
Exploring the Influence of Chalcogens on Metalloporphyrins: A DFT Study

Abstract

1. Introduction
2. Results
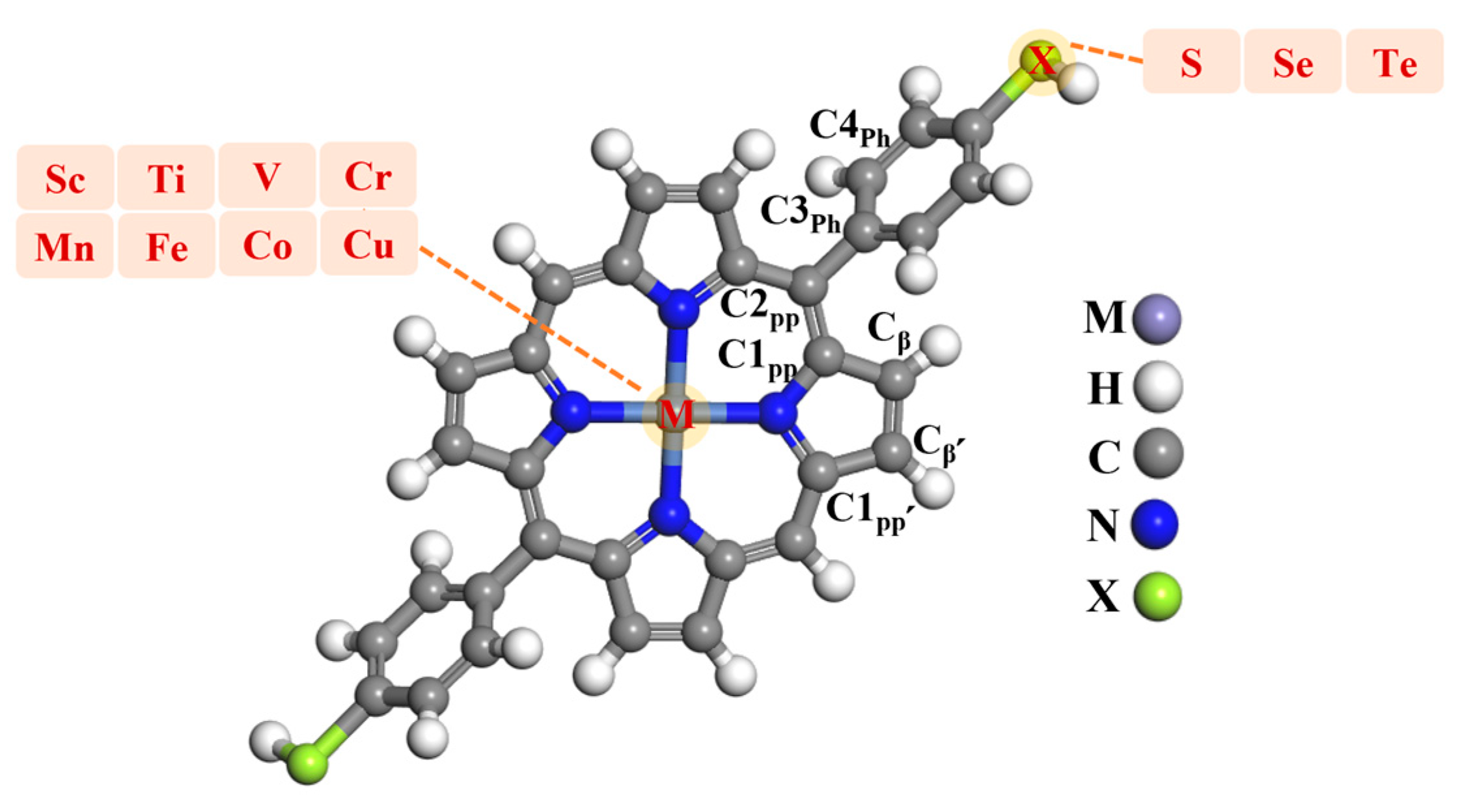
2.1. Metalloporphyrins Composition
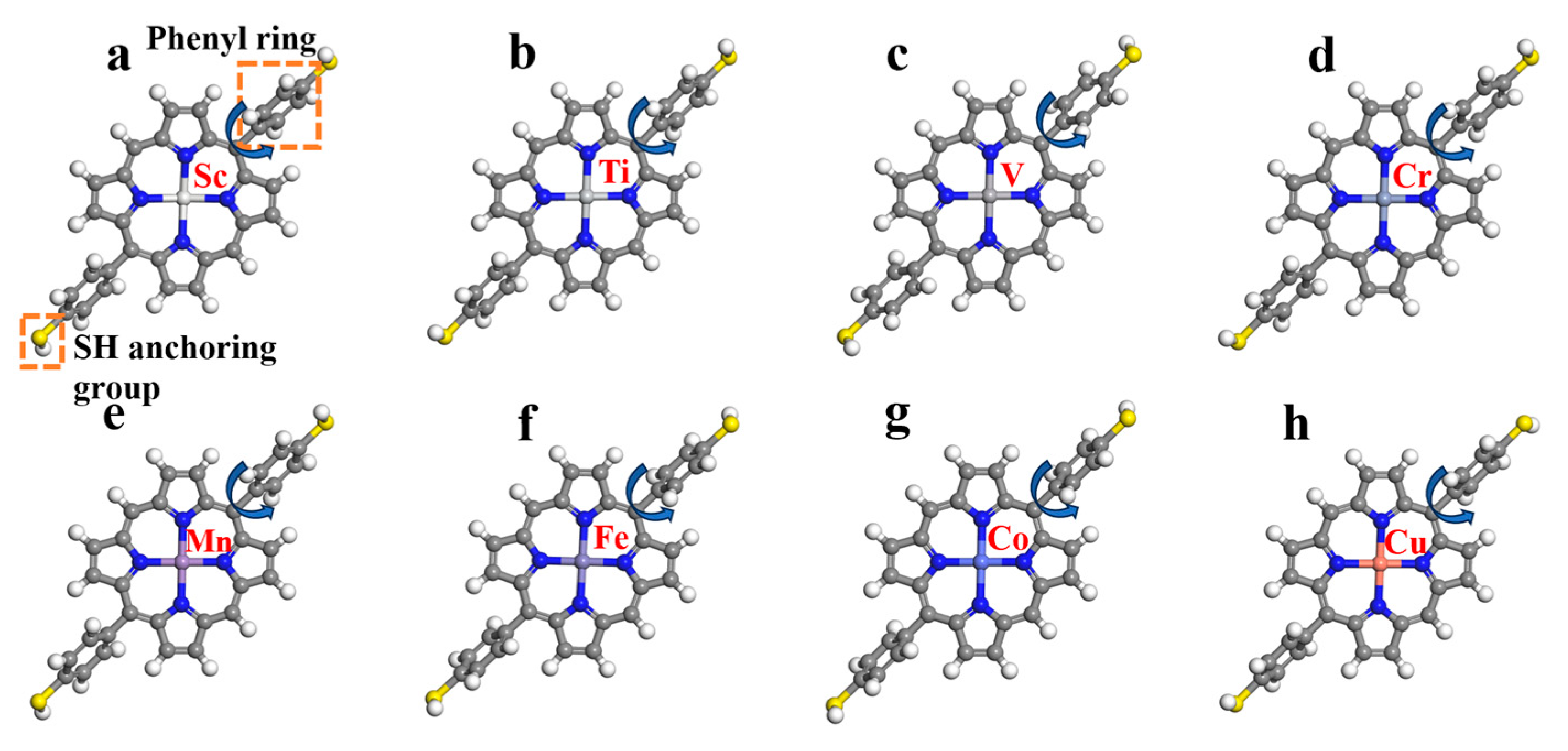
2.2. Structural Analysis of Metalloporphyrins with Thiol Termination
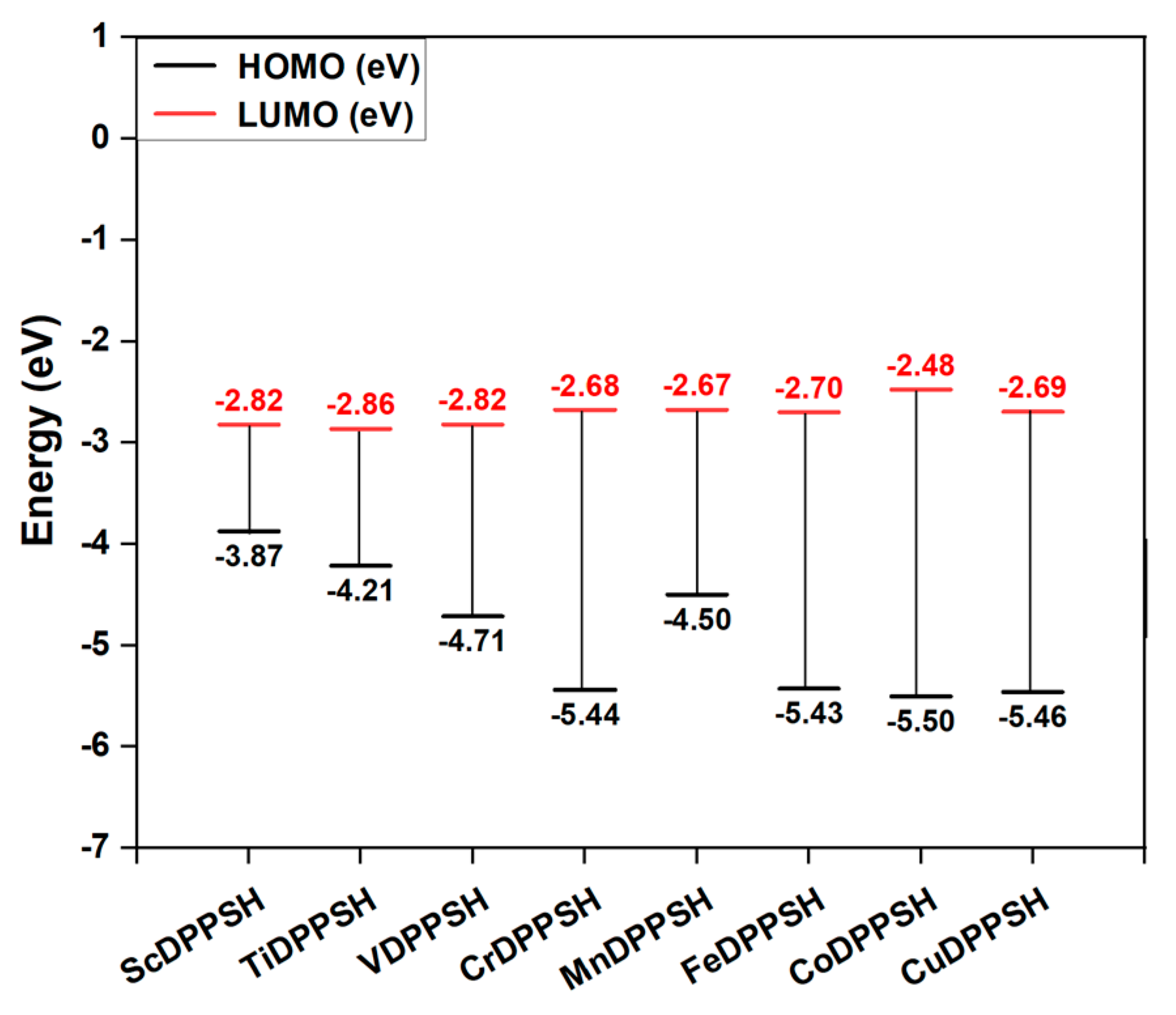
2.3. Electronic Structure Properties
2.4. Conceptual DFT-Based Electronic Structure Reactivity Descriptors for Thiol-Terminated Metalloporphyrins
2.5. Comparison of Thiol-Terminated Metalloporphryins to -SeH- and -TeH-Terminated Metalloporphyrins
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
References
- Zhan, X.; Kim, D.; Ullah, Z.; Lee, W.; Gross, Z.; Churchill, D.G. Photophysics of corroles and closely related systems for emergent solar energy, medicinal, and materials science applications. Coord. Chem. Rev. 2023, 495, 215363. [Google Scholar] [CrossRef]
- Deng, D.; Chang, Y.; Liu, W.; Ren, M.; Xia, N.; Hao, Y. Advancements in Biosensors Based on the Assembles of Small Organic Molecules and Peptides. Biosensors 2023, 13, 773. [Google Scholar] [CrossRef] [PubMed]
- Arnaut, L.G.; Pereira, M.M. Overcoming the challenges of infrared photosensitizers in photodynamic therapy: The making of redaporfin. ChemComm. 2023, 59, 9457–9468. [Google Scholar] [CrossRef]
- Lee, H.; Park, H.; Ryu, D.Y.; Jang, W.-D. Porphyrin-based supramolecular polymers. Chem. Soc. Rev. 2023, 52, 1947–1974. [Google Scholar] [CrossRef]
- Monteiro, C.J.; Faustino, M.A.F.; Serpa, C. Porphyrin-Based Compounds: Synthesis and Application. Molecules 2023, 28, 7108. [Google Scholar] [CrossRef]
- Ostovan, A.; Papior, N.; Naghavi, S.S. Half-metallic porphyrin-based molecular junctions for spintronic applications. Phys. Rev. B 2021, 104, 235435. [Google Scholar] [CrossRef]
