Abstract
Single-ring aromatic compounds including BTX (benzene, toluene, xylene) serve as essential building blocks for high-performance fuels and specialty chemicals, with extensive applications spanning polymer synthesis, pharmaceutical manufacturing, and aviation fuel formulation. Current industrial production predominantly relies on non-renewable petrochemical feedstocks, posing the dual challenges of resource depletion and environmental sustainability. The catalytic hydrodeoxygenation (HDO) of lignin-derived phenolic substrates emerges as a technologically viable pathway for sustainable aromatic hydrocarbon synthesis, offering critical opportunities for lignin valorization and biorefinery advancement. This article reviews the relevant research on the conversion of lignin-derived phenolic compounds’ HDO to benzene and aromatic hydrocarbons, systematically categorizing and summarizing the different types of catalysts and their reaction mechanisms. Furthermore, we propose a strategic framework addressing current technical bottlenecks, highlighting the necessity for the synergistic development of robust heterogeneous catalysts with tailored active sites and energy-efficient process engineering to achieve scalable biomass conversion systems.
1. Introduction
The progressive depletion of conventional hydrocarbon reserves coupled with escalating ecological degradation necessitates paradigm-shifting innovations in sustainable energy systems. Within this context, lignocellulosic biomass has emerged as a strategic carbon-neutral feedstock, with its cascade conversion technologies attracting concerted research efforts across global scientific and industrial communities. Particularly, the inherent aromaticity of lignin derivatives positions them as biogenic precursors for synthetic aromatics’ production—a critical pathway for circular carbon economy implementation [1,2,3,4].
As the predominant biomass reservoir on Earth, lignocellulosic materials yield an annual global output exceeding 170 billion metric tons [5,6,7,8]. Structurally characterized by a heterogeneous macromolecular matrix, these materials are constituted by three interwoven biopolymers: (1) cellulose (30–50%), a crystalline glucose polymer; (2) hemicellulose (20–30%), an amorphous heteropolysaccharide; and (3) lignin (15–30%), a methoxylated phenylpropanoid network (Figure 1) [9,10,11]. The structurally well-defined carbohydrate polymers (cellulose/hemicellulose) have been extensively valorized through established biorefinery protocols, serving as feedstocks for second-generation bioethanol and platform chemicals [12,13,14,15,16]. Conversely, lignin’s highly branched polyphenolic architecture—featuring C-C/C-O-C interunit linkages—creates a persistent technological bottleneck for selective depolymerization [1,4,17]. This structural recalcitrance confines over 98% of industrial lignin to low-value thermal applications [18,19,20], where its combustion not only exhibits suboptimal energy efficiency (~50% lower calorific value than coal) but also exacerbates carbon sequestration deficits through non-circular CO2 emissions [21,22]. Such systemic underutilization underscores the imperative for advanced catalytic strategies to unlock lignin’s latent potential as a renewable aromatic reservoir.
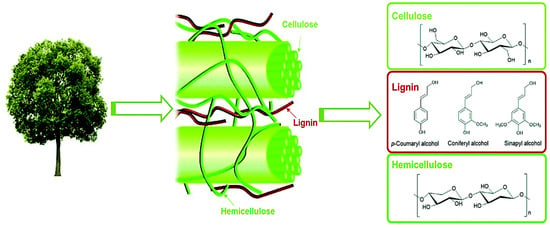
Figure 1.
The schematic diagram of the composition of lignocellulosic biomass [23]. Copyright 2016 Royal Society of Chemistry.
Contemporary lignin valorization strategies predominantly employ a two-stage cascade biorefinery paradigm: initial depolymerization of the macromolecular matrix into phenolic monomers, followed by precision HDO catalysis to yield premium aromatic hydrocarbons [24,25]. Mechanistic studies reveal two competing reaction coordinates in phenolic HDO processes: (i) a C-O bond scission with aromatic ring preservation via selective hydrogenolysis and (ii) ring hydrogenation concurrent with oxygen removal. The former pathway proves particularly advantageous for synthesizing drop-in fuel components, given aromatic hydrocarbons’ superior octane ratings (e.g., toluene: ~120 RON) and direct compatibility with existing fuel infrastructure [26]. Notably, the aromatic-selective routes demonstrate enhanced process economics, with hydrogen consumption metrics reduced by 30–50% compared to saturation pathways—a critical efficiency parameter for industrial scalability.
The technological impasse resides in achieving kinetically favored C-O cleavage over thermodynamically preferential C-C bond rupture. This selectivity conundrum necessitates atomic-level catalyst engineering to manipulate adsorption configurations and transition-state energetics. Recent breakthroughs in adsorption configuration engineering (e.g., tilted vs. flat phenol adsorption modes) and electronic perturbation strategies (metal–support charge-transfer effects) have opened new frontiers in aromatic yield optimization [27,28].
Improving the selectivity and yield of phenol HDO for the preparation of BTX (benzene, toluene, xylene) faces multiple challenges, with the core issue being how to precisely regulate the dynamic competition between catalyst active sites and reaction pathways [29,30,31,32]. Firstly, catalyst designs need to balance the dual-functional requirements of deoxygenation and aromatic ring stabilization: metal sites need to provide moderate hydrogenation capacity to avoid oversaturation of aromatic rings, while acidic supports need to promote deoxygenation but avoid strong acid-induced side reactions. Secondly, selective control of the reaction pathway is particularly crucial. Phenol HDO may be carried out through direct deoxygenation or hydrogenation dehydration pathways. The former directly generates aromatic hydrocarbons but requires inhibition of the deep hydrogenation of intermediates such as cyclohexanone, while the latter can easily lead to aromatic ring saturation and reduce BTX yield. In addition, the complexity of lignin-derived phenolic substrates, such as substituent positions and methoxy steric hindrance, can significantly interfere with deoxygenation efficiency.
Accordingly, a great deal of exceedingly excellent articles and reviews concerning the HDO of lignin-derived phenolic compounds have been published due to the importance of the HDO reaction in biomass conversion and upgradation. Seminal reviews by Zhong et al. established the methodological compendium for phenolic HDO across heterogeneous, homogeneous, and enzymatic systems [33]. Subsequent analyses by Wang’s team systematically deconstructed the multifunctional synergy in metal–acid bifunctional catalysts [34], while Sreedhar’s work mapped the reaction landscape for cycloalkane intermediates’ production [35]. Collectively, these intellectual foundations provide the crucial guides required for next-generation catalyst architectures [2,33,34,35].
At present, a large number of excellent articles have been published on the conversion of phenolic compounds’ HDO into aromatic hydrocarbons, among which benzene, toluene, and xylene (BTX) are three typical aromatic compounds [36]. In the current research field, there are few systematic reviews on the development of catalysts for the selective HDO of phenols to prepare aromatic hydrocarbons. In view of this, we did not focus on precious metal/non-precious metal catalysts or general lignin conversion pathways as in previous reviews but comprehensively reviewed the latest research results on the catalytic system and related strategies of lignin-derived phenolic compounds for the sustainable production of aromatic hydrocarbons through HDO catalysis. At the same time, a summary and explanation of the reaction pathways and mechanisms involved were provided to help deepen the understanding of the basic characteristics of catalysts. Finally, we put forward some preliminary suggestions for future research, hoping to promote the industrialization process of synthesizing high-value-added aromatic hydrocarbons and other chemicals from renewable lignin resources.
2. Catalytic Conversion of Phenolics into Aromatics
Benzene–toluene–xylene (BTX) constitutes the principal aromatic triad in petrochemical manufacturing, commanding an annual global output exceeding 10,000 tons through conventional naphtha reforming and steam-cracking processes [37]. They are currently used in a wide range of applications. In addition to being used as fuel additives and solvents, they can also be considered as the starting materials for the manufacture of various chemicals and polymers [25,26,37,38,39,40,41]. With escalating decarbonization mandates, biogenic BTX production via lignin-derived phenolic HDO has emerged as a carbon-negative alternative to fossil-derived routes—demonstrating lower lifecycle greenhouse gas emissions [42]. The evolving frontier in catalyst engineering has witnessed remarkable progress in designing heterogeneous catalytic systems for aromatic compound production. Advanced material platforms spanning loaded metal to transition metal compounds (sulfides, carbides, nitrides, phosphides) now demonstrate tailored efficacy in transforming lignocellulosic phenolic derivatives into benzene–toluene–xylene (BTX). This diversification in catalytic design strategies enables precise control over deoxygenation pathways and aromatic stabilization mechanisms.
2.1. Noble Metal Catalysts
Noble metals dominate phenolic HDO research due to their unparalleled hydrogen dissociation efficiency and oxygen-removal capabilities. Currently, three major noble metals, including palladium, platinum, and ruthenium, were studied extensively and considered as the most suitable metal catalysts in the BTX production from lignin-derived phenolic compounds.
2.1.1. Palladium (Pd)-Based Catalysts
Palladium (Pd), a precious and costly metal, plays a significant role in the hydrodeoxygenation (HDO) of phenolics. Hong et al. explored the synergistic catalytic effects of Pd and Fe in the HDO of m-cresol using Pd/Fe2O3 catalysts [43]. Their study revealed that the addition of Pd enhances the reduction of Fe under an H2 atmosphere, and the reduced catalyst demonstrates resistance to the Fe surface oxidation caused by reactants and water during the HDO process. Through characterization and kinetic studies, the synergistic effect between Pd and Fe was identified, being attributed to three key factors: the promotion of H2 activation by Pd, the stabilization of metallic Fe, and the enhancement of product desorption by Pd. The mechanism of Pd-Fe synergy is illustrated in Figure 2a. It involves H2 preferentially adsorbing and dissociating on Pd entities attached to the Fe surface, followed by spillover to metallic Fe sites where m-cresol adsorbs and activates [34]. The unique adsorption pattern of m-cresol on the Fe surface contributes to the high selectivity of BTX as direct HDO products. Additionally, Pd serves as the active site for H2 activation, maintaining high hydrogen coverage on the metallic Fe surface. These findings suggest that the proposed synergistic catalysis could be extended to other noble metal-promoted Fe catalysts, offering a promising strategy for designing highly active HDO catalysts in the future.
The choice of load material significantly influences both the activity and product distribution in HDO reactions. Barrios and colleagues investigated the performance of Pd loaded on SiO2 and Nb2O5 [44,45]. Their results showed that the reaction rate for phenol HDO over Pd/Nb2O5 was 90 times higher than that of the SiO2-loaded catalyst. Furthermore, the main product varied depending on the support: cyclohexanone was predominantly formed over Pd/SiO2, while benzene was the primary product over Pd/Nb2O (Figure 2b). The high activity and selectivity toward benzene for Pd/Nb2O were likely due to the strong interaction between the oxygenophilic Nb5+/Nb4+ cations and the oxygen in the phenol molecule [44]. Similarly, De Souza and colleagues compared the catalytic performance of Pd supported on various carriers, including SiO2, Al2O3, TiO2, ZrO2, CeO2, and CeZrO2, for phenol HDO [46]. For instance, benzene was the main product for Pd/TiO2 and Pd/ZrO2, whereas cyclohexanone predominated for Pd/SiO2, Pd/Al2O3, Pd/CeO2, and Pd/CeZrO2. The high selectivity toward benzene for the Pd/TiO2 and Pd/ZrO2 catalysts may be attributed to the oxophilic sites represented by the incompletely coordinated Ti4+ and Zr4+ cations near the metal particle periphery. In contrast, the weaker interaction between the metal cations of other supports and oxygen favored the hydrogenation of phenol to cyclohexanone.
Recent studies further explored the reaction pathways for the HDO of phenolics using Pd supported on different carriers in a fixed-bed reactor at 573 K [47]. The results demonstrated that the support type significantly influences the product distribution. For example, Pd supported on SiO2 and CeO2 favored ring hydrogenation, leading to oxidation products such as cyclohexanone. In contrast, the use of ZrO2, TiO2, and Nb2O5 as supports promoted the formation of benzene and toluene through either the carbonyl hydrogenation or direct deoxygenation of reciprocal isomeric intermediates (Figure 2c). Additionally, the reaction pathway for the removal of the methoxy group in the HDO of anisole was found to depend on the support. For all catalysts except Pd/Nb2O5, the preferred pathway involved demethylation to produce phenol, followed by further deoxygenation to benzene. However, the superior oxygenophilicity of Nb cations in Pd/Nb2O5 favored direct deoxygenation, resulting in the formation of benzene and methanol.
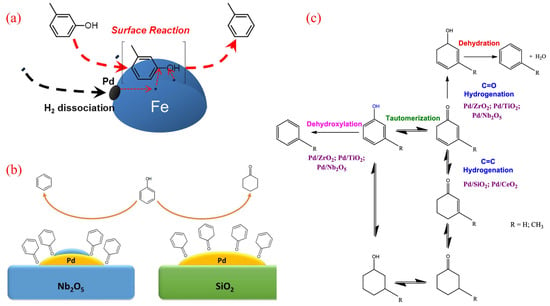
Figure 2.
(a) Mechanism of Pd-Fe synergy in HDO of m-cresol [43]. Copyright 2014 American Chemical Society. (b) The HDO reaction of phenol over Pd/SiO2 and Pd/Nb2O5 [44]. Copyright 2017 Elsevier. (c) Reaction scheme for the HDO of phenolics over Pd supported on various oxides [47]. Copyright 2021 American Chemical Society.
2.1.2. Ruthenium (Ru)-Based Catalysts
Ruthenium-based catalysts have demonstrated exceptional performance in the hydrodeoxygenation (HDO) of lignin-derived phenolics, particularly in enhancing BTX (benzene, toluene, xylene) formation. A breakthrough study by Wang et al. revealed that Ru/Nb2O5 systems achieved 64% BTX selectivity with a 35.5% yield during 20 h organosolv lignin conversion, substantially outperforming Ru catalysts supported on ZrO2, Al2O3, and TiO2 [48]. Notably, this configuration exhibited 80% toluene selectivity in p-cresol conversion through optimized C-O bond scission. Mechanistic investigations combining spectroscopic characterization and density functional theory (DFT) analyses elucidated Nb2O5’s dual functionality: a strong phenol adsorption capacity and remarkable reduction in the activation energy for aromatic C-O cleavage. The catalytic synergy arises from Ru’s hydrogenolytic activity, where surface-adsorbed H* species facilitate aryl-O bond dissociation. Crucially, the weak π-complex interaction between the generated aromatics and Nb2O5 surface enables rapid product desorption, effectively suppressing overhydrogenation side reactions. This mechanistic framework aligns with recent advancements in Au-Nb2O5 nanocomposites, which demonstrated analogous C-O bond activation efficiency during lignin hydrogenolysis [30].
