
Influence of Hydrolysis on Non-Isothermal Crystallization of Poly(Butylene Succinate-Co-Adipate) (PBSA)


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Hydrolysis
Compost
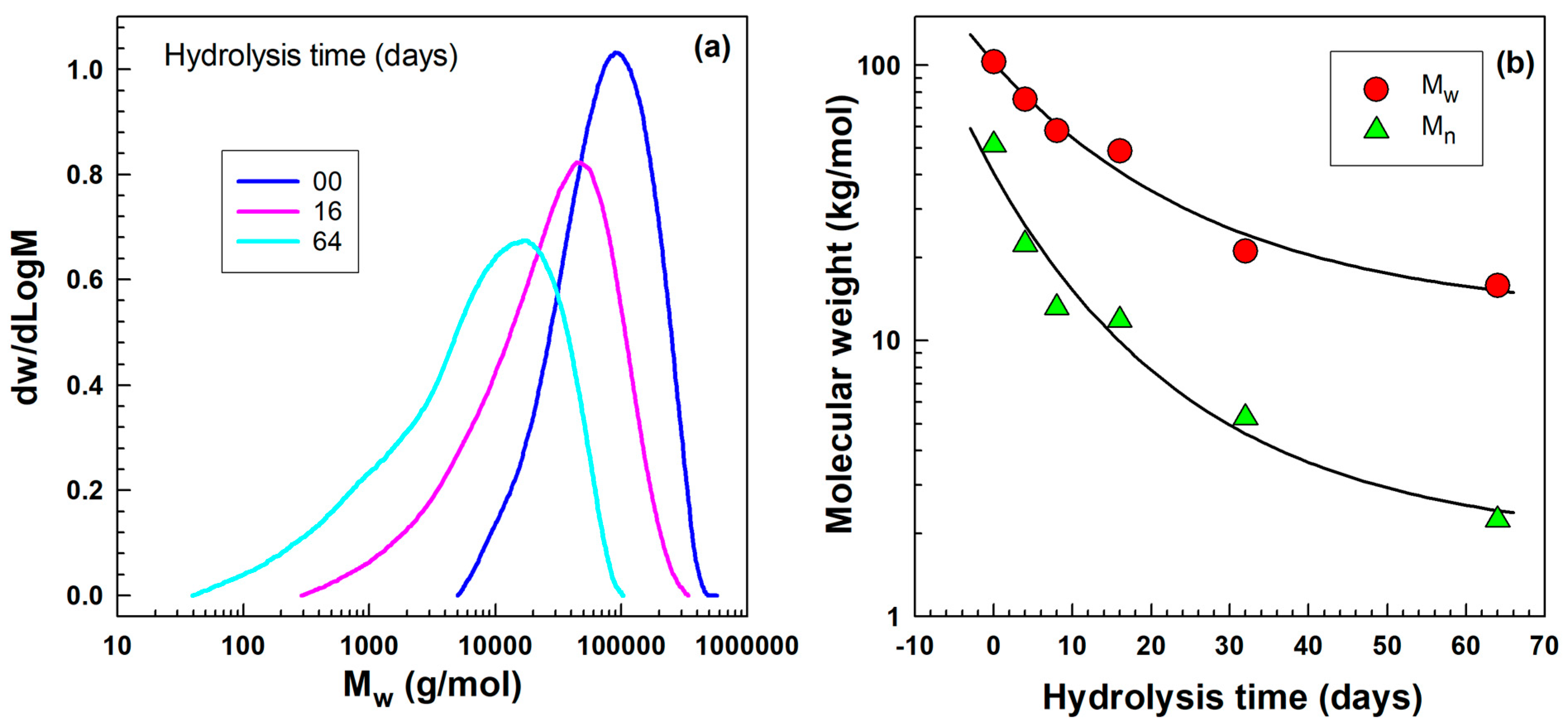
2.2. Molecular Weight by Gel Permeation Chromatography (GPC)
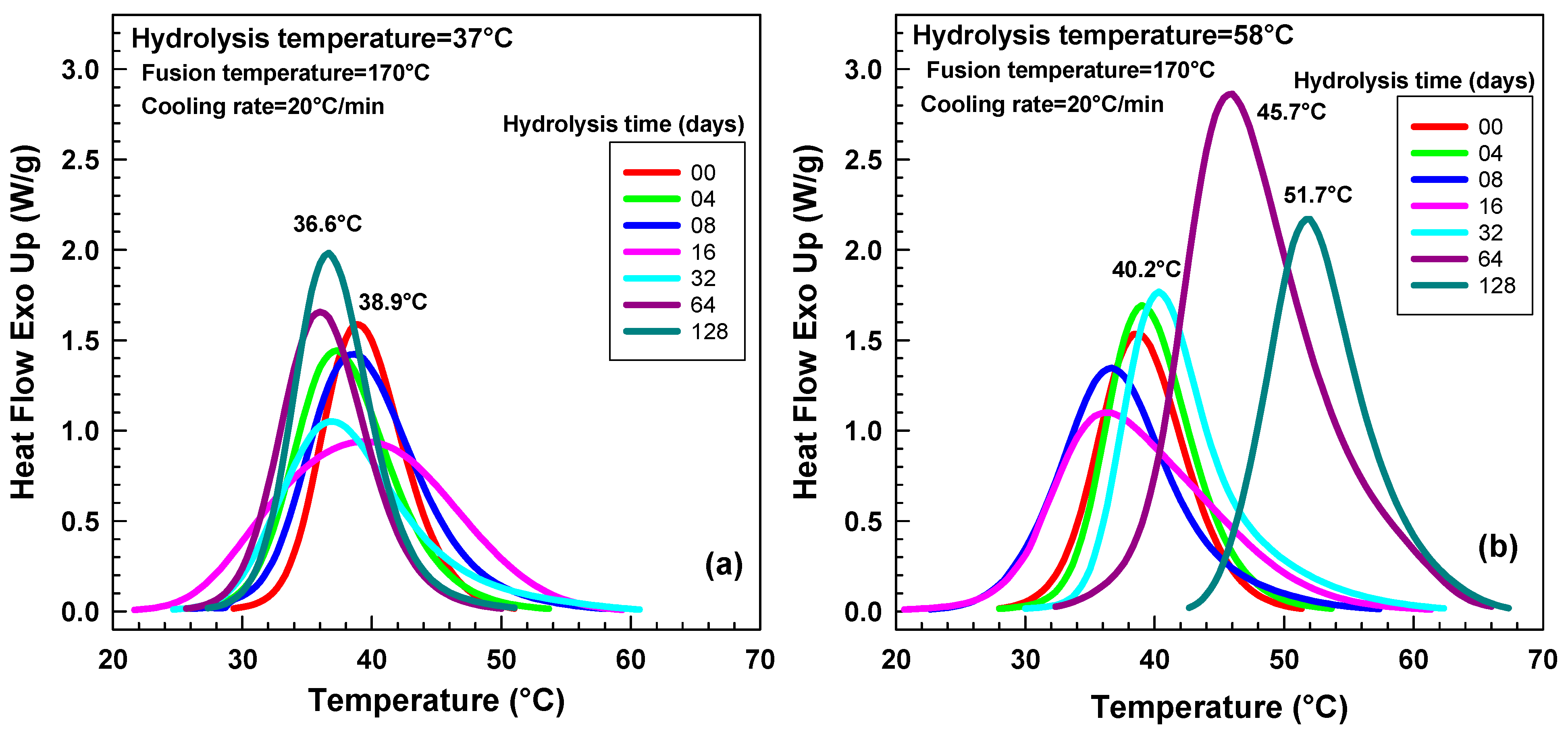
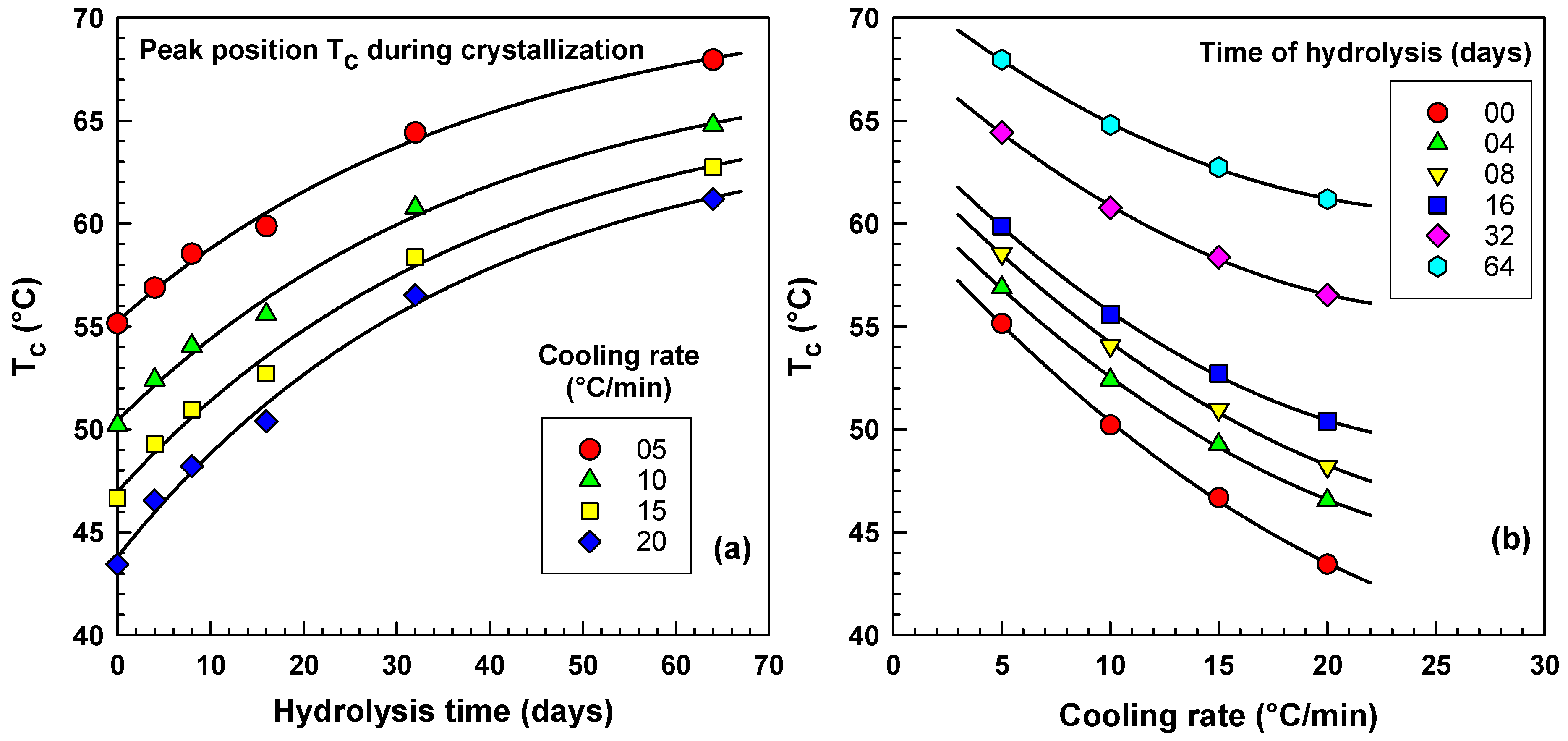
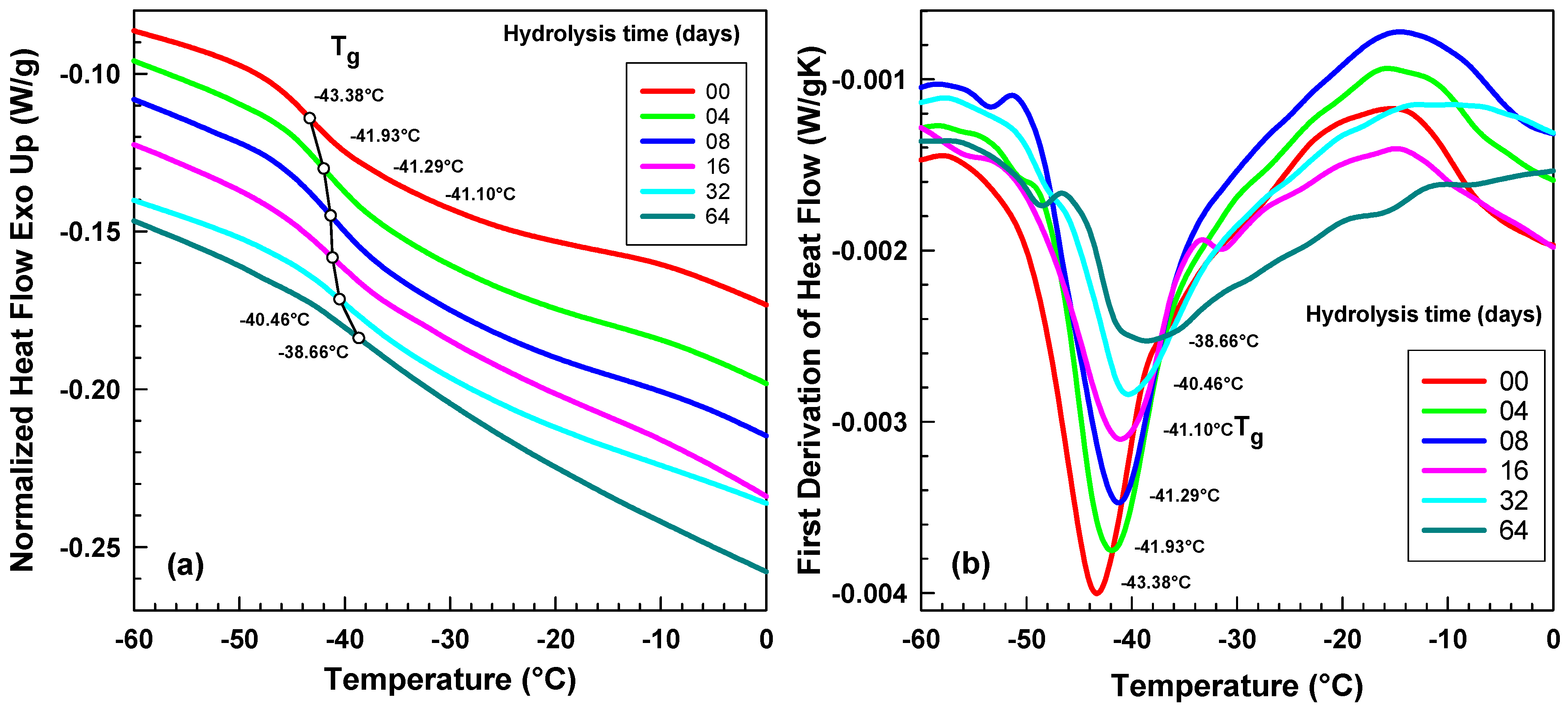
2.3. DSC Analysis
2.3.1. Initial Screening Experiments
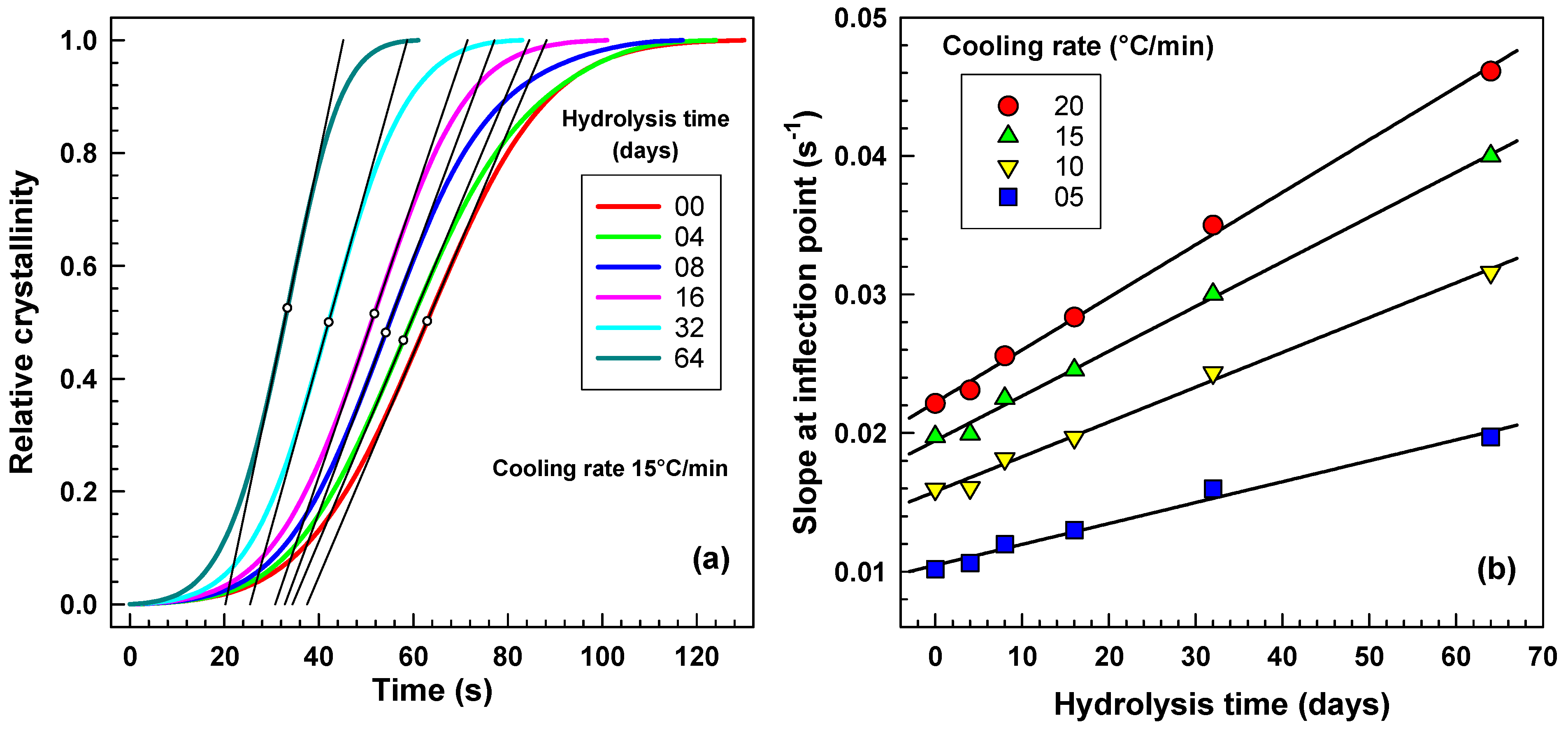
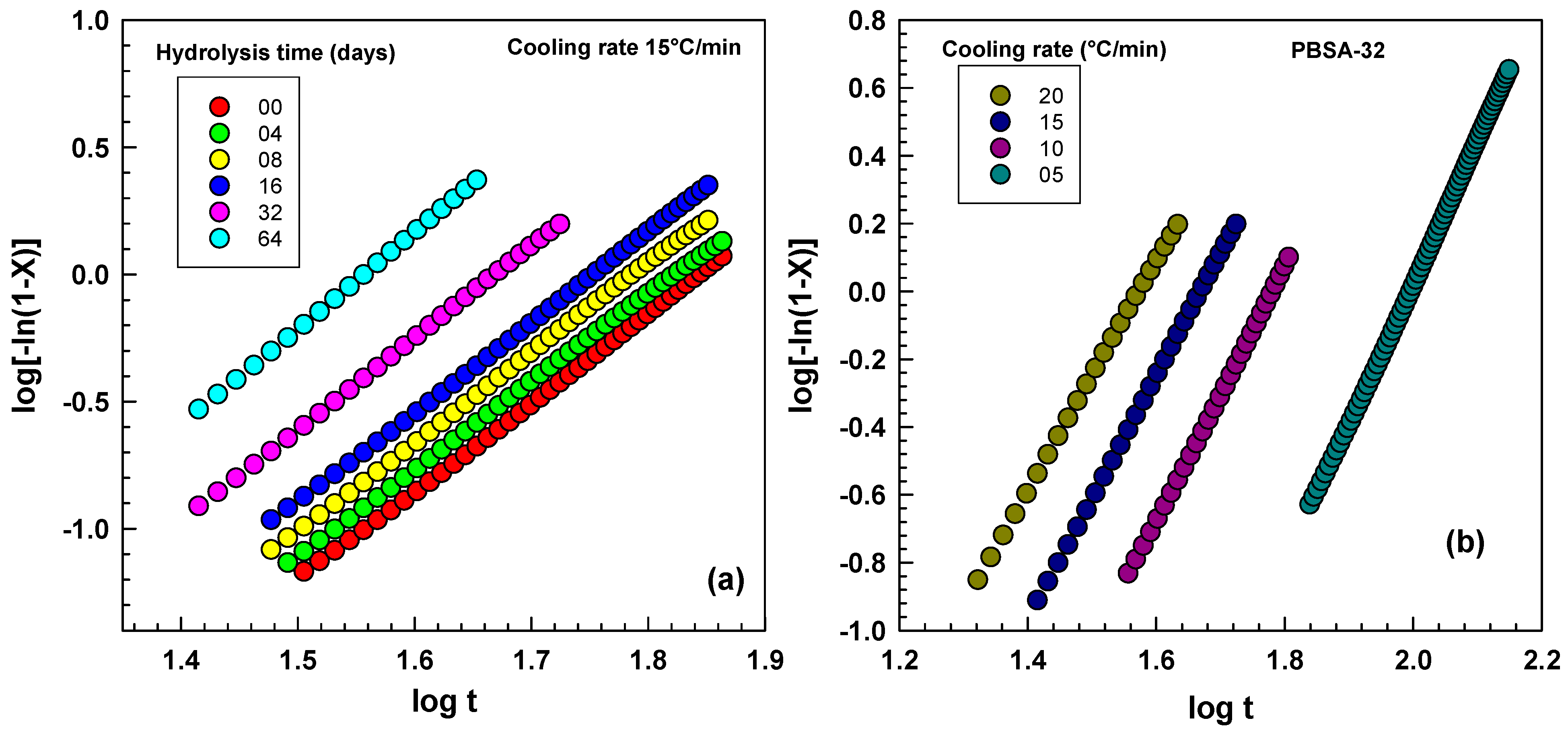
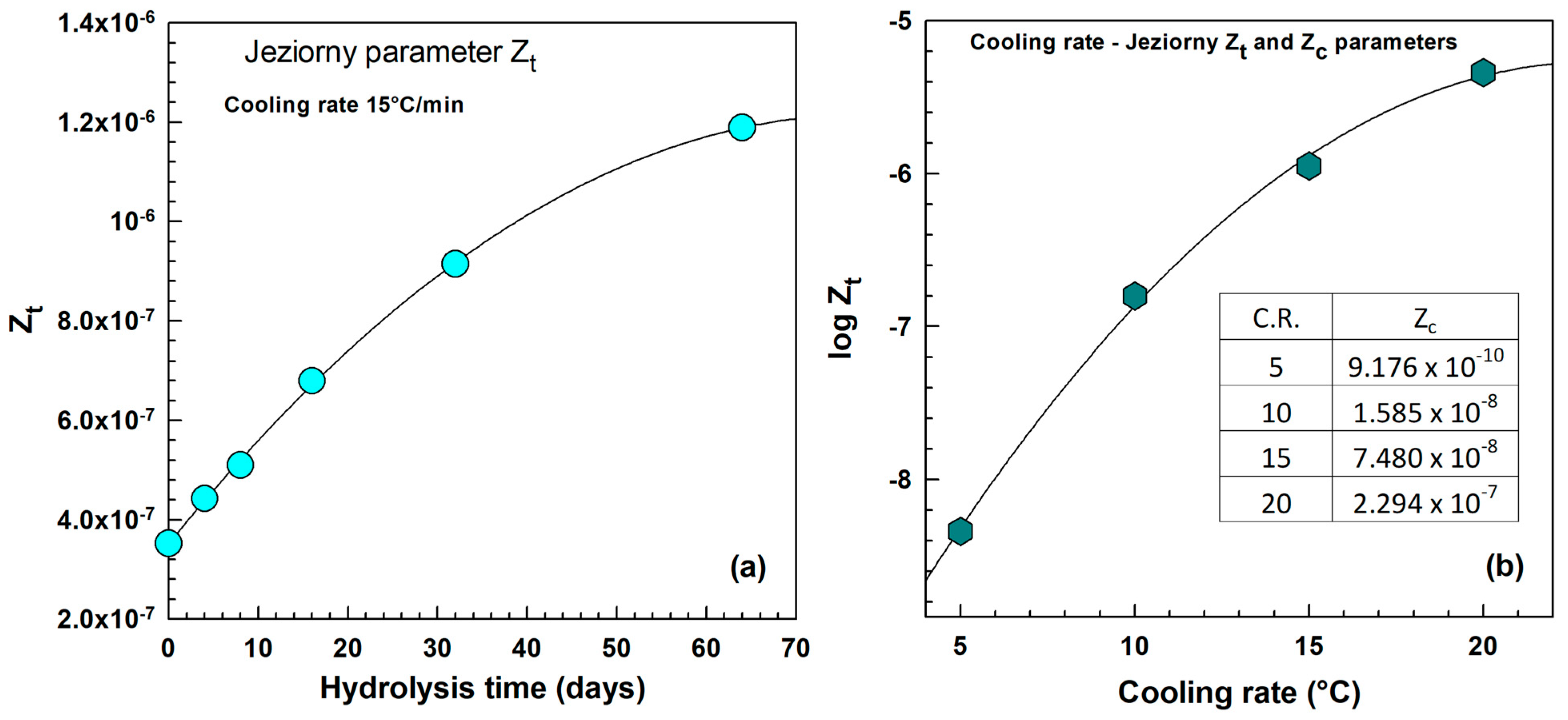
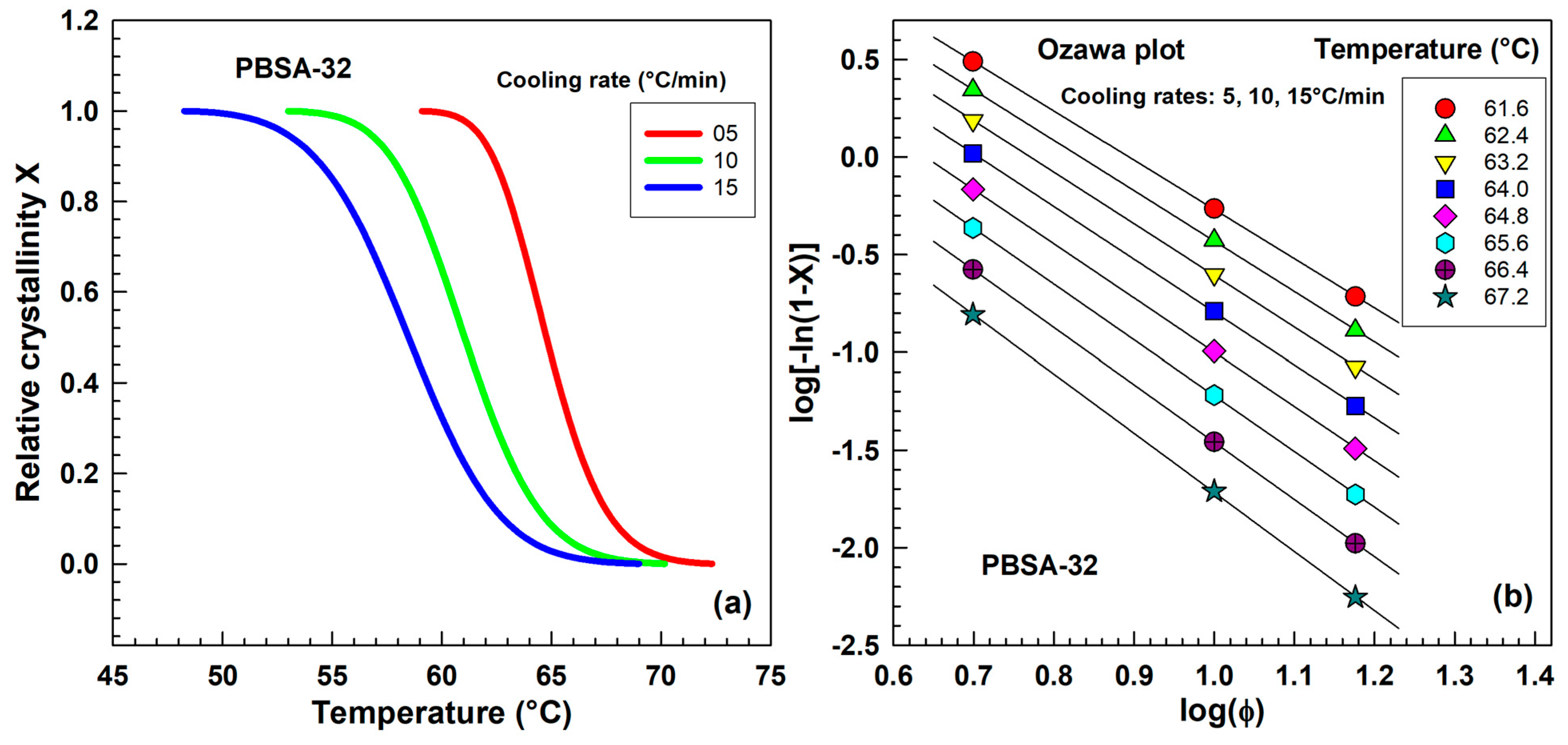
2.3.2. The Avrami Analysis of Non-Isothermal Crystallization
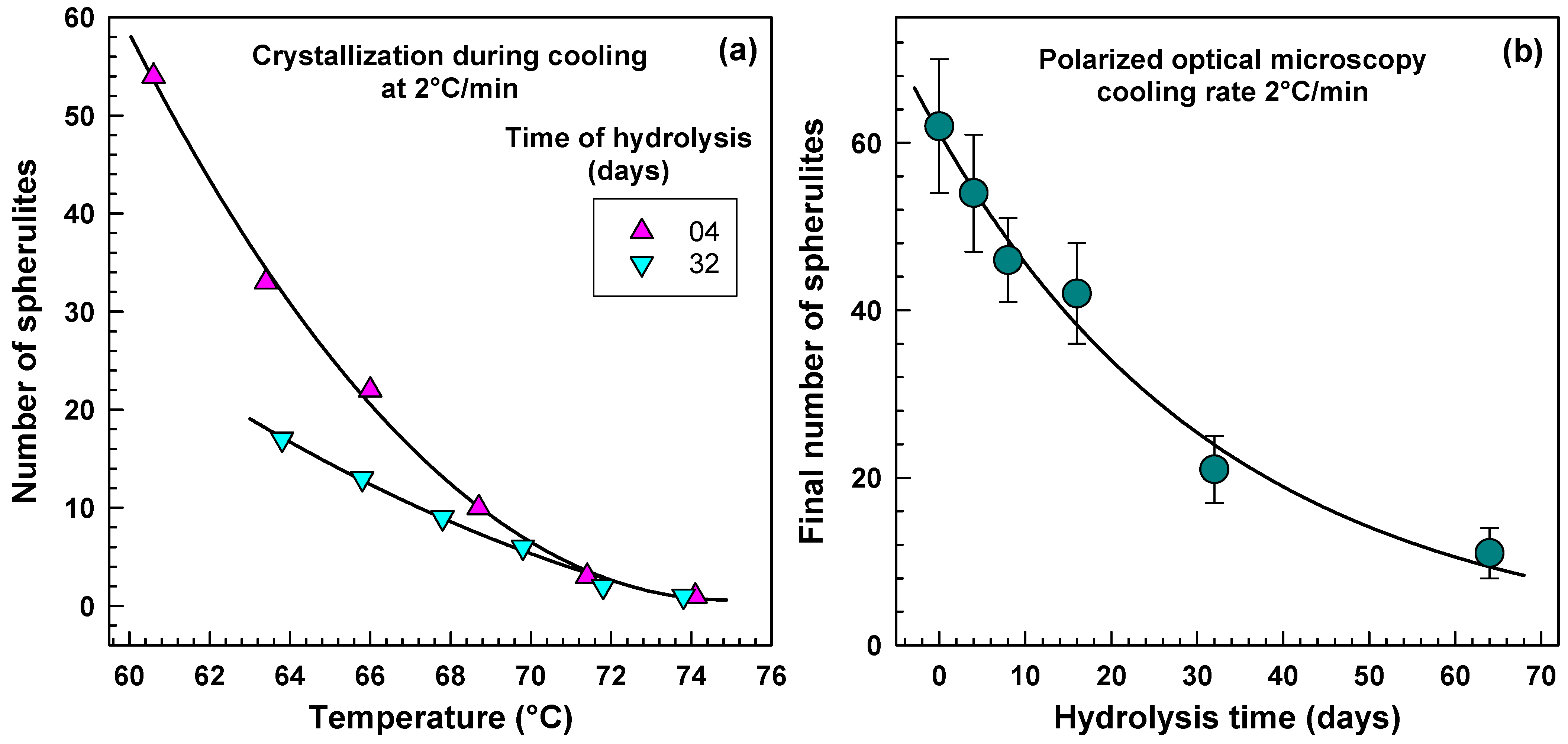
2.4. Polarized Optical Microscopy Results
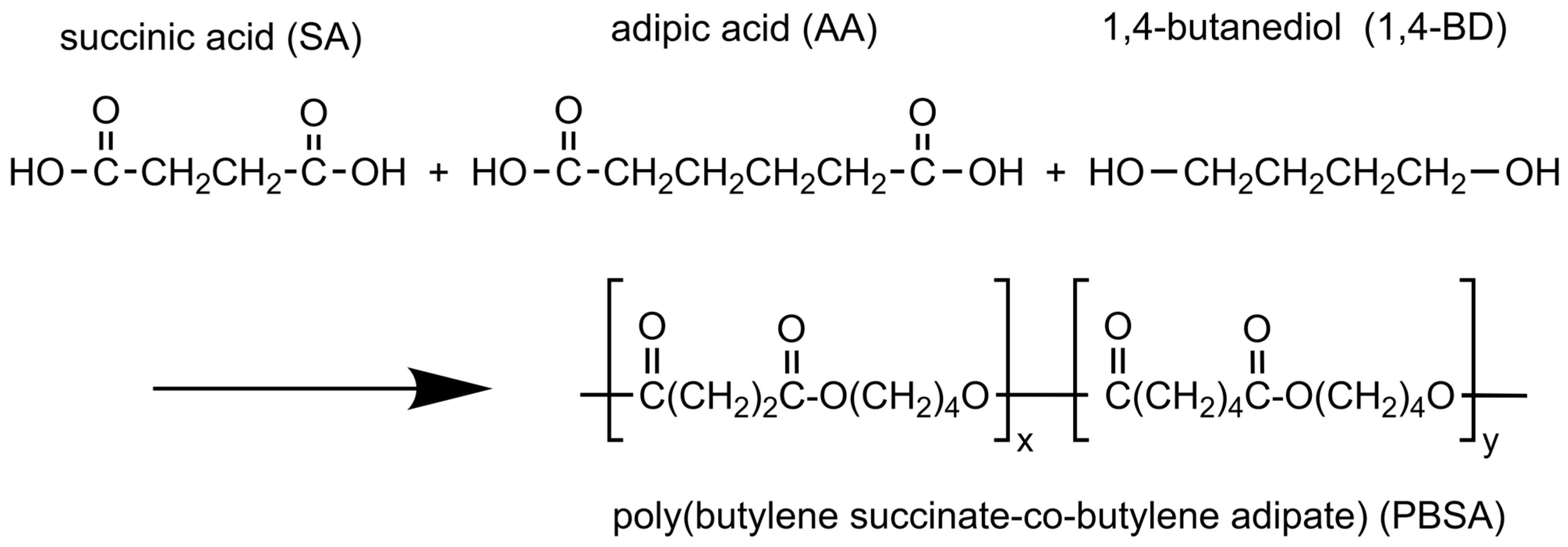
3. Materials and Methods
3.1. Materials
3.2. Compression Molding
3.3. Hydrolysis in Phosphate Buffered Saline
Hydrolysis at Elevated Temperature
- is degree of hydrolysis PBSA [%];
- total organic carbon in liquid after hydrolysis [mg/L];
- the weight of the PBSA sample used in the hydrolysis [mg];
- theoretical carbon content in the tested film PBSA film [%];
- the total volume of the liquid phase in the bottle [L].
3.4. Gel Permeation Chromatography (GPC)
3.5. Differential Scanning Calorimetry (DSC)
3.6. Polarized Optical Microscopy (POM)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gammino, M.; Gioia, C.; Maio, A.; Scaffaro, R.; Lo Re, G. Chemical-free Reactive Melt Processing of Biosourced Poly(butylene-succinate-adipate) for Improved Mechanical Properties and Recyclability. ACS Appl. Polym. Mater. 2024, 6, 5866–5877. [Google Scholar] [CrossRef]
- Nomadolo, N.; Dada, O.E.; Swanepoel, A.; Mokhena, T.; Muniyasamy, S. A Comparative Study on the Aerobic Biodegradation of the Biopolymer Blends of Poly(butylene succinate), Poly(butylene adipate terephthalate) and Poly(lactic acid). Polymers 2022, 14, 1894. [Google Scholar] [CrossRef]
- Padermshoke, A.; An, Y.J.; Kajiwara, T.; Masunaga, H.; Kobayashi, Y.; Ito, H.; Sasaki, S.; Noguchi, H.; Kusuno, A.; Takahara, A. Photooxidation-induced conformational changes and degradation behaviors of poly(butylene succinate) and poly(butylene succinate-adipate). J. Polym. Sci. 2024, 62, 3794–3807. [Google Scholar] [CrossRef]
- Verma, S.K.; Prasad, A.; Sonika; Katiyar, V. State of art review on sustainable biodegradable polymers with a market overview for sustainability packaging. Mater. Today Sustain. 2024, 26, 100776. [Google Scholar] [CrossRef]
- Prieur, M.; Sudre, G.; Gouanvé, F.; Fulchiron, R.; Espuche, E. Effect of mechanical stretching and low amount of silica adding on gas transport properties of poly(butylene succinate-co-adipate) films. Polymer 2024, 304, 127135. [Google Scholar] [CrossRef]
- Nath, D.; Misra, M.; Al-Daoud, F.; Mohanty, A.K. Studies on poly(butylene succinate) and poly(butylene succinate-adipate)-based biodegradable plastics for sustainable flexible packaging and agricultural applications: A comprehensive review. RSC Sustain. 2025, 3, 1267–1302. [Google Scholar] [CrossRef]