- Zhou, X.-Y.; He, B.; Zhang, Y.; Ni, J.-Y.; Liu, Q.-P.; Wang, M.; Shen, H.-M.; She, Y.-B. Selective and Efficient Catalytic Oxygenation of Alkyl Aromatics Employing H2O2 Catalyzed by Simple Porphyrin Iron (II) under Mild Conditions. Processes 2023, 11, 1187. [Google Scholar] [CrossRef]
- Martin, D.J.; Mercado, B.Q.; Mayer, J.M. All Four Atropisomers of Iron Tetra (o-N, N, N-Trimethylanilinium) Porphyrin in Both the Ferric and Ferrous States. Inorg. Chem. 2021, 60, 5240–5251. [Google Scholar] [CrossRef]
- Jurow, M.; Schuckman, A.E.; Batteas, J.D.; Drain, C.M. Porphyrins as molecular electronic components of functional devices. Coord. Chem. Rev. 2010, 254, 2297–2310. [Google Scholar] [CrossRef]
- Rask, A.; Zimmerman, P.M. The many-body electronic interactions of Fe (II)–porphyrin. J. Chem. Phys. 2022, 156, 094110. [Google Scholar] [CrossRef]
- Gross, A.J.; Bucher, C.; Coche-Guerente, L.; Labbé, P.; Downard, A.J.; Moutet, J.-C. Nickel (II) tetraphenylporphyrin modified surfaces via electrografting of an aryldiazonium salt. Electrochem. Commun. 2011, 13, 1236–1239. [Google Scholar] [CrossRef]
- Chen, D.; Jin, Z.; Xing, H. Titanium–porphyrin metal–organic frameworks as visible-light-driven catalysts for highly efficient sonophotocatalytic reduction of Cr (VI). Langmuir 2022, 38, 12292–12299. [Google Scholar] [CrossRef]
- Chang, I.J.; Jeon, Y.S.; Hwang, K.J. Synthesis and band gap analysis of designed porphyrin derivatives containing electron donating and accepting group. Bull. Korean Chem. Soc. 2019, 40, 173–179. [Google Scholar] [CrossRef]
- Sanders, J.K.; Bampos, N.; Clyde-Watson, Z.; Darling, S.L.; Hawley, J.C.; Kim, H.J.; Mak, C.C.; Webb, S.J. Axial coordination chemistry of metalloporphyrins. ChemInform 2003, 34. [Google Scholar] [CrossRef]
- Windischbacher, A.; Puschnig, P. Computational study on the adsorption of small molecules to surface-supported Ni-porphyrins. Inorganica Chim. Acta 2023, 558, 121719. [Google Scholar] [CrossRef]
