

Recent Progress in Heteroatom-Containing Metalloporphyrin-Based Catalysts for CO2 Reduction


Abstract

1. Introduction
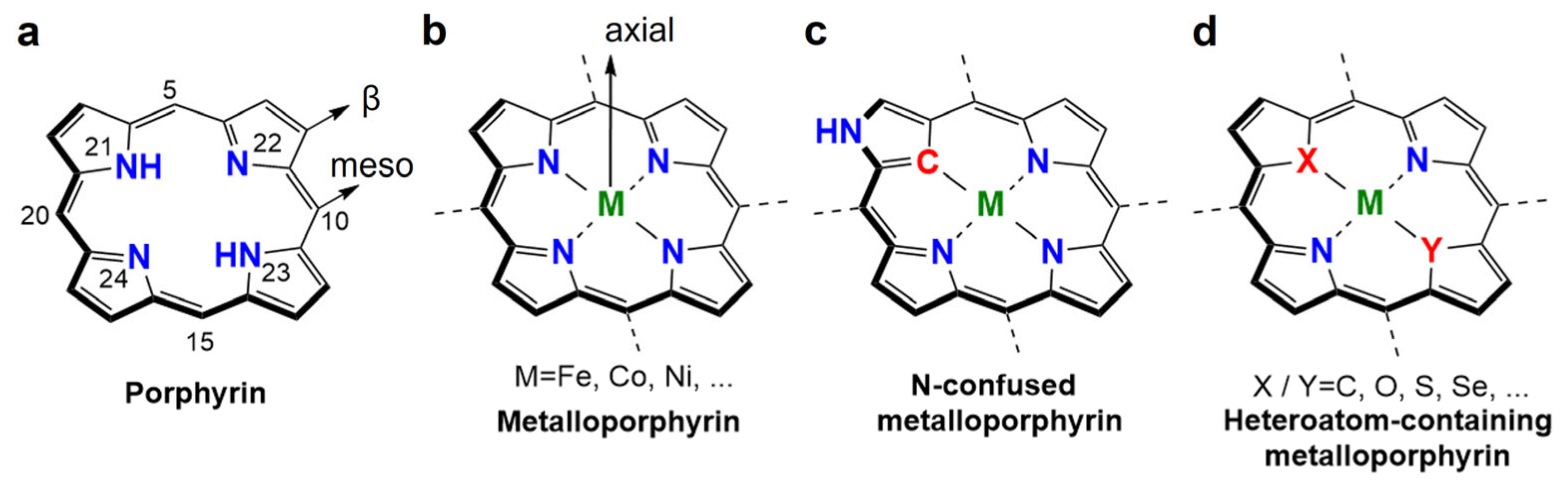
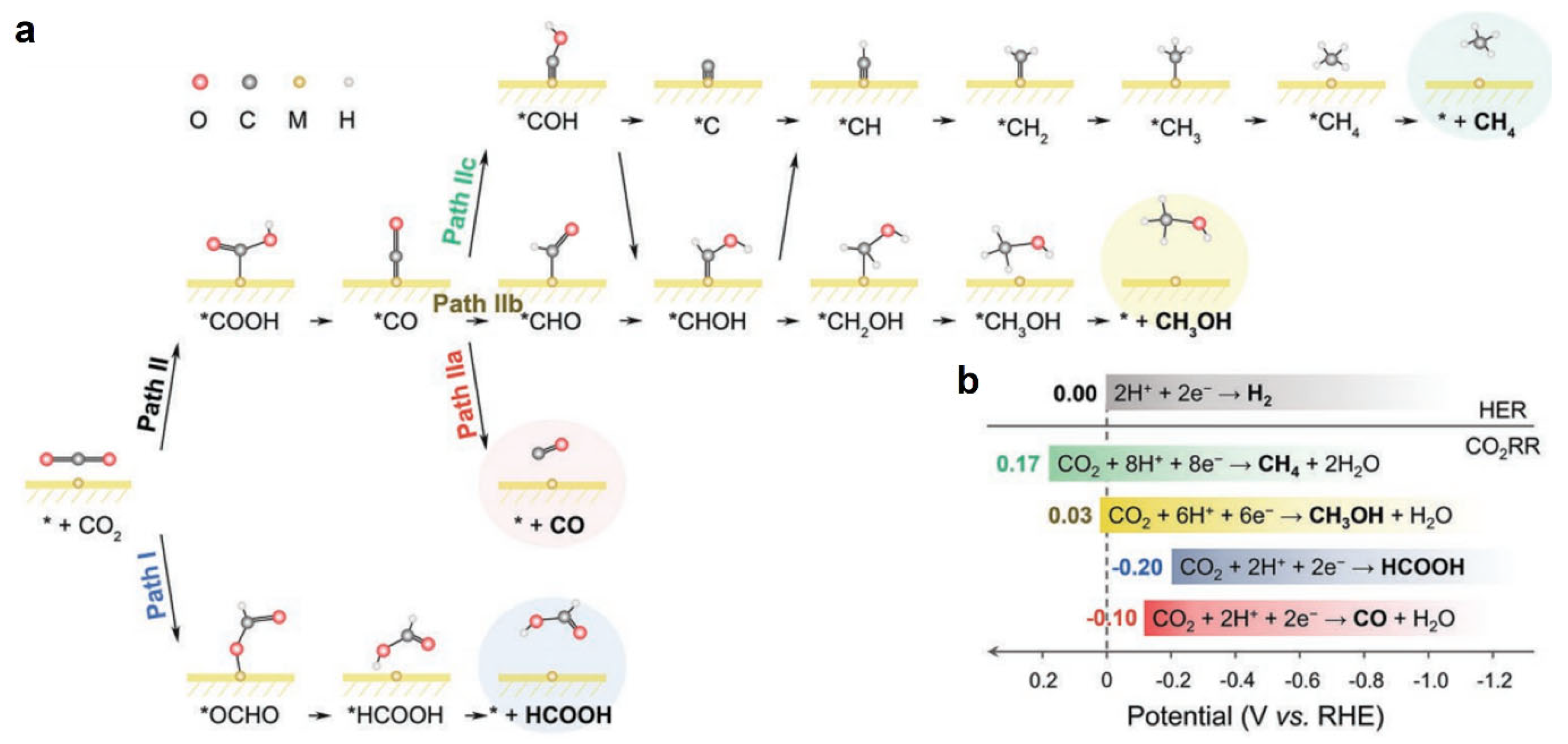
2. Metalloporphyrins for CO2RR
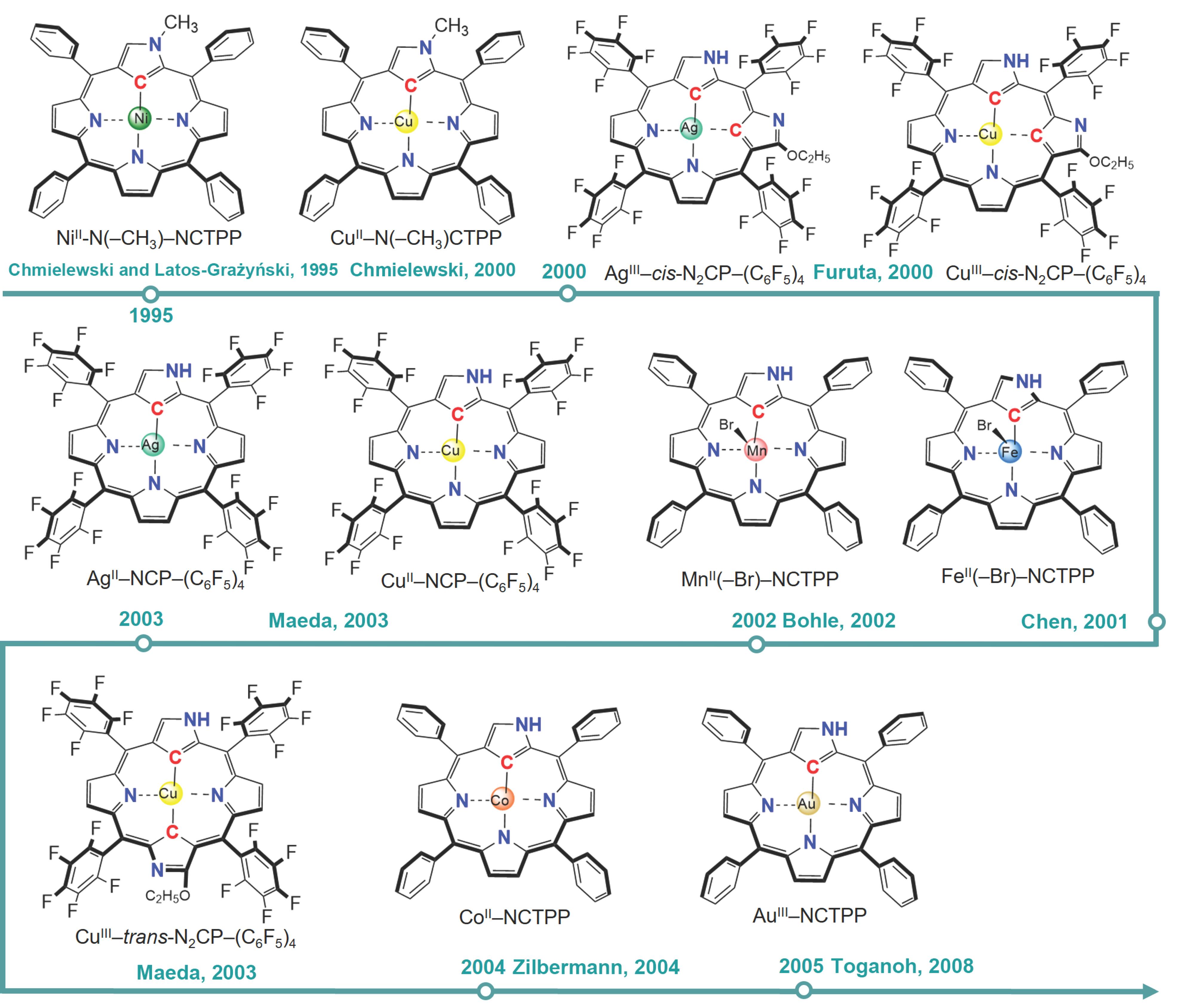
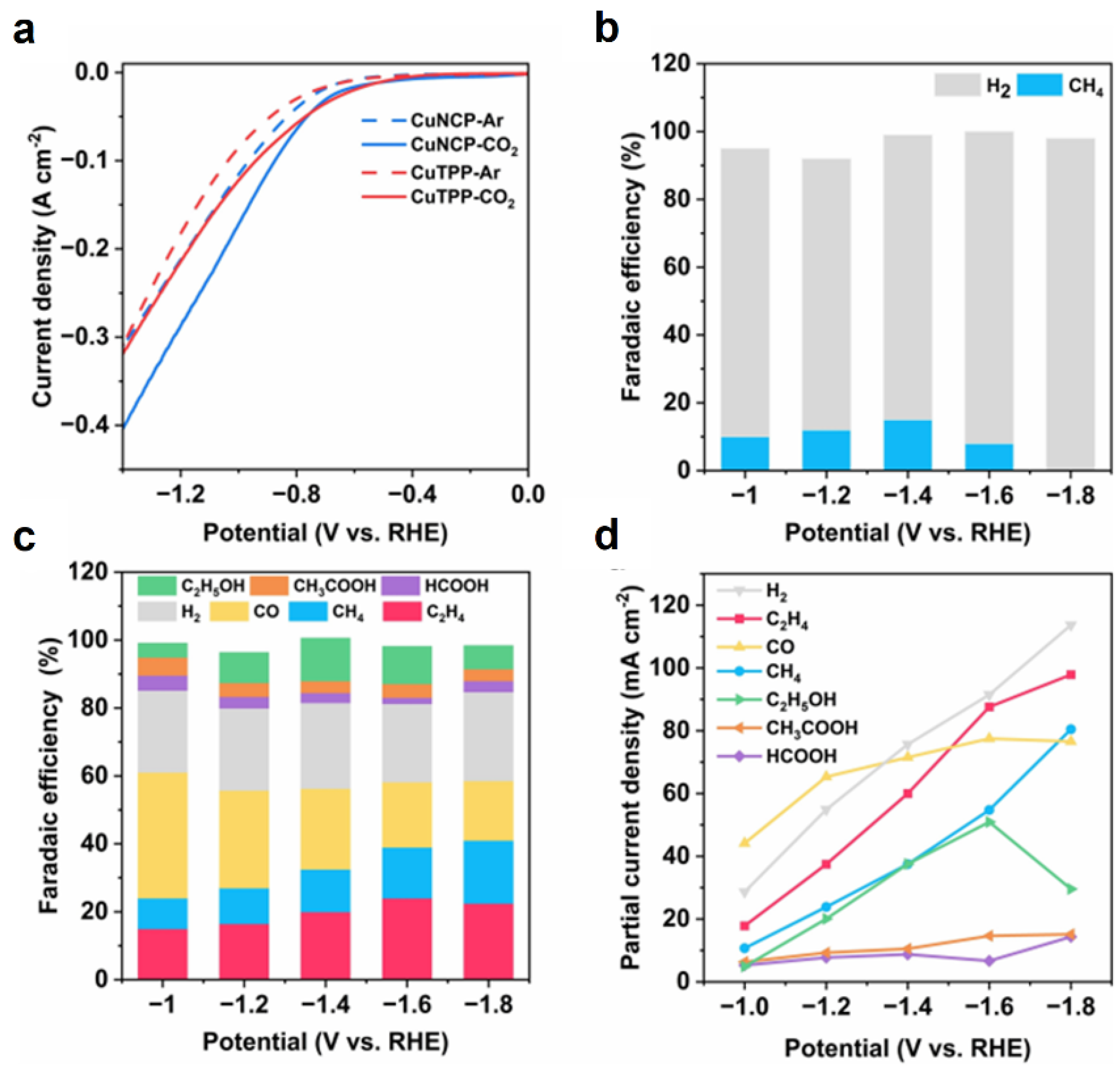
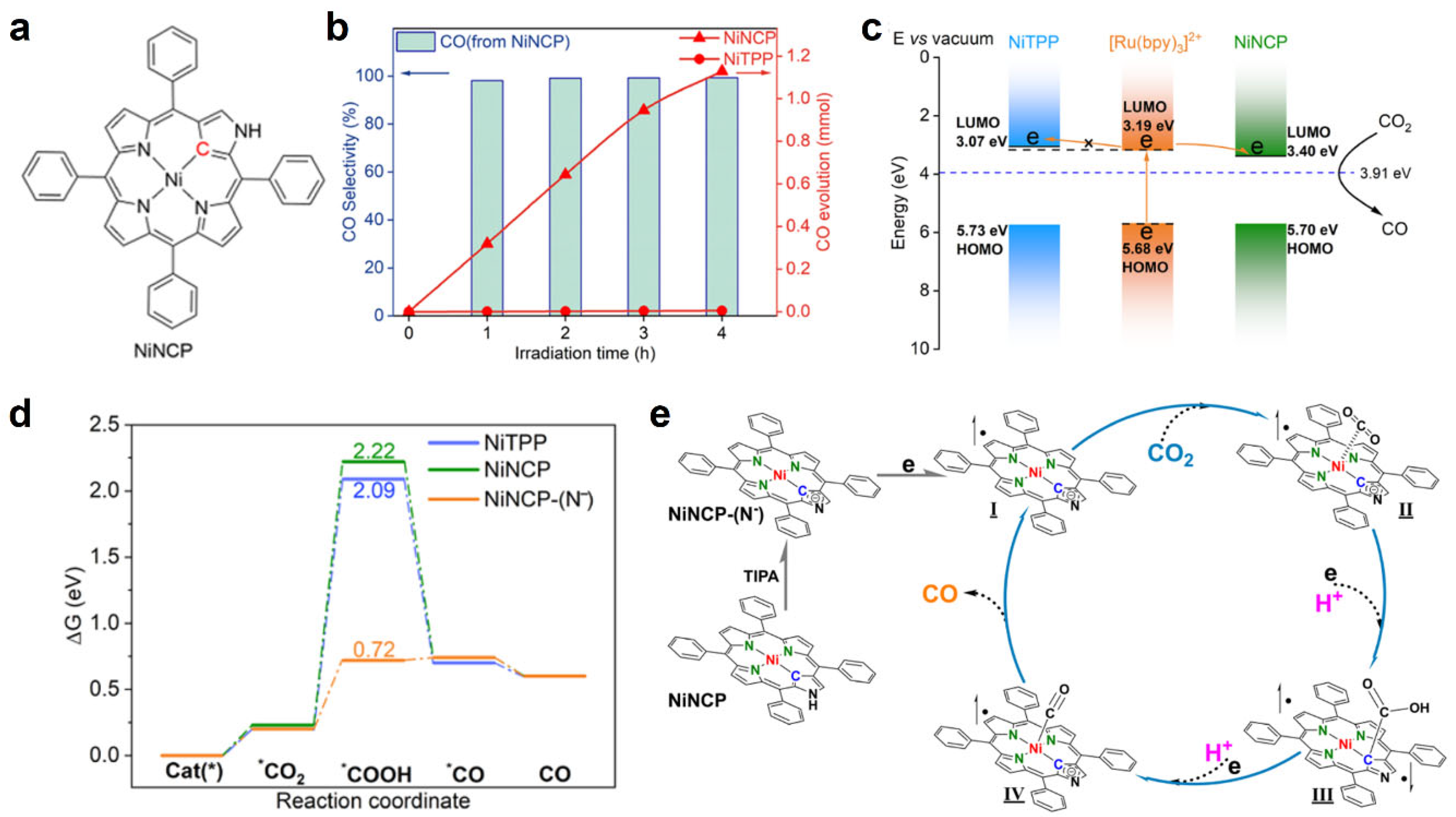
3. N-Confused Metalloporphyrin
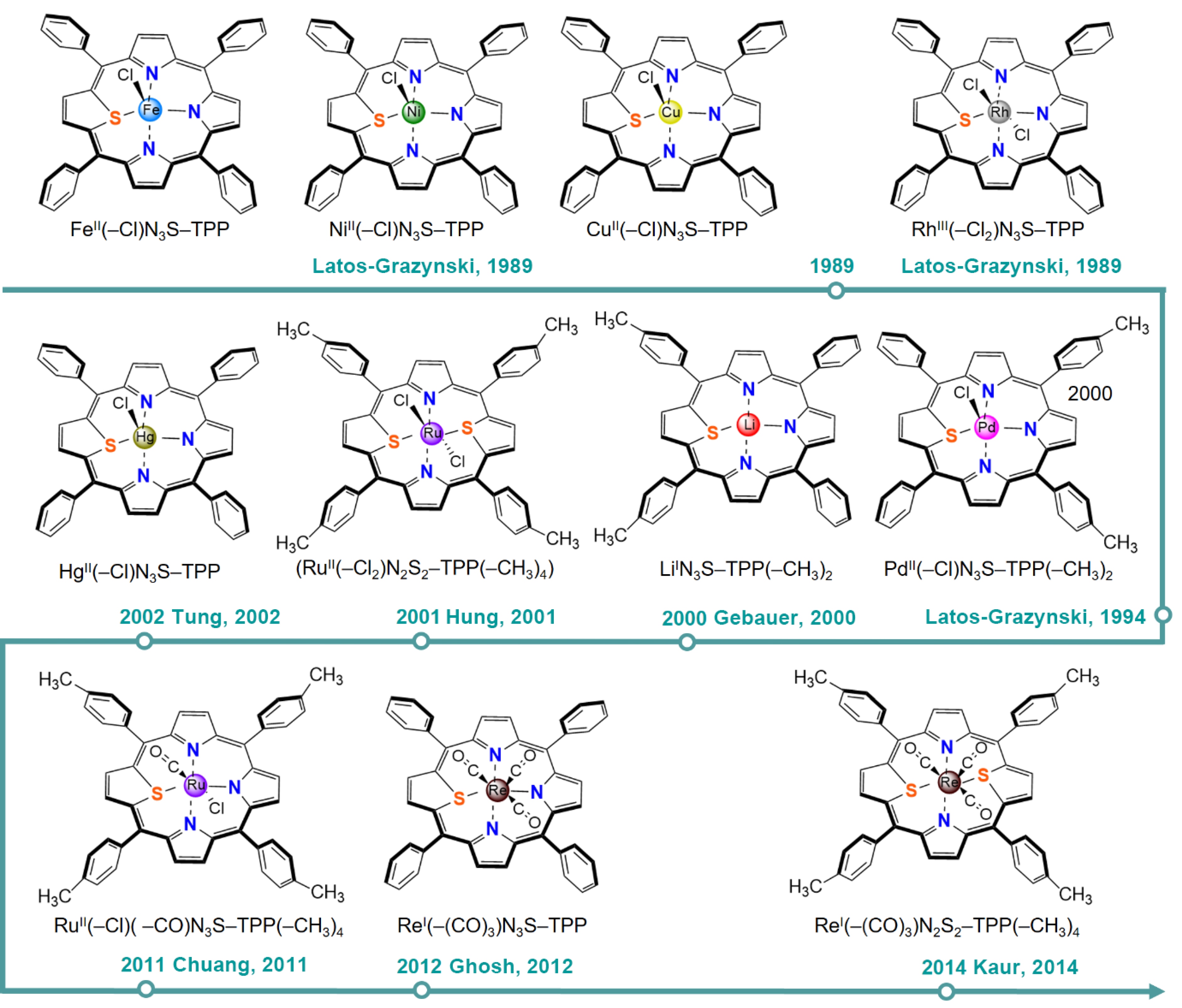
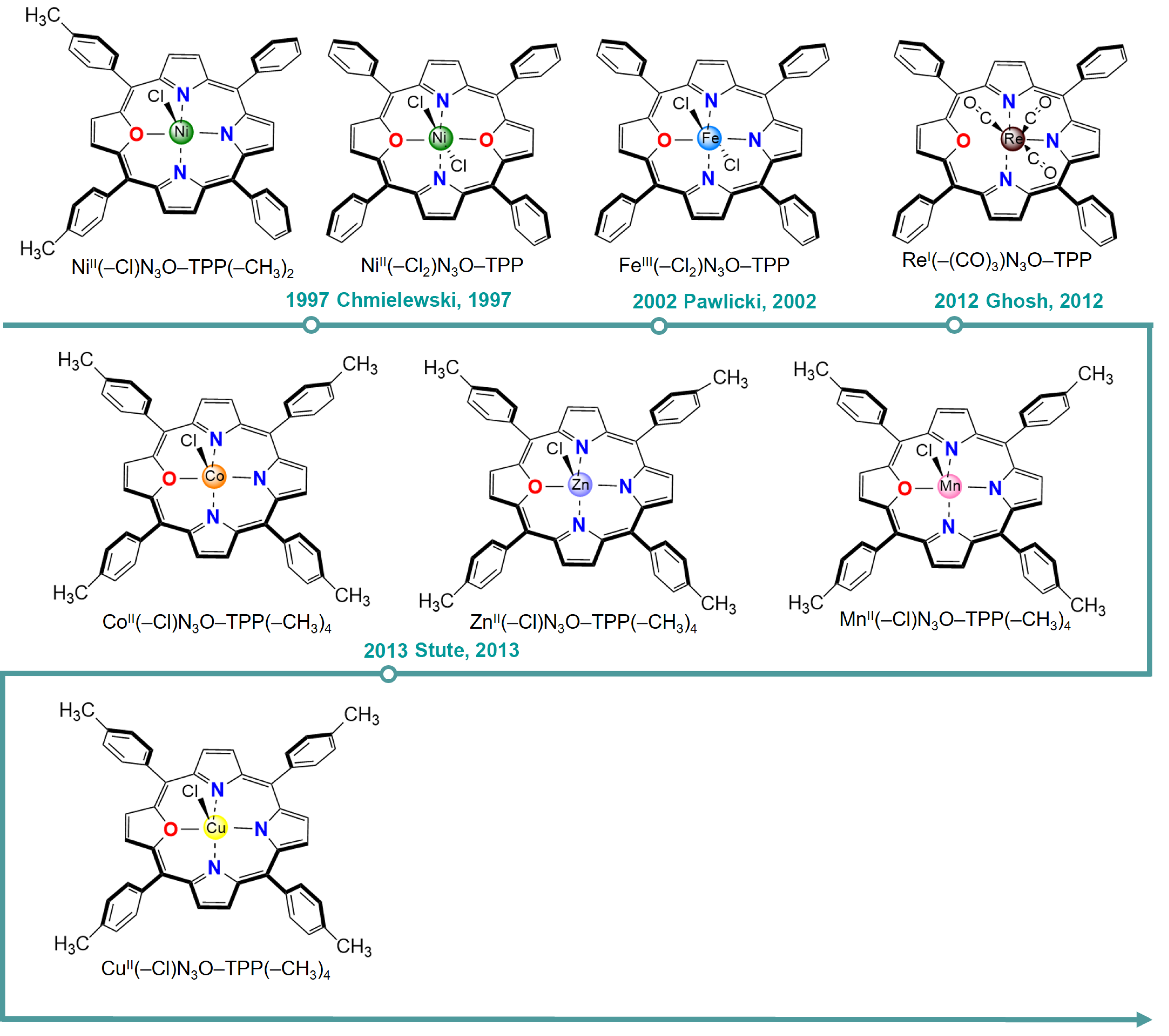
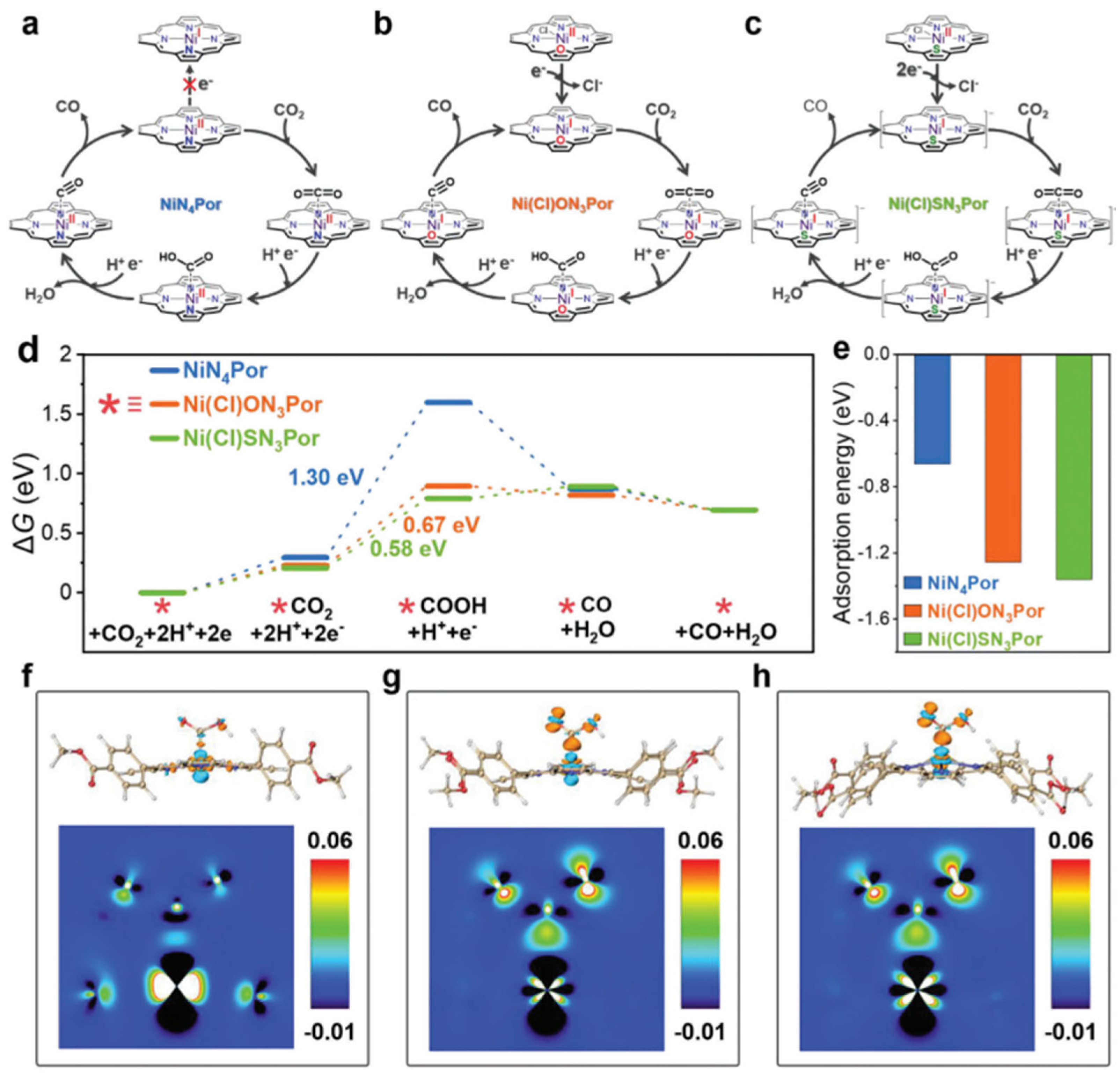
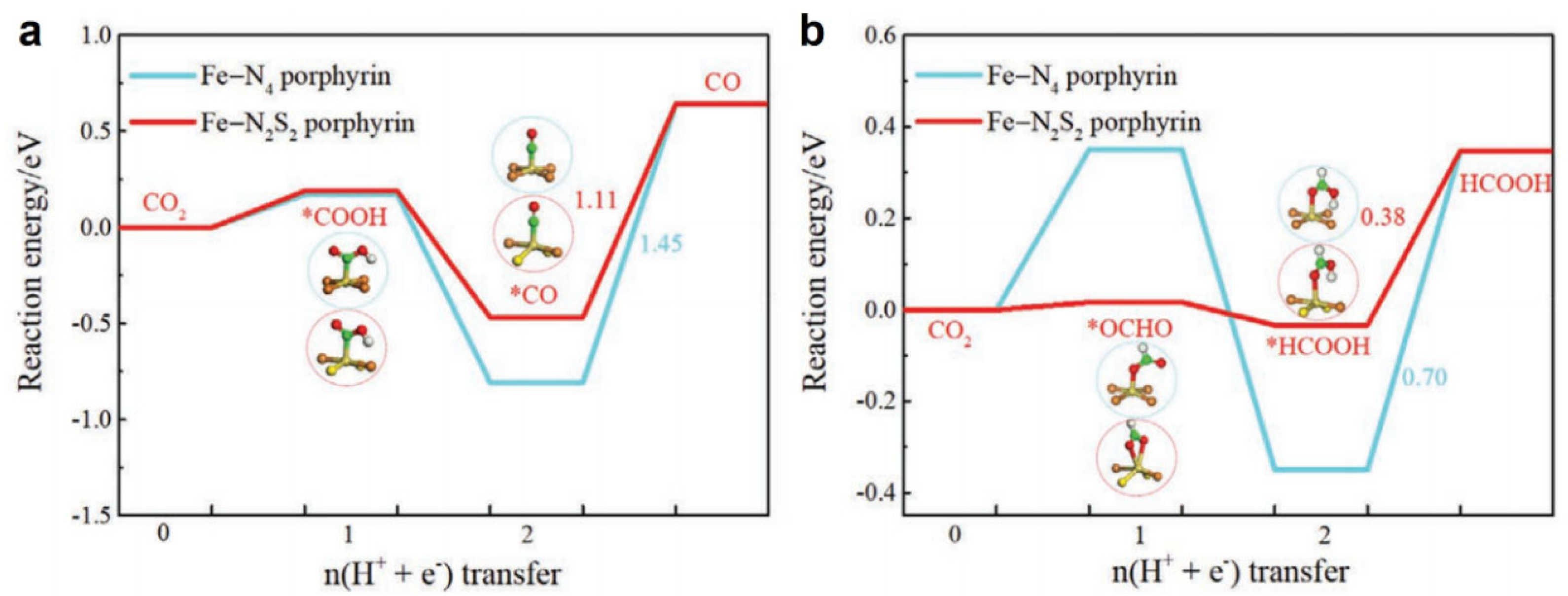
4. O/S-Substituted Metalloporphyrin
5. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chu, S.; Majumdar, A. Opportunities and Challenges for a Sustainable Energy Future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Callahan, C.W. Present and Future Limits to Climate Change Adaptation. Nat. Sustain. 2025, 8, 336–342. [Google Scholar] [CrossRef]
- Suri, D.; Das, S.; Choudhary, S.; Venkanna, G.; Sharma, B.; Afroz, M.A.; Tailor, N.K.; Joshi, R.; Satapathi, S.; Tripathi, K. Enigma of Sustainable CO2 Conversion to Renewable Fuels and Chemicals Through Photocatalysis, Electrocatalysis, and Photoelectrocatalysis: Design Strategies and Atomic Level Insights. Small 2025, 21, 2408981. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A Review of Catalysts for the Electroreduction of Carbon Dioxide to Produce Low-Carbon Fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef]
- Birdja, Y.Y.; Pérez-Gallent, E.; Figueiredo, M.C.; Göttle, A.J.; Calle-Vallejo, F.; Koper, M.T.M. Advances and Challenges in Understanding the Electrocatalytic Conversion of Carbon Dioxide to Fuels. Nat. Energy 2019, 4, 732–745. [Google Scholar] [CrossRef]
- Jin, S.; Hao, Z.; Zhang, K.; Yan, Z.; Chen, J. Advances and Challenges for the Electrochemical Reduction of CO2 to CO: From Fundamentals to Industrialization. Angew. Chem. Int. Ed. 2021, 60, 20627–20648. [Google Scholar] [CrossRef]
- She, X.; Wang, Y.; Xu, H.; Chi Edman Tsang, S.; Ping Lau, S. Challenges and Opportunities in Electrocatalytic CO2 Reduction to Chemicals and Fuels. Angew. Chem. Int. Ed. 2022, 61, e202211396. [Google Scholar] [CrossRef]
- Grim, R.G.; Huang, Z.; Guarnieri, M.T.; Ferrell, J.R.; Tao, L.; Schaidle, J.A. Transforming the Carbon Economy: Challenges and Opportunities in the Convergence of Low-Cost Electricity and Reductive CO2 Utilization. Energy Environ. Sci. 2020, 13, 472–494. [Google Scholar] [CrossRef]
- Xia, J.; Wang, B.; Di, J.; Li, Y.; Yang, S.-Z.; Li, H.; Guo, S. Construction of Single-Atom Catalysts for Electro-, Photo- and Photoelectro-Catalytic Applications: State-of-the-Art, Opportunities, and Challenges. Mater. Today 2022, 53, 217–237. [Google Scholar] [CrossRef]
- Ali, S.A.; Sadiq, I.; Estrader, M.; Ahmad, T. Atomic-Level Tuning Strategies in Designing Active Catalysts for Heterogeneous CO2 Conversion into Chemical Feedstock. ChemCatChem 2025, 17, e202500032. [Google Scholar] [CrossRef]
- Zhong, S.; Bird, A.; Kopec, R.E. The Metabolism and Potential Bioactivity of Chlorophyll and Metallo-chlorophyll Derivatives in the Gastrointestinal Tract. Mol. Nutr. Food Res. 2021, 65, e2000761. [Google Scholar] [CrossRef] [PubMed]
- Osman, D.; Cooke, A.; Young, T.R.; Deery, E.; Robinson, N.J.; Warren, M.J. The Requirement for Cobalt in Vitamin B12: A Paradigm for Protein Metalation. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2021, 1868, 118896. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, N.; Lei, H.; Li, X.; Zheng, H.; Wang, H.; Zhang, W.; Cao, R. Bioinspired N4-Metallomacrocycles for Electrocatalytic Oxygen Reduction Reaction. Coord. Chem. Rev. 2021, 442, 213996. [Google Scholar] [CrossRef]
- Zhang, R.; Warren, J.J. Recent Developments in Metalloporphyrin Electrocatalysts for Reduction of Small Molecules: Strategies for Managing Electron and Proton Transfer Reactions. ChemSusChem 2021, 14, 293–302. [Google Scholar] [CrossRef]