Solvent engineering significantly regulates the efficiency and product distribution of hydrogenolysis deoxygenation (HDO) reactions through key parameters such as solubility, polarity, and hydrogen supply capacity. Currently, biomass-catalyzed HDO mainly uses water, alcohols, and their composite solvent systems [49,50]. Based on this, the research team developed the Ru/Nb2O5-MC catalyst through an initial wet impregnation method and systematically investigated its solvent effect in phenolic compound HDO [51]. Under optimized conditions of 250 °C and 2-bar H2, the biphasic catalytic system achieved the complete conversion of phenol and obtained 80% benzene selectivity. Compared to a single decahydronaphthalene or aqueous system, the decahydronaphthalene/water biphasic system increases benzene selectivity by 15–20%. Its advantage lies in the metal–acid dual-functional synergistic mechanism: Ru nanoparticles activate hydrogen molecules through dissociation adsorption, while the acidic sites of the Nb2O5-MC carrier can specifically adsorb oxygen-containing intermediates, promoting deoxygenation pathways through polarized Caryl-OH bonds and stable dehydration transition states. The water phase at the biphasic interface effectively suppresses the benzene ring hydrogenation side reaction through thermodynamic regulation, resulting in a 30–40% increase in selectivity. The kinetic isotope effect experiment confirms that this process mainly follows the direct deoxygenation mechanism. This solvent-mediated catalytic strategy achieved the efficient synthesis of benzene with low hydrogen consumption under mild conditions, providing a new engineering solution for the conversion of lignin-derived phenolic compounds into high-value chemicals.
The innovative application of hydrogen donor reagents provides an effective strategy for the directed conversion of phenolic compounds into BTX, replacing traditional hydrogen gas. Guo’s research team recently developed the Ru/Nb2O5-SiO2 catalytic system, which achieved the efficient conversion of p-cresol to toluene in a 2-propanol hydrogen-transfer medium [52]. The system achieved a toluene yield of 84.0% under reaction conditions at 230 °C, and its superiority lies in the significant reduction in the C-O bond dissociation energy by the NbOx species and the synergistic effect of the moderate hydrogen-transfer activity of Ru nanoparticles. The analysis of reaction pathways (Figure 3a) shows that in the presence of 2-propanol, the system mainly follows the direct deoxygenation pathway (DDO, route 1), generating only trace amounts of methylcyclohexanone/alcohol intermediates (route 2, total selectivity of 5.2%), which can be further converted to toluene through subsequent dehydrogenation reactions. It is worth noting that compared to the molecular hydrogen system, the hydrogen-transfer strategy increases the selectivity of aromatic hydrocarbons, which is attributed to the steric hindrance effect of the donor alcohol effectively suppressing excessive hydrogenation of aromatic rings. This breakthrough work establishes a new paradigm for the preparation of BTX from lignin-derived phenols through a nonhydrogen atmosphere catalytic conversion, providing important theoretical support for the optimization of the hydrogen economy in biomass-refining processes.
The study of ruthenium-based catalytic systems has been further expanded to titanium dioxide supports, and the mechanism of Ru/TiO2 in the direct hydrogenation deoxygenation (HDO) of phenol has revealed a unique catalytic pathway mediated by water [53]. The initial benzene selectivity of the system was only 38% at 573 K and 3.79 MPa, while the introduction of 10 wt% water significantly increased the selectivity to 95%, attributed to the dynamic proton network formed by water molecules on the TiO2 surface. As shown in Figure 3b, the water molecules adsorbed on the surface of hydroxylated TiO2 achieve bidirectional proton transfer through the Ru/TiO2 interface. During the extraction of phenolic hydroxyl groups, the C-O bond dissociation energy is reduced through a proton synergistic transfer mechanism. The synchronous hydrogen overflow effect induces the generation of oxygen vacancies in TiO2, and its amphiphilic properties promote the cleavage of H2 to generate active hydrogen species, forming a continuous deoxygenation–dehydrogenation cycle. The latest research progress indicates that reactive atmosphere engineering can further optimize the performance of the system. The Duan team achieved the efficient conversion of cresol to toluene by introducing 6-bar N2 at 160 °C and 1-bar H2 (Figure 3c) [54]. Nitrogen forms hydrogenated nitrogen species on the Ru metal surface through chemical adsorption, and the activation energy of the Caryl-O bond is reduced through a proton-relay mechanism. At the same time, the nitrogen coating dynamically regulates the distribution of active sites, increasing the adsorption rate of cresol and reducing the apparent activation energy of the reaction. This gas–solid interface regulation strategy provides a new process enhancement approach for lignin catalytic conversion.
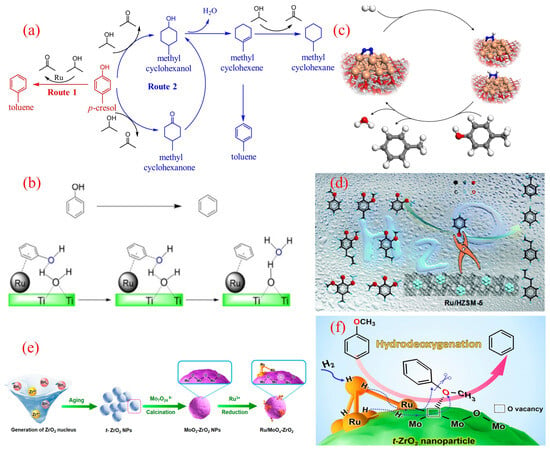
Figure 3.
(a) Reaction pathway of p-cresol HDO over the Ru/Nb2O5-SiO2 catalyst [52]. Copyright 2017 Elsevier. (b) Water-assisted HDO of phenol over the Ru/TiO2 catalyst [53]. Copyright 2015 American Chemical Society. (c) The combination of N2 activation and an HDO reaction over the Ru/TiO2 catalyst [54]. Copyright 2019 Springer Nature. (d) The selective HDO of phenolics to BTX over Ru/HZSM-5 in water [55]. Copyright 2016 Royal Society of Chemistry. (e) Schematic synthetic process for MoOx-decorated ZrO2-supported Ru nanocluster catalysts; (f) The selective HDO of anisole to benzene over the Ru/MoOx-ZrO2 catalyst [56]. Copyright 2021 American Chemical Society.
The research on ruthenium-based catalyst systems has further extended to the field of molecular sieve supports, and Ru/HZSM-5 has demonstrated excellent performance in the directional conversion of phenolic monomers (phenol, anisole, guaiacol, and eugenol) to benzene derivatives (BTX) [55]. In an aqueous reaction system composed of 240 °C, 2-bar H2, and 6-bar N2, the catalyst achieved 100% BTX selectivity for the conversion of guaiacol. In-depth research has shown that the topology of HZSM-5 has a decisive impact on catalytic performance: the cross-shaped HZSM-5-supported ruthenium catalyst (Ru/c-HZSM-5) achieved a 97% benzene yield under the same conditions. X-ray photoelectron spectroscopy (XPS) and infrared spectroscopy (IR) analysis revealed that the special pore structure of the cross-shaped molecular sieve induces the generation of electron-deficient ruthenium species, whose strong electronegativity significantly enhances the adsorption capacity of reactants. Additionally, the adsorption of guaiacol and hydrogen on cross-shaped Ru/HZSM-5 was more significant than other catalysts, which may be due to the abundant Lewis acid sites on cross-shaped Ru/HZSM-5, thus promoting the adsorption of oxygen atoms inside guaiacol (Figure 3d). The synergistic effect of electronic structure adsorption performance reduces the C-O bond dissociation energy, ultimately achieving the high-fidelity deoxygenation of aromatic ring structures [55].
The latest research breakthrough has achieved a performance leap of ruthenium-based catalysts in the aqueous hydrogenation deoxygenation (HDO) of benzyl ether through an interface engineering strategy [56]. The research team innovatively constructed a MoO3 modification layer on the surface of tetragonal zirconia (t-ZrO2) and formed a Ruδ+-Ov-Mo5+ ternary active interface by precisely loading Ru nanoclusters (Figure 3e). Under mild reaction conditions, the benzene selectivity of the catalyst reached 84.7%. Its performance leap is attributed to a unique electronic synergistic effect: MoOx defect clusters induce Ru nanoclusters to exhibit partial oxidation states through charge transfer, significantly enhancing the chemical adsorption energy of methoxy groups (Figure 3f). Advanced microscopic and spectroscopic analyses revealed a precisely engineered architecture where atomic-scale Ru clusters coexist with defective MoOx moieties on tetragonal zirconia (t-ZrO2) substrates. This unique configuration induces charge redistribution at metal–oxide interfaces, generating the electron-deficient Ruδ+ sites and coordinatively unsaturated oxygen vacancies. The established SMS creates robust electronic coupling between metallic Ru and MoOx species. Such synergistic interactions not only optimize active-site configuration but also endow the hybrid system with exceptional thermal stability during prolonged catalytic operation [56].
2.1.3. Platinum (Pt)-Based Catalysts
The platinum-based catalytic system exhibits unique advantages in the field of directed deoxygenation of phenolic compounds through precise interface regulation. The Pt/HBeta bifunctional catalyst developed by the Resasco team achieved the efficient conversion of meta cresol and benzyl ether under low-pressure conditions at 673 K [57,58]. The catalytic conversion frequency of this system was three times higher than that of a pure HBeta molecular sieve, and the BTX selectivity exceeded 90%. Mechanistic studies have shown that Pt nanoparticles and Brønsted acid sites form a dual-functional synergistic mechanism: the metal site selectively activates the adjacent aromatic ring of Caryl-OH to generate a hexadienol intermediate, and the adjacent acid site immediately triggers a dehydration reaction to complete the deoxygenation cycle (Figure 4a) [59]. Zhu’s research group further expanded the application of this system in the conversion of guaiacol and found that Pt/HBeta was regulated by a unique adsorption configuration—the aromatic ring plane adsorbed on the Pt crystal surface and the oxygen atom anchored acid site—resulting in a direct deoxygenation pathway contribution rate of over 85%, successfully inhibiting competitive reactions such as demethylation and decarbonylation [60].
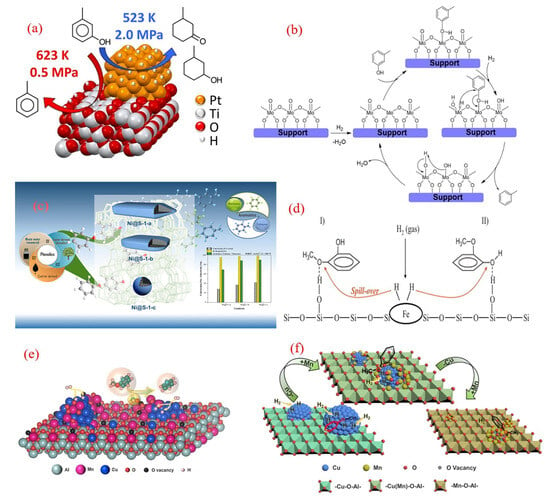
Figure 4.
(a) The HDO reaction of m-cresol over the Pt/TiO2 catalyst [61]. Copyright 2016 American Chemical Society. (b) Mechanism of the direct deoxygenation route of m-cresol on a schematic molybdenum oxide-site species [62]. Copyright 2017 Elsevier. (c) The upgrading of phenolics to BTX over the different Ni@silicalite-1 catalysts [63]. Copyright 2021 American Chemical Society. (d) Possible reaction mechanism of the HDO conversion of guaiacol over the Fe/SiO2 catalyst [39]. Copyright 2012 Elsevier. (e) The HDO of anisole to benzene over the Mn-doped Cu/Al2O3 catalysts; (f) the mechanism for the anisole HDO on Cu/MnAlOx with the Mn/Cu molar ratio increasing [64]. Copyright 2021 Elsevier.
A significant breakthrough has been made in the study of the structure–activity relationship between the carrier effect and reaction pathway of platinum-based catalysts. Griffin’s team systematically analyzed the performance differences between Pt/C and Pt/TiO2 in the hydrogenation deoxygenation (HDO) of meta cresol using an experimental computational strategy [61]. This study used a gas-phase flow reactor to simulate two process conditions, catalytic rapid pyrolysis (CFP: 623 K, 0.5 MPa) and hydroprocessing (HT: 523 K, 2.0 MPa), and conducted comparative analysis under a unified control of a 35% conversion rate. The experimental data show that under CFP conditions, the toluene carbon selectivity of Pt/TiO2 reaches 78%, which is significantly higher than that of Pt/C at 46%. Under HT conditions, both types of catalysts mainly generate 3-methylcyclohexanone/alcohol (selectivity >95%), which is highly consistent with Ruddy’s thermodynamic regulation law [65]: aromatic ring hydrogenation tends to be in the low-temperature region, while direct deoxygenation and acid-catalyzed reactions dominate the high-temperature region (>623 K). Mechanistic studies have shown (Figure 4a) that the Pt metal phase dominates the hydrogenation pathway at 523 K, while the oxygen vacancy effect of TiO2 support is significantly enhanced at 623 K, resulting in a decrease in deoxidation activation energy through metal–carrier electronic interaction. This temperature-dependent synergistic mechanism is reflected in the participation of TiO2 surface hydroxyl groups in proton-transfer networks at high temperatures, optimizing the Caryl-O bond dissociation pathway and increasing toluene selectivity.
Noble metal catalysts (e.g., Pd, Pt, Ru) exhibit unparalleled efficiency in phenolic HDO, primarily due to their exceptional hydrogen dissociation capabilities and tailored interactions with oxygen-containing functional groups. Despite their superior performance, noble metals face critical limitations, including high cost, susceptibility to coking during prolonged operation, and limited availability, which restrict their large-scale industrial adoption.
2.2. Non-Noble Metal Catalysts
Due to the high cost of noble metal-based catalysts, which makes them less viable for large-scale industrial applications, researchers have been developing cost-effective, non-precious metal-loaded catalysts as alternative systems for the conversion of phenolic compounds to benzene, toluene, and xylene (BTX). These systems have shown great promise in hydrodeoxygenation (HDO) processes.
The group of Prasomsri explored the use of a homogeneous MoO3 catalyst, under reaction conditions of 593 K, 3 h, and 0.1 MPa H2, and they achieved 94% overall conversion with high yields of benzene and toluene [66]. The success of the MoO3 catalyst was primarily attributed to its ability to selectively cleave the phenolic Ph-OMe bond over the weaker aliphatic Ph-O-Me bond. Advancing this research direction, Zhang’s team conducted a systematic evaluation of defect-engineered MoO3 catalysts for aromatic production under lower H2 environments [62]. Their work demonstrated effective phenol-to-benzene transformation (98.1% conversion, 99.5% selectivity) under mild hydrotreatment conditions (340 °C, H2/N2 = 1:6 v/v at 3.5 MPa total pressure). Mechanistic studies revealed that coordinatively unsaturated sites generated by surface oxygen vacancies served as active centers, facilitating selective C-O bond scission while inhibiting aromatic ring hydrogenation. Specifically, the oxygen vacancies (Mo5+ sites) acted as the active centers for phenol HDO, facilitating the adsorption of lone-pair electrons from phenol and enabling the hydrolysis of carbonyl intermediates to form benzene. Additionally, the introduction of N2 into the reactor prevented the over-reduction of MoO3, maintaining a high-oxygen-vacancy content on the catalyst surface and enhancing the HDO conversion of phenol to benzene.