- Zhou, Q.; Yamada, A.; Feng, Q.; Hoskins, A.; Dunietz, B.D.; Lewis, K.M. Modification of molecular conductance by in situ deprotection of thiol-based porphyrin. ACS Appl. Mater. Interfaces 2017, 9, 15901–15906. [Google Scholar] [CrossRef]
- Bashir, B.; Alotaibi, M.M.; Clayborne, A.Z. Computational investigation of structural, electronic, and spectroscopic properties of Ni and Zn metalloporphyrins with varying anchoring groups. J. Chem. Phys. 2024, 160, 134305. [Google Scholar] [CrossRef]
- Sarmah, A.; Hobza, P. Directly linked metalloporphyrins: A quest for bio-inspired materials. Mater. Adv. 2020, 1, 1895–1908. [Google Scholar] [CrossRef]
- Schuth, N.; Mebs, S.; Huwald, D.; Wrzolek, P.; Schwalbe, M.; Hemschemeier, A.; Haumann, M. Effective intermediate-spin iron in O2-transporting heme proteins. Proc. Natl. Acad. Sci. USA 2017, 114, 8556–8561. [Google Scholar] [CrossRef]
- Cao, M.; Gao, A.; Liu, Y.; Zhou, Y.; Sun, Z.; Li, Y.; He, F.; Li, L.; Mo, L.; Liu, R. Theoretical study on electronic structural properties of catalytically reactive metalloporphyrin intermediates. Catalysts 2020, 10, 224. [Google Scholar] [CrossRef]
- Balducci, L.; Bianchi, D.; Bortolo, R.; D’Aloisio, R.; Ricci, M.; Tassinari, R.; Ungarelli, R. Direct oxidation of benzene to phenol with hydrogen peroxide over a modified titanium silicalite. Angew. Chem. 2003, 115, 5087–5090. [Google Scholar] [CrossRef]
- Reimers, J.; Lü, T.; Crossley, M.; Hush, N. Molecular electronic properties of fused rigid porphyrin-oligomer molecular wires. Chem. Phys. Lett. 1996, 256, 353–359. [Google Scholar] [CrossRef]
- Ayalew, M.E. DFT studies on molecular structure, thermodynamics parameters, HOMO-LUMO and spectral analysis of pharmaceuticals compound quinoline (Benzo [b] Pyridine). J. Biophys. Chem. 2022, 13, 29–42. [Google Scholar] [CrossRef]
