechanical properties. Colloids Surf. A Physicochem. Eng. Asp. 2024, 692, 133988. [Google Scholar] [CrossRef]
- Birdja, Y.Y.; Pérez-Gallent, E.; Figueiredo, M.C.; Göttle, A.J.; Calle-Vallejo, F.; Koper, M.T.M. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 2019, 4, 732–745. [Google Scholar] [CrossRef]
- Xie, W.F.; Li, B.K.; Liu, L.; Li, H.; Yue, M.Z.; Niu, Q.M.; Liang, S.Y.; Shao, X.D.; Lee, H.Y.Y.; Lee, J.Y.; et al. Advanced systems for enhanced CO2 electroreduction. Chem. Soc. Rev. 2025, 54, 898–959. [Google Scholar] [CrossRef]
- Guo, W.W.; Liu, S.J.; Tan, X.X.; Wu, R.Z.; Yan, X.P.; Chen, C.J.; Zhu, Q.G.; Zheng, L.R.; Ma, J.Y.; Zhang, J.; et al. Highly Efficient CO2 Electroreduction to Methanol through Atomically Dispersed Sn Coupled with Defective CuO Catalysts. Angew. Chem. Int. Ed. 2021, 60, 21979–21987. [Google Scholar] [CrossRef]
- He, X.Y.; Lin, L.; Li, X.Y.; Zhu, M.Z.; Zhang, Q.H.; Xie, S.J.; Mei, B.B.; Sun, F.F.; Jiang, Z.; Cheng, J.; et al. Roles of copper(I) in water-promoted CO2 electrolysis to multi-carbon compounds. Nat. Commun. 2024, 15, 9923. [Google Scholar] [CrossRef]
- Wang, Q.; Wei, H.; Liu, P.; Su, Z.; Gong, X.-Q. Recent advances in copper-based catalysts for electrocatalytic CO2 reduction toward multi-carbon products. Nano Res. Energy 2024, 3, e9120112. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Koper, M.T.M. Theoretical Considerations on the Electroreduction of CO to C2 Species on Cu(100) Electrodes. Angew. Chem. Int. Ed. 2013, 52, 7282–7285. [Google Scholar] [CrossRef]
- Li, M.; Hu, Y.; Wu, T.; Sumboja, A.; Geng, D. How to enhance the C2 products selectivity of copper-based catalysts towards electrochemical CO2 reduction?—A review. Materials Today 2023, 67, 320–343. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, Z.; Lai, W.; Wang, Q.; Qiao, Y.; Tao, H.; Lian, C.; Liu, M.; Ma, C.; Pan, A.; et al. CO2 electroreduction to multicarbon products in strongly acidic electrolyte via synergistically modulating the local microenvironment. Nat. Commun. 2022, 13, 7596. [Google Scholar] [CrossRef]
- Pan, F.; Duan, X.; Fang, L.; Li, H.; Xu, Z.; Wang, Y.; Wang, T.; Li, T.; Duan, Z.; Chen, K.-J. Long-Range Confinement-Driven Enrichment of Surface Oxygen-Relevant Species Promotes C−C Electrocoupling in CO2 Reduction. Adv. Energy Mater. 2024, 14, 2303118. [Google Scholar] [CrossRef]
- Huang, P.; He, B.; Dong, Y.; Zhou, J.; Xu, J.; Pan, C.; Lou, Y.; Wang, Y.; Zhang, Y.; Huang, H.; et al. Preferentially Stabilizing the Watershed Intermediates by Adsorbate-adsorbate Interaction to Accelerate CO2 Electroreduction to Ethanol. Adv. Funct. Mater. 2024, 25, 2424583. [Google Scholar] [CrossRef]
- De Gregorio, G.L.; Burdyny, T.; Loiudice, A.; Iyengar, P.; Smith, W.A.; Buonsanti, R. Facet-Dependent Selectivity of Cu Catalysts in Electrochemical CO2 Reduction at Commercially Viable Current Densities. ACS Catal. 2020, 10, 4854–4862. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, Z.-J.; Cheng, D.; Li, H.; Yu, J.; Wang, Q.; Gao, H.; Guo, J.; Wang, H.; Ozin, G.A.; et al. Efficient CO2 electroreduction on facet-selective copper films with high conversion rate. Nat. Commun. 2021, 12, 5745. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, W.; Choi, C.; Kim, G.; Cho, K.M.; Lim, J.; Kim, S.J.; Al-Saggaf, A.; Gereige, I.; Lee, H.; et al. High Facets on Nanowrinkled Cu via Chemical Vapor Deposition Graphene Growth for Efficient CO2 Reduction into Ethanol. ACS Catal. 2021, 11, 5658–5665. [Google Scholar] [CrossRef]
