

Push-Pull OPEs in Blue-Light Anticancer Photodynamic Therapy

 ,
,  ,
,  ,
,  , ,
, ,  , and
, and 
Abstract

1. Introduction
2. Results and Discussion
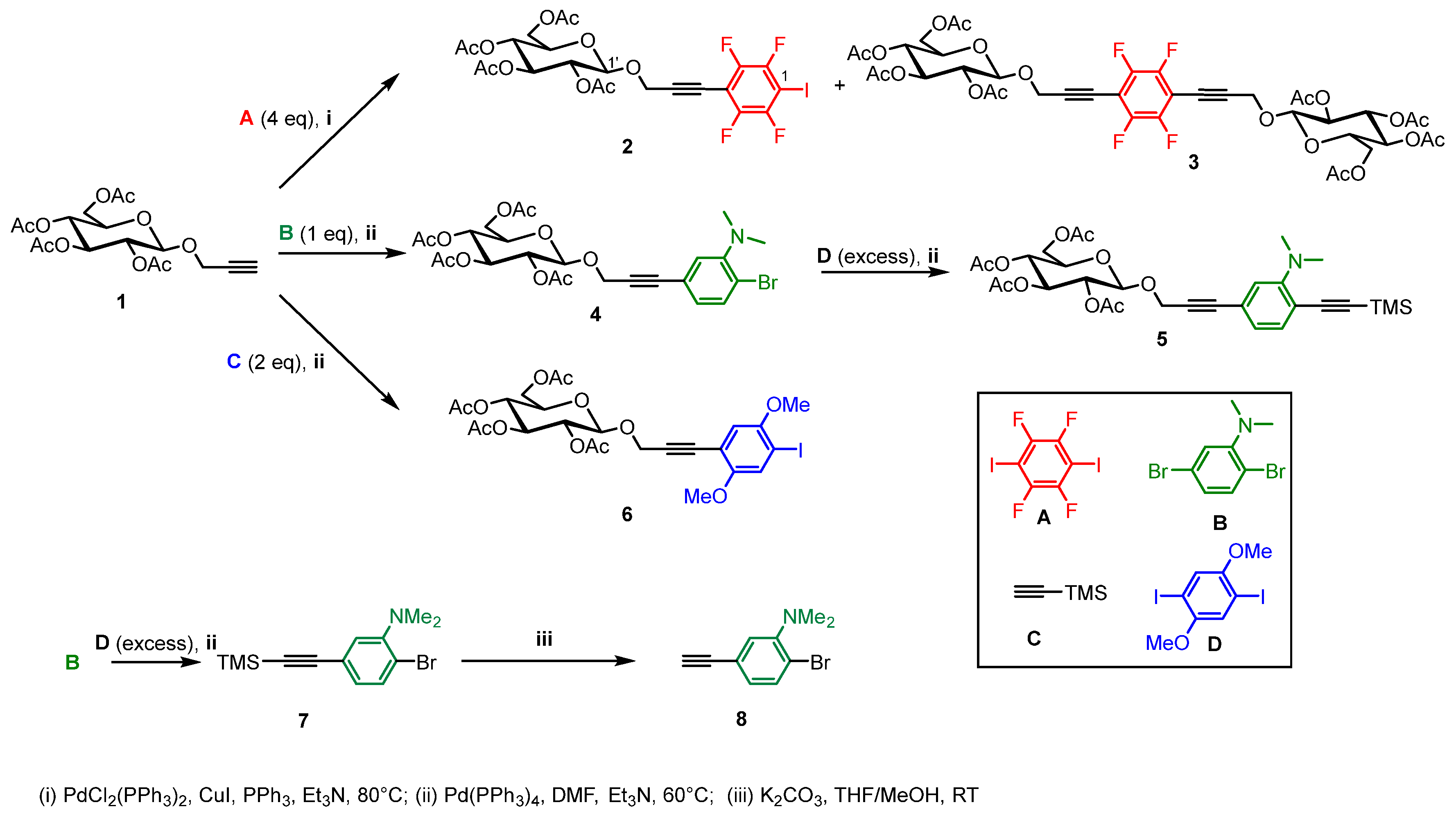
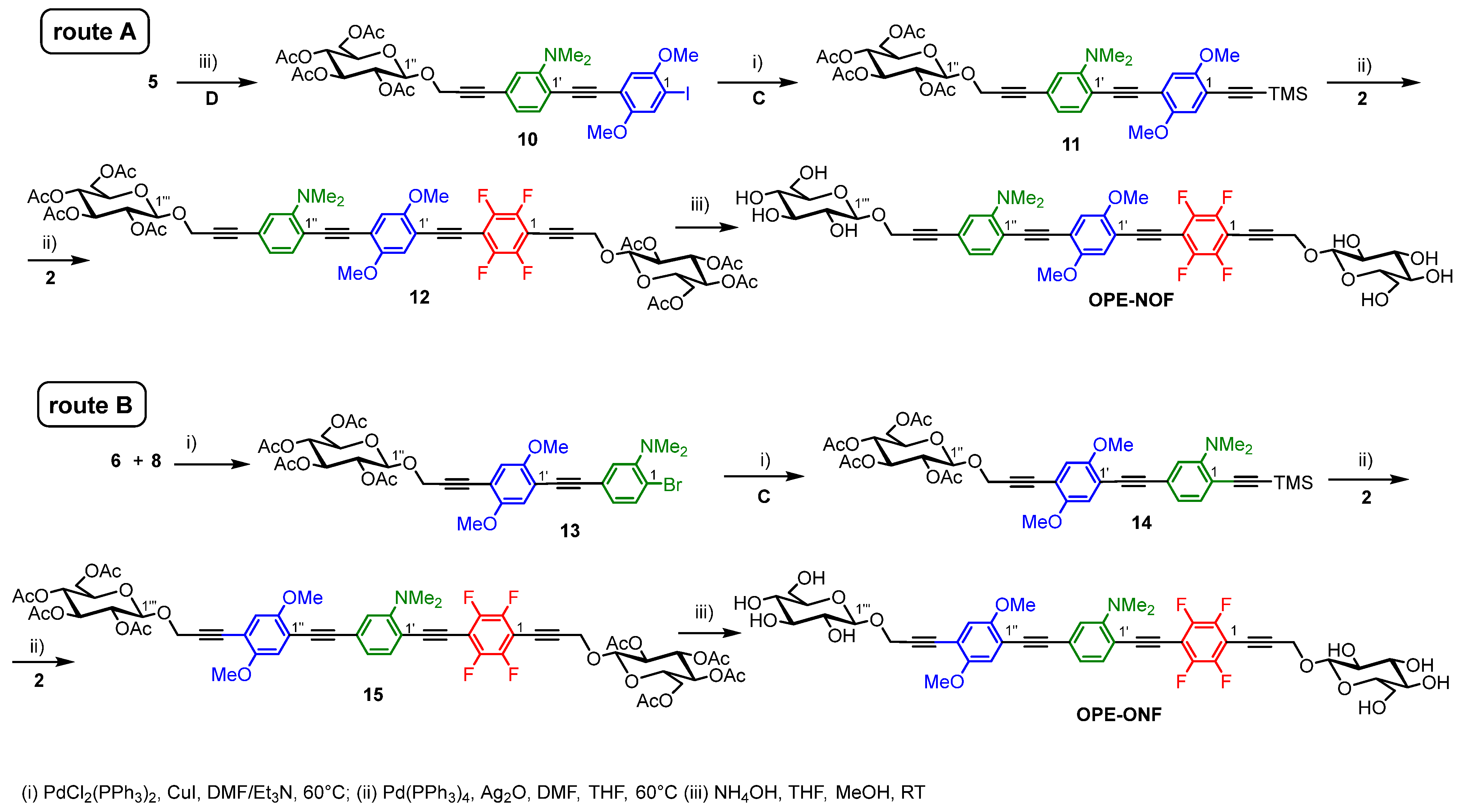
2.1. Synthesis
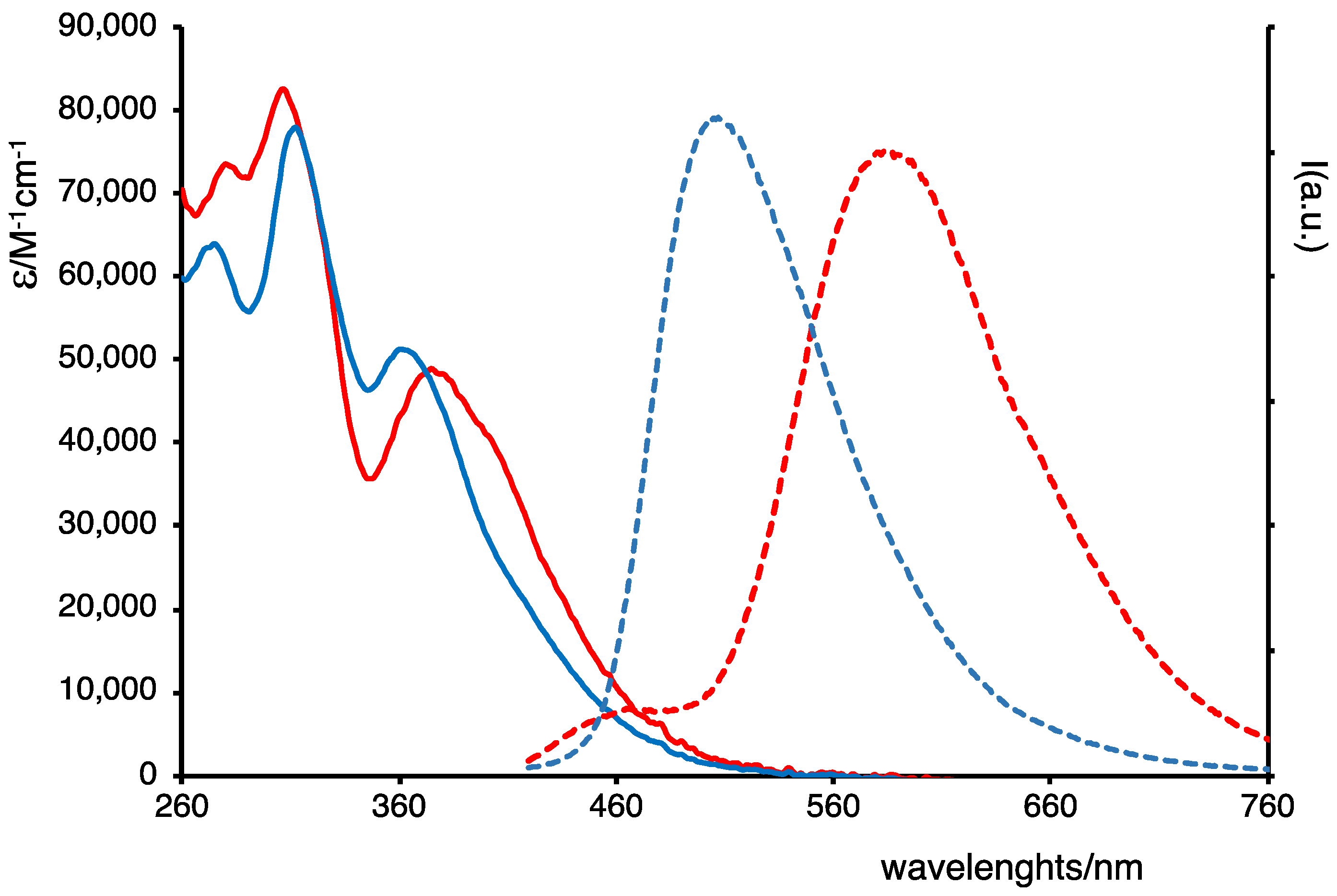
2.2. Photophysical Properties
2.3. Biological Study
2.3.1. Dark Toxicity
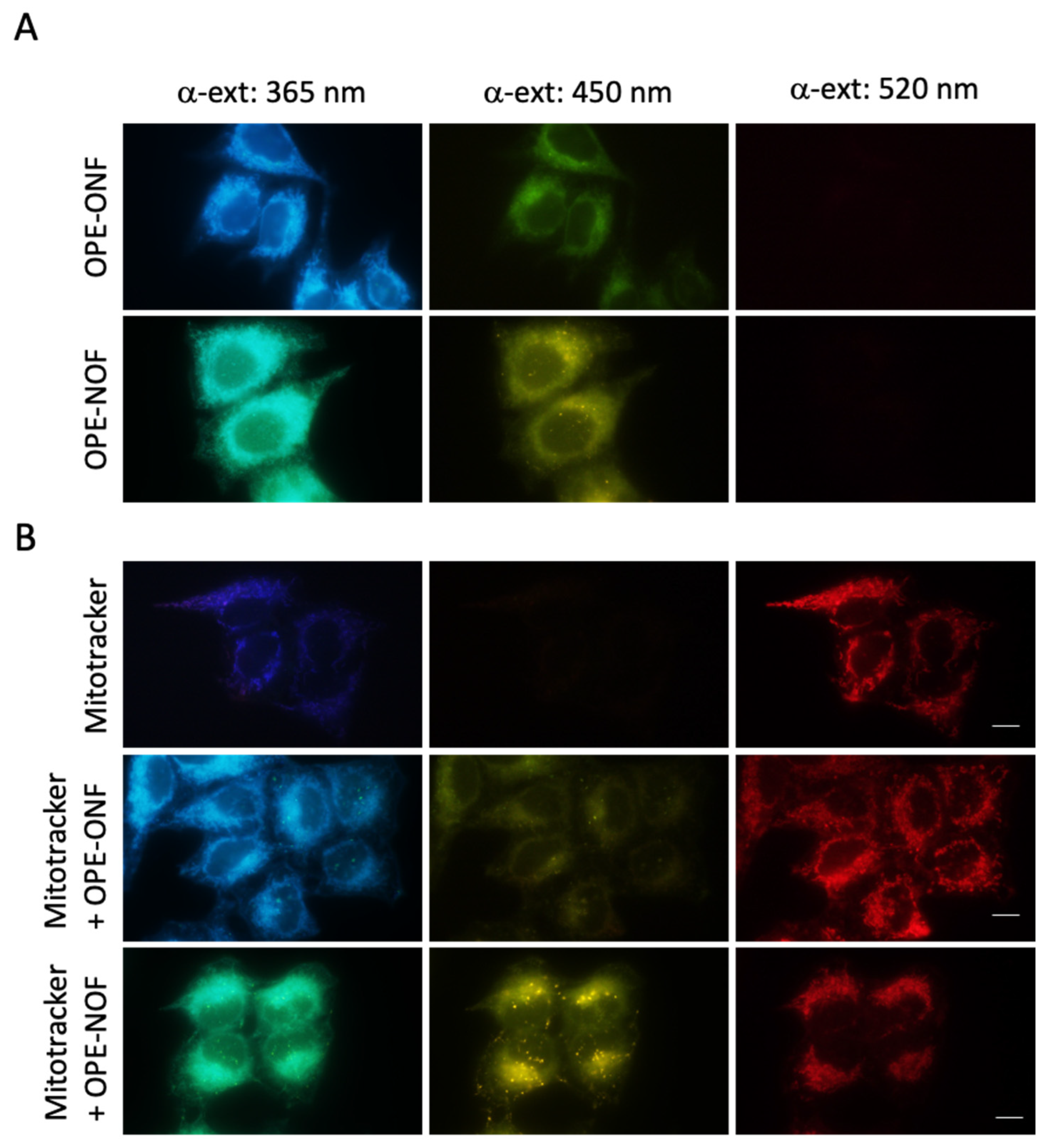
2.3.2. Subcellular Localization
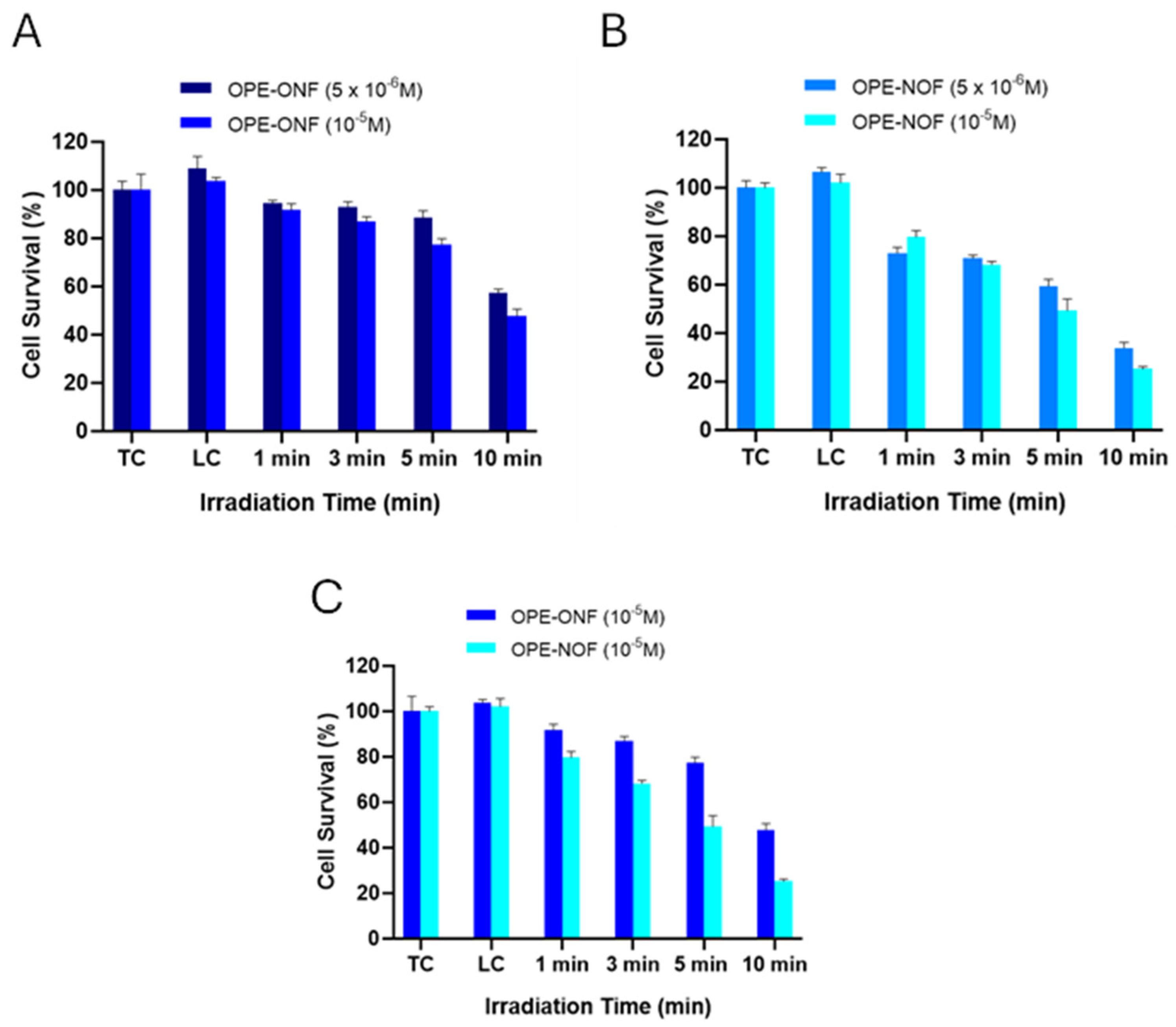
2.3.3. Photodynamic Effects of OPE-ONF and OPE-NOF
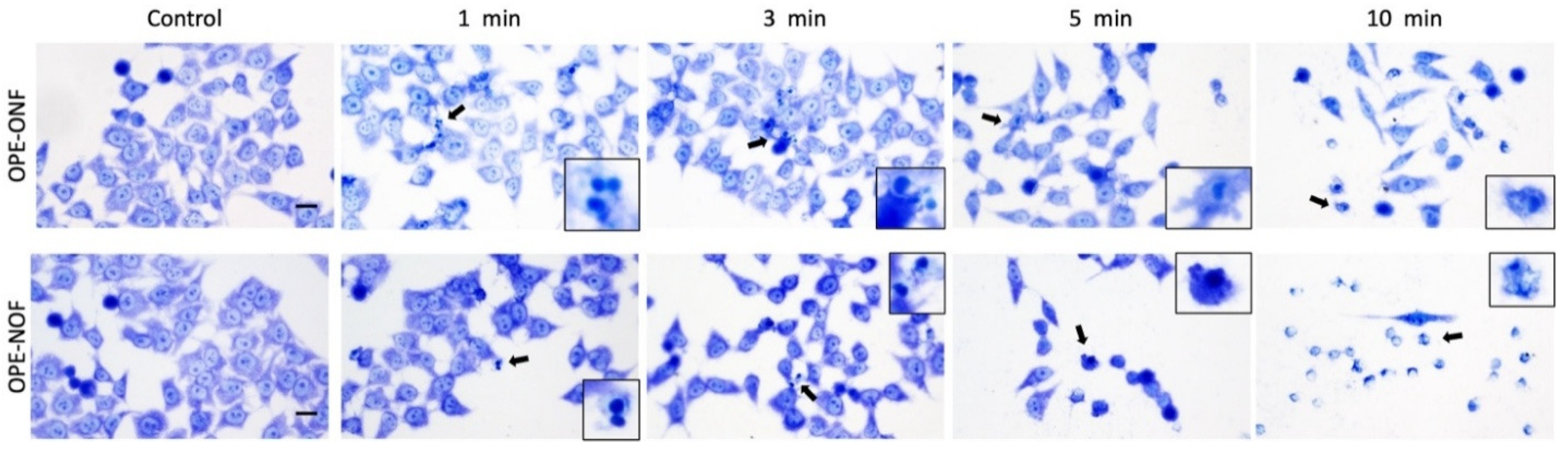
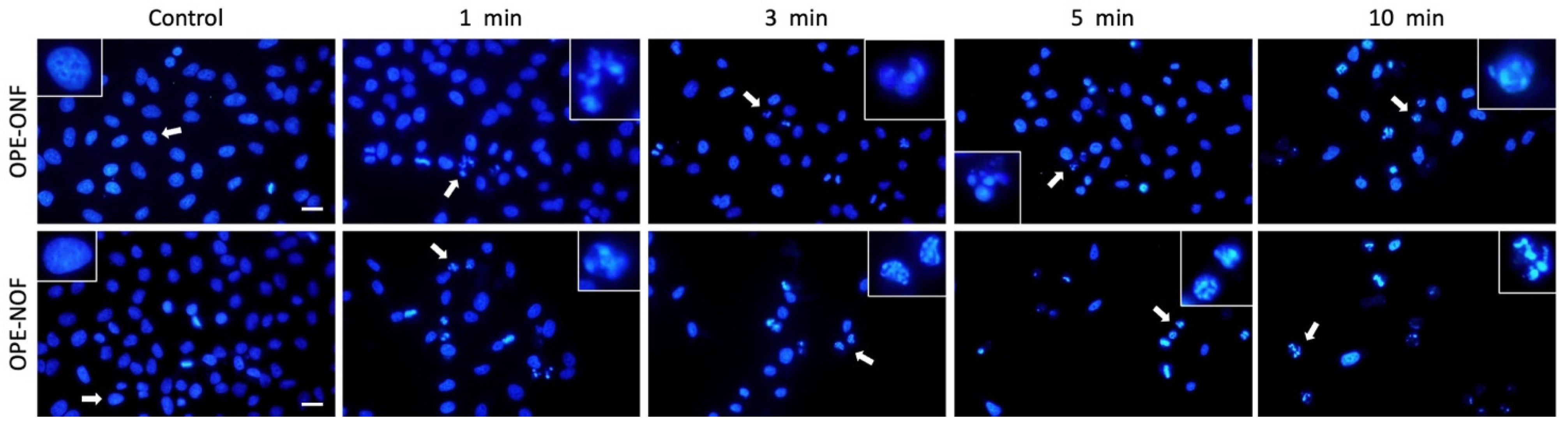
2.3.4. Morphological Evaluation
3. Experimental
3.1. Chemistry
3.2. Cell Culture
3.3. Intracellular Localization of OPE-ONF and OPE-NOF
3.4. Photodynamic Treatment In Vitro
3.5. Viability Assay
3.6. Cell Morphology
3.7. DAPI Staining
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Juarranz, A.; Jaén, P.; Sanz-Rodríguez, F.; Cuevas, J.; González, S. Photodynamic therapy of cancer. Basic principles and applications. Clin. Transl. Oncol. 2008, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Yao, Y.; Liu, Q.; Guo, Z.; Yan, C.; Zhu, W.-H. Self-adaptive photodynamic therapy for boosting therapeutic efficiency in tumor. Sci. China Chem. 2025, 2025, 1–10. [Google Scholar] [CrossRef]