The hydrodeoxygenation (HDO) of m-cresol to toluene was investigated using MoO3-based catalysts supported on various materials, including SiO2, Al2O3, TiO2, ZrO2, and CeO2 [67]. The catalytic systems selectively exhibited C-O bond scission capabilities while preserving aromatic unsaturation, enabling the effective transformation of m-cresol into toluene with notable conversion efficiency and exceptional selectivity. Supporting materials play a crucial role in molybdenum-based catalysts, especially in stabilizing specific low-oxidation-state molybdenum ions and affecting their reduction performance, with TiO2 and ZrO2 identified as the most effective supports for enhancing activity and stability. The reaction mechanism remained inconclusive, but the rate of reaction appeared to be influenced by ligand-unsaturated Mo sites, which are essential for the oxygen-vacancy-driven mechanism. Further studies by Vinicius et al. explored the impact of different supports on MoOx-based catalysts for m-cresol HDO at 340 °C and a total pressure of 4 MPa (Figure 4b) [68]. Regardless of the catalyst used, the selectivity for m-cresol to toluene consistently exceeded 80%, with the catalytic activity influenced by the nature of the support, following the order as follows: MoOx/Al2O3 > MoOx/SBA-15 > MoOx/SiO2. This variation was attributed to differences in the reducibility of Mo species, which could be enhanced by using acidic carriers like Al2O3 or mesoporous materials like SBA-15 compared to commercial SiO2. The proposed reaction mechanism involves the adsorption of oxygen from the reactant onto oxygen vacancies, followed by the heterolytic dissociation of H2 into proton and hydride species. The hydride adds to a carbon atom bearing an OH group, leading to the cleavage of the C-O bond and the formation of toluene. Oxygen vacancies are restored by releasing water during the process [67,69]. Mo species are present as polymolybdate phases, including Mo6+, which reacts with H2 to form MoOx species (Mo IV) and release water, facilitating the breaking of C-O bonds [70]. This study highlights the importance of support materials in optimizing MoO3 catalysts for HDO processes, offering insights into the development of cost-effective and efficient catalytic systems for industrial applications [68].
However, despite these advantages, MoO3-based catalysts face challenges such as deactivation due to coke deposition and over-reduction, which can lead to the formation of inactive Mo species [62,68]. These limitations have prompted researchers to explore alternative non-precious metal catalysts for HDO processes.
Nickel-based catalysts have emerged as a promising alternative for the HDO of phenolics to BTX [71,72,73]. Researchers have investigated a wide range of Ni-containing catalysts supported on diverse carriers [74]. The comparative evaluation of nickel-based catalytic systems revealed significant carrier-dependent performance in aromatic production. When supported on microporous carbon matrices, Ni nanoparticles achieved an 80% benzene yield under optimized conditions (310 °C, 3-bar H2). The superior performance of the Ni/C catalyst was attributed to its strong acidic sites and excellent metal dispersion, which favored the selective cleavage of the C-O bond. Similarly, the Ni/TiO2 catalyst demonstrated high selectivity for benzene, albeit with limited anisole conversion, due to strong metal–support interactions [75]. These findings suggest that the catalytic activity and selectivity of Ni-based catalysts are significantly influenced by the nature of the support material.
The catalytic mechanism of Ni-based catalysts involves the adsorption of reactants onto ligand-unsaturated Ni sites, which promote the activation of hydrogen and stabilize transition states, leading to the cleavage of the C-O bond. The size of the Ni particles also plays a crucial role in determining reaction pathways and product selectivity [76]. Smaller Ni particles tend to enhance the conversion rate by providing more active sites, while larger particles may influence the selectivity toward specific products. This highlights the importance of optimizing the Ni particle size and support properties to achieve desired catalytic outcomes.
This study further explores the optimization of Ni-based catalysts for the hydrodeoxygenation (HDO) of phenolics to BTX. Notably, Ni/Ce1−xNbxO2 catalysts were developed, demonstrating exceptional performance in the HDO of phenol. At 300 °C, the Ni/CeO2 catalyst achieved a remarkable 92% yield of benzene [77]. The reaction mechanism involves phenol tautomerization followed by hydrogenation to cyclohexanone and hydrolysis to methane [74]. The addition of Nb significantly alters the product distribution, with the increased Nb content enhancing benzene selectivity while reducing the formation of hydrogenation and hydrolysis products. This improvement is attributed to the niobium-rich surface and the strong interaction between phenol’s oxygen and the oxygenophilic Nb5+ sites, which promotes carbonyl hydrogenation.
In another approach, Ni@S-1 catalysts with varying crystal sizes were synthesized using an in situ encapsulation method and applied to m-cresol conversion (Figure 4c) [63]. These catalysts exhibited outstanding stability over 300 h, achieving 78.4% phenol conversion and a 73.1% BTX yield. The encapsulation of Ni nanoparticles within the zeolite matrix modulates reactant adsorption patterns and leverages the shape-selective properties of silicalite-1, enhancing aromatic selectivity and catalyst stability. The reduction in the Ni@silicalite-1 crystal size is further improved by selectively eliminating the hydroxyl groups that hindered the further hydrogenation of aromatics [63]. This design also prevents the migration and aggregation of Ni nanoparticles, maintaining catalyst performance even during long-term runs and avoiding undesired benzene ring reactions.
The exploration of cost-effective and environmentally friendly catalysts for the hydrodeoxygenation (HDO) of phenolics to BTX has recently expanded to include iron (Fe)- and manganese–copper (Mn-Cu)-based systems. In a notable study, Fe/SiO2 catalysts were investigated for the gas-phase HDO of guaiacol, achieving 74% conversion and a 38% BTX yield at 400 °C and 10 bar [39]. These Fe-based catalysts demonstrated advantages such as minimal aromatic ring hydrogenation and low-coking tendencies. The mechanism involves the adsorption of guaiacol’s oxygen atoms at weak acidic OH sites on the silica surface, favoring C-O bond cleavage over C-C bond breakage [64]. Active hydrogen species, derived from H2 dissociation on Fe particles, play a critical role in facilitating the reaction (Figure 4d). This work highlights Fe as a promising, sustainable catalyst for lignin-to-BTX production via rapid pyrolysis and the subsequent HDO of lignin vapors.
In another development, Mn-doped Cu/Al2O3 catalysts, synthesized from layered double hydroxides, were tested for the liquid-phase HDO of anisole (Figure 4e) [78]. The addition of Mn significantly enhanced benzene selectivity, reaching ~65% for the 4Cu/8Mn4AlOx catalyst—six times higher than that of undoped Cu/Al2O3. Structural studies revealed that MnOx doping optimized the surface structure of Cu particles and generated a high density of oxygen vacancies (OVs). These OV sites, along with metallic Cu, synergistically activated the C-O bond in anisole, improving HDO activity. However, excessive Mn/Cu ratios led to MnOx encapsulating Cu particles, reducing the active sites for H2 activation and suppressing catalytic performance. This study demonstrates the potential of Mn-Cu/Al2O3 catalysts for selective and efficient HDO processes.
Non-noble metal catalysts (e.g., Mo, Ni, Fe, Mn-Cu) offer cost-effective alternatives for HDO, leveraging earth-abundant elements to achieve selective deoxygenation. However, these catalysts often suffer from lower intrinsic activity compared to noble metals, as well as challenges like metal leaching (Ni) and redox instability (Mo), requiring further advancements in stability and regeneration protocols.
2.3. Bimetallic Catalysts
The addition of a second metal can modify the electronic or structural properties of the primary-metal-loaded catalyst compared to its monometallic counterpart, potentially enhancing catalytic activity, selectivity, and durability. This modification can occur through changes in the d-band electron density of the primary metal or by altering its surface configuration [34,79,80,81,82]. Additionally, the two metals may fulfill distinct roles during the reaction process [83].
2.3.1. Fe-Containing Bimetallic Catalysts
The oxophilic nature of iron (Fe) makes it highly effective for deoxygenation processes, particularly due to its strong affinity for oxygen-containing groups in phenolic compounds. However, Fe has a limitation: its poor ability to dissociate H2 [34]. To overcome this, researchers developed a carbon-supported PdFe bimetallic catalyst, which enhances the overall hydrodeoxygenation (HDO) rate while maintaining efficient oxygen removal during the HDO of guaiacol [84]. This PdFe/C catalyst demonstrated superior performance, achieving an 83.2% benzene yield at 450 °C, significantly outperforming the monometallic Fe/C catalyst, which yielded only 43.3%. The possible structures of the bimetallic catalyst, as shown in Figure 5a, include a monolayer of Pd on the Fe (110) surface or a mixed surface of Pd and Fe atoms. Characterization and density functional theory (DFT) studies reveal that the Pd-Fe alloy surface is enriched with Pd nanodots, while Fe particles dominate the subsurface, indicating that Fe serves as the primary active site for the reaction. Meanwhile, Pd modifies the electronic structure, enhancing catalytic activity. Figure 5b revealed three distinct adsorption configurations on the catalyst surface. Adsorption energetics analysis demonstrated a positive correlation between the binding strength and proximity to Pd active centers, with the third adsorption position exhibiting the most thermodynamically favorable adsorption energy, conclusively establishing preferential phenol adsorption at the Fe surface sites. As phenol interacts strongly with the Fe surface, the C-O bond weakens, making the HDO of phenol more susceptible.
The addition of a second metal can create additional active sites, often acidic in nature, which can significantly enhance deoxygenation [34,85]. Phan et al. demonstrated that incorporating Fe into a Ru/meso-TiO2 catalyst drastically alters its surface properties and catalytic performance, achieving over 80% benzene selectivity in the HDO of anisole at 250 °C and 1 MPa of H2 [86]. In this system, Ru particles efficiently dissociate H2, while the oxygenophilic Fe sites strengthen interactions between the oxygen-containing functional groups and the carrier surface. The synergistic interaction between Ru and Fe enhances C-O bond cleavage without ring hydrogenation, thereby minimizing H2 consumption (Figure 5c). Additionally, the bimetallic catalyst’s improved activity is linked to an increase in oxygen vacancies on the carrier surface. Similarly, a bimetallic FeReOx/ZrO2 catalyst was investigated for the mild HDO of various phenolic compounds [26]. Notably, at 250 °C, it delivered a BTX yield of 50.5% in the HDO of m-cresol, along with high stability, retaining 93.7% activity after 160 consecutive reactions. The catalyst’s mesoporous structure, oxygenophilic nature, and balanced acidity—induced by rhenium oxide and zirconium oxide carriers—contribute to its high dehydration efficiency. This system offers a cost-effective, sustainable route for BTX production under atmospheric pressure at 350 °C, bringing functional chemicals from lignin biorefining closer to commercialization.
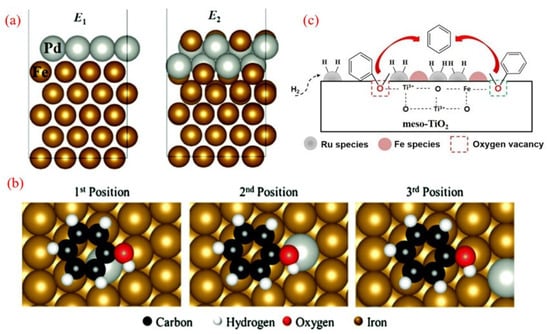
Figure 5.
(a) The structure of the Pd layer in the matrix on the Fe (110) surface. (b) Adsorption conformation of phenol on the Fe (110) surface Pd atom. The distance from the adsorbate to the palladium atom increases from position 1 to position 3 [84]. Copyright 2013 Elsevier. (c) The possible mechanism of the anisole HDO over the RuFe/meso-TiO2 catalyst [86]. Copyright 2018 Elsevier.
The key role of the hydrogen overflow effect in the heterogeneous catalytic hydrogenation deoxygenation (HDO) process has been experimentally verified [87]. Taking the bimetallic Pt@Fe@SiO2 system as an example [88], the catalyst accurately constructs a hierarchical pore structure through a dual-mode plate method: Pt nanoparticles are located in SiO2 micropores, and Fe nanoparticles are distributed in macropores. In the HDO reaction of guaiacol, although it is difficult to achieve surface hydrogen migration with SiO2 as a non-reducible carrier, the introduction of C1 to C3 carbonyl compounds or ester oxygen-containing additives allows the active hydrogen atoms generated at Pt sites to migrate across scales to Fe active sites, improving the migration efficiency (Figure 6a). This molecular-mediated hydrogen-transfer mechanism significantly enhances the deoxygenation activity of bimetallic systems compared to single-metal Pt/Fe catalysts.
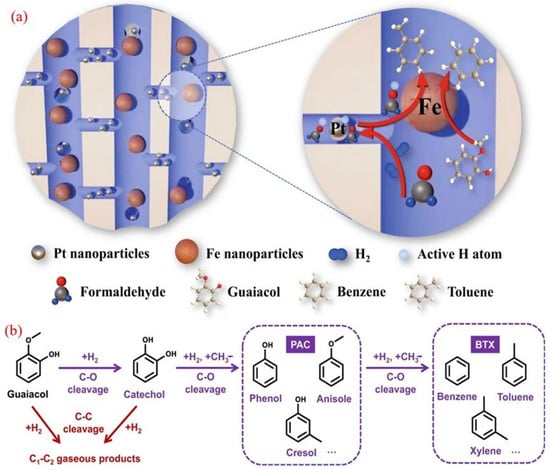
Figure 6.
(a) Hydrogen spillover enhanced by oxygenate additives during catalysis; (b) the suggested HDO route of guaiacol to produce BTX [88]. Copyright 2022 Springer Nature.
Dynamics analysis shows that oxygen-containing additives reduce the activation energy of the key hydrogenation steps on the Fe surface by stabilizing transition-state hydrogen species. It is worth noting that such hydrogen carriers can be generated through the in situ dehydrogenation or steam reforming of alcohol precursors, simultaneously providing H2 sources for BTX synthesis. This study has pioneered a new strategy of “molecular assisted hydrogen overflow”, providing a universal solution for the synergistic catalysis of spatially isolated active sites on inert carriers.