- Apicella, A.; Molinari, G.; Gigante, V.; Pietrosanto, A.; Incarnato, L.; Aliotta, L.; Lazzeri, A. Poly(lactic acid) (PLA)/poly(butylene succinate adipate) (PBSA) films with Micro fibrillated cellulose (MFC) and cardanol for packaging applications. Cellulose 2024, 31, 9173–9190. [Google Scholar] [CrossRef]
- Champa-Bujaico, E.; Díez-Pascual, A.M.; Garcia-Diaz, P.; Sessini, V.; Mosquera, M.E.G. Machine learning algorithms to optimize the properties of bio-based poly (butylene succinate-co- butylene adipate) nanocomposites with carbon nanotubes. Ind. Crop Prod. 2024, 219, 119018. [Google Scholar] [CrossRef]
- Possari, L.T.; Cosas-Fernandes, J.P.; Bettini, S.H.P. Compatibility and interphase properties of poly(butylene succinate-co-adipate) (PBSA)/Kraft lignin films assessed by nanomechanical analyses. Polymer 2022, 254, 125065. [Google Scholar] [CrossRef]
- Niang, B.; Schiavone, N.; Askanian, H.; Verney, V.; Ndiaye, D.; Diop, A.B. Development and Characterization of PBSA-Based Green Composites in 3D-Printing by Fused Deposition Modelling. Materials 2022, 15, 7570. [Google Scholar] [CrossRef]
- Delorme, A.E.; Radusin, T.; Myllytie, P.; Verney, V.; Askanian, H. Enhancement of Gas Barrier Properties and Durability of Poly(butylene succinate-co-butylene adipate)-Based Nanocomposites for Food Packaging Applications. Nanomaterials 2022, 12, 978. [Google Scholar] [CrossRef] [PubMed]
- Aliotta, L.; Gigante, V.; Dal Pont, B.; Miketa, F.; Coltelli, M.B.; Lazzeri, A. Tearing fracture of poly(lactic acid) (PLA)/poly(butylene succinate-co-adipate) (PBSA) cast extruded films: Effect of the PBSA content. Eng. Fract. Mech. 2023, 289, 109450. [Google Scholar] [CrossRef]
- Malz, F.; Arndt, J.H.; Balko, J.; Barton, B.; Büsse, T.; Imhof, D.; Pfaendner, R.; Rode, K.; Brüll, R. Analysis of the molecular heterogeneity of poly(lactic acid)/poly(butylene succinate-co-adipate) blends by hyphenating size exclusion chromatography with nuclear magnetic resonance and infrared spectroscopy. J. Chromatogr. A 2021, 1638, 461819. [Google Scholar] [CrossRef] [PubMed]
- Puekpoonpoal, N.; Phattarateera, S.; Kerddonfag, N.; Aht-Ong, D. Morphology development of PLAs with different stereo-regularities in ternary blend PBSA/PBS/PLA films. Polym.-Plast. Technol. Mater. 2021, 60, 1672–1685. [Google Scholar] [CrossRef]
- Altieri, R.; Seggiani, M.; Esposito, A.; Cinelli, P.; Stanzione, V. Thermoplastic Blends Based on Poly(Butylene Succinate-co-Adipate) and Different Collagen Hydrolysates from Tanning Industry-II: Aerobic Biodegradation in Composting Medium. J. Polym. Environ. 2021, 29, 3375–3388. [Google Scholar] [CrossRef]
- Strangis, G.; Rossi, D.; Cinelli, P.; Seggiani, M. Seawater Biodegradable Poly(butylene succinate-adipate)-Wheat Bran Biocomposites. Materials 2023, 16, 2593. [Google Scholar] [CrossRef]
- Cappello, M.; Rossi, D.; Filippi, S.; Cinelli, P.; Seggiani, M. Wood Residue-Derived Biochar as a Low-Cost, Lubricating Filler in Poly(butylene succinate-adipate) Biocomposites. Materials 2023, 16, 570. [Google Scholar] [CrossRef]
- Thummarungsan, N.; Sirivat, A. Magneto-Responsive Biopolymer Composite Based on Plasticized Poly(Butylene Succinate-Co-Butylene Adipate) and Fe3O4 for Flexible Actuator Application. J. Polym. Environ. 2024, 32, 3246–3257. [Google Scholar] [CrossRef]