- Yao, Y.; Chen, S.; Yan, C.; Wang, J.; Liu, J.; Zhu, W.-H.; Fan, C.; Guo, Z. Photo-Triggered Fluorescence Polyelectrolyte Nanoassemblies: Manipulate and Boost Singlet Oxygen in Photodynamic Therapy. Angew. Chem. Int. Ed. 2025, 64, e202416963. [Google Scholar] [CrossRef]
- Fan, W.; Huang, P.; Chen, X. Overcoming the Achilles’ heel of photodynamic therapy. Chem. Soc. Rev. 2016, 45, 6488–6519. [Google Scholar] [CrossRef]
- Filatov, M.A. Heavy-atom-free BODIPY photosensitizers with intersystem crossing mediated by intramolecular photoinduced electron transfer. Org. Biomol. Chem. 2020, 18, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Huo, Y.; Yao, G.; Feng, Y.; Weng, J.; Zhao, W.; Guo, W. Heavy Atom-Free, Mitochondria-Targeted, and Activatable Photosensitizers for Photodynamic Therapy with Real-Time In-Situ Therapeutic Monitoring. Angew. Chem. Int. Ed. 2022, 61, e202201815. [Google Scholar] [CrossRef]
- Nguyen, V.-N.; Yan, Y.; Zhao, J.; Yoon, J. Heavy-Atom-Free Photosensitizers: From Molecular Design to Applications in the Photodynamic Therapy of Cancer. Acc. Chem. Res. 2021, 54, 207–220. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Z.; Hou, Y.; Yan, Y.; Zhao, J.; Dick, B. Recent development of heavy-atom-free triplet photosensitizers: Molecular structure design, photophysics and application. J. Mater. Chem. C 2021, 9, 11944–11973. [Google Scholar] [CrossRef]
- Bureš, P. Fundamental aspects of property tuning in push–pull molecules. RSC Adv. 2014, 4, 58826–58851. [Google Scholar] [CrossRef]
- Gautam, P.; Yu, C.P.; Zhang, G.; Hillier, V.E.; Chan, J.M.W. Pulling with the pentafluorosulfanyl acceptor in push–pull dyes. J. Org. Chem. 2017, 82, 11008–11020. [Google Scholar] [CrossRef]
- Niu, X.; Gautam, P.; Kuang, Z.; Yu, C.P.; Guo, Y.; Song, H.; Guo, Q.; Chan, J.M.W.; Xia, A. Intramolecular charge transfer and solvation dynamics of push–pull dyes with different π-conjugated linkers. Phys. Chem. Chem. Phys. 2019, 21, 17323–17331. [Google Scholar] [CrossRef] [PubMed]
- Balakirev, D.O.; Solodukhin, A.N.; Peregudova, S.M.; Svidchenko, E.A.; Surin, N.M.; Fedorov, Y.V.; Ponomarenko, S.A.; Luponosov, Y.N. Luminescent push-pull triphenylamine-based molecules end-capped with various electron-withdrawing groups: Synthesis and properties. Dyes Pigm. 2023, 208, 110777. [Google Scholar] [CrossRef]