2.3.2. Ni-Containing Bimetallic Catalysts
Ni-based catalysts possess superior hydrogenation abilities for biomass upgrading. However, their limited propensity for oxygen removal restricts their application in HDO reactions [89,90]. As a result, numerous bimetallic Ni catalysts have been developed to enhance their deoxygenation efficiency. Tao and colleagues developed highly reactive NiMo bimetallic catalysts [91]. Interestingly, the synthesized Ni-Mo/SiO2 catalyst demonstrated remarkable selectivity (>96%) for BTX and achieved a 99% conversion rate under low-H2 partial-pressure conditions. Additionally, the catalyst’s plentiful acid sites significantly promoted the methyl-transfer reaction, leading to reduced carbon losses. Similarly, Yang et al. reported that the bimetallic Ni-Mo/SiO2 catalyst exhibited high activity and selectivity (>80%) for the HDO of m-cresol to toluene across a broad temperature range (250–350 °C) under atmospheric hydrogen pressure. Further confirmation revealed that the interaction between the Ni core surface and the optimal MoOx species enhancing the Ni surface is crucial for attaining high activity and selectivity [92]. This study offers novel insights into cost-effective HDO processes for products derived from lignin thermal degradation.
Wang’s group studied the influence of indium on the catalytic performance of Ni/SiO2 in the hydrodeoxygenation (HDO) [93]. Under conditions of 300 °C, 0.1 MPa, and a H2/anisole ratio of 25, the Ni40In/SiO2 catalyst with 40% Ni content achieved a higher BTX yield of 60.4%, compared to that of 51.6% from the Ni/SiO2 catalyst (Figure 7a). They found that the size of the Ni-In bimetallic microcrystals was similar to that of monometallic Ni with the same Ni content. However, the bimetallic Ni-In catalyst absorbed significantly less H2 because the In atoms diluted the Ni atoms.
Charge transfer from In to Ni was observed in the bimetallic Ni-In catalysts, indicating a close interaction between the Ni and In atoms, along with geometrical and electronic modifications of Ni by In. In anisole HDO, the Ni-In bimetallic catalysts showed lower activity than monometallic Ni but exhibited higher selectivity for BTX. Furthermore, the bimetallic catalysts demonstrated reduced methanation activity, leading to higher carbon yields and decreased hydrogen consumption [94]. Additionally, a lower Ni/In ratio resulted in a greater impact of In on catalytic performance, with BTX selectivity primarily determined by the Ni/In ratio and nearly independent of the Ni content [93].
Furthermore, the geometric and electronic effects of bimetallic Ni-Re catalysts for the selective HDO of m-cresol to toluene were further investigated [95]. The incorporation of Re into the Ni/SiO2 catalyst stabilizes highly dispersed NiO through robust Ni-O-Re interactions. As anticipated, the bimetallic catalysts exhibit both geometric and electronic effects: (1) Re disperses the Ni surface into smaller aggregates and creates Ni-Re adjacent sites, and (2) the proximity between Ni and Re reduces the occupancy of the Ni d-band. Unlike the adsorption of phenol on the bare Ni (111) surface via the benzene ring (with O pointing away from the surface), the adsorption of phenol on the (Re)Ni (111) surface occurs via the benzene ring on Ni and the O atom on Re at Ni-Re adjacent sites, which facilitates C-O bond cleavage (Figure 7c). The incorporation of Re in the surface alloy modifies the Ni surface by breaking it into smaller, discrete regions (geometric effect) and reduces the d-band electron density of Ni (electronic effect) [95]. DFT calculations indicate that the Ni-Re neighboring site facilitates the cleavage of the C-O bond by adsorbing the O atom on Re and the benzene ring on the adjacent Ni atom, thus enhancing deoxygenation and promoting toluene production. At the same time, the reduced electron density in the Ni d-band weakens the adsorption of benzene rings on the surface, preventing the C-C hydrogenolysis of aromatic products.
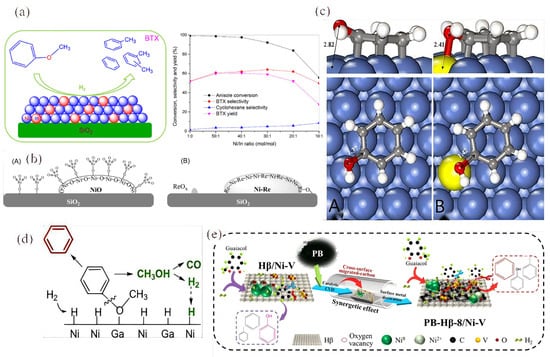
Figure 7.
(a) The performance of the In on Ni/SiO2 catalyst in the HDO of anisole with different products [93]. Copyright 2017 Elsevier. (b) Schematic representation of the structure of the bimetallic Ni-Re catalyst before (left) and after (right) reduction at 450 °C [95]. Copyright 2017 Elsevier. (c) Phenol adsorption onto the Ni (A) surface and (B) phenol adsorption onto the (Re)Ni (111) surface [95]. Copyright 2017 Elsevier. (d) The plausible reaction mechanism in the HDO of anisole on NixGa/SiO2 [96]. Copyright 2017 Elsevier. (e) The proposed mechanism of modification between aromatic selectivity, oxygen vacancies, and H2 absorption [97]. Copyright 2021 American Chemical Society.
The bimetallic NixGa/SiO2 catalysts were synthesized using the impregnation method followed by reduction at 550 °C and were evaluated for the vapor hydrodeoxygenation (HDO) of anisole at 0.1 MPa and 300 °C [96]. The results demonstrated that NixGa/SiO2 exhibited higher anisole conversion and benzene selectivity compared to the monometallic Ni/SiO2, while also showing lower selectivity for phenol. Specifically, at the same anisole conversion rate of 31%, the selectivities for benzene were 75.2%, 83.0%, and 92.6% for Ni/SiO2, Ni6Ga/SiO2, and Ni3Ga/SiO2, respectively.
The preferential cleavage of the CAr-O bond on the Ni-Ga alloy and the Ni3Ga intermetallic compound (IMC) results from the synergistic effects between adjacent Ni and Ga sites, as illustrated in Figure 7d. In the case of the Ni-Ga alloy and Ni3Ga IMC, the oxygen in anisole is more likely to be adsorbed at Ga sites, where Ga shows a stronger affinity for O compared to Ni, as inferred from H2 studies. This sequence of interactions ultimately leads to the cleavage of the CAr-O bond, allowing for the formation of benzene. In summary, the NixGa/SiO2 bimetallic catalyst achieved a higher benzene yield while significantly lowering hydrogen consumption, marking a crucial advancement for the rational design of future catalysts in biomass upgrading applications.
In an innovative approach, biochar-modified Hβ/Ni-V catalysts were developed and tested for the atmospheric hydrodeoxygenation (HDO) of guaiacol to produce benzene, toluene, and xylene (BTX) [97]. Among these, the catalyst modified with pine nut shell biochar (PB), specifically PB-Hβ-8/Ni-V, achieved the highest selectivity for benzene and toluene at 69.17%. This enhancement in selectivity was attributed to the synergistic effects between the biochar and the Hβ/Ni-V catalyst. The mechanism behind this improvement involves the formation of a stable carbon layer on the Hβ/Ni-V catalyst, created through a metal-catalyzed chemical vapor deposition process utilizing volatiles from pyrolyzed PB (as illustrated in Figure 7e). In addition to forming this carbon layer, the thermal reduction of the carbon effectively decorated the surface metal, increasing the availability of active sites, particularly Ni0 and V3+. This increase in Ni0 sites enhances the catalyst’s ability to adsorb and dissociate hydrogen, thereby boosting its hydrogenation activity.
Moreover, the presence of biochar enhances the affinity of the catalyst surface for reactants, along with creating more oxygen vacancies [98]. This combination contributes to the improved selective adsorption of oxygen-containing groups and facilitates the breaking of CAr-OH bonds, leading to enhanced deoxygenation activity [97]. Overall, the incorporation of biochar modifications significantly improved the HDO activity of the catalysts, resulting in a greater yield of BTX. The use of pyrolytic biochar also presents a promising and cost-effective alternative to traditional zeolite–metal catalysts, making it a viable option for sustainable biomass conversion strategies.
2.3.3. Re- and W-Based Bimetallic Catalysts
In addition to Ni- and Fe-based bimetallic catalysts, a variety of other bimetallic combinations, including Pd-Re, Re-Mo, Re-V, Ru-W, and Pt-Wu, have been developed and utilized for converting various phenolic compounds into benzene, toluene, and xylene (BTX) [99,100,101,102,103,104]. For instance, research conducted by Pouya et al. found that ZrO2 showed better catalytic performance compared to cerium dioxide, attributed to its higher acid density, which enhances catalytic activity. All the hydrogenated metals were effective in promoting the hydrogenation of carbonyl groups, leading to selective BTX production. Among the developed catalysts, PdReOx/ZrO2 exhibited the highest BTX yield of 77.2%. This high activity is partially due to the weak acid strength contributed by the zirconium oxide carrier and rhenium oxide, which prevents phenolic adsorption and trapping at lower temperatures [99].
In another study focusing on anisole HDO, bimetallic Re-MoOx/TiO2 and Re-VOx/TiO2 catalysts were evaluated under conditions of 300 °C and 3 MPa of H2 [101]. Re-based catalysts exhibited a unique ability to favor the production of aromatic compounds due to their oxygenophilic properties. When combined with Mo and V cations in lower oxidation states, the selectivity for desired aromatic products like benzene and toluene was significantly improved. This enhanced selectivity can be attributed to the stronger adsorption of anisole on the surface oxygen vacancy sites of MoOx or VOx. However, the extent of this improvement depended on the specific combination of Re with Mo or V, highlighting the synergistic interactions present between the Re and MoOx species in particular. The catalytic activity, measured in terms of the intrinsic reaction rate, was determined by the nature of the specific surface species present. For instance, in the Re-MoOx/TiO2 catalyst, the activity was primarily linked to the exposed Mo5+ sites, while in the Re-VOx/TiO2 catalyst, the activity was mainly influenced by the Re4+ sites [101]. This research underscores the critical importance of designing catalysts with specific active sites tailored for the formation of targeted products, facilitating more efficient pathways for the transformation of lignin-derived phenolic compounds into valuable aromatic products like BTX.
Bimetallic tungsten (W)-based catalysts have shown remarkable catalytic performance for hydrodeoxygenation (HDO). Notably, the Pt-WOx/C catalyst exhibited exceptional activity and selectivity for the HDO of m-cresol (Figure 8a) [103]. Under various reaction conditions, this catalyst achieved over 94% selectivity for the desired products and demonstrated high stability, showing resistance to deactivation from coking. In contrast, the Pt/C catalyst showed lower selectivity for toluene production during the HDO process of m-cresol.
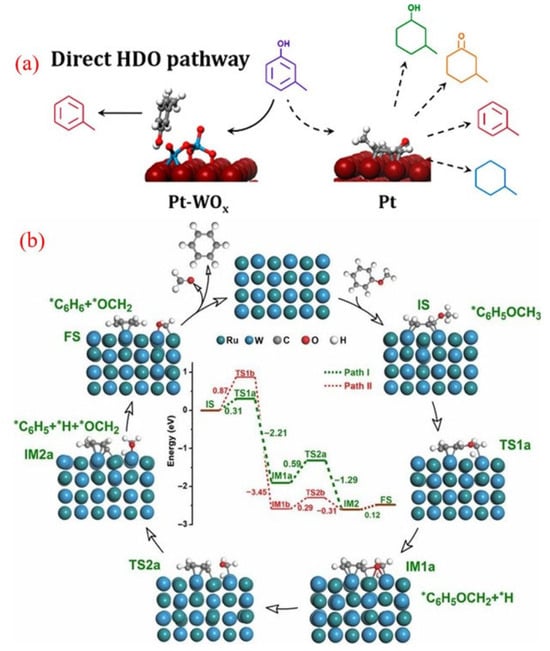
Figure 8.
(a) The direct HDO of m-cresol over WOx-decorated Pt/C catalysts [103]. Copyright 2018 American Chemical Society. (b) The tentative reaction mechanism for the SSH reaction of anisole on a RuW/SiO2 catalyst [104]. Copyright 2019 Springer science.
Reactivity studies, along with density functional theory (DFT) calculations, revealed that the HDO reaction on the Pt-WOx/C catalyst occurs via the direct hydrogenolysis of the C-O bond in m-cresol, which is adsorbed onto the oxygen vacancy sites present on the WOx species. The interaction between Pt and the WOx complexes not only stabilizes the oxide but also reduces the energy barrier for the formation of oxygen vacancies. These vacancies are crucial for creating redox sites on WOx that actively participate in the direct C-O bond hydrogenolysis of adsorbed phenolics [103]. Moreover, the selective adsorption at these sites reduces the interaction of the aromatic ring with the Pt surface, thus minimizing the occurrence of side reactions.
Additionally, pioneering work by Meng and colleagues recently introduced a novel self-supported hydrogenolysis (SSH) process for converting aromatic ethers to BTX, utilizing hydrogen generated from the reactants themselves [104]. The RuW alloy nanoparticles served as efficient catalysts for these reactions. Notably, this method is unique because it eliminates the need for external hydrogen or other reducing agents and avoids the hydrogenation of the aromatic ring.
Mechanistic studies suggest that the adjacent Ru and W species in the RuW alloy nanoparticles work synergistically to enhance the SSH reaction. DFT studies (Figure 8b) identified two primary reaction pathways for the SSH of anisole with benzene: In path I, the aliphatic C-H (CAl-H) bond is first activated to produce a *C6H5OCH2 + *H intermediate (IM1a), followed by the hydrogenolysis of the CAr-O bond in *C6H5OCH2 to generate benzene and formaldehyde (path I, green dashed lines). Path II involves initial cleavage of the CAr-O bond to a *C6H5 + *OCH3 intermediate (IM1b), followed by the activation and dissociation of the CAl-H bond in *OCH3 to formaldehyde and benzene (Path II, red dashed line). This innovative, hydrogen-free, and efficient approach presents significant potential for producing functional chemicals from renewable biomass resources. The discovery of this method has opened avenues for exploring numerous other reactions and optimizing biomass conversion technologies.
Bimetallic catalysts enhance HDO performance through synergistic interactions between two metals, optimizing active-site configuration and reaction pathways. These systems balance activity, selectivity, and stability while reducing noble metal loading, but their complex synthesis and thermal management requirements pose challenges for industrial scaling.
2.4. Transition Metal-Based Catalysts
2.4.1. Metal Sulfide Catalysts
Transition metal sulfides represent classic catalysts widely researched for hydrogenation reactions, demonstrating superior activity compared to transition metal oxides. They are extensively utilized in the hydrogenation of petroleum and coal-based liquid fuels, offering seamless compatibility with large-scale industrial hydrogenation units and catalyst production facilities [2,105,106,107].