- Gershoni-Poranne, R.; Rahalkar, A.P.; Stanger, A. The predictive power of aromaticity: Quantitative correlation between aromaticity and ionization potentials and HOMO–LUMO gaps in oligomers of benzene, pyrrole, furan, and thiophene. Phys. Chem. Chem. Phys. 2018, 20, 14808–14817. [Google Scholar] [CrossRef]
- Ballabio, M.; Cánovas, E. Electron Transfer at Quantum Dot–Metal Oxide Interfaces for Solar Energy Conversion. ACS Nanoscience Au 2022, 2, 367–395. [Google Scholar] [CrossRef]
- Gujarathi, P.B. Recent emerging applications of porphyrins and Metalloporphyrins and their analogue in diverse areas. Pharma. Innov. 2020, 9, 80–86. [Google Scholar] [CrossRef]
- Yang, X.F.; Dong, Y.J.; Yu, H.L.; Tao, X.X.; Liu, Y.S. Rotating single molecule-based devices: Single-spin switching, negative differential electrical and thermoelectric resistance. Chem. Phys. 2024, 577, 112131. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Goerigk, L. How do DFT-DCP, DFT-NL, and DFT-D3 compare for the description of London-dispersion effects in conformers and general thermochemistry? J. Chem. Theory Comput. 2014, 10, 968–980. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Revealing the nature of intermolecular interaction and configurational preference of the nonpolar molecular dimers (H2)2,(N2)2, and (H2)(N2). J. Mol. Model. 2013, 19, 5387–5395. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Pearson, R.G. The electronic chemical potential and chemical hardness. J. Mol. Struc. THEOCHEM 1992, 255, 261–270. [Google Scholar] [CrossRef]
- Bhatia, M. An overview of conceptual-DFT based insights into global chemical reactivity of volatile sulfur compounds (VSCs). Comput. Toxicol. 2023, 29, 100295. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Parthasarathi, R.; Elango, M.; Subramanian, V.; Krishnamoorthy, B.; Gutierrez-Oliva, S.; Toro-Labbé, A.; Roy, D.; Chattaraj, P. Multiphilic descriptor for chemical reactivity and selectivity. J. Phys. Chem. A 2007, 111, 9130–9138. [Google Scholar] [CrossRef]