- Thummarungsan, N.; Rotjanasuworapong, K.; Pattavarakorn, D.; Sirivat, A. Poly(o-phenylenediamine)/poly(butylene succinate-co-butylene adipate) blend as electroactive material for actuator application. Mater. Chem. Phys. 2023, 298, 127484. [Google Scholar] [CrossRef]
- Soulenthone, P.; Suzuki, M.; Tachibana, Y.; Furukori, M.; Saito, T.; Kawamura, R.; Bankole, P.O.; Kasuya, K.I. A marine-derived bacterium capable of degrading poly(butylene succinate-co-adipate), poly(e-caprolactone), and poly(butylene adipate-co-terephthalate) in marine ecosystems. Polym. Degrad. Stabil. 2025, 232, 111161. [Google Scholar] [CrossRef]
- Rossi, D.; Cappello, M.; Filippi, S.; Cinelli, P.; Seggiani, M. Potential of Lauryl Gallate as Stability and Recyclability Improver of Poly (Butylene succinate-adipate). J. Polym. Environ. 2024, 32, 1202–1216. [Google Scholar] [CrossRef]
- Chien, H.L.; Tsai, Y.T.; Tseng, W.S.; Wu, J.A.; Kuo, S.L.; Chang, S.L.; Huang, S.J.; Liu, C.T. Biodegradation of PBSA Films by Elite Isolates and Farmland Soil. Polymers 2022, 14, 1320. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Camargo, R.A.; Liu, G.M.; Cavallo, D.; Wang, D.J.; Müller, A.J. Effect of the Crystallization Conditions on the Exclusion/Inclusion Balance in Biodegradable Poly(butylene succinate-butylene adipate) Copolymers. Biomacromolecules 2020, 21, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Galyon, H.; Vibostok, S.; Duncan, J.; Ferreira, G.; Whittington, A.; Havens, K.; McDevitt, J.; Cockrum, R. Digestibility Kinetics of Polyhydroxyalkanoate and Poly(butylene succinate-adipate) after In Vitro Fermentation in Rumen Fluid. Polymers 2022, 14, 2103. [Google Scholar] [CrossRef]
- La Fuente, C.I.A.; Maniglia, B.C.; Tadini, C.C. Biodegradable polymers: A review about biodegradation and its implications and applications. Packag. Technol. Sci. 2023, 36, 81–95. [Google Scholar] [CrossRef]
- Elenskaya, N.; Vindokurov, I.; Sadyrin, E.; Nikolaev, A.; Tashkinov, M. Experimental Evaluation of the Effect of Degradation on the Mechanical Behavior and Morphometric Characteristics of Functionally Graded Polymer Scaffolds. Polymers 2024, 16, 3474. [Google Scholar] [CrossRef]
- Yao, X.; Yang, X.; Lu, Y.S.; Qiu, Y.Y.; Zeng, Q.D. Review of the Synthesis and Degradation Mechanisms of Some Biodegradable Polymers in Natural Environments. Polymers 2025, 17, 66. [Google Scholar] [CrossRef]
- Oleksy, M.; Dynarowicz, K.; Aebisher, D. Advances in Biodegradable Polymers and Biomaterials for Medical Applications-A Review. Molecules 2023, 28, 6213. [Google Scholar] [CrossRef]
- Sabalina, A.; Platnieks, O.; Gaidukova, G.; Aunins, A.; Eiduks, T.V.; Gaidukovs, S. Thermomechanical and mechanical analysis of polylactic acid/polyhydroxyalkanoate/poly(butylene succinate-adipate) binary and ternary blends. RSC Adv. 2025, 15, 501–512. [Google Scholar] [CrossRef]
- Baidurah, S.; Takada, S.; Shimizu, K.; Yasue, K.; Arimoto, S.; Ishida, Y.; Yamane, T.; Ohtani, H. Evaluation of Biodegradability of Poly(Butylene Succinate-Butylene Adipate) on the Basis of Copolymer Composition Determined by Thermally Assisted Hydrolysis and Methylation-Gas Chromatography. Int. J. Polym. Anal. Charact. 2012, 17, 29–37. [Google Scholar] [CrossRef]