- Verbitskiy, E.V.; Gennady, L.; Rusinov, O.N.; Chupakhin, V.N. Charushin Design of fluorescent sensors based on azaheterocyclic push-pull systems towards nitroaromatic explosives and related compounds: A review. Dyes Pigm. 2020, 180, 108414. [Google Scholar] [CrossRef]
- Rosadoni, E.; Bellina, F.; Lessi, M.; Micheletti, C.; Ventura, F.; Pucci, A. Y-shaped alkynylimidazoles as effective push-pull fluorescent dyes for luminescent solar concentrators (LSCs). Dyes Pigm. 2022, 201, 110262. [Google Scholar] [CrossRef]
- Nosova, E.V.; Lipunova, G.N.; Zyryanov, G.V.; Charusin, V.N.; Chupakhin, O.N. Functionalized 1,3,5-triazine derivatives as components for photo- and electroluminescent materials. Org. Chem. Front. 2022, 9, 6646–6683. [Google Scholar] [CrossRef]
- Zhao, X.; Du, J.; Sun, W.; Fan, J.; Peng, X. Regulating charge transfer in cyanine dyes: A universal methodology for enhancing cancer phototherapeutic efficacy. Acc. Chem. Res. 2024, 57, 2582–2593. [Google Scholar] [CrossRef]