- Piqué, O.; Low, Q.H.; Handoko, A.D.; Yeo, B.S.; Calle-Vallejo, F. Selectivity Map for the Late Stages of CO and CO2 Reduction to C2 Species on Copper Electrodes. Angew. Chem. Int. Ed. 2021, 60, 10784–10790. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yan, X.; Wu, Y.; Liu, S.; Sun, X.; Zhu, Q.; Feng, R.; Wu, T.; Qian, Q.; Liu, H.; et al. The in situ study of surface species and structures of oxide-derived copper catalysts for electrochemical CO2 reduction. Chem. Sci. 2021, 12, 5938–5943. [Google Scholar] [CrossRef]
- Dutta, A.; Montiel, I.Z.; Erni, R.; Kiran, K.; Rahaman, M.; Drnec, J.; Broekmann, P. Activation of bimetallic AgCu foam electrocatalysts for ethanol formation from CO2 by selective Cu oxidation/reduction. Nano Energy 2020, 68, 104331. [Google Scholar] [CrossRef]
- Chen, C.; Li, Y.; Yu, S.; Louisia, S.; Jin, J.; Li, M.; Ross, M.B.; Yang, P. Cu-Ag Tandem Catalysts for High-Rate CO2 Electrolysis toward Multicarbons. Joule 2020, 4, 1688–1699. [Google Scholar] [CrossRef]
- Herzog, A.; Bergmann, A.; Jeon, H.S.; Timoshenko, J.; Kühl, S.; Rettenmaier, C.; Lopez Luna, M.; Haase, F.T.; Roldan Cuenya, B. Operando Investigation of Ag-Decorated Cu2O Nanocube Catalysts with Enhanced CO2 Electroreduction toward Liquid Products. Angew. Chem. Int. Ed. 2021, 60, 7426–7435. [Google Scholar] [CrossRef]
- Huang, T.-T.; Ning, K.; Zhao, B. Two birds, one stone: Enhancement of flame retardancy and antibacterial property of viscose fabric using an aminoazole-based cyclotriphosphazene. Int. J. Biol. Macromol. 2023, 253, 126875. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhu, C.; Wu, Z.; Xuan, J.; Francisco, J.S.; Wang, H. In Situ Observation of the pH Gradient near the Gas Diffusion Electrode of CO2 Reduction in Alkaline Electrolyte. J. Am. Chem. Soc. 2020, 142, 15438–15444. [Google Scholar] [CrossRef]
- Monteiro, M.C.O.; Dattila, F.; Hagedoorn, B.; García-Muelas, R.; López, N.; Koper, M.T.M. Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat. Catal. 2021, 4, 654–662. [Google Scholar] [CrossRef]
- Li, M.; Garg, S.; Chang, X.; Ge, L.; Li, L.; Konarova, M.; Rufford, T.E.; Rudolph, V.; Wang, G. Toward Excellence of Transition Metal-Based Catalysts for CO2 Electrochemical Reduction: An Overview of Strategies and Rationales. Small Methods 2020, 4, 2000033. [Google Scholar] [CrossRef]
- Lei, Q.; Zhu, H.; Song, K.; Wei, N.; Liu, L.; Zhang, D.; Yin, J.; Dong, X.; Yao, K.; Wang, N.; et al. Investigating the Origin of Enhanced C2+ Selectivity in Oxide-/Hydroxide-Derived Copper Electrodes during CO2 Electroreduction. J. Am. Chem. Soc. 2020, 142, 4213–4222. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; He, B.; Liu, X.; Sun, J. Ionic Liquids-Promoted Electrocatalytic Reduction of Carbon Dioxide. Ind. Eng. Chem. Res. 2020, 59, 20235–20252. [Google Scholar] [CrossRef]