- Lucas, D.Z.; Isaltino, A.B.; Paulo, C.d.S.F.; Fabricio, B.Z.; Lucas, B.B.; Osvaldo, A.S.; Yassuko, I. Metalloporphyrins in Drug and Pesticide Catalysis as Powerful Tools to Elucidate Biotransformation Mechanisms. Mini-Rev. Org. Chem. 2016, 13, 281–288. [Google Scholar] [CrossRef]
- Bonin, J.; Maurin, A.; Robert, M. Molecular Catalysis of the Electrochemical and Photochemical Reduction of CO2 with Fe and Co Metal Based Complexes. Recent Advances. Coord. Chem. Rev. 2017, 334, 184–198. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and Perspective of Electrocatalytic CO2 Reduction for Renewable Carbonaceous Fuels and Chemicals. Adv. Sci. 2017, 5, 1700275. [Google Scholar] [CrossRef]
- Costentin, C.; Passard, G.; Robert, M.; Savéant, J.M. Ultraefficient Homogeneous Catalyst for the CO2-to-CO Electrochemical Conversion. Proc. Natl. Acad. Sci. USA 2014, 111, 14990. [Google Scholar] [CrossRef]
- Abdinejad, M.; Wilm, L.F.B.; Dielmann, F.; Kraatz, H.B. Electroreduction of CO2 Catalyzed by Nickel Imidazolin-2-ylidenamino-Porphyrins in Both Heterogeneous and Homogeneous Molecular Systems. ACS Sustain. Chem. Eng. 2020, 9, 521–530. [Google Scholar] [CrossRef]
- Lin, S.; Diercks, C.S.; Zhang, Y.-B.; Kornienko, N.; Nichols, E.M.; Zhao, Y.; Paris, A.R.; Kim, D.; Yang, P.; Yaghi, O.M.; et al. Covalent Organic Frameworks Comprising Cobalt Porphyrins for Catalytic CO2 Reduction in Water. Science 2015, 349, 1208–1213. [Google Scholar] [CrossRef]
- Kornienko, N.; Zhao, Y.; Kley, C.S.; Zhu, C.; Kim, D.; Lin, S.; Chang, C.J.; Yaghi, O.M.; Yang, P. Metal–Organic Frameworks for Electrocatalytic Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2015, 137, 14129–14135. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Xie, R.-K.; Mao, M.-J.; Chai, G.-L.; Yi, J.-D.; Zhao, S.-S.; Huang, Y.-B.; Cao, R. Integration of Strong Electron Transporter Tetrathiafulvalene into Metalloporphyrin-Based Covalent Organic Framework for Highly Efficient Electroreduction of CO2. ACS Energy Lett. 2020, 5, 1005–1012. [Google Scholar] [CrossRef]
- Gao, F.; Wu, Y.-P.; Wu, X.-Q.; Li, D.-S.; Yang, G.; Wang, Y.-Y. Transition-Metal Porphyrin-Based MOFs In Situ-Derived Hybrid Catalysts for Electrocatalytic CO2 Reduction. Inorg. Chem. 2024, 63, 8948–8957. [Google Scholar] [CrossRef]
- Younis, S.A.; Lim, D.-K.; Kim, K.-H.; Deep, A. Metalloporphyrinic metal-organic frameworks: Controlled synthesis for catalytic applications in environmental and biological media. Adv. Colloid Interface Sci. 2020, 277, 102108. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Diao, Y.; Qin, X.; Xu, Z.; Ke, H.; Zhu, X. Donor–Acceptor Covalent Organic Frameworks of Nickel(II) Porphyrin for Selective and Efficient CO2 Reduction into CO. Dalton Trans. 2020, 49, 15587–15591. [Google Scholar] [CrossRef]
- Domingo-Tafalla, B.; Chatterjee, T.; Palomares, E. Recent Advances in the Rational Designing of Metalloporphyrinoid-Based CO2 Reduction Catalysts: From Molecular Structural Tuning to the Application in Catalysis. J. Porphyr. Phthalocyanines 2023, 27, 23–46. [Google Scholar] [CrossRef]
- Kim, H.; Shin, D.; Yang, W.; Won, D.H.; Oh, H.-S.; Chung, M.W.; Jeong, D.; Kim, S.H.; Chae, K.H.; Ryu, J.Y.; et al. Identification of Single-Atom Ni Site Active toward Electrochemical CO2 Conversion to CO. J. Am. Chem. Soc. 2021, 143, 925–933. [Google Scholar] [CrossRef]
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) The Porphyrin Handbook; Academic Press: New York, NY, USA, 2000; Volume 1. [Google Scholar]
- Kuznetsov, A.E. Core-modified porphyrins: Novel building blocks in chemistry. Phys. Sci. Rev. 2023, 8, 1513–1543. [Google Scholar] [CrossRef]
- Yong, C.; Song, C.; Lu, G.; Wang, X.; Song, G.; Shi, G.; Wang, Y.; Xie, X.; Sun, J. Effect of Axial Ligands on Photocatalytic CO2 Reduction of Zirconium-Based Porphyrins. ACS Appl. Energy Mater. 2025, 8, 4379–4386. [Google Scholar] [CrossRef]
- He, H.; Qiu, Z.-Y.; Yin, Z.; Kong, J.; Dang, J.-S.; Lei, H.; Zhang, W.; Cao, R. The meso-substituent electronic effect of Fe porphyrins on the electrocatalytic CO2 reduction reaction. Chem. Commun. 2024, 60, 5916–5919. [Google Scholar] [CrossRef]