- Wang, X.; Song, Y.; Pan, G.; Han, W.; Wang, B.; Cui, L.; Ma, H.; An, H.; Xie, Z.; Xu, B.; et al. Exploiting radical-pair intersystem crossing for maximizing singlet oxygen quantum yields in pure organic fluorescent photosensitizers. Chem. Sci. 2020, 11, 10921. [Google Scholar] [CrossRef]
- Bunz, U.H.F. Poly(aryleneethynylene)s: syntheses, properties, structures, and applications. Chem. Rev. 2000, 100, 1605–1644. [Google Scholar] [CrossRef]
- Thomas, S.W., III; Joly, G.D.; Swager, T.M. Chemical sensors based on amplifying fluorescent conjugated polymers. Chem. Rev. 2007, 107, 1339–1386. [Google Scholar] [CrossRef]
- Gorenskaia, E.; Low, P.J. Methods for the analysis, interpretation, and prediction of single-molecule junction conductance behavior. Chem. Sci. 2024, 15, 9510–9556. [Google Scholar] [CrossRef]
- Kaya, K.; Khalil, M.; Chi, E.Y.; Whitten, D.G. An effective approach to the disinfection of pathogens: Cationic conjugated polyelectrolytes and oligomers. ACS Appl. Bio Mater. 2023, 6, 2916–2924. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, C.M.A.; Barattucci, A.; Bonaccorsi, P.M. A Portrait of the OPE as a biological agent. Molecules 2021, 26, 3088. [Google Scholar] [CrossRef] [PubMed]