- Ruggero, F.; Carretti, E.; Gori, R.; Lotti, T.; Lubello, C. Monitoring of degradation of starch-based biopolymer film under different composting conditions, using TGA, FTIR and SEM analysis. Chemosphere 2020, 246, 125770. [Google Scholar] [CrossRef]
- Sessa, V.; Bhandari, R. Composting Heat Recovery for Residential Consumption: An Assessment of Viability. Sustainability 2023, 15, 4006. [Google Scholar] [CrossRef]
- Apicella, A.; Malafeev, K.V.; Scarfato, P.; Incarnato, L. Generation of Microplastics from Biodegradable Packaging Films Based on PLA, PBS and Their Blend in Freshwater and Seawater. Polymers 2024, 16, 2268. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, C.; Yi, X.; Li, L.; Zhou, J.; Han, X.; Gao, Y. Research and Evaluation of a New Autogenic Acid System Suitable for Acid Fracturing of a High-Temperature Reservoir. ACS Omega 2020, 5, 20734–20738. [Google Scholar] [CrossRef]
- Wang, Y.; Murcia Valderrama, M.A.; van Putten, R.-J.; Davey, C.J.E.; Tietema, A.; Parsons, J.R.; Wang, B.; Gruter, G.-J.M. Biodegradation and Non-Enzymatic Hydrolysis of Poly(Lactic-co-Glycolic Acid) (PLGA12/88 and PLGA6/94). Polymers 2022, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Shimizu, K.; Sato, Y. Hydrolytic degradation of poly(L-lactic acid): Combined effects of UV treatment and crystallization. J. Appl. Polym. Sci. 2012, 125, 2394–2406. [Google Scholar] [CrossRef]
- Bikiaris, D.N.; Papageorgiou, G.Z.; Achilias, D.S. Synthesis and comparative biodegradability studies of three poly(alkylene succinate)s. Polym. Degrad. Stabil. 2006, 91, 31–43. [Google Scholar] [CrossRef]
- Marten, E.; Müller, R.J.; Deckwer, W.D. Studies on the enzymatic hydrolysis of polyesters I.: Low molecular mass model esters and aliphatic polyesters. Polym. Degrad. Stabil. 2003, 80, 485–501. [Google Scholar] [CrossRef]
- Tsuji, H.; Okumura, A. Crystallization and hydrolytic/thermal degradation of a novel stereocomplexationable blend of poly(L-2-hydroxybutyrate) and poly(D-2-hydroxybutyrate). Polym. J. 2011, 43, 317–324. [Google Scholar] [CrossRef]
- Rangari, D.; Vasanthan, N. Study of Strain-Induced Crystallization and Enzymatic Degradation of Drawn Poly(L-lactic acid) (PLLA) Films. Macromolecules 2012, 45, 7397–7403. [Google Scholar] [CrossRef]
- Muthuraj, R.; Misra, M.; Mohanty, A.K. Hydrolytic degradation of biodegradable polyesters under simulated environmental conditions. J. Appl. Polym. Sci. 2015, 132, 42189. [Google Scholar] [CrossRef]
- Rabello, M.S.; White, J.R. Crystallization and melting behaviour of photodegraded polypropylene. 2. Re-crystallization of degraded molecules. Polymer 1997, 38, 6389–6399. [Google Scholar] [CrossRef]
- Somrang, N.; Nithitanakul, M.; Grady, B.P.; Supaphol, P. Non-isothermal melt crystallization kinetics for ethylene-acrylic acid copolymers and ethylene-methyl acrylate-acrylic acid terpolymers. Eur. Polym. J. 2004, 40, 829–838. [Google Scholar] [CrossRef]
- Liu, F.Y.; Xu, C.L.; Zeng, J.B.; Li, S.L.; Wang, Y.Z. Non-isothermal crystallization kinetics of biodegradable poly(butylene succinate-co-diethylene glycol succinate) copolymers. Thermochim. Acta 2013, 568, 38–45. [Google Scholar] [CrossRef]