The cobalt molybdenum sulfur (CoMoS) catalyst system has made significant progress in expanding its application from traditional petroleum refining and hydrogenation desulfurization (HDS) to biomass catalytic deoxygenation [108,109]. The Bui team conducted the first systematic study on the carrier effect of a CoMoS catalyst in the guaiacol HDO reaction [110] and found that compared with the commonly used Al2O3 carrier in industry, the CoMoS system loaded with ZrO2 increased the benzene selectivity to 42%. Its superiority lies in the strong electronic interaction between the CoMoS active phase and the carrier. However, the CoMoS catalyst obtained by traditional preparation methods has bottleneck problems such as high-temperature sulfur loss and rapid deactivation [111].
In response to the above challenges, the Liu team innovatively developed a novel catalyst that anchors isolated Co atoms with single-layer MoS2 [111]. By using H2 high-temperature treatment to construct sulfur vacancy defects on the MoS2 substrate, Co atoms are covalently bonded with vacancies to form Co-S-Mo active sites, exhibiting excellent activity, selectivity, and stability without sulfur loss in the 4-methylphenol conversion reaction. This breakthrough provides a new paradigm for the design of highly stable sulfide catalysts for biomass HDO processes.
Song et al. demonstrated a surface atom engineering strategy that significantly enhanced the catalytic activity and selectivity of sulfide catalysts by achieving well-dispersed Co-doped MoS2 nanomaterials, maximizing the Co-Mo-S phase for efficient phenolic conversion to BTX (Figure 9a) [112]. Their self-induced method optimally modulates Co-substituted S sites, allowing Co atoms to bond well to the upper surface of the MoS2 nanosheets while preserving their structure. The characterization results showed a high density of Co-Mo-S phases on the catalyst’s surface, contributing to accelerated HDO reactions. As a result, using the engineered CoMoS catalyst led to the effective conversion of mixed phenols to BTX, achieving high yields exceeding 85% (Figure 9b). These findings may inspire further surface engineering of transition metal-doped catalytic nanomaterials.
Liu’s group utilized zeolitic imidazolate framework-67 as a cobalt precursor and template, employing a simple hydrothermal method to create CoMoS catalysts by depositing MoS2 ultrathin sheets inside the framework [113]. The optimized CoMoS-0.18 catalyst achieved a 92.4% conversion of cresol and a 95.5% selectivity for toluene at 250 °C. This high performance was linked to the development of accessible surface CoMoS phases. Additionally, H2 preactivation facilitated the creation of sulfur vacancies in MoS2, enhancing CoMoS interfaces through surface recombination. As illustrated in Figure 9c, the in situ generation of the CoMoS phase highlights the importance of H2 preactivation in boosting HDO activity. Both showed improved p-cresol conversion and toluene selectivity after H2 pre-reduction, suggesting a significant enhancement of the HDO pathway’s activity.
In another approach, Zhang and colleagues introduced a straightforward H2O2 etching method to modify the concentration of acidic sites on the CoMoS catalyst surface (Figure 9d) [114]. By adjusting the stoichiometric ratio of H2O2 to MoS2, the researchers optimized sulfur defects on the MoS2 surface, thereby stabilizing cobalt species. This process culminated in the formation of active CoMoS catalytic sites. The optimized Co-MoS2-2 catalyst exhibited the highest density of acidic sites, which translated into a 3.4-fold enhancement in catalytic activity for the hydrodeoxygenation (HDO) of toluene derived from p-cresol, compared to the baseline Co-MoS2 sample. Furthermore, a direct correlation was observed between the HDO activity and the surface acid content (encompassing both Lewis and Brønsted acidity) within the Co-MoS2 catalysts. This relationship is crucial for effectively cleaving C-O bonds, highlighting the importance of surface acidity in determining catalytic performance. Overall, enhancing surface acidic sites represents a promising strategy for designing more efficient catalytic systems.
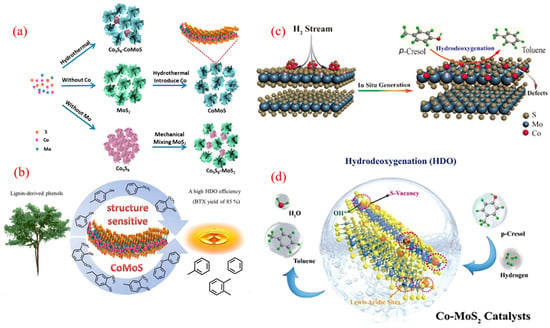
Figure 9.
(a) Surface engineering of the CoMoS catalyst for the conversion of the phenolic HDO to the product BTX; (b) synthesis diagram of cobalt-doped MoS2 nanohybrids [112]. Copyright 2019 American Chemical Society. (c) Illustration of the hydrogen-flow-induced in situ formation of active CoMoS sites for the selective HDO of p-cresol to toluene [104]. Copyright 2020 American Chemical Society. (d) Tailoring of surface acidic sites in Co-MoS2 catalysts for the HDO of p-cresol to toluene [114]. Copyright 2021 American Chemical Society.
A robust catalyst composed of Mo-doped Co9S8 nanoparticles supported on an Al2O3 matrix was successfully developed through a simple method using CoAl-hydrotalcite as a precursor. This multifunctional Mo-Co9S8/Al2O3 catalyst demonstrates exceptional hydrodeoxygenation (HDO) activity and selectivity in converting lignin to benzene, as illustrated in Figure 10a [115]. The catalyst facilitates the electron transfer from the Co to Mo sites within the Mo-Co9S8 structure, enabling the effective adsorption of oxidized compounds and contributing to its outstanding HDO performance. Notably, during the HDO of diphenyl ethers (DPEs), the catalyst achieved an impressive 99.8% conversion and a 91% yield of benzene, maintaining stability over at least 10 reaction cycles at 265 °C. The Mo-Co9S8 structure forms the covalent Mo-S-Co bonds on the Co9S8 surface, enhancing its ability to adsorb and activate oxygen-containing substrates. This feature promotes the efficient cleavage of C-O bonds while minimizing the unwanted hydrogenation of benzene rings, further highlighting its catalytic efficiency and selectivity.
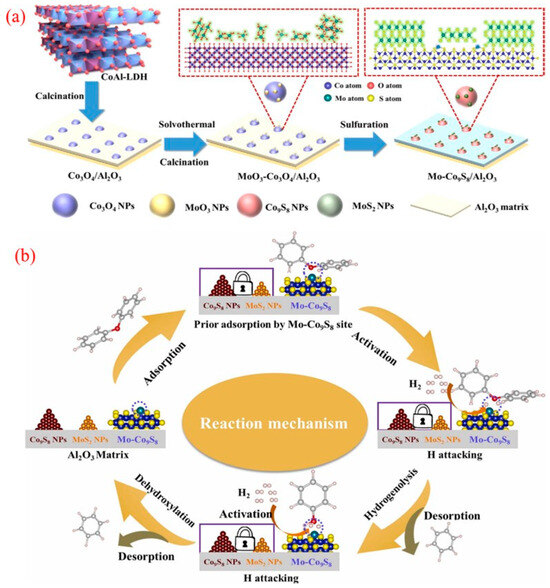
Figure 10.
(a) Schematic illustration of the preparation of the Mo-Co9S8/Al2O3 catalyst; (b) illustration on the catalytic mechanism of the DPE HDO reaction over the eMo-Co9S8 site [115]. Copyright 2022 Elsevier.
The proposed catalytic mechanism for HDO over Mo-Co9S8/Al2O3, as depicted in Figure 10b, involves several critical steps. Initially, the negatively charged carbonyl oxygen in DPEs is adsorbed by the unsaturated Mo sites on the catalyst surface, which weakens the C-O bond. The electron transfer from Co to Mo enhances hydrogen dissociation activity, enabling dissociated hydrogen species at the Mo-Co9S8 site to attack the C-O group. This results in the effective cleavage of the 4-O-5 bond, ultimately producing phenol and benzene [116]. Subsequently, phenol undergoes a similar adsorption and activation process to convert into benzene through deoxygenation. This strategy highlights an innovative approach for rationally designing efficient and stable sulfide catalysts suitable for high-temperature reactions.
In addition to cobalt, nickel (Ni) serves as another promoter in sulfide catalysts primarily to enhance activity, although it often negatively affects aromatic selectivity [117,118]. Wang and colleagues explored the effectiveness of NiMoS2 catalysts in the hydrodeoxygenation (HDO) of cresol and found that adding Ni increases the catalyst’s activity [106]. However, they observed a decline in the selectivity for toluene as the Ni content increased. To further investigate, the authors synthesized NiSx using a microwave-assisted hydrothermal method, which was then mixed with MoS2 to promote cresol HDO at 300 °C and 40 bar [119]. The results indicated that the combination of NiSx with MoS2 improved conversion rates but did not alter product distribution. Comparisons between Ni-Mo-S, NiSx/MoS2, and physically mixed NiSx + MoS2 suggested that synergies between NiSx and MoS2—beyond merely forming a Ni-Mo-S phase—were key to enhancing activity [106].
The researchers proposed a remote-control model to explain their findings: cresols predominantly adsorb on MoS2 sites and are subsequently transferred to MoS2, where they react with hydrogen atoms that are dissociated on NiSx sites. This interaction highlights the importance of using NiSx in conjunction with MoS2 to optimize performance in cresol HDO reactions, balancing high activity with product selectivity [107,109,120].
2.4.2. Transition Metal Phosphides
Transition metal phosphides, such as molybdenum phosphide (MoP), have emerged as highly effective catalysts for the hydrodeoxygenation (HDO) of phenolic compounds. MoP has demonstrated superior performance compared to its sulfide and oxide counterparts, exhibiting higher catalytic activity, lower activation energy, and greater selectivity towards aromatic compounds in the HDO conversion of p-cresols [121]. The enhanced catalytic performance of MoP is primarily attributed to the increased electron density of molybdenum in its phosphide form. This higher electron density around the molybdenum center likely enhances its nucleophilic properties, which can improve the catalyst’s ability to bind reactants and facilitate bond cleavage. Consequently, this leads to faster reaction kinetics and a lower activation energy, making the catalytic process more efficient.
According to molecular orbital theory, the lowest unoccupied molecular orbital (LUMO) of the C-O bond is antibonding, meaning that electrons donated from the catalyst to this orbital can facilitate C-O bond dissociation. Consequently, MoP, possessing the highest electron density among the catalysts studied, shows the strongest activity [122]. In experiments, MoP achieved a toluene selectivity of 60% and a conversion of 58% at 623 K and 4.4 MPa. To improve the selectivity for benzene in BTX production, Rensel and colleagues incorporated oxophilic iron (Fe) into the MoP catalyst, resulting in the bimetallic FeMoP catalyst. This bimetallic formulation produced benzene with up to 90% selectivity during the conversion of phenol at 673 K and 2.1 MPa of hydrogen [123]. Similarly, the conversion of anisole using the FeMoP catalyst also reached 90% selectivity for benzene. Further investigations of the FeMoP catalysts revealed the presence of two distinct metal sites: the metal site itself and a coordination unsaturation site (CUS) [124]. The CUS site likely arises from Moδ+ species on the catalyst surface and is proposed to act as the active site for binding the hydroxyl group in phenol, destabilizing the Caryl-O bond. This bond is subsequently cleaved by hydrogen dissociated from the metal site, enhancing the overall HDO activity and selectivity of the catalyst [125].
A series of phosphide catalysts, including MoP, Fe2P, Co2P, WP, and Ni2P, have been utilized for the hydrodeoxygenation (HDO) of phenolics into BTX [126,127,128]. Among these, Ni2P-based catalysts demonstrated superior conversion of phenolics and higher selectivity for BTX, attributed to their desirable stability and deoxygenation capabilities [34]. The Ni2P/SiO2 catalyst achieved the highest turnover frequency and a benzene selectivity of 60%. Kinetic studies revealed an apparent activation energy of 40 kJ/mol for Ni2P/SiO2, lower than the activation energy for direct C-O bond cleavage. This suggests that increased benzene production is primarily due to the hydrogenation of the C=C double bond in the aromatic ring, followed by dehydration.
Further investigation into the HDO properties of Ni2P/SiO2 with guaiacol under varying reaction conditions showed that the reaction pressure significantly influenced the reaction pathways and product distribution [129]. At 1 bar and 300 °C, the direct deoxygenation (DDO) pathway to benzene was favored, achieving 62% selectivity. Conversely, at 8 bar, the prehydrogenation (HYD) pathway led to cyclohexane as the major product, resulting in only 8% benzene selectivity (Figure 11a). This finding aligns with previous studies where cyclohexane was the primary O-free product in the HDO of anisole, emphasizing that low temperatures and higher pressures favor ring saturation [130]. X-ray Absorption Fine Structure (XAFS) measurements and density functional theory (DFT) calculations indicated that both the atomic hydrogen (H) and hydroxyl (OH) groups could adsorb on the unsaturated triple hollow (TFH) Ni sites. These results suggest that the DDO pathway is facilitated by surface OH groups, whereas the HYD pathway is promoted by more reduced surfaces. Consequently, the selectivity for BTX products can be modulated by adjusting the balance of H and OH groups on the catalyst surface.
Additionally, variations in product distribution may also result from different active sites on the Ni2P phases and their adsorption behaviors [131,132]. Ni2P crystals, characterized by a rhombohedral structure, comprise two types of nickel sites: Ni(1) and Ni(2) [133]. The Ni(1) site, with a sub-tetrahedral structure terminated by Ni3P2, serves as an active site for H2 dissociation and can adsorb phenolic molecules through the oxygen atom of the C-OH bond [132]. After a nucleophilic attack by hydride species, the adsorbed C-OH bonds are cleaved, forming aromatic products (Figure 11b). In contrast, the Ni(2) site may facilitate the flat adsorption of phenolic substrates, promoting hydrogenation of the aromatic rings.
In recent years, Lan and colleagues have further studied the performance of the Ni2P/SiO2 catalyst in HDO reaction, specifically investigating the removal of cinnamic acid and cresol by the catalyst under 300 °C and 1-bar conditions [134]. The results are encouraging, as it was found that the selectivity of cinnamic acid and cresol for the oxidation of benzene reached 86% and 81%, respectively. However, as the reaction time prolongs, the selectivity of benzene gradually decreases, while the selectivity of phenol significantly increases. This change is closely related to the changes in the surface properties of the catalyst. Specifically, the content of Niδ+ and Ni0 on the catalyst surface increases, while the P/Ni ratio decreases. Research suggests that Niδ+, as a Lewis acid site, tends to catalyze demethylation reactions rather than demethoxylation reactions, while Ni0 only exhibits low levels of dehydroxylation activity. In addition, the decrease in the P/Ni ratio reduces the number of Brønsted acid sites, thereby inhibiting the dehydroxylation reaction. These changes resulted in a transition from a phosphorus-rich environment on the catalyst surface to a nickel-rich environment, causing the HDO product of cinnamic acid to change from benzene to phenol.