- Meier, H.; Mühling, B.; Kolshorn, H. Red- and blue-shifts in oligo(1,4-phenylene ethynylene)s having terminal donor−acceptor substitutions. Eur. J. Org. Chem. 2004, 2004, 1033–1042. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Tanaka, T.; Kobayashi, S.; Wakamiya, T.; Matsubara, Y.; Yoshida, Z.-I. Light-emitting efficiency tuning of rod-shaped π conjugated systems by donor and acceptor groups. J. Am. Chem. Soc. 2005, 127, 9332–9333. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Ochi, T.; Matsubara, Y.; Yoshida, Z.-I. Highly emissive whole rainbow fluorophores consisting of 1,4-bis(2-phenylethynyl)benzene core skeleton: Design, synthesis, and light-emitting characteristics. J. Phys. Chem. A 2015, 119, 8630–8642. [Google Scholar] [CrossRef]
- Lara-Pardo, A.; Mancuso, A.; Simón-Fuente, S.; Bonaccorsi, P.M.; Gangemi, C.M.A.; Moliné, M.A.; Puntoriero, F.; Ribagorda, M.; Barattucci, A.; Sanz-Rodriguez, F. Amino-OPE glycosides and blue light: A powerful synergy in photodynamic therapy. Org. Biomol. Chem. 2023, 21, 386–396. [Google Scholar] [CrossRef]
