
Identification of a Non-Retinoid Opsin Ligand Through Pharmacophore-Guided Virtual Screening—A Novel Potential Rhodopsin-Stabilizing Compound
 , , ,
, , , 
 and
and
Abstract
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Pharmacophore Model Generation
3.2. Database Generation and Pharmacophore Screening
3.3. Hierarchical Consensus Docking Analysis
3.4. Molecular Dynamics Simulations
3.5. Biological Evaluation of Identified Compounds
3.6. Binding Energy Evaluations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GPCR | G-protein-coupled receptor |
| RHO | rhodopsin |
| RP | retinitis pigmentosa |
| 11-cis-RAL | 11-cis-retinal |
| 9-cis-RAL | 9-cis-retinal |
| RPE | retinal pigment epithelium |
| MD | molecular dynamics |
| MM-PBSA | molecular mechanics/Poisson–Boltzmann surface area |
| AUC | area under the curve |
| GAFF | general Amber force field |
| PME | Particle Mesh Ewald |
| PBS | phosphate-buffered saline |
References
- Kiser, P.D.; Golczak, M.; Palczewski, K. Chemistry of the retinoid (visual) cycle. Chem. Rev. 2014, 114, 194–232. [Google Scholar] [CrossRef] [PubMed]
- Kiser, P.D.; Palczewski, K. Pathways and disease-causing alterations in visual chromophore production for vertebrate vision. J. Biol. Chem. 2021, 296, 100072. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, K.M.; Rivolta, C. Genes Associated with Retinitis Pigmentosa and Allied Diseases Are Frequently Mutated in the General Population. PLoS ONE 2012, 7, e41902. [Google Scholar] [CrossRef] [PubMed]
- Mendes, H.F.; van der Spuy, J.; Chapple, J.P.; Cheetham, M.E. Mechanisms of cell death in rhodopsin retinitis pigmentosa: Implications for therapy. Trends Mol. Med. 2005, 11, 177–185. [Google Scholar] [CrossRef]
- Sahel, J.-A.; Marazova, K.; Audo, I. Clinical Characteristics and Current Therapies for Inherited Retinal Degenerations. Cold Spring Harb. Perspect. Med. 2015, 5, a017111. [Google Scholar] [CrossRef] [PubMed]
- Diakatou, M.; Manes, G.; Bocquet, B.; Meunier, I.; Kalatzis, V. Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 20, 2542. [Google Scholar] [CrossRef] [PubMed]
- Mendes, H.F.; Cheetham, M.E. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2008, 17, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Jastrzebska, B.; Golczak, M.; Gulati, S.; Tang, H.; Seibel, W.; Li, X.; Jin, H.; Han, Y.; et al. A novel small molecule chaperone of rod opsin and its potential therapy for retinal degeneration. Nat. Commun. 2018, 9, 1976. [Google Scholar] [CrossRef] [PubMed]
- Mattle, D.; Kuhn, B.; Aebi, J.; Bedoucha, M.; Kekilli, D.; Grozinger, N.; Alker, A.; Rudolph, M.G.; Schmid, G.; Schertler, G.F.X.; et al. Ligand channel in pharmacologically stabilized rhodopsin. Proc. Natl. Acad. Sci. USA 2018, 115, 3640–3645. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Martinelli, A.; Tuccinardi, T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J. Enzyme Inhib. Med. Chem. 2016, 31, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, M.; Galati, S.; Piazza, L.; Gado, F.; Granchi, C.; Macchia, M.; Giordano, A.; Tuccinardi, T.; Poli, G. Watermelon: Setup and validation of an in silico fragment-based approach. J. Enzyme Inhib. Med. Chem. 2024, 39, 2356179. [Google Scholar] [CrossRef] [PubMed]
- Åbacka, H.; Masoni, S.; Poli, G.; Huang, P.; Gusso, F.; Granchi, C.; Minutolo, F.; Tuccinardi, T.; Hagström-Andersson, A.K.; Lindkvist-Petersson, K. SMS121, a new inhibitor of CD36, impairs fatty acid uptake and viability of acute myeloid leukemia. Sci. Rep. 2024, 14, 9104. [Google Scholar] [CrossRef] [PubMed]
- Galati, S.; Di Stefano, M.; Macchia, M.; Poli, G.; Tuccinardi, T. MolBook UNIPI─Create, Manage, Analyze, and Share Your Chemical Data for Free. J. Chem. Inf. Model. 2023, 63, 3977–3982. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Wu, B.X.; Sarna, T.; Rohrer, B.; Redmond, T.M.; Crouch, R.K. 9-cis Retinal Increased in Retina of RPE65 Knockout Mice with Decrease in Coat Pigmentation†. Photochem. Photobiol. 2006, 82, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Seidel, T.; Langer, T. Conformational Sampling of Small Molecules with iCon: Performance Assessment in Comparison with OMEGA. Front. Chem. 2018, 6, 229. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz., K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Sainas, S.; Giorgis, M.; Circosta, P.; Gaidano, V.; Bonanni, D.; Pippione, A.C.; Bagnati, R.; Passoni, A.; Qiu, Y.; Cojocaru, C.F.; et al. Targeting Acute Myelogenous Leukemia Using Potent Human Dihydroorotate Dehydrogenase Inhibitors Based on the 2-Hydroxypyrazolo[1,5-a]pyridine Scaffold: SAR of the Biphenyl Moiety. J. Med. Chem. 2021, 64, 5404–5428. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Structure | AUC ± SD |
|---|---|---|
| VS1 |  | 0.78 ± 0.15 |
| VS3 |  | 0.92 ± 0.17 |
| VS5 | 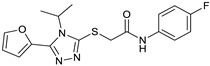 | 0.90 ± 0.10 |
| β-ionone |  | 0.73 ± 0.27 |
| 9-cis-RAL |  | 1 ± 0.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Stefano, M.; Ghilardi, M.; Poles, C.; Piazza, L.; Demontis, G.C.; Poli, G.; Tuccinardi, T.; Macchia, M. Identification of a Non-Retinoid Opsin Ligand Through Pharmacophore-Guided Virtual Screening—A Novel Potential Rhodopsin-Stabilizing Compound. Molecules 2025, 30, 2328. https://doi.org/10.3390/molecules30112328
Di Stefano M, Ghilardi M, Poles C, Piazza L, Demontis GC, Poli G, Tuccinardi T, Macchia M. Identification of a Non-Retinoid Opsin Ligand Through Pharmacophore-Guided Virtual Screening—A Novel Potential Rhodopsin-Stabilizing Compound. Molecules. 2025; 30(11):2328. https://doi.org/10.3390/molecules30112328
Chicago/Turabian StyleDi Stefano, Miriana, Maria Ghilardi, Clarissa Poles, Lisa Piazza, Gian Carlo Demontis, Giulio Poli, Tiziano Tuccinardi, and Marco Macchia. 2025. "Identification of a Non-Retinoid Opsin Ligand Through Pharmacophore-Guided Virtual Screening—A Novel Potential Rhodopsin-Stabilizing Compound" Molecules 30, no. 11: 2328. https://doi.org/10.3390/molecules30112328
APA StyleDi Stefano, M., Ghilardi, M., Poles, C., Piazza, L., Demontis, G. C., Poli, G., Tuccinardi, T., & Macchia, M. (2025). Identification of a Non-Retinoid Opsin Ligand Through Pharmacophore-Guided Virtual Screening—A Novel Potential Rhodopsin-Stabilizing Compound. Molecules, 30(11), 2328. https://doi.org/10.3390/molecules30112328