- Sonea, A.; Warren, J.J. Pendant Catechol Group Improves the Performance of Iron Porphyrin CO2 Reduction Catalysts. ACS Catal. 2025, 15, 1444–1454. [Google Scholar] [CrossRef]
- Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 2016, 138, 16639–16644. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Yang, D.-T.; Ye, R.; Zeng, J.; Corbin, N.; Manthiram, K. Inductive and electrostatic effects on cobalt porphyrins for heterogeneous electrocatalytic carbon dioxide reduction. Catal. Sci. Technol. 2019, 9, 974–980. [Google Scholar] [CrossRef]
- Zhong, Y.H.; Wang, Y.; Zhao, S.Y.; Xie, Z.X.; Chung, L.H.; Liao, W.M.; Yu, L.; Wong, W.Y.; He, J. Regulating the Electronic Configuration of Ni Sites by Breaking Symmetry of Ni-Porphyrin to Facilitate CO2 Photocatalytic Reduction. Adv. Funct. Mater. 2024, 34, 2316199. [Google Scholar] [CrossRef]
- Yuan, H.; Krishna, A.; Wei, Z.; Su, Y.; Chen, J.; Hua, W.; Zheng, Z.; Song, D.; Mu, Q.; Pan, W.; et al. Ligand-Bound CO2 as a Nonclassical Route toward Efficient Photocatalytic CO2 Reduction with a Ni N-Confused Porphyrin. J. Am. Chem. Soc. 2024, 146, 10550–10558. [Google Scholar] [CrossRef]
- Cao, S.; Wei, S.; Wei, X.; Zhou, S.; Chen, H.; Hu, Y.; Wang, Z.; Liu, S.; Guo, W.; Lu, X. Can N, S Cocoordination Promote Single Atom Catalyst Performance in CO2RR? Fe-N2S2 Porphyrin versus Fe-N4 Porphyrin. Small 2021, 17, e2100949. [Google Scholar] [CrossRef]
- Hou, X.; Ding, J.; Liu, W.; Zhang, S.; Luo, J.; Liu, X. Asymmetric Coordination Environment Engineering of Atomic Catalysts for CO2 Reduction. Nanomaterials 2023, 13, 309. [Google Scholar] [CrossRef]
- Yang, F.; Han, H.; Duan, H.; Fan, F.; Chen, S.; Yu Xia, B.; He, Y.-L. A Review on Single Site Catalysts for Electrochemical CO2 Reduction. Adv. Energy Mater. 2025, 15, 2405726. [Google Scholar] [CrossRef]
- Tang, T.; Wang, Z.; Guan, J. Structural Optimization of Carbon-Based Diatomic Catalysts towards Advanced Electrocatalysis. Coord. Chem. Rev. 2023, 492, 215288. [Google Scholar] [CrossRef]
- Verma, S.; Yadav, A. Emerging Single-Atom Catalysts and Nanomaterials for Photoelectrochemical Reduction of Carbon Dioxide to Value-Added Products: A Review of the Current State-of-the-Art and Future Perspectives. Energy Fuels 2023, 37, 5712–5742. [Google Scholar] [CrossRef]
- Cui, J.; Yao, Z.; Zhu, B.; Tang, J.; Sun, J.; Xiong, P.; Fu, Y.; Zhang, W.; Zhu, J. Coordination Microenvironment Regulation in Carbon−Based Atomically Dispersed Metal Catalysts for Clean Energy−Conversion. Adv. Funct. Mater. 2024, 34, 2405328. [Google Scholar] [CrossRef]
- Manbeck, G.F.; Fujita, E. A Review of Iron and Cobalt Porphyrins, Phthalocyanines and Related Complexes for Electrochemical and Photochemical Reduction of Carbon Dioxide. J. Porphyr. Phthalocyanines 2015, 19, 45–64. [Google Scholar] [CrossRef]
- Gotico, P.; Halime, Z.; Aukauloo, A. Recent Advances in Metalloporphyrin-Based Catalyst Design towards Carbon Dioxide Reduction: From Bio-Inspired Second Coordination Sphere Modifications to Hierarchical Architectures. Dalton Trans. 2020, 49, 2381–2396. [Google Scholar] [CrossRef] [PubMed]
- Dedic, D.; Dorniak, A.; Rinner, U.; Schofberger, W. Recent Progress in (Photo-)-Electrochemical Conversion of CO2 With Metal Porphyrinoid-Systems. Front. Chem. 2021, 9, 685619. [Google Scholar] [CrossRef]
- Chatterjee, T.; Shetti, V.S.; Sharma, R.; Ravikanth, M. Heteroatom-Containing Porphyrin Analogues. Chem. Rev. 2017, 117, 3254. [Google Scholar] [CrossRef]
- Gupta, I.; Ravikanth, M. Recent Developments in Heteroporphyrins and Their analogues. Coord. Chem. Rev. 2006, 250, 468–518. [Google Scholar] [CrossRef]
- Szyszko, B.; Latos-Grażyński, L. Core chemistry and skeletal rearrangements of porphyrinoids and metalloporphyrinoids. Chem. Soc. Rev. 2015, 44, 3588–3616. [Google Scholar] [CrossRef]
- Takahashi, K.; Hiratsuka, K.; Sasaki, H.; Toshima, S. Electrocatalytic Behavior of Metal Porphyrins in the Reduction of Carbon Dioxide. Chem. Lett. 1979, 8, 305–308. [Google Scholar] [CrossRef]