- Barattucci, A.; Deni, E.; Bonaccorsi, P.M.; Ceraolo, M.G.; Papalia, T.; Santoro, A.; Sciortino, M.T.; Puntoriero, F. Oligo (phenylene ethynylene) glucosides: Modulation of cellular uptake capacity preserving light ON. J. Org. Chem. 2014, 79, 5113–5120. [Google Scholar] [CrossRef] [PubMed]
- Deni, E.; Zamarrón, A.; Bonaccorsi, P.M.; Carreño, M.C.; Juarranz, A.; Puntoriero, F.; Sciortino, M.T.; Ribagorda, M.; Barattucci, A. Glucose-functionalized amino-OPEs as biocompatible photosensitizers in PDT. Eur. J. Med. Chem. 2016, 11, 58–71. [Google Scholar] [CrossRef]
- Mancuso, A.; Barattucci, A.; Bonaccorsi, P.; Giannetto, A.; La Ganga, G.; Musarra-Pizzo, M.; Sciortino, T.M.G.; Sciortino, M.T.; Puntoriero, F.; Di Pietro, M.L. Carbohydrates and charges on oligo(phenylene ethynylenes): Towards the design of cancer bullets. Chem. Eur. J. 2018, 24, 16972–16976. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. The Sonogashira reaction: A booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef]
- Mancuso, A. Synthetic Strategies, Photophysical and First Biological Applications of New Glycoamino OPEs. Ph.D. Thesis, University of Messina, Messina, Italy, 2019. Available online: https://hdl.handle.net/11570/3147364 (accessed on 21 May 2025).