- Ratti, C.; Richard, P.; Tabard, A.; Guilard, R. Synthesis and characterization of a new series of titanium (IV) porphyrins co-ordinated to a disulphur or a diselenium ligand. J. Chem. Soc. Chem. Commun. 1989, 2, 69–70. [Google Scholar] [CrossRef]
- Scheidt, W.R.; Reed, C.A. Stereochemistry of the toluene solvate of. alpha,. beta,. gamma,. delta.-tetraphenylporphinatochromium (II). Inorg. Chem. 1978, 17, 710–714. [Google Scholar] [CrossRef]
- Kirner, J.F.; Reed, C.A.; Scheidt, W.R. Stereochemistry of manganese porphyrins. 2. The toluene solvate of. alpha,. beta,. gamma,. delta.-tetraphenylporphinatomanganese (II) at 20 and-175. degree. C. J. Am. Chem. Soc. 1977, 99, 1093–1101. [Google Scholar] [CrossRef]
- Wondimagegn, T.; Rauk, A. The structures and stabilities of the complexes of biologically available ligands with Fe (III)–porphine: An ab initio study. J. Phys. Chem. B 2011, 115, 569–579. [Google Scholar] [CrossRef]
- Collman, J.P.; Hoard, J.; Kim, N.; Lang, G.; Reed, C.A. Synthesis, stereochemistry, and structure-related properties of. alpha,. beta,. gamma,. delta.-tetraphenylporphinatoiron (II). J. Am. Chem. Soc. 1975, 97, 2676–2681. [Google Scholar] [CrossRef]
- Scheidt, W.R. Stereochemistry of low-spin cobalt porphyrins. III. Crystal structure and molecular stereochemistry of bis (piperidine)-. alpha,. beta,. gamma,. delta.-tetraphenylporphinatocobalt (II). J. Am. Chem. Soc. 1974, 96, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, E.B.; Miller, C.K.; Webb, L.E. Crystal and molecular structures of some metal tetraphenylporphines. J. Am. Chem. Soc. 1964, 86, 2342–2347. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian, Version 16, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2016.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | M-N | N-C1pp | C1pp-Cβ | Cβ-Cβ′ | C1pp-C2pp-C3Ph-C4Ph |
|---|---|---|---|---|---|
| ScDPPSH | 2.09 | 1.38 | 1.43 | 1.37 | 69.23 |