- Call, A.; Cibian, M.; Yamamoto, K.; Nakazono, T.; Yamauchi, K.; Sakai, K. Highly Efficient and Selective Photocatalytic CO2 Reduction to CO in Water by a Cobalt Porphyrin Molecular Catalyst. ACS Catal. 2019, 9, 4867–4874. [Google Scholar] [CrossRef]
- Yuan, H.; Yu, Y.; Yang, S.; Lei, Q.; Yang, Z.; Lan, B.; Han, Z. Photocatalytic CO2 Reduction with Iron Porphyrin Catalysts and Anthraquinone Dyes. Chem. Commun. 2024, 60, 6292–6295. [Google Scholar] [CrossRef]
- Shen, J.; Kortlever, R.; Kas, R.; Birdja, Y.Y.; Diaz-Morales, O.; Kwon, Y.; Ledezma-Yanez, I.; Schouten, K.J.P.; Mul, G.; Koper, M.T.M. Electrocatalytic Reduction of Carbon Dioxide to Carbon Monoxide and Methane at an Immobilized Cobalt Protoporphyrin. Nat. Commun. 2015, 6, 8177. [Google Scholar] [CrossRef]
- Amanullah, S.; Saha, P.; Dey, A. Activating the Fe(I) State of Iron Porphyrinoid with Second-Sphere Proton Transfer Residues for Selective Reduction of CO2 to HCOOH via Fe(III/II)–COOH Intermediates. J. Am. Chem. Soc. 2021, 143, 13579–13592. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Sun, Q.; Wang, Z.; Huang, B.; Dai, Y.; Qin, X.; Zhang, X. Chemical Adsorption Enhanced CO2 Capture and Photoreduction over a Copper Porphyrin Based Metal Organic Framework. ACS Appl. Mater. Interfaces 2013, 5, 7654–7658. [Google Scholar] [CrossRef]
- Abdinejad, M.; Seifitokaldani, A.; Dao, C.; Sargent, E.H.; Zhang, X.-a.; Kraatz, H.B. Enhanced Electrochemical Reduction of CO2 Catalyzed by Cobalt and Iron Amino Porphyrin Complexes. ACS Appl. Energy Mater. 2019, 2, 1330–1335. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Li, M.; Liao, R.-Z. Mechanistic Insights into Photochemical CO2 Reduction to CH4 by a Molecular Iron–Porphyrin Catalyst. Inorg. Chem. 2023, 62, 9400–9417. [Google Scholar] [CrossRef]
- Ifraemov, R.; Mukhopadhyay, S.; Hod, I. Photo-Assisted Electrochemical CO2 Reduction to CH4 Using a Co-Porphyrin-Based Metal–Organic Framework. Sol. RRL 2023, 7, 2201068. [Google Scholar] [CrossRef]
- Han, J.; An, P.; Liu, S.; Zhang, X.; Wang, D.; Yuan, Y.; Guo, J.; Qiu, X.; Hou, K.; Shi, L.; et al. Reordering d Orbital Energies of Single-Site Catalysts for CO2 Electroreduction. Angew. Chem. Int. Ed. 2019, 58, 12711–12716. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Zhu, Y.-N.; Long, X.; Li, X.-B.; Zhang, Q.; Yang, G.; Du, S.; Liu, Z.; Liu, Z.; Peng, F. Theoretical Calculations and Experimental Verification of Carbon Dioxide Reduction Electrocatalyzed by Metalloporphyrin. J. Colloid Interface Sci. 2024, 668, 366–374. [Google Scholar] [CrossRef]
- Nielsen, I.M.B.; Leung, K. Cobalt−Porphyrin Catalyzed Electrochemical Reduction of Carbon Dioxide in Water. 1. A Density Functional Study of Intermediates. J. Phys. Chem. A 2010, 114, 10166–10173. [Google Scholar] [CrossRef]
- Shen, J.; Kolb, M.J.; Göttle, A.J.; Koper, M.T.M. DFT Study on the Mechanism of the Electrochemical Reduction of CO2 Catalyzed by Cobalt Porphyrins. J. Phys. Chem. C 2016, 120, 15714–15721. [Google Scholar] [CrossRef]
- Miyamoto, K.; Asahi, R. Water Facilitated Electrochemical Reduction of CO2 on Cobalt-Porphyrin Catalysts. J. Phys. Chem. C 2019, 123, 9944–9948. [Google Scholar] [CrossRef]
- Bochlin, Y.; Ben-Eliyahu, Y.; Kadosh, Y.; Kozuch, S.; Zilbermann, I.; Korin, E.; Bettelheim, A. DFT and Empirical Considerations on Electrocatalytic Water/Carbon Dioxide Reduction by CoTMPyP in Neutral Aqueous Solutions**. ChemPhysChem 2020, 21, 2644–2650. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Chen, J.-Y.; Siegbahn, P.E.M.; Liao, R.-Z. Harnessing Noninnocent Porphyrin Ligand to Circumvent Fe-Hydride Formation in the Selective Fe-Catalyzed CO2 Reduction in Aqueous Solution. ACS Catal. 2020, 10, 6332–6345. [Google Scholar] [CrossRef]
- Cao, Y.C.; Shi, L.L.; Li, M.; You, B.; Liao, R.Z. Deciphering the Selectivity of the Electrochemical CO2 Reduction to CO by a Cobalt Porphyrin Catalyst in Neutral Aqueous Solution: Insights from DFT Calculations. ChemistryOpen 2023, 12, e202200254. [Google Scholar] [CrossRef] [PubMed]