- Le Delliou, B.; Vitrac, O.; Castro, M.; Bruzaud, S.; Domenek, S. Characterization of a new bio-based and biodegradable blends of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and poly(butylene-co-succinate-co-adipate). J. Appl. Polym. Sci. 2022, 139, 52124. [Google Scholar] [CrossRef]
- Ren, M.Q.; Song, J.B.; Song, C.L.; Zhang, H.L.; Sun, X.H.; Chen, Q.Y.; Zhang, H.F.; Mo, Z.S. Crystallization kinetics and morphology of poly(butylene succinate-co-adipate). J. Polym. Sci. Part B-Polym. Phys. 2005, 43, 3231–3241. [Google Scholar] [CrossRef]
- Papadimitriou, S.A.; Papageorgiou, G.Z.; Bikiaris, D.N. Crystallization and enzymatic degradation of novel poly(epsilon-caprolactone-co-propylene succinate) copolymers. Eur. Polym. J. 2008, 44, 2356–2366. [Google Scholar] [CrossRef]
- Kratochvíl, J.; Kelnar, I. A simple method of evaluating non-isothermal crystallization kinetics in multicomponent polymer systems. Polym. Test. 2015, 47, 79–86. [Google Scholar] [CrossRef]
- Hoffman, J.D. Role of Reptation in the Rate of Crystallization of Polyethylene Fractions from the Melt. Polymer 1982, 23, 656–670. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Miller, R.L. Test of the Reptation Concept—Crystal-Growth Rate as a Function of Molecular-Weight in Polyethylene Crystallized from the Melt. Macromolecules 1988, 21, 3038–3051. [Google Scholar] [CrossRef]
- Avella, M.; Dellerba, R.; Martuscelli, E. Fiber reinforced polypropylene: Influence of IPP molecular weight on morphology, crystallization, and thermal and mechanical properties. Polym. Compos. 1996, 17, 288–299. [Google Scholar] [CrossRef]
- Skoglund, P.; Fransson, A. Continuous cooling and isothermal crystallization of polycaprolactone. J. Appl. Polym. Sci. 1996, 61, 2455–2465. [Google Scholar] [CrossRef]
- Alizadeh, A.; Sohn, S.; Quinn, J.; Marand, H.; Shank, L.C.; Iler, H.D. Influence of structural and topological constraints on the crystallization and melting behavior of polymers: 3. Bisphenol A polycarbonate. Macromolecules 2001, 34, 4066–4078. [Google Scholar] [CrossRef]
- Chae, H.G.; Kim, B.C.; Im, S.S.; Han, Y.K. Effect of molecular weight and branch structure on the crystallization and rheological properties of poly(butylene adipate). Polym. Eng. Sci. 2001, 41, 1133–1139. [Google Scholar] [CrossRef]
- Chen, X.D.; Hou, G.; Chen, Y.J.; Yang, K.; Dong, Y.P.; Zhou, H. Effect of molecular weight on crystallization, melting behavior and morphology of poly(trimethylene terephalate). Polym. Test. 2007, 26, 144–153. [Google Scholar] [CrossRef]
- He, Y.; Fan, Z.Y.; Hu, Y.F.; Wu, T.; Wei, J.; Li, S.M. DSC analysis of isothermal melt-crystallization, glass transition and melting behavior of poly(L-lactide) with different molecular weights. Eur. Polym. J. 2007, 43, 4431–4439. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Bikiaris, D.N.; Achilias, D.S. Effect of molecular weight on the cold-crystallization of biodegradable poly(ethylene succinate). Thermochim. Acta 2007, 457, 41–54. [Google Scholar] [CrossRef]