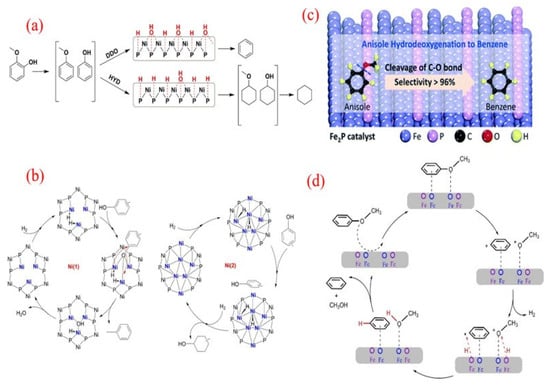
Figure 11.
(a) The proposed reaction routes of the HDO of guaiacol over the Ni2P catalyst [129]. Copyright 2014 Elsevier. (b) Different Ni sites on Ni2P for the deoxygenation of cresol [132]. Copyright 2017 Elsevier. (c) The HDO of anisole to benzene over the Fe2P catalyst; (d) the possible reaction route of anisole HDO over Fe2P [135]. Copyright 2020 Royal Society of Chemistry.
In addition to the Ni2P catalyst, the Fe2P single-metal catalyst also exhibits high selectivity for cresol in HDO reactions [135]. At 200 °C and atmospheric hydrogen conditions, Fe2P can convert cresol to benzene, ultimately achieving a benzene yield of 96% (Figure 11c). It is worth noting that even when the conversion rate is increased to 97.3%, the high selectivity of benzene remains unchanged, indicating that Fe2P has unique advantages in Caryl-O bond cleavage. In-depth DFT calculations support this observation, indicating that the oxygen atoms in cresol form a stable conformation with the Fe atoms on the Fe2P surface, making the breaking of Caryl-OCH3 bonds thermodynamically more favorable (Figure 11d). The calculation results show that compared with other possible cleavage pathways, the adsorption energy for Caryl-OCH3 bond cleavage is higher, at −2.9 eV. This further confirms the superiority of Fe2P in promoting specific bond cleavage.
The specific mechanism of the reaction can be summarized as follows: the phenol molecule first adsorbs on the Fe2P surface, and the benzene ring and oxygen atoms form coordinated bonds with the Fe sites on the surface. Subsequently, the Caryl-O bond is broken, resulting in the formation of C6H5* and CH3O* intermediates. These intermediates further bind with hydrogen atoms dissociated from adjacent Fe sites, ultimately forming benzene. This study not only deepens the understanding of iron-based phosphide catalysts but also broadens the application scope of transition metal phosphides in HDO reactions, providing new ideas for future catalyst designs.
Transition metal phosphide catalysts, such as nickel phosphide (Ni2P) and molybdenum phosphide (MoP), have shown remarkable potential in the hydrodeoxygenation (HDO) of phenolic compounds [59,136]. Their superior catalytic activity and selectivity compared to traditional catalysts can be attributed to their unique electronic properties. Moreover, these phosphide catalysts demonstrate higher selectivity towards aromatic compounds, likely due to their ability to stabilize specific intermediates and provide active sites that favor aromatic product formation [137]. However, their poor stability and tendency to deactivate in aqueous environments pose significant challenges for industrial applications.
2.4.3. Transition Metal Carbides, Nitrides
Transition metal carbonitrides exhibit noble metal-like catalytic behavior in the field of directed deoxygenation of phenolic compounds due to their unique electronic structural properties [138,139]. In this type of material, the insertion of carbon/nitrogen atoms into the metal lattice triggers a d-band contraction effect, resulting in an increase in the metal bond length and an increase in the surface electron density, thereby optimizing the C-O bond activation ability [140]. Although the selective regulation law of BTX in this system has not been fully elucidated, the groundbreaking work of Lee’s team has confirmed that the Mo2C catalyst can achieve the efficient conversion of benzyl ether under normal temperature and pressure conditions, with BTX selectivity exceeding 90% [141]. Its superiority lies in the selective passivation of hydrogenation sites by oxygen-containing intermediates generated in situ, which has been further validated in the guaiacol conversion system (benzene selectivity >90%) [142].
Reaction pathway analysis reveals that the lower dissociation energy of the Caryl-OCH3 bond compared to Caryl-OH promotes the preferential removal of methoxy groups [142]. Synchronous characterization confirmed that phenolic compounds form strong electronic coupling with the Mo2C surface through Caryl-O bonds, and this specific adsorption configuration promotes weaker CarylO-CH3 bonds to preferentially break over C-OH bonds. In addition, methanol/water titration experiments showed that the dynamic modification of surface-oxygen-containing species can inhibit aromatic ring hydrogenation activity, increasing aromatic selectivity [122].
In recent years, in-depth research has been conducted on the surface chemical properties of Mo2C catalysts in HDO reactions, especially when using the operating state NAP-XPS technology [143]. It was found that the Mo3+ phase on the surface of Mo2C is dominant, and there is also a small amount of high-valence molybdenum ions (Mo5+ and Mo6+) on the surface. It is worth noting that even in the presence of oxygenated compound feeds, the oxidation state and oxygen coverage of molybdenum remain constant throughout the entire reaction process. Meanwhile, the dominant position of Mo2+ and the slight changes in its high-valence-state molybdenum ions (Mo5+/Mo6+) indicate that the carbonyl phase is the main active site of Mo2C in the HDO reaction.
In this regard, Wang et al. further investigated the performance of a Mo2C-MoCxOy composite catalyst on activated carbon for the HDO reaction of meta-methylphenol [144]. At a 623 K and 4.3 MPa H2 pressure, the catalyst can selectively generate toluene with 75% selectivity. Through detailed study of kinetics, it was found that catalysts with different Mo2C-MoCxOy ratios exhibit consistent apparent activation energies on the direct oxidation and hydrogenation reaction pathways. This indicates that the active sites of the catalyst are determined by their own state and may be related to the adsorption or exchange of oxygen on the catalyst surface. From this, it can be inferred that both the Mo2C and MoCxOy phases may promote the oxidation reaction.
In addition, the ordered mesoporous Mo2C and W2C catalysts were prepared by the hard-template method, and their performance under reaction conditions also varied depending on the type of metal [145]. At atmospheric pressure and 443 K, the selectivity of the W2C catalyst for converting cresol to benzene reached 96%, while the selectivity of the Mo2C catalyst for benzene was slightly lower at 423 K, at 80%. This difference is mainly attributed to the higher oxide affinity of tungsten compared to that of molybdenum. Dynamics studies have shown that the production rate of benzene catalyzed by W2C and Mo2C is almost independent of the pressure of cresol, but the dependence on H2 pressure is close to half order, indicating that the generation of benzene requires two different active sites. Although the apparent activation energies of the two catalysts are similar, the formation frequency of benzene catalyzed by W2C is only 20% of that of Mo2C, indicating that the essential differences in the epitopes of tungsten carbide and molybdenum are not determined by the identity of the epitopes. This may be because the excessively strong W-O bond reduces the oxidation efficiency, thereby affecting the rate of benzene formation.
The dispersion of MoCx nanoclusters into the micropores of faujasite zeolite (FAU) also significantly improved the stability of the catalyst [146]. The catalyst prepared by this strategy can promote an alkylation reaction with high aromaticity selectivity, thereby retaining carbon in the desired final product (Figure 12). This high selectivity is mainly attributed to the Brønsted acid sites in the MoCx/FAU catalyst transferring the methoxy group of one molecule of cresol to another molecule. Compared with the physical mixture of Mo2C and FAU, the bifunctional reaction stability of MoCx/FAU is significantly improved within 20 h. This is because in the Mo2C + FAU mixture, the physical distance between the Brønsted acid site and the Mo2C site is relatively long, resulting in the rapid desorption of intermediates before continuous reaction steps, thereby forming high BTX selectivity [146]. This discovery not only demonstrates the important advantages of introducing transition metal carbonyl nanoclusters into zeolite structures but also opens up new pathways for other chemical reactions that require bifunctional metals and acid sites. In addition to gas-phase reactions, MoCx is also used in liquid-phase reactions. The Smirnov team has developed MoCx-SiO2 and bimetallic NiMoCx-SiO2 catalysts for the HDO reaction of cresol [147]. At 320 °C and 60-bar pressure, the MoCx-SiO2 catalyst can selectively convert cresol to benzene with approximately 70% selectivity. Dynamics studies have shown that the reaction pathway involves direct cleavage of C-O bonds to form benzene, rather than through phenolic intermediates. Meanwhile, the selectivity of benzene depends on the variation in the Ni content. The use of Ni as a promoter increases the activity of ring saturation and reduces the selectivity of bimetallic catalysts for benzene.
Figure 12.
The HDO of anisole to BTX by a MoCx-encapsulated FAU zeolite catalyst [146]. Copyright 2017 American Chemical Society.
Recent advancements reveal that constructing bimetallic carbide systems (e.g., Mo-W carbides) significantly enhances the catalytic performance in lignin valorization. Tran’s team conducted a pioneering comparison between MoWC composites and their monometallic counterparts (Mo2C, WC) for guaiacol hydrodeoxygenation (HDO) [148]. The experimental data demonstrated the superior activity of MoWC, achieving high benzene selectivity at 92% conversion, starkly contrasting with WC’s mere 23% conversion. Notably, product profiles diverged substantially: Mo2C predominantly yielded cyclohexene (59.1%), whereas WC favored phenol formation (69%). This performance disparity originates from the dual-active-site configuration in MoWC systems’ oxygen-affinic centers and hydrogen-activation sites [149]. The incorporation of tungsten enhances surface oxophilicity, preferentially adsorbing oxygen-containing moieties (hydroxyl/methoxy groups) over aromatic π-electrons, thereby suppressing cycloaddition side reactions. XPS characterization confirmed electron migration from W to Mo atoms, inducing distinct electronic configurations compared to monometallic carbides. The synergistic interplay in MoWC enables effective H2 activation while maintaining strong reactant adsorption through oxygen functionalities, collectively promoting selective oxygen removal during HDO processes [148].
Some research has investigated nitride catalysts, such as MoNx and CoMoNx, using guaiacol as a typical model compound to partially deoxygenate it to other phenols [150,151]. However, this makes it a challenge to directly compare BTX selectivity. Likewise, the group of Ga-synthesized Mo2N and CoMoNx catalysts in the HDO of guaiacol [152]. These nitriles directly cleave the Caromatic–OCH3 bond in guaiacol to form phenol and then saturate the ring in the HDO process to form cyclohexene and cyclohexane at 300 °C and 50-bar H2 pressure. Notably, the product distribution can be adjusted by applying the oxophilic support. As an example, when Mo2N is loaded on TiO2, the catalyst exhibits more than 90% BTX selectivity in phenol HDO at 623 K and 2.5 MPa [153]. The high activity might be attributed to TiO2 as an oxygenophilic support that promotes the selective cleavage of Caryl-O bond similarly to metal catalysts. Therefore, further in-depth studies on it are needed in the future.
In general, carbide and nitride catalysts, including molybdenum carbide (Mo2C), exhibit promising catalytic performance in the hydrodeoxygenation (HDO) of phenolic compounds. However, these catalysts face limitations such as susceptibility to oxidation, which compromises their stability. Additionally, the reaction mechanisms underlying their catalytic activity remain poorly understood. To address these challenges, further investigations are required to enhance the stability of carbide catalysts and elucidate their reaction mechanisms.
3. Conclusions and Perspectives
With the increasing demand for fuels and the heavy reliance on non-renewable petroleum, the sustainable production of chemicals and fuels from renewable resources has emerged as a critical scientific and engineering objective for modern society. Substantial global research efforts are focused on identifying and developing renewable energy sources to replace traditional fossil feedstocks in our current energy framework. Among various biomass constituents, lignin stands out as a particularly promising candidate due to its unique aromatic architecture and high energy density [39,154,155]. The catalytic hydrodeoxygenation (HDO) of lignin-derived phenolics has emerged as a transformative approach for generating value-added aromatic commodities, offering a sustainable pathway to mitigate fossil resource depletion [156,157,158,159]. This review systematically examines recent breakthroughs in the catalytic transformation of representative lignin-derived phenolic compounds into functionalized aromatics, including benzene and its derivatives. And the conversion rates and selectivity of different catalysts are shown in Table 1. Nevertheless, several fundamental challenges persist, encompassing both technological limitations and mechanistic uncertainties, which must be resolved to advance this field toward practical implementation.

Table 1.
Performances of different catalysts with BTX selectivity in the HDO of lignin-derived phenolic compounds.
(i) Catalyst deactivation and stability remain critical challenges in HDO transformations, significantly impacting industrial applicability. Common deactivation mechanisms include coking (accumulation of polyaromatic species on active sites), metal sintering (particle agglomeration under high temperature/hydrogen pressure), and oxidation (loss of active metal states in oxygen-containing environments). Stability issues are further exacerbated in aqueous-phase HDO due to metal–support interactions and hydrolysis. Future research must prioritize (1) developing anti-coking catalyst architectures (e.g., mesoporous supports, core–shell structures) to mitigate pore blockage, (2) engineering robust metal–support interfaces (e.g., strong metal–support interactions, oxide-stabilized metal nanoparticles) to suppress sintering, (3) exploring redox-active additives to maintain metal reducibility and oxygen vacancy stability, and (4) implementing in situ regeneration protocols (e.g., periodic oxidation–reduction cycles) to restore catalyst activity. Addressing these challenges will be pivotal for translating lab-scale HDO efficiency into industrially viable, long-duration catalytic processes.
(ii) Green solvent engineering in phenolic hydrogenation–deoxygenation processes. In modern sustainable chemistry, the development of eco-friendly reaction media represents a critical challenge for industrial applications. Water has been widely validated as a green reaction medium for the selective HDO of phenolic compounds, offering advantages such as high activity and product specificity. However, its inherent volatility at reaction temperatures (typically >150 °C) leads to significant solvent loss and corrosion risks in reactor systems. To address these limitations, recent research has explored the use of low-volatility solvents with tunable polarity. Ionic liquids (ILs), with their customizable anion–cation interactions, have shown potential for improving product selectivity through solvation effects. Concurrently, deep eutectic solvents (DESs) composed of hydrogen bond donors and acceptors have emerged as promising alternatives, exhibiting a comparable catalytic performance to that of ILs while offering lower toxicity and higher biodegradability. Despite these advances, current solvent optimization studies are primarily empirical, lacking systematic mechanistic analysis linking solvent properties to reaction outcomes. Therefore, computational modeling combined with in situ characterization techniques should be prioritized to establish structure–activity relationships, enabling rational designs of next-generation solvent systems.