- Göttle, A.J.; Koper, M.T.M. Proton-Coupled Electron Transfer in the Electrocatalysis of CO2 Reduction: Prediction of Sequential vs. Concerted Pathways using DFT. Chem. Sci. 2017, 8, 458–465. [Google Scholar] [CrossRef]
- Ren, Z.; Gong, K.; Zhao, B.; Chen, S.-L.; Xie, J. Boosting the Catalytic Performance of Metalloporphyrin-based Covalent Organic Frameworks via Coordination Engineering for CO2 and O2 Reduction. Mater. Chem. Front. 2024, 8, 1958–1970. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Huang, Q.; He, C.-T.; Chen, Y.; Liu, J.; Shen, F.-C.; Lan, Y.-Q. Oriented Eectron Transmission in Polyoxometalate-Metalloporphyrin Organic Framework for Highly Selective Electroreduction of CO2. Nat. Commun. 2018, 9, 4466. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Warburton, R.E.; Soudackov, A.V.; Hammes-Schiffer, S. Theoretical Modeling of Electrochemical Proton-Coupled Electron Transfer. Chem. Rev. 2022, 122, 10599–10650. [Google Scholar] [CrossRef]
- Ren, Z.; Zhao, B.; Xie, J. Designing N-Confused Metalloporphyrin-Based Covalent Organic Frameworks for Enhanced Electrocatalytic Carbon Dioxide Reduction. Small 2023, 19, e2301818. [Google Scholar] [CrossRef]
- Furuta, H.; Asano, T.; Ogawa, T. “N-Confused Porphyrin”: A New Isomer of Tetraphenylporphyrin. J. Am. Chem. Soc. 1994, 116, 767–768. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Latos-Grażyński, L. N-Methyltetraphenylporphyrin with an Inverted N-Methylpyrrole Ring: The First Isomer of N-Methyltetraphenylporphyrin. J. Chem. Soc. Perkin Trans. II Phys. Org. Chem. 1995, 24, 503–509. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Latos-Grażyński, L.; Schmidt, I. Copper(II) Complexes of Inverted Porphyrin and Its Methylated Derivatives. Inorg. Chem. 2000, 39, 5475. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Hung, C.H. Synthesis and Characterization of Iron N-Confused Porphyrins: Structural Evidences of Agostic Interaction. Inorg. Chem. 2001, 40, 5070–5071. [Google Scholar] [CrossRef]
- Bohle, D.S.; Chen, W.C.; Hung, C.H. Metal Oxidation Promoted C-H Activation in Manganese Complexes of N-Confused Porphyrin. Inorg. Chem. 2002, 41, 3334. [Google Scholar] [CrossRef]
- Maeda, H.; Osuka, A.; Ishikawa, Y.; Aritome, I.; Hisaeda, Y.; Furuta, H. N-Confused Porphyrin Bearing meso-Perfluorophenyl Groups: A Potential Agent that Forms Stable Square Planar Complexes with Cu(II) and Ag(III). Org. Lett. 2003, 5, 1293. [Google Scholar] [CrossRef]
- Zilbermann, I.; Maimon, E.; Ydgar, R.; Shames, A.I.; Korin, E.; Soifer, L.; Bettelheim, A. Spectroscopic and Electrochemical Characterization of Solutions and Films of a New Redox Couple: Co(II)/Co(III) N-Confused Porphyrin. Inorg. Chem. Commun. 2004, 7, 1238. [Google Scholar] [CrossRef]
- Harvey, J.D.; Shaw, J.L.; Herrick, R.S.; Ziegler, C.J. The Synthesis of Isostructural Mo2+ Porphyrin and N-Confused Porphyrin Complexes. Chem. Commun. 2005, 10, 4663. [Google Scholar] [CrossRef]
- Srinivasan, A.; Toganoh, M.; Niino, T.; Osuka, A.; Furuta, H. Synthesis of N-Confused Tetraphenylporphyrin Rhodium Complexes Having Versatile Metal Oxidation States. Inorg. Chem. 2008, 47, 11305. [Google Scholar] [CrossRef]
- Toganoh, M.; Ikeda, S.; Furuta, H. Synthesis, Reactivity, and Properties of N-Fused Porphyrin Rhenium(I) Tricarbonyl Complexes. Inorg. Chem. 2007, 46, 10003. [Google Scholar] [CrossRef]
- Furuta, H.; Kubo, N.; Maeda, H.; Ishizuka, T.; Osuka, A.; Nanami, H.; Ogawa, T. N-Confused Double-Decker Porphyrins. Inorg. Chem. 2000, 39, 5424. [Google Scholar] [CrossRef] [PubMed]
- Toganoh, M.; Niino, T.; Furuta, H. Luminescent Au(III) Organometallic Complex of N-Confused Tetraphenylporphyrin. Chem. Commun. 2008, 13, 4070. [Google Scholar] [CrossRef]
- Peng, C.C.; Yang, F.-A.; Chen, J.-H.; Wang, S.-S.; Tung, J.-Y. Mercury(II) Complex of Inverted N-Methylated Porphyrin: HgPh(2-NCH3NCTPP). Polyhedron 2008, 27, 2309–2314. [Google Scholar] [CrossRef]