- Yang, J.J.; Pan, P.J.; Dong, T.; Inoue, Y. Crystallization kinetics and crystalline structure of biodegradable Poly(ethylene adipate). Polymer 2010, 51, 807–815. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of Phase Change. II Transformation-Time Relations for Random Distribution of Nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Fang, H.; Wu, F.J. Nonisothermal Crystallization Kinetics of Poly(butylene terephthalate)/Multiwalled Carbon Nanotubes Nanocomposites Prepared by Polymerization. J. Appl. Polym. Sci. 2014, 131, 40849. [Google Scholar] [CrossRef]
- Svoboda, P.; Dvorackova, M.; Svobodova, D. Influence of biodegradation on crystallization of poly (butylene adipate-co-terephthalate). Polym. Adv. Technol. 2019, 30, 552–562. [Google Scholar] [CrossRef]
- Jiang, G.; Wang, H.T.; Yu, L.; Li, H.L. Improving crystallization properties of PBSA by blending PBS as a polymeric nucleating agent to prepare high-performance PPC/PBSA/AX8900 blown films. Polym. Eng. Sci. 2022, 62, 1166–1177. [Google Scholar] [CrossRef]
- Bandyopadhyay, J.; Ray, S.S. Effect of Nanoclay on the Nonisothermal Crystallization of Poly(propylene) and its Blend with Poly[(butylene succinate)-co-adipate]. Mol. Cryst. Liq. Cryst. 2012, 556, 176–190. [Google Scholar] [CrossRef]
- Chen, C.; Fei, B.; Peng, S.W.; Zhuang, Y.G.; Dong, L.S.; Feng, Z.L. Nonisothermal crystallization and melting behavior of poly(3-hydroxybutyrate) and maleated poly(3-hydroxybutyrate). Eur. Polym. J. 2002, 38, 1663–1670. [Google Scholar] [CrossRef]
- Xu, G.; Shi, W.F.; Hu, P.; Mo, S.P. Crystallization kinetics of polypropylene with hyperbranched polyurethane acrylate being used as a toughening agent. Eur. Polym. J. 2005, 41, 1828–1837. [Google Scholar] [CrossRef]
- Huang, J.W.; Wen, Y.L.; Kang, C.C.; Tseng, W.J.; Yeh, M.Y. Nonisothermal crystallization of high density polyethylene and nanoscale calcium carbonate composites. Polym. Eng. Sci. 2008, 48, 1268–1278. [Google Scholar] [CrossRef]
- Huang, J.W.; Hung, Y.C.; Wen, Y.L.; Kang, C.C.; Yeh, M.Y. Polylactide/Nano- and Micro-Scale Silica Composite Films. II. Melting Behavior and Cold Crystallization. J. Appl. Polym. Sci. 2009, 112, 3149–3156. [Google Scholar] [CrossRef]
- Papageorgiou, D.G.; Papageorgiou, G.Z.; Bikiaris, D.N.; Chrissafis, K. Crystallization and melting of propylene-ethylene random copolymers. Homogeneous nucleation and β-nucleating agents. Eur. Polym. J. 2013, 49, 1577–1590. [Google Scholar] [CrossRef]
- Wellen, R.M.R.; Canedo, E.L. On the Kissinger equation and the estimate of activation energies for non-isothermal cold crystallization of PET. Polym. Test. 2014, 40, 33–38. [Google Scholar] [CrossRef]
- Shao, Y.T.; Wu, C.G.; Cheng, S.Y.; Zhou, F.; Yan, H.B. Effects of toughening propylene/ethylene graft copolymer on the crystallization behavior and mechanical properties of polypropylene random-copolymerized with a small amount of ethylene. Polym. Test. 2015, 41, 252–263. [Google Scholar] [CrossRef]
- Somsunan, R.; Mainoiy, N. Isothermal and non-isothermal crystallization kinetics of PLA/PBS blends with talc as nucleating agent. J. Therm. Anal. Calorim. 2020, 139, 1941–1948. [Google Scholar] [CrossRef]
- Ozawa, T. Kinetics of Non-Isothermal Crystallization. Polymer 1971, 12, 150–158. [Google Scholar] [CrossRef]