- Mu, S.; Li, L.; Zhao, R.; Lu, H.; Dong, H.; Cui, C. Molecular-Scale Insights into Electrochemical Reduction of CO2 on Hydrophobically Modified Cu Surfaces. ACS Appl. Mater. Interfaces 2021, 13, 47619–47628. [Google Scholar] [CrossRef]
- Wang, W.; Ma, Z.; Fei, X.; Wang, X.; Yang, Z.; Wang, Y.; Zhang, J.; Ning, H.; Tsubaki, N.; Wu, M. Joint tuning the morphology and oxygen vacancy of Cu2O by ionic liquid enables high-efficient CO2 reduction to C2 products. Chem. Eng. J. 2022, 436, 135029. [Google Scholar] [CrossRef]
- Cai, H.; Yang, H.; Feng, J.; Zhou, K.; Liu, C.; Hu, Q.; He, C. Ionic Liquid-Induced Product Switching in CO2 Electroreduction on Copper Reaction Interface. Adv. Funct. Mater. 2024, 34, 2404102. [Google Scholar] [CrossRef]
- Zhang, X.-D.; Liu, T.; Liu, C.; Zheng, D.-S.; Huang, J.-M.; Liu, Q.-W.; Yuan, W.-W.; Yin, Y.; Huang, L.-R.; Xu, M.; et al. Asymmetric Low-Frequency Pulsed Strategy Enables Ultralong CO2 Reduction Stability and Controllable Product Selectivity. J. Am. Chem. Soc. 2023, 145, 2195–2206. [Google Scholar] [CrossRef]
- Coskun, O.K.; Dongare, S.; Doherty, B.; Klemm, A.; Tuckerman, M.; Gurkan, B. Tailoring Electrochemical CO2 Reduction on Copper by Reactive Ionic Liquid and Native Hydrogen Bond Donors. Angew. Chem. Int. Ed. 2024, 63, e202312163. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, A.R.; Vidal-López, A.; Brotons-Rufes, A.; Pajski, J.J.; Zafar, S.; Mahmood, R.A.; Khan, M.U.; Poater, A.; Chawla, M.; Cavallo, L. Amino acid ionic liquids as efficient catalysts for CO2 capture: A combined static and dynamic approach. Results Surf. Interfaces 2024, 14, 100175. [Google Scholar] [CrossRef]
- Wang, R.; Qi, C.; Jian, Z.; Zhao, H.; Zhang, P.; An, S.; Li, Q.; Wang, L. Development of dual-functionalized ionic liquids for biphasic solvents: Enhancing the CO2 absorption through two-stage reaction and promoting the energy-saving regeneration. Fuel 2024, 359, 130382. [Google Scholar] [CrossRef]
- Dai, C.; Yang, Y.; Fisher, A.; Liu, Z.; Cheng, D. Interaction of CO2 with metal cluster-functionalized ionic liquids. J. CO2 Util. 2016, 16, 257–263. [Google Scholar] [CrossRef]
- Wang, Y.; Hatakeyama, M.; Ogata, K.; Wakabayashi, M.; Jin, F.; Nakamura, S. Activation of CO2 by ionic liquid EMIM–BF4 in the electrochemical system: A theoretical study. Phys. Chem. Chem. Phys. 2015, 17, 23521–23531. [Google Scholar] [CrossRef]
- Chang, Q.; Lee, J.H.; Liu, Y.; Xie, Z.; Hwang, S.; Marinkovic, N.S.; Park, A.-H.A.; Kattel, S.; Chen, J.G. Electrochemical CO2 Reduction Reaction over Cu Nanoparticles with Tunable Activity and Selectivity Mediated by Functional Groups in Polymeric Binder. JACS Au 2021, 2, 214–222. [Google Scholar] [CrossRef]