- Shao, P.; Ren, Z.; Zhao, B.; Wang, X.; Li, J.; Xie, J.; Wang, B.; Feng, X. Theory-Guided Design of N-Confused Porphyrinic Covalent Organic Frameworks for Oxygen Reduction Reaction. J. Am. Chem. Soc. 2025, 147, 8769–8777. [Google Scholar] [CrossRef]
- Furuta, H.; Maeda, H.; Osuka, A. Doubly N-Confused Porphyrin: A New Complexing Agent Capable of Stabilizing Higher Oxidation States. J. Am. Chem. Soc. 2000, 122, 803–807. [Google Scholar] [CrossRef]
- Maeda, H.; Osuka, A.; Furuta, H. Trans Doubly N-Confused Porphyrins: Cu(III) Complexation and Formation of Rodlike Hydrogen-Bonding Networks. J. Am. Chem. Soc. 2003, 125, 15690–15691. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Tranca, D.; Rodriguez-Hernandez, F.; Zhang, J.; Lu, C.; Zhu, J.; Liang, H.W.; Zhuang, X. Well-defined N3C1 -anchored Single-Metal-Sites for Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 2024, 63, e202314833. [Google Scholar] [CrossRef]
- Hua, W.; Liu, T.; Zheng, Z.; Yuan, H.; Xiao, L.; Feng, K.; Hui, J.; Deng, Z.; Ma, M.; Cheng, J.; et al. Pulse Electrolysis Turns on CO2 Methanation through N-Confused Cupric Porphyrin. Angew. Chem. Int. Ed. 2024, 63, e202315922. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Latos-Grazynski, L.; Olmstead, M.M.; Balch, A.L. Nickel Complexes of 21-Oxaporphyrin and 21, 23-Dioxaporphyrin. Chemistry 1997, 3, 268–278. [Google Scholar] [CrossRef]
- Pawlicki, M.; Latos-Grażyński, L. Iron Complexes of 5,10,15,20-Tetraphenyl-21-Oxaporphyrin. Inorg. Chem. 2002, 41, 5866–5873. [Google Scholar] [CrossRef]
- Pawlicki, M.; Latos-Grażyński, L. Reactivity of Iron(II) 5,10,15,20-Tetraaryl-21-Oxaporphyrin with Arylmagnesium Bromide: Formation of Paramagnetic Six-Coordinate Complexes with Two Axial Aryl Groups. Inorg. Chem. 2004, 43, 5564–5571. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Ravikanth, M. Rhenium(I) Tricarbonyl Complexes of 5,10,15,20-Tetraphenyl-21-Thia and 21-Oxaporphyrins. Inorg. Chem. 2012, 51, 6700–6709. [Google Scholar] [CrossRef] [PubMed]
- Stute, S.; Götzke, L.; Meyer, D.; Merroun, M.L.; Rapta, P.; Kataeva, O.; Seichter, W.; Gloe, K.; Dunsch, L.; Gloe, K. Molecular Structure, UV/Vis Spectra, and Cyclic Voltammograms of Mn(II), Co(II), and Zn(II) 5,10,15,20-Tetraphenyl-21-oxaporphyrins. Inorg. Chem. 2013, 52, 1515–1524. [Google Scholar] [CrossRef]
- Latos-Grazynski, L.; Lisowski, J.; Olmstead, M.M.; Balch, A.L. Five-coordinate Complexes of 21-Thiaporphyrin. Preparations, Spectra, and Structures of Iron(II), Nickel(II), and Copper(II) Complexes. Inorg. Chem. 1989, 28, 1183–1188. [Google Scholar] [CrossRef]
- Latos-Grazynski, L.; Lisowski, J.; Olmstead, M.M.; Balch, A.L. Preparation and Structural Characterization of a Six-Coordinate 21-Thiaporphyrin Complex: RhIII(STPP)Cl2 (STPP = tetraphenyl-21-thiaporphyrin anion). Inorg. Chem. 1989, 28, 3328–3331. [Google Scholar] [CrossRef]
- Latos-Grazynski, L.; Lisowski, J.; Chmielewski, P.; Grzeszczuk, M.; Olmstead, M.M.; Balch, A.L. Palladium Complexes of 21-Thiaporphyrin: Syntheses and Characterization. Inorg. Chem. 1994, 33, 192–197. [Google Scholar] [CrossRef]
- Gebauer, A.; Schmidt, J.A.R.; Arnold, J. Synthesis, Characterization, and Properties of a Lithium 21-Thiaporphyrin Complex. Inorg. Chem. 2000, 39, 3424–3427. [Google Scholar] [CrossRef]
- Tung, J.-Y.; Liau, B.-C.; Elango, S.; Chen, J.-H.; Hsieh, H.-Y.; Liao, F.-L.; Wang, S.-L.; Hwang, L.-P. Preparation and Structural Characterization of Mercury 21-Thiaporphyrin Complex: HgII(STPP)Cl (STPP=tetraphenyl-21-thiaporphyrin anion). Inorg. Chem. Commun. 2002, 5, 150–155. [Google Scholar] [CrossRef]
- Chuang, C.-H.; Ou, C.-K.; Liu, S.-T.; Kumar, A.; Ching, W.-M.; Chiang, P.-C.; dela Rosa, M.A.C.; Hung, C.-H. Ruthenium Complexes of Thiaporphyrin and Dithiaporphyrin. Inorg. Chem. 2011, 50, 11947–11957. [Google Scholar] [CrossRef]
- Hung, C.-H.; Ou, C.-K.; Lee, G.-H.; Peng, S.-M. Structure and Characterization of the First Metal Complex of Dithiaporphyrin: Ru(S2TTP)Cl2. Inorg. Chem. 2001, 40, 6845–6847. [Google Scholar] [CrossRef]