| TiDPPSH | 2.07 | 1.39 | 1.44 | 1.37 | 67.93 |
| VDPPSH | 2.06 | 1.38 | 1.43 | 1.36 | 68.26 |
| CrDPPSH | 2.05 | 1.38 | 1.44 | 1.36 | 69.09 |
| MnDPPSH | 2.03 | 1.38 | 1.44 | 1.36 | 69.64 |
| FeDPPSH | 2.01 | 1.38 | 1.44 | 1.36 | 70.63 |
| CoDPPSH | 1.99 | 1.37 | 1.43 | 1.36 | 71.29 |
| CuDPPSH | 2.03 | 1.37 | 1.44 | 1.36 | 69.97 |
| System | M | N | C2pp | SH |
|---|---|---|---|---|
| ScDPPSH | 0.99 | 0.01 | 0.06 | −0.50 |
| TiDPPSH | 0.50 | 0.20 | 0.08 | −0.50 |
| VDPPSH | 0.58 | 0.22 | 0.20 | −0.50 |
| CrDPPSH | 0.53 | 0.26 | 0.29 | −0.50 |
| MnDPPSH | 0.70 | 0.25 | 0.24 | −0.50 |
| FeDPPSH | 0.76 | 0.26 | 0.61 | −0.50 |
| CoDPPSH | 0.87 | 0.32 | 0.63 | −0.50 |
| CuDPPSH | 0.60 | 0.27 | 0.61 | −0.50 |
| System | µ (eV) | η (eV) | σ (eV) | ω (eV) |
|---|---|---|---|---|
| ScDPPSH | −3.35 | 1.05 | 0.48 | 5.33 |
| TiDPPSH | −3.54 | 1.35 | 0.37 | 4.64 |
| VDPPSH | −3.77 | 1.89 | 0.26 | 3.75 |
| CrDPPSH | −4.06 | 2.76 | 0.18 | 2.98 |
| MnDPPSH | −3.59 | 1.82 | 0.27 | 3.53 |
| FeDPPSH | −4.06 | 2.73 | 0.18 | 30.3 |
| CoDPPSH | −3.99 | 3.03 | 0.17 | 2.63 |
| CuDPPSH | −4.08 | 2.76 | 0.18 | 3.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bashir, B.; Clayborne, A.Z. Exploring the Influence of Chalcogens on Metalloporphyrins: A DFT Study. Molecules 2025, 30, 2254. https://doi.org/10.3390/molecules30112254
Bashir B, Clayborne AZ. Exploring the Influence of Chalcogens on Metalloporphyrins: A DFT Study. Molecules. 2025; 30(11):2254. https://doi.org/10.3390/molecules30112254
Chicago/Turabian StyleBashir, Beenish, and Andre Z. Clayborne. 2025. "Exploring the Influence of Chalcogens on Metalloporphyrins: A DFT Study" Molecules 30, no. 11: 2254. https://doi.org/10.3390/molecules30112254
APA StyleBashir, B., & Clayborne, A. Z. (2025). Exploring the Influence of Chalcogens on Metalloporphyrins: A DFT Study. Molecules, 30(11), 2254. https://doi.org/10.3390/molecules30112254