- Li, X.-Q.; Duan, G.-Y.; Chen, J.-W.; Han, L.-J.; Zhang, S.-J.; Xu, B.-H. Regulating electrochemical CO2RR selectivity at industrial current densities by structuring copper@poly(ionic liquid) interface. Appl. Catal. B Environ. 2021, 297, 120471. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- He, R.; Huang, K.; Yang, X.; Xu, J.; Tong, Z. Copper electrocatalyst modified with pyridinium-based ionic liquids for the efficient synthesis of ethylene through electrocatalytic CO2 reduction reaction. Chem. Eng. J. 2024, 502, 158067. [Google Scholar] [CrossRef]
- Sha, Y.; Zhang, J.; Cheng, X.; Xu, M.; Su, Z.; Wang, Y.; Hu, J.; Han, B.; Zheng, L. Anchoring Ionic Liquid in Copper Electrocatalyst for Improving CO2 Conversion to Ethylene. Angew. Chem. Int. Ed. 2022, 61, e202200039. [Google Scholar] [CrossRef]
- Duan, G.Y.; Li, X.Q.; Ding, G.R.; Han, L.J.; Xu, B.H.; Zhang, S.J. Highly Efficient Electrocatalytic CO2 Reduction to C2+ Products on a Poly(ionic liquid)-Based Cu0–Cu1 Tandem Catalyst. Angew. Chem. Int. Ed. 2022, 61, e202110657. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Kwon, S.; Cheng, T.; Xu, M.; Tieu, P.; Lee, C.; Cai, J.; Lee, H.M.; Pan, X.; Duan, X.; et al. Highly active and stable stepped Cu surface for enhanced electrochemical CO2 reduction to C2H4. Nat. Catal. 2020, 3, 804–812. [Google Scholar] [CrossRef]
- Zhong, D.; Zhao, Z.-J.; Zhao, Q.; Cheng, D.; Liu, B.; Zhang, G.; Deng, W.; Dong, H.; Zhang, L.; Li, J.; et al. Coupling of Cu(100) and (110) Facets Promotes Carbon Dioxide Conversion to Hydrocarbons and Alcohols. Angew. Chem. Int. Ed. 2021, 60, 4879–4885. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Raybaud, P.; Hafner, J.; Kresse, G.; Kasztelan, S.; Toulhoat, H. Structure, Energetics, and Electronic Properties of the Surface of a Promoted MoS2 Catalyst: An ab Initio Local Density Functional Study. J. Catal. 2000, 190, 128–143. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis as Projected from Plane-Wave Basis Sets. J. Phys. Chem. A 2011, 115, 5461–5466. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | IS | TS | FS | |
|---|---|---|---|---|
| Sample | ||||
| Cu | 1.999 Å | 1.972 Å | 2.025 Å | |
| Cu–IL (SH) | 1.975 Å | 1.967 Å | 2.021 Å | |
| Cu–IL (COOH) | 1.990 Å | 1.975 Å | 2.026 Å | |
| Cu–IL (CH3) | 2.016 Å | 1.979 Å | 2.029 Å | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Zhou, W.; Ma, J.; Wang, Z.; Zhang, C. Electronic Modulation of Cu Catalytic Interfaces by Functionalized Ionic Liquids for Enhanced CO2 Reduction. Molecules 2025, 30, 2352. https://doi.org/10.3390/molecules30112352
Wang C, Zhou W, Ma J, Wang Z, Zhang C. Electronic Modulation of Cu Catalytic Interfaces by Functionalized Ionic Liquids for Enhanced CO2 Reduction. Molecules. 2025; 30(11):2352. https://doi.org/10.3390/molecules30112352
Chicago/Turabian StyleWang, Chuanhui, Wei Zhou, Jiamin Ma, Zhi Wang, and Congyun Zhang. 2025. "Electronic Modulation of Cu Catalytic Interfaces by Functionalized Ionic Liquids for Enhanced CO2 Reduction" Molecules 30, no. 11: 2352. https://doi.org/10.3390/molecules30112352
APA StyleWang, C., Zhou, W., Ma, J., Wang, Z., & Zhang, C. (2025). Electronic Modulation of Cu Catalytic Interfaces by Functionalized Ionic Liquids for Enhanced CO2 Reduction. Molecules, 30(11), 2352. https://doi.org/10.3390/molecules30112352