(iii) The current state of the art in phenolic HDO catalysis relies predominantly on miniaturized steel reactors or batch-type systems for catalyst development and kinetic studies, leveraging their ease of operation and process simplicity. While these platforms enable high-throughput catalyst evaluation and kinetic parameter determination, large-scale industrial implementation necessitates fixed-bed reactors due to their superior continuous processing capabilities and potential for higher space–time yields. To bridge the gap between lab-scale success and commercial deployment, future research should focus on process intensification strategies for continuous-flow systems, integrating catalyst design with reactor engineering. This approach would allow precise control over residence time distribution, reactant concentration gradients, and thermal management, thereby improving product selectivity. However, scaling up HDO processes faces significant challenges, including heterogeneous reaction dynamics at biomass–catalyst interfaces, multiphase mass-transfer limitations, and reactor fouling mitigation. Computational fluid dynamics (CFD) modeling combined with in situ characterization techniques will be essential for optimizing reactor hydrodynamics and heat-transfer coefficients. Additionally, techno-economic analysis should be conducted to evaluate the feasibility of integrating HDO units with existing biorefinery infrastructure, ensuring compatibility with downstream processing operations. The current literature lacks comprehensive studies on reactor performance stability over extended operation periods, which is critical for industrial viability.
(iv) Considering the biomass-derived nature of phenolic compounds, future research should shift focus toward valorizing lignin as a sustainable feedstock for producing platform chemicals and renewable fuels. Central to this effort is the design of one-pot catalytic systems that couple lignin depolymerization with in situ hydrogenation–deoxygenation (HDO), enabling the direct conversion of native lignin into high-value products. Such integrated processes offer dual advantages: minimizing post-depolymerization stabilization steps while reducing energy-intensive separation operations through enhanced product selectivity. However, realizing this potential requires resolving fundamental gaps in understanding lignin’s structural transformation during HDO, particularly regarding its macromolecular architecture, solubility behavior, and thermal stability. Recent advancements in advanced characterization tools, including heteronuclear single-quantum coherence (HSQC) spectroscopy, three-dimensional nuclear magnetic resonance (3D NMR), and long-range correlation (HMBC) techniques, have enabled unprecedented insights into lignin’s chemical composition. Complementary in situ techniques, such as synchrotron-based X-ray absorption spectroscopy (XAS) and time-resolved infrared (TRIR) spectroscopy, are now being employed to monitor dynamic structural changes during HDO. The thermodynamic modeling of lignin-derived phenolic intermediates and their HDO pathways is also gaining traction, with computational methods like density functional theory (DFT) providing atomistic-level mechanistic insights. Future breakthroughs in this field will likely arise from integrating experimental characterization with multi-scale modeling approaches to design tailored catalysts capable of selectively targeting lignin’s recalcitrant linkages while maintaining high stability under industrially relevant conditions.
In conclusion, this review aims to offer a thorough understanding of the current state of knowledge regarding the production and innovative catalytic upgrading of functional chemicals. These insights are applicable to the catalytic hydrodeoxygenation (HDO) upgrading pathways of various biomass-derived platform model compounds, whether originating from lignin or carbohydrates. Furthermore, the integrated approach combining lignin depolymerization with downstream processes such as HDO and etherification (ECH) presents a promising pathway for the sustainable production of high-value functional chemicals in the future. This research field is bound to greatly expand and provide a green future for our society with sustainable chemical products.
Author Contributions
S.D.: writing—review and editing, writing—original draft, conceptualization. G.F.: writing—review and editing, supervision, methodology, investigation. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Scientific Research Projects of Hebei Education Department (QN2019050) and the Natural Science Foundation of Hebei Province (B2020202004).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ye, K.; Liu, Y.; Wu, S.; Zhuang, J. A review for lignin valorization: Challenges and perspectives in catalytic hydrogenolysis. Ind. Crops Prod. 2021, 172, 114008. [Google Scholar] [CrossRef]
- Shu, R.; Li, R.; Lin, B.; Wang, C.; Cheng, Z.; Chen, Y. A review on the catalytic hydrodeoxygenation of lignin-derived phenolic compounds and the conversion of raw lignin to hydrocarbon liquid fuels. Biomass Bioenergy 2020, 132, 105432. [Google Scholar] [CrossRef]
- Zhou, C.-H.; Xia, X.; Lin, C.-X.; Tong, D.-S.; Beltramini, J. Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels. Chem. Soc. Rev. 2011, 40, 5588–5617. [Google Scholar] [CrossRef]
- Sun, Z.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef]
- Amidon, T.E.; Liu, S. Water-based woody biorefinery. Biotechnol. Adv. 2009, 27, 542–550. [Google Scholar] [CrossRef]
- Martínez Angel, T. How to break down crystalline cellulose. Science 2016, 352, 1050–1051. [Google Scholar] [CrossRef]
- Lang, M.; Li, H. Sustainable Routes for the Synthesis of Renewable Adipic Acid from Biomass Derivatives. ChemSusChem 2022, 15, e202101531. [Google Scholar] [CrossRef]
- Árvai, C.; Medgyesi, Z.; LUI, M.; Mika, L. The Chemistry of Levulinic Acid: Its Potential in the Production of Biomass—Based Chemicals. Adv. Synth. Catal. 2024, 366, 4846–4888. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, L.; Wang, T.; Zhang, Q.; Wang, C.; Yan, J.; Ma, L. One-Pot Catalytic Conversion of Raw Lignocellulosic Biomass into Gasoline Alkanes and Chemicals over LiTaMoO6 and Ru/C in Aqueous Phosphoric Acid. ACS Sustain. Chem. Eng. 2015, 3, 1745–1755. [Google Scholar] [CrossRef]
- Sun, Z.; Bottari, G.; Afanasenko, A.; Stuart, M.C.A.; Deuss, P.J.; Fridrich, B.; Barta, K. Complete lignocellulose conversion with integrated catalyst recycling yielding valuable aromatics and fuels. Nat. Catal. 2018, 1, 82–92. [Google Scholar] [CrossRef]
- Wijaya, Y.P.; Smith, K.J.; Kim, C.S.; Gyenge, E.L. Electrocatalytic hydrogenation and depolymerization pathways for lignin valorization: Toward mild synthesis of chemicals and fuels from biomass. Green Chem. 2020, 22, 7233–7264. [Google Scholar] [CrossRef]
- Carpenter, D.; Westover, T.L.; Czernik, S.; Jablonski, W. Biomass feedstocks for renewable fuel production: A review of the impacts of feedstock and pretreatment on the yield and product distribution of fast pyrolysis bio-oils and vapors. Green Chem. 2014, 16, 384–406. [Google Scholar] [CrossRef]
- Akhtari, S.; Sowlati, T.; Day, K. Economic feasibility of utilizing forest biomass in district energy systems—A review. Renew. Sust. Energ. Rev. 2014, 33, 117–127. [Google Scholar] [CrossRef]
- Ruppert, A.M.; Weinberg, K.; Palkovits, R. Hydrogenolysis Goes Bio: From Carbohydrates and Sugar Alcohols to Platform Chemicals. Angew. Chem. Internat. Edit. 2012, 51, 2564–2601. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, C.; Li, D.; Liu, Q.; Tan, J.; Wang, C.; Cai, C.; Ma, L. Recent advances in catalytic conversion of biomass to 5-hydroxymethylfurfural and 2, 5-dimethylfuran. Renew. Sust. Energ. Rev. 2019, 103, 227–247. [Google Scholar] [CrossRef]
- Liao, Y.; Liu, Q.; Wang, T.; Long, J.; Zhang, Q.; Ma, L.; Liu, Y.; Li, Y. Promoting Hydrolytic Hydrogenation of Cellulose to Sugar Alcohols by Mixed Ball Milling of Cellulose and Solid Acid Catalyst. Energ. Fuels 2014, 28, 5778–5784. [Google Scholar] [CrossRef]
- Achyuthan, K.E.; Achyuthan, A.M.; Adams, P.D.; Dirk, S.M.; Harper, J.C.; Simmons, B.A.; Singh, A.K. Supramolecular self-assembled chaos: Polyphenolic lignin’s barrier to cost-effective lignocellulosic biofuels. Molecules 2010, 15, 8641–8688. [Google Scholar] [CrossRef]
- Mateo, S.; Fabbrizi, G.; Moya, A. Lignin from Plant—Based Agro—Industrial Biowastes: From Extraction to Sustainable Applications. Polymers 2025, 17, 952. [Google Scholar] [CrossRef]
- Shu, R.; Zhang, Q.; Ma, L.; Xu, Y.; Chen, P.; Wang, C.; Wang, T. Insight into the solvent, temperature and time effects on the hydrogenolysis of hydrolyzed lignin. Bioresour. Technol. 2016, 221, 568–575. [Google Scholar] [CrossRef]
- Ma, H.; Li, H.; Zhao, W.; Li, L.; Liu, S.; Long, J.; Li, X. Selective depolymerization of lignin catalyzed by nickel supported on zirconium phosphate. Green Chem. 2019, 21, 658–668. [Google Scholar] [CrossRef]
- Shu, R.; Long, J.; Yuan, Z.; Zhang, Q.; Wang, T.; Wang, C.; Ma, L. Efficient and product-controlled depolymerization of lignin oriented by metal chloride cooperated with Pd/C. Bioresour. Technol. 2015, 179, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Yu, I.; Liu, Y.; Ruan, X.; Tsang, D.; Hunt, A.; Ok, Y.; Song, H.; Zhang, S. Lignin valorization for the production of renewable chemicals: State-of-the-art review and future prospects. Bioresour. Technol. 2018, 269, 465–475. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, H.; Chen, S.; Wu, J. Catalytic co-pyrolysis of lignocellulosic biomass with polymers: A critical review. Green Chem. 2016, 18, 4145–4169. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.; Zhang, T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chem. Rev. 2015, 115, 11559–12090. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
- Sirous-Rezaei, P.; Jae, J.; Ha, J.-M.; Ko, C.H.; Kim, J.M.; Jeon, J.-K.; Park, Y.-K. Mild hydrodeoxygenation of phenolic lignin model compounds over a FeReOx/ZrO2 catalyst: Zirconia and rhenium oxide as efficient dehydration promoters. Green Chem. 2018, 20, 1472–1483. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, Z.; Wu, Y.; Tu, R.; Yang, H.; Jiang, E.; Xu, X. Smoke calcination induced C-Ni/VOX triphase interface micro-environment Controls selectivity in guaiacol hydrodeoxygenation. Chem. Eng. J. 2025, 505, 159229. [Google Scholar] [CrossRef]
- LV, W.; Jiang, Y.; Tian, K.; Wang, X.; Li, X.; Tong, J.; Chen, A.; Chen, X. Constructing Ni-MgO wrapped by carbon sphere to prepare a defect-rich catalyst for the conversion of lignin-derived oligomers into hydrocarbons under mild conditions. Chem. Eng. J. 2025, 509, 161057. [Google Scholar]
- Lonchay, W.; Bagnato, G.; Sanna, A. Highly selective hydropyrolysis of lignin waste to benzene, toluene and xylene in presence of zirconia supported iron catalyst. Bioresour. Technol. 2022, 361, 127727. [Google Scholar] [CrossRef]
- Dong, L.; Xin, Y.; Liu, X.; Guo, Y.; Pao, C.; Chen, J.; Wang, Y. Selective hydrodeoxygenation of lignin oil to valuable phenolics over Au/Nb2O5 in water. Green Chem. 2019, 21, 3081–3090. [Google Scholar] [CrossRef]
- Liu, B.; Qi, Y.; Qiu, X.; Zou, H.; Lin, X. Photoelectrocatalytic Pathway for the Preparation of Power-Effective Aviation Fuel Precursors from Lignin. Adv. Funct. Mater. 2025, 35, 2421552. [Google Scholar] [CrossRef]
- Hu, L.; Wei, X.; Xu, M.; Kang, Y.; Guo, X.; Zhang, F.; Zong, Z.; Bai, H. Selective organic phase hydrodeoxygenation of typical phenolic monomers and two lignin oils over highly active Pd/Hβ catalyst for high-grade bio-fuel production. J. Environ. Chem. Eng. 2021, 9, 106599. [Google Scholar] [CrossRef]
- Zhong, J.; Chen, J.; Chen, L. Selective hydrogenation of phenol and related derivatives. Catal. Sci. Technol. 2014, 4, 3555–3569. [Google Scholar] [CrossRef]
- Wang, X.; Arai, M.; Wu, Q.; Zhang, C.; Zhao, F. Hydrodeoxygenation of lignin-derived phenolics—A review on the active sites of supported metal catalysts. Green Chem. 2020, 22, 8140–8168. [Google Scholar] [CrossRef]
- Gundekari, S.; Karmee, S.K. Recent Catalytic Approaches for the Production of Cycloalkane Intermediates from Lignin-Based Aromatic Compounds: A Review. Review 2021, 6, 1715–1733. [Google Scholar] [CrossRef]