- Nie, W.C.; Xiao, Q.; Wu, J.M.; Song, F.; Wang, X.L.; Wang, Y.Z. Dendritic crystallization and morphology control of random poly(p-dioxanone-co-butylene-co-succinate) copolyesters. Eur. Polym. J. 2018, 108, 76–84. [Google Scholar] [CrossRef]
- Qiu, S.T.; Su, Z.Q.; Qiu, Z.B. Crystallization Kinetics, Morphology, and Mechanical Properties of Novel Biodegradable Poly(ethylene succinate-co-ethylene suberate) Copolyesters. Ind. Eng. Chem. Res. 2016, 55, 10286–10293. [Google Scholar] [CrossRef]
- Xue, P.; Qiu, Z.B. Synthesis, thermal properties, and crystallization kinetics of novel biodegradable poly(ethylene succinate-co-diethylene glycol succinate) copolyesters. Thermochim. Acta 2015, 606, 45–52. [Google Scholar] [CrossRef]
- Liu, G.C.; Zeng, J.B.; Huang, C.L.; Jiao, L.; Wang, X.L.; Wang, Y.Z. Crystallization Kinetics and Spherulitic Morphologies of Biodegradable Poly(butylene succinate-co-diethylene glycol succinate) Copolymers. Ind. Eng. Chem. Res. 2013, 52, 1591–1599. [Google Scholar] [CrossRef]
- Yang, Y.; Qiu, Z.B. Crystallization kinetics and morphology of biodegradable poly(butylene succinate-co-ethylene succinate) copolyesters: Effects of comonomer composition and crystallization temperature. Crystengcomm 2011, 13, 2408–2417. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Bikiaris, D.N. Synthesis, cocrystallization, and enzymatic degradation of novel poly(butylene-co-propylene succinate) copolymers. Biomacromolecules 2007, 8, 2437–2449. [Google Scholar] [CrossRef]
- Crist, B.; Schultz, J.M. Polymer spherulites: A critical review. Prog. Polym. Sci. 2016, 56, 1–63. [Google Scholar] [CrossRef]
- Jariyavidyanont, K.; Schick, C.; Janke, A.; Androsch, R. Homogeneous Crystal Nucleation in Poly (butylene succinate-butylene adipate): Challenging the Nuclei-Transfer Step in Tammann’s Method. J. Phys. Chem. B 2024, 128, 12008–12021. [Google Scholar] [CrossRef]
- Chen, M.; Chung, C.T. Analysis of crystallization kinetics of poly(ether ether ketone) by a nonisothermal method. J. Polym. Sci. Part B-Polym. Phys. 1998, 36, 2393–2399. [Google Scholar] [CrossRef]
- Nouira, S.; Hassine, T.; Fitoussi, J.; Shirinbayan, M.; Gamaoun, F.; Tcharkhtchi, A. Non-isothermal crystallization kinetics and its effect on the mechanical properties of homopolymer isotactic polypropylene. J. Polym. Res. 2022, 29, 26. [Google Scholar] [CrossRef]
- Jasiurkowska-Delaporte, M.; Rozwadowski, T.; Juszynska-Galazka, E. Kinetics of Non-Isothermal and Isothermal Crystallization in a Liquid Crystal with Highly Ordered Smectic Phase as Reflected by Differential Scanning Calorimetry, Polarized Optical Microscopy and Broadband Dielectric Spectroscopy. Crystals 2019, 9, 205. [Google Scholar] [CrossRef]
- Dobrzynska-Mizera, M.; Dutkiewicz, M.; Sterzynski, T.; Di Lorenzo, M.L. Polypropylene-based composites containing sorbitol-based nucleating agent and siloxane-silsesquioxane resin. J. Appl. Polym. Sci. 2016, 133, 43476. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Cimmino, S.; Silvestre, C. Nonisothermal crystallization of isotactic polypropylene blended with poly(alpha-pinene). 2. Growth rates. Macromolecules 2000, 33, 3828–3832. [Google Scholar] [CrossRef]