- Kaur, T.; Ghosh, A.; Rajakannu, P.; Ravikanth, M. Synthesis and Crystal Structure of the Rhenium(I) Tricarbonyl Complex of 5,10,15,20-Tetra-p-tolyl-21,23-dithiaporphyrin. Inorg. Chem. 2014, 53, 2355–2357. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, P.; Grzeszczuk, M.; Latos-Grazynski, L.; Lisowski, J. Studies of the Reduction of the Nickel(II) Complex of 5,10,15,20-Tetraphenyl-21-Thiaporphyrin to Form Corresponding Nickel(I) Complexes. Inorg. Chem. 1989, 28, 3546–3552. [Google Scholar] [CrossRef]
- Chmielewski, P.J.; Latos-Grażyński, L. Core Modified Porphyrins—A Macrocyclic Platform for Organometallic Chemistry. Coord. Chem. Rev. 2005, 249, 2510–2533. [Google Scholar] [CrossRef]
- Pareek, Y.; Ravikanth, M. Thiaporphyrins: From Building Blocks to Multiporphyrin Arrays. RSC Adv. 2014, 4, 7851–7880. [Google Scholar] [CrossRef]
- Ding, X.; Jin, Y.; Wang, H.; Qi, D. Molecular Modification of Planar Four-Coordinated Cobalt Active Site for the Electrochemical Reduction of Carbon Dioxide: A Density Functional Theory Study. Inorg. Chem. Front. 2023, 10, 7054–7063. [Google Scholar] [CrossRef]
- Göttle, A.J.; Koper, M.T.M. Determinant Role of Electrogenerated Reactive Nucleophilic Species on Selectivity during Reduction of CO2 Catalyzed by Metalloporphyrins. J. Am. Chem. Soc. 2018, 140, 4826–4834. [Google Scholar] [CrossRef]
- Costentin, C.; Savéant, J.-M. Towards an Intelligent Design of Molecular Electrocatalysts. Nat. Rev. Chem. 2017, 1, 0087. [Google Scholar] [CrossRef]
- Han, J.; Li, N.; Chen, D.; Xu, Q.; Lu, J. Boosting Photocatalytic Activity for Porphyrin-based D-A Conjugated Polymers via Dual Metallic Sites Regulation. Appl. Catal. B Environ. 2022, 317, 121724. [Google Scholar] [CrossRef]
- Toganoh, M.; Furuta, H. Creation from Confusion and Fusion in the Porphyrin World—The Last Three Decades of N-Confused Porphyrinoid Chemistry. Chem. Rev. 2022, 122, 8313–8437. [Google Scholar] [CrossRef]
- Dong, H.; Fang, L.; Chen, K.-X.; Wei, J.-X.; Li, J.-X.; Qiao, X.; Wang, Y.; Zhang, F.-M.; Lan, Y.-Q. Dual Metallosalen-Based Covalent Organic Frameworks for Artificial Photosynthetic Diluted CO2 Reduction. Angew. Chem. Int. Ed. 2025, 64, e202414287. [Google Scholar] [CrossRef]
- Abdinejad, M.; Tang, K.; Dao, C.; Saedy, S.; Burdyny, T. Immobilization Strategies for Porphyrin-Based Molecular Catalysts for the Electroreduction of CO2. J. Mater. Chem. A 2022, 10, 7626–7636. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Lu, J.; Ren, X.; Wu, N.; Wang, D.; Fang, J.; Zhong, J. Nitrogen-Rich Carbon Nanomaterials Embedded with Ni Nanoparticles for Electrochemical Carbon Dioxide Reduction. ACS Appl. Nano Mater. 2025, 8, 4649–4657. [Google Scholar] [CrossRef]
- Cheng, K.; Kong, S.; Wang, J.; Wang, Q.; Yuan, S.; Li, P.-Z.; Zhao, Y. Integrating Multifunctionalities into a 3D Covalent Organic Framework for Efficient CO2 Photoreduction. Angew. Chem. Int. Ed. 2025, 64, e202504772. [Google Scholar] [CrossRef]
- Fan, S.-C.; Yuan, W.; Wang, J.-W.; Xing, C.; Li, D.; Wang, Y.; Zhai, Q.-G. Metallic Engineering in Metal-Porphyrin Frameworks for Highly Efficient CO2 Photoreduction. Chem. Eng. J. 2024, 501, 157716. [Google Scholar] [CrossRef]
- Han, H.; Cui, H.; Wang, J.; Ge, K.; Yang, Y. Photocatalytic Reduction of CO2 by Multimetallic 2D MOF-Co/Cu/Fe. ChemistrySelect 2024, 9, e202403927. [Google Scholar] [CrossRef]
- Zaar, F.; Emanuelsson, R.; Gaiser, P.; Strømme, M.; Sjödin, M. Characterization and Catalytic Prospects of Metalloporphyrin-Functionalized Conducting Polymers. Electrochim. Acta 2023, 467, 143003. [Google Scholar] [CrossRef]
- Zhang, H.; Gu, C.; Fan, M.; Zhao, Z.; Kong, X.; Geng, Z. Applications of In-Situ Spectroscopic Techniques towards CO2 Electroreduction. Sci. China Chem. 2024, 67, 3925–3933. [Google Scholar] [CrossRef]
- Salamé, A.; Hon Cheah, M.; Bonin, J.; Robert, M.; Anxolabéhère-Mallart, E. Operando Spectroelectrochemistry Unravels the Mechanism of CO2 Electrocatalytic Reduction by an Fe Porphyrin. Angew. Chem. Int. Ed. 2024, 63, e202412417. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2 | CO | HCOOH | CH3OH | CH4 | DFT Method | Ref. | |