- Giovenzana, G.B.; Monti, D.; Palmisano, G.; Panza, L. Synthesis of carboranyl derivatives of alkynyl glycosides as potential BNCT agents. Tetrahedron 1999, 55, 14123–14136. [Google Scholar] [CrossRef]
- Mancuso, A.; Massaro, M.; Federica Leone, F.; Bonaccorsi, P.M.; Compagnini, G.; Gangemi, C.M.A.; Puntoriero, F.; Ribagorda, M.; Scardaci, V.; Viseras, C.; et al. Glucosyl OPE-modified halloysite nanotubes and their potential as phototherapy agents for bacterial infections. Surf. Interf. 2025, 62, 106207. [Google Scholar] [CrossRef]
- Mori, A.; Kondo, T.; Kato, T.; Nishihara, Y. Palladium-catalyzed cross-coupling polycondensation of bisalkynes with dihaloarenes activated by tetrabutylammonium hydroxide or silver (I) oxide. Chem. Lett. 2001, 30, 286–287. [Google Scholar] [CrossRef]
- Moan, J.; Berg, K. The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochem. Photobiol. 1991, 53, 549–553. [Google Scholar] [CrossRef]
- Ji, Z.; Yang, G.; Vasovic, V.; Cunderlikova, B.; Suo, Z.; Nesland, J.M.; Peng, Q. Subcellular localization pattern of protoporphyrin IX is an important determinant for its photodynamic efficiency of human carcinoma and normal cell lines. J. Photochem. Photobiol. B: Biol. 2006, 84, 213–220. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HeLa | ||
|---|---|---|
| [PS] | OPE-ONF | OPE-NOF |
| Control | 100 ± 3.2 | 100 ± 0.88 |
| DMSO | 94.60 ± 3.26 * | 94.60 ± 3.26 * |
| 10−6 M | 103.16 ± 5.65 | 101.29 ± 4.95 |
| 2.5 × 10−6 M | 102.33 ± 1.78 | 100.67 ± 4.78 |
| 5 × 10−6 M | 102.89 ± 2.00 | 100.69 ± 5.72 |
| 10−5 M | 104.32 ± 2.44 | 100.97 ± 6.66 |
| 2 × 10−5 M | 96.14 ± 0.97 | 86.05 ± 2.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lameiro, A.; Gangemi, C.M.A.; Mancuso, A.; Bonaccorsi, P.M.; Di Pietro, M.L.; Gómez-Pastor, S.; Puntoriero, F.; Sanz-Rodríguez, F.; Barattucci, A. Push-Pull OPEs in Blue-Light Anticancer Photodynamic Therapy. Molecules 2025, 30, 2310. https://doi.org/10.3390/molecules30112310
Lameiro A, Gangemi CMA, Mancuso A, Bonaccorsi PM, Di Pietro ML, Gómez-Pastor S, Puntoriero F, Sanz-Rodríguez F, Barattucci A. Push-Pull OPEs in Blue-Light Anticancer Photodynamic Therapy. Molecules. 2025; 30(11):2310. https://doi.org/10.3390/molecules30112310
Chicago/Turabian StyleLameiro, Ana, Chiara M. A. Gangemi, Aurora Mancuso, Paola Maria Bonaccorsi, Maria Letizia Di Pietro, Silvia Gómez-Pastor, Fausto Puntoriero, Francisco Sanz-Rodríguez, and Anna Barattucci. 2025. "Push-Pull OPEs in Blue-Light Anticancer Photodynamic Therapy" Molecules 30, no. 11: 2310. https://doi.org/10.3390/molecules30112310
APA StyleLameiro, A., Gangemi, C. M. A., Mancuso, A., Bonaccorsi, P. M., Di Pietro, M. L., Gómez-Pastor, S., Puntoriero, F., Sanz-Rodríguez, F., & Barattucci, A. (2025). Push-Pull OPEs in Blue-Light Anticancer Photodynamic Therapy. Molecules, 30(11), 2310. https://doi.org/10.3390/molecules30112310