- Lindstrom, A.; Albertsson, A.C.; Hakkarainen, M. Development of a solid-phase extraction method for simultaneous extraction of adipic acid, succinic acid and 1,4-butanediol formed during hydrolysis of poly(butylene adipate) and poly(butylene succinate). J. Chromatogr. A 2004, 1022, 171–177. [Google Scholar] [CrossRef]
- Ahn, B.D.; Kim, S.H.; Kim, Y.H.; Yang, J.S. Synthesis and characterization of the biodegradable copolymers from succinic acid and adipic acid with 1,4-butanediol. J. Appl. Polym. Sci. 2001, 82, 2808–2826. [Google Scholar] [CrossRef]
- Perez-Camargo, R.K.; Fernandez-d’Arlas, B.; Cavallo, D.; Debuissy, T.; Pollet, E.; Averous, L.; Muller, A.J. Tailoring the Structure, Morphology, and Crystallization of Isodimorphic Poly(butylene succinate-ran-butylene adipate) Random Copolymers by Changing Composition and Thermal History. Macromolecules 2017, 50, 597–608. [Google Scholar] [CrossRef]
- Baidurah, S.; Takada, S.; Shimizu, K.; Ishida, Y.; Yamane, T.; Ohtani, H. Evaluation of biodegradation behavior of poly(butylene succinate-co-butylene adipate) with lowered crystallinity by thermally assisted hydrolysis and methylation-gas chromatography. J. Anal. Appl. Pyrolysis 2013, 103, 73–77. [Google Scholar] [CrossRef]
- Tserki, V.; Matzinos, P.; Pavlidou, E.; Panayiotou, C. Biodegradable aliphatic polyesters.: Part II.: Synthesis and characterization of chain extended poly(butylene succinate-butylene adipate). Polym. Degrad. Stabil. 2006, 91, 377–384. [Google Scholar] [CrossRef]
- Tserki, V.; Matzinos, P.; Pavlidou, E.; Vachliotis, D.; Panayiotou, C. Biodegradable aliphatic polyesters. Part I. Properties and biodegradation of poly(butylene succinate-co-butylene adipate). Polym. Degrad. Stabil. 2006, 91, 367–376. [Google Scholar] [CrossRef]
- ISO 527-3:2018; Plastics—Determination of tensile properties—Part 3: Test conditions for films and sheets. International Organization for Standardization: Geneva, Switzerland, 2018.
- D4703-24; Standard Practice for Compression Molding Thermoplastic Materials into Test Specimens, Plaques, or Sheets. ASTM International: West Conshohocken, PA, USA, 2024.







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svarcova, A.; Dvorackova, M.; Svoboda, P. Influence of Hydrolysis on Non-Isothermal Crystallization of Poly(Butylene Succinate-Co-Adipate) (PBSA). Molecules 2025, 30, 2252. https://doi.org/10.3390/molecules30112252
Svarcova A, Dvorackova M, Svoboda P. Influence of Hydrolysis on Non-Isothermal Crystallization of Poly(Butylene Succinate-Co-Adipate) (PBSA). Molecules. 2025; 30(11):2252. https://doi.org/10.3390/molecules30112252
Chicago/Turabian StyleSvarcova, Anna, Marie Dvorackova, and Petr Svoboda. 2025. "Influence of Hydrolysis on Non-Isothermal Crystallization of Poly(Butylene Succinate-Co-Adipate) (PBSA)" Molecules 30, no. 11: 2252. https://doi.org/10.3390/molecules30112252
APA StyleSvarcova, A., Dvorackova, M., & Svoboda, P. (2025). Influence of Hydrolysis on Non-Isothermal Crystallization of Poly(Butylene Succinate-Co-Adipate) (PBSA). Molecules, 30(11), 2252. https://doi.org/10.3390/molecules30112252