|---|---|---|---|---|---|---|---|
| FeN4-TPP-Por | −0.59 | −1.45 | −0.70 | −0.90 | −0.73 | Cluster PBE-D3 | [37] |
| FeN2S2-TPP-Por | −0.26 | −1.11 | −0.38 | −0.40 | −0.56 | ||
| FeN4-Por-COF | −1.25 | −1.25 | / | / | / | ||
| FeN3S-Por-COF | −1.25 | −1.18 | / | / | / | Periodic PBE-D3 | |
| FeN2S2-Por-COF | −1.03 | −0.91 | / | / | / | [67] | |
| FeN3O-Por-COF | −1.50 | −1.24 | / | / | / | ||
| FeN2O2-Por-COF | −1.26 | −0.66 | / | / | / | ||
| CoN4-Por | / | −1.27 | / | −0.80 | −0.80 | [106] | |
| CoN3S-Por | / | −0.58 | / | −0.91 | −0.91 | Cluster ωB97XD-D3BJ | |
| CoN2S2-Por | / | −1.17 | / | −0.66 | −1.20 | ||
| CoN3O-Por | / | −1.11 | / | −1.04 | −1.04 | ||
| CoN2O2-Por | / | −2.12 | / | −0.96 | −1.40 | ||
| CoN4-Por-COF | −0.91 | −0.89 | −1.36 | −1.31 | −1.63 | [71] | |
| CoN3C1-Por-COF | −0.79 | −0.76 | / | −0.76 | −0.99 | Periodic PBE-D3 | |
| CoN2C2-Por-COF | −0.63 | −0.60 | / | −0.60 | −1.44 | ||
| CoN3S-Por-COF | −0.75 | −0.74 | / | / | / | Periodic PBE-D3 | |
| CoN2S2-Por-COF | −0.84 | −0.88 | / | / | / | [67] | |
| CoN3O-Por-COF | −0.64 | −0.67 | / | / | / | ||
| CoN2O2-Por-COF | −0.79 | −0.58 | / | / | / | ||
| NiN4-Por-COF | −1.52 | −1.52 | / | / | / | ||
| NiN3S-Por-COF | −1.16 | −1.08 | / | / | / | ||
| NiN2S2-Por-COF | −1.28 | −1.25 | / | / | / | Periodic PBE-D3 | [67] |
| NiN3O-Por-COF | −0.97 | −1.35 | / | / | / | ||
| NiN2O2-Por-COF | −1.23 | −0.98 | / | / | / |
| Catalysts | Cores | Product, FE | Potential | Method | Cat. Type | Solvent | Ref. |
|---|---|---|---|---|---|---|---|
| Iron-TPP | FeN4 | HCOOH | −0.70 VRHE (UL) | DFT | EC | H2O | [37] |
| Iron-21,23-dithia-TPP | FeN2S2 | HCOOH CH3OH | −0.38 VRHE (UL) −0.40 VRHE (UL) | ||||
| Cobalt-TPP | CoN4 | CO, 95% | −0.60 VRHE (Ered) | Expt. | EC | 0.5 M NaHCO3 | [34] |
| Cobalt 21-thia-Por | CoN3S | CO | −0.58 VRHE (UL) | DFT | EC | H2O | [106] |
| Cobalt 21,23-dithia-Por | CoN2S2 | CH3OH | −0.66 VRHE (UL) | ||||
| Cobalt 21-oxa-Por | CoN3O | CH3OH, CH4 | −1.04 VRHE (UL) | ||||
| Cobalt 21,23-dioxa-Por | CoN2O2 | CH3OH | −0.96 VRHE (UL) | ||||
| CoN4-Por-COFs | CoN4 | CO | −0.89 VRHE (UL) | DFT | EC | H2O | [71] |
| CoN3C-Por-COFs | CoN3C | CO, CH3OH | −0.76 VRHE (UL) | ||||
| CoN2C2-Por-COFs | CoN2C2 | CO, CH3OH | −0.60 VRHE (UL) | ||||
| Nickel-TPP | NiN4 | CO, 2% CO, 29% | −0.75 VRHE (Ered) −1.10 VRHE (Ered) | Expt.+DFT Expt. | EC | 0.5 M KHCO3 DMF | [27] [19] |
| Nickel-21-oxa-TPP | NiN3O | CO, 80% | −0.65 VRHE (Ered) | Expt.+DFT | EC | 0.5 M KHCO3 | [27] |
| Nickel-TPP(-COOH)4 | NiN4 | CO, 82.8% | −1.52 VFc+/Fc(Ered)/−1.29 VRHE | Expt.+DFT | PC | CH3CN/H2O | [35] |
| Nickel-21-oxa-TPP(-COOH)4 | NiN3O | CO, 94.0% | −1.41 VFc+/Fc(Ered)/−1.18 VRHE | ||||
| Nickel-21-thia-TPP(-COOH)4 | NiN3S | CO, 96.4% | −0.82 VFc+/Fc(Ered)/−0.60 VRHE | ||||
| Nickel-TPP | NiN4 | CO, <5% | / | Expt.+DFT | PC | CH3CN | [36] |
| Nickel-NCTPP | NiN3C | CO, 98% | −1.41 VAg/Ag+(Ered)/−1.26 VRHE | ||||
| Copper-TPP | CuN4 | CO, <15% | −1.8 VRHE(Ered) | Expt. | EC | 1 M KOH | [89] |
| Copper-NCTPP | CuN3C | CH4, >60% | −1.6 VRHE(Ered) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Wei, Q.; Ren, Z.; Xie, J. Recent Progress in Heteroatom-Containing Metalloporphyrin-Based Catalysts for CO2 Reduction. Molecules 2025, 30, 2287. https://doi.org/10.3390/molecules30112287
Li Z, Wei Q, Ren Z, Xie J. Recent Progress in Heteroatom-Containing Metalloporphyrin-Based Catalysts for CO2 Reduction. Molecules. 2025; 30(11):2287. https://doi.org/10.3390/molecules30112287
Chicago/Turabian StyleLi, Zhuo, Qianqian Wei, Zhixin Ren, and Jing Xie. 2025. "Recent Progress in Heteroatom-Containing Metalloporphyrin-Based Catalysts for CO2 Reduction" Molecules 30, no. 11: 2287. https://doi.org/10.3390/molecules30112287
APA StyleLi, Z., Wei, Q., Ren, Z., & Xie, J. (2025). Recent Progress in Heteroatom-Containing Metalloporphyrin-Based Catalysts for CO2 Reduction. Molecules, 30(11), 2287. https://doi.org/10.3390/molecules30112287