- Yan, J.; Li, Z.; Zhang, Y.; Liu, R.; Zhou, L.; Fu, P. Hydrodeoxygenation of lignin phenolic derivatives to aromatics: A review of catalyst functionalization for targeted deoxygenation and active site modification strategies. Fuel Process. Technol. 2023, 250, 107914. [Google Scholar] [CrossRef]
- Wong, S.S.; Shu, R.; Zhang, J.; Liu, H.; Yan, N. Downstream processing of lignin derived feedstock into end products. Chem. Soc. Rev. 2020, 49, 5510–5560. [Google Scholar] [CrossRef]
- Verma, J.; Petru, M.; Goel, S. Cellulose based materials to accelerate the transition towards sustainability. Ind. Crop. Prod. 2024, 210, 118078. [Google Scholar] [CrossRef]
- Olcese, R.N.; Bettahar, M.; Petitjean, D.; Malaman, B.; Giovanella, F.; Dufour, A. Gas-phase hydrodeoxygenation of guaiacol over Fe/SiO2 catalyst. Appl. Catal. B-Environ. 2012, 115–116, 63–73. [Google Scholar] [CrossRef]
- Holladay, J.E.; White, J.F.; Bozell, J.J.; Johnson, D.J.B.F. Top Value-Added Chemicals from Biomass—Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin. Biomass Fuels 2007, 2, 263–275. [Google Scholar]
- Ponnusamy, V.K.; Nguyen, D.D.; Dharmaraja, J.; Shobana, S.; Banu, J.R.; Saratale, R.G.; Chang, S.W.; Kumar, G. A review on lignin structure, pretreatments, fermentation reactions and biorefinery potential. Bioresour. Technol. 2019, 271, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jiang, E.; Du, Y.; Li, B. BTX from the gas-phase hydrodeoxygenation and transmethylation of guaiacol at room pressure. Renew. Energy 2016, 96, 458–468. [Google Scholar]
- Hong, Y.; Zhang, H.; Sun, J.; Ayman, K.M.; Hensley, A.J.R.; Gu, M.; Engelhard, M.H.; McEwen, J.-S.; Wang, Y. Synergistic Catalysis between Pd and Fe in Gas Phase Hydrodeoxygenation of m-Cresol. ACS Catal. 2014, 4, 3335–3345. [Google Scholar] [CrossRef]
- Barrios, A.M.; Teles, C.A.; de Souza, P.M.; Rabelo-Neto, R.C.; Jacobs, G.; Davis, B.H.; Borges, L.E.P.; Noronha, F.B. Hydrodeoxygenation of phenol over niobia supported Pd catalyst. Catal. Today 2018, 302, 115–124. [Google Scholar] [CrossRef]
- Ji, N.; Yin, J.; Rong, Y.; Li, H.; Yu, Z.; Lei, Y.; Wang, S.; Diao, X. More than a support: The unique role of Nb2O5 in supported metal catalysts for lignin hydrodeoxygenation. Catal. Sci. Technol. 2022, 12, 3751–3766. [Google Scholar] [CrossRef]
- de Souza, P.M.; Rabelo-Neto, R.C.; Borges, L.E.P.; Jacobs, G.; Davis, B.H.; Resasco, D.E.; Noronha, F.B. Hydrodeoxygenation of Phenol over Pd Catalysts. Effect of Support on Reaction Mechanism and Catalyst Deactivation. ACS Catal. 2017, 7, 2058–2073. [Google Scholar] [CrossRef]
- Teles, C.A.; de Souza, P.M.; Rabelo-Neto, R.C.; Teran, A.; Jacobs, G.; Resasco, D.E.; Noronha, F.B. Hydrodeoxygenation of Lignin-Derived Compound Mixtures on Pd-Supported on Various Oxides. ACS Sustain. Chem. Eng. 2021, 9, 12870–12884. [Google Scholar] [CrossRef]
- Shao, Y.; Xia, Q.; Dong, L.; Liu, X.; Han, X.; Parker, S.F.; Cheng, Y.; Daemen, L.L.; Ramirez-Cuesta, A.J.; Yang, S.; et al. Selective production of arenes via direct lignin upgrading over a niobium-based catalyst. Nat. Commun. 2017, 8, 16104. [Google Scholar] [CrossRef]
- Hongkailers, S.; Phumpradit, S.; Phanpa, C.; Pattiya, A.; Ngamcharussrivichai, C.; Yokoi, T.; Hinchiranan, N. Bio-phenols production via hydrodeoxygenation of lignin-derived guaiacol andbio-oil over high water-tolerant NiMo/Al2O3-ZrO2 catalysts. Clean. Eng. Technol. 2024, 23, 100858. [Google Scholar]
- Wei, H.; Wang, Z.; Li, H. Sustainable biomass hydrodeoxygenation in biphasic systems. Green Chem. 2022, 24, 1930–1950. [Google Scholar] [CrossRef]
- Zhang, C.; Jia, C.; Cao, Y.; Yao, Y.; Xie, S.; Zhang, S.; Lin, H. Water-assisted selective hydrodeoxygenation of phenol to benzene over the Ru composite catalyst in the biphasic process. Green Chem. 2019, 21, 1668–1679. [Google Scholar] [CrossRef]
- Guo, T.; Xia, Q.; Shao, Y.; Liu, X.; Wang, Y. Direct deoxygenation of lignin model compounds into aromatic hydrocarbons through hydrogen transfer reaction. Appl. Catal. A 2017, 547, 30–36. [Google Scholar] [CrossRef]
- Nelson, R.C.; Baek, B.; Ruiz, P.; Goundie, B.; Brooks, A.; Wheeler, M.C.; Frederick, B.G.; Grabow, L.C.; Austin, R.N. Experimental and Theoretical Insights into the Hydrogen-Efficient Direct Hydrodeoxygenation Mechanism of Phenol over Ru/TiO2. ACS Catal. 2015, 5, 6509–6523. [Google Scholar] [CrossRef]
- Duan, H.; Liu, J.-C.; Xu, M.; Zhao, Y.; Ma, X.-L.; Dong, J.; Zheng, X.; Zheng, J.; Allen, C.S.; Danaie, M.; et al. Molecular nitrogen promotes catalytic hydrodeoxygenation. Nat. Catal. 2019, 2, 1078–1087. [Google Scholar] [CrossRef]
- Luo, Z.; Zheng, Z.; Wang, Y.; Sun, G.; Jiang, H.; Zhao, C. Hydrothermally stable Ru/HZSM-5-catalyzed selective hydrogenolysis of lignin-derived substituted phenols to bio-arenes in water. Green Chem. 2016, 18, 5845–5858. [Google Scholar] [CrossRef]
- Xiang, L.; Liu, M.; Fan, G.; Yang, L.; Li, F. MoOx-Decorated ZrO2 Nanostructures Supporting Ru Nanoclusters for Selective Hydrodeoxygenation of Anisole to Benzene. ACS Appl. Nano Mater. 2021, 4, 12588–12599. [Google Scholar] [CrossRef]
- Zhu, X.; Nie, L.; Lobban, L.L.; Mallinson, R.G.; Resasco, D.E. Efficient Conversion of m-Cresol to Aromatics on a Bifunctional Pt/HBeta Catalyst. Energy Fuels 2014, 28, 4104–4111. [Google Scholar] [CrossRef]
- Zhu, X.; Lobban, L.L.; Mallinson, R.G.; Resasco, D.E. Bifunctional transalkylation and hydrodeoxygenation of anisole over a Pt/HBeta catalyst. J. Catal. 2011, 281, 21–29. [Google Scholar] [CrossRef]
- Golubeva, M.; Maximov, A. Transition metal compounds in the hydrodeoxygenation of biomass derivatives. Renew. Sustain. Energy Rev. 2025, 210, 115153. [Google Scholar] [CrossRef]
- Nie, L.; Peng, B.; Zhu, X. Vapor-Phase Hydrodeoxygenation of Guaiacol to Aromatics over Pt/HBeta: Identification of the Role of Acid Sites and Metal Sites on the Reaction Pathway. ChemCatChem 2018, 10, 1064–1074. [Google Scholar] [CrossRef]
- Griffin, M.B.; Ferguson, G.A.; Ruddy, D.A.; Biddy, M.J.; Beckham, G.T.; Schaidle, J.A. Role of the Support and Reaction Conditions on the Vapor-Phase Deoxygenation of m-Cresol over Pt/C and Pt/TiO2 Catalysts. ACS Catal. 2016, 6, 2715–2727. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, J.; Zhang, Q.; Liu, Q.; Li, Y.; Chen, L.; Wang, C.; Ma, L. Hydrodeoxygenation of lignin-derived phenolic compounds into aromatic hydrocarbons under low hydrogen pressure using molybdenum oxide as catalyst. Catal. Today 2019, 319, 41–47. [Google Scholar] [CrossRef]
- Zhang, J.; Duan, F.; Xie, Y.; Ning, P.; Zhao, H.; Shi, Y. Encapsulated Ni Nanoparticles within Silicalite-1 Crystals for Upgrading Phenolic Compounds to Arenes. Ind. Eng. Chem. Res. 2021, 60, 13790–13801. [Google Scholar] [CrossRef]
- Popov, A.; Kondratieva, E.; Goupil, J.M.; Mariey, L.; Bazin, P.; Gilson, J.-P.; Travert, A.; Maugé, F. Bio-oils Hydrodeoxygenation: Adsorption of Phenolic Molecules on Oxidic Catalyst Supports. J. Phys. Chem. C 2010, 114, 15661–15670. [Google Scholar] [CrossRef]
- Nie, L.; de Souza, P.M.; Noronha, F.B.; An, W.; Sooknoi, T.; Resasco, D.E. Selective conversion of m-cresol to toluene over bimetallic Ni–Fe catalysts. J. Mol. Catal. A Chem. 2014, 388–389, 47–55. [Google Scholar] [CrossRef]
- Prasomsri, T.; Shetty, M.; Murugappan, K.; Román-Leshkov, Y. Insights into the catalytic activity and surface modification of MoO3 during the hydrodeoxygenation of lignin-derived model compounds into aromatic hydrocarbons under low hydrogen pressures. Energy Environ. Sci. 2014, 7, 2660–2669. [Google Scholar] [CrossRef]
- Shetty, M.; Murugappan, K.; Prasomsri, T.; Green, W.H.; Román-Leshkov, Y. Reactivity and stability investigation of supported molybdenum oxide catalysts for the hydrodeoxygenation (HDO) of m-cresol. J. Catal. 2015, 331, 86–97. [Google Scholar] [CrossRef]
- Gonçalves, V.O.O.; Ciotonea, C.; Arrii-Clacens, S.; Guignard, N.; Roudaut, C.; Rousseau, J.; Clacens, J.-M.; Royer, S.; Richard, F. Effect of the support on the hydrodeoxygenation of m-cresol over molybdenum oxide based catalysts. Appl. Catal. B Environ. 2017, 214, 57–66. [Google Scholar] [CrossRef]
- Christodoulakis, A.; Boghosian, S. Molecular structure and activity of molybdena catalysts supported on zirconia for ethane oxidative dehydrogenation studied by operando Raman spectroscopy. J. Catal. 2008, 260, 178–187. [Google Scholar] [CrossRef]
- Prasomsri, T.; Nimmanwudipong, T.; Román-Leshkov, Y. Effective hydrodeoxygenation of biomass-derived oxygenates into unsaturated hydrocarbons by MoO3 using low H2 pressures. Energy Environ. Sci. 2013, 6, 1732–1738. [Google Scholar] [CrossRef]
- Lin, Y.; Shu, R.; Yin, T.; Tian, Z.; Wang, C.; Xu, Y. Catalytic hydrodeoxygenation of lignin-derived phenolic compounds with Ni-based aluminum phosphate catalyst. Mol. Catal. 2024, 567, 114444. [Google Scholar] [CrossRef]
- Diao, X.; Hao, L.; Shi, Y.; Zhang, S.; Ji, N. Boosted hydrodeoxygenation of lignin and its derivatives to cycloalkanes over Ni catalysts with surface decoration of AlPO4 species. J. Energy Chem. 2025, 104, 360–371. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, G.; Dou, S.; Dong, Y.; Kong, X. WOx boosted hollow Ni nanoreactors for the hydrodeoxygenation of lignin derivatives. Int. J. Biol. Macromol. 2024, 269, 132156. [Google Scholar] [CrossRef]
- Yang, Y.; Ochoa-Hernández, C.; de la Peña O’Shea, V.A.; Pizarro, P.; Coronado, J.M.; Serrano, D.P. Effect of metal–support interaction on the selective hydrodeoxygenation of anisole to aromatics over Ni-based catalysts. Appl. Catal. B-Environ. 2014, 145, 91–100. [Google Scholar] [CrossRef]
- Yang, F.; Liu, D.; Zhao, Y.; Wang, H.; Han, J.; Ge, Q.; Zhu, X. Size Dependence of Vapor Phase Hydrodeoxygenation of m-Cresol on Ni/SiO2 Catalysts. ACS Catal. 2018, 8, 1672–1682. [Google Scholar] [CrossRef]
- Luo, B.; Chen, X.; Guo, Y.; Shu, R.; Wang, C.; Li, J.; Wang, J.; Tian, Z.; Chen, Y. Efficient hydrodeoxygenation of lignin-derived oxygenates over Ni/Al2O3-C catalyst under mild conditions. J. Energy Inst. 2025, 118, 101944. [Google Scholar] [CrossRef]
- Resende, K.A.; Braga, A.H.; Noronha, F.B.; Hori, C.E. Hydrodeoxygenation of phenol over Ni/Ce1-xNbxO2 catalysts. Appl. Catal. B-Environ. 2019, 245, 100–113. [Google Scholar] [CrossRef]
- Wang, X.; Feng, S.; Wang, Y.; Zhao, Y.; Huang, S.; Wang, S.; Ma, X. Enhanced hydrodeoxygenation of lignin-derived anisole to arenes catalyzed by Mn-doped Cu/Al2O3. Green Energy Environ. 2021, 8, 927–937. [Google Scholar] [CrossRef]
- Alonso, D.M.; Wettstein, S.G.; Dumesic, J.A. Bimetallic catalysts for upgrading of biomass to fuels and chemicals. Chem. Soc. Rev. 2012, 41, 8075–8098. [Google Scholar] [CrossRef]
- Rebelli, J.; Detwiler, M.; Ma, S.; Williams, C.T.; Monnier, J.R. Synthesis and characterization of Au–Pd/SiO2 bimetallic catalysts prepared by electroless deposition. J. Catal. 2010, 270, 224–233. [Google Scholar] [CrossRef]
- Lv, Y.; Yang, K.; Lin, X.; Zhou, K.; Liu, Y.; Ye, X.; Song, L.; Lin, C.; Yang, G.; Liu, M. Selective production of cycloalkanes through the catalytic hydrodeoxygenation of lignin with CoNi2@BTC catalysts without external hydrogen. Int. J. Biol. Macromol. 2025, 303, 140496. [Google Scholar] [CrossRef] [PubMed]
- Mennani, M.; Abdellaoui, Y.; Benhamou, A.; Lopez-Maldonado, E.; Kasbaji, M.; Achaby, M.; Moubarik, A.; Kassab, Z. FeNi bimetallic functionalized lignin catalyst for sustainable oxidation processes. Sustain. Mater. Technol. 2025, 43, e01267. [Google Scholar] [CrossRef]
- Robinson, A.M.; Hensley, J.E.; Medlin, J.W. Bifunctional Catalysts for Upgrading of Biomass-Derived Oxygenates: A Review. ACS Catal. 2016, 6, 5026–5043. [Google Scholar] [CrossRef]
- Sun, J.; Karim, A.M.; Zhang, H.; Kovarik, L.; Li, X.S.; Hensley, A.J.; McEwen, J.-S.; Wang, Y. Carbon-supported bimetallic Pd–Fe catalysts for vapor-phase hydrodeoxygenation of guaiacol. J. Catal. 2013, 306, 47–57. [Google Scholar] [CrossRef]
- Li, J.; Xia, S. Bimetallic NiMo Using MOF-Derived Carbon-Supported Catalysts for the Reaction of Lauric Acid to Alkane. Catal. Surv. Asia 2024, 28, 269–282. [Google Scholar] [CrossRef]
- Phan, T.N.; Ko, C.H. Synergistic effects of Ru and Fe on titania-supported catalyst for enhanced anisole hydrodeoxygenation selectivity. Catal. Today 2018, 303, 219–226. [Google Scholar] [CrossRef]
- Khoobiar, S. Particle to Particle Migration of Hydrogen Atoms on Platinum—Alumina Catalysts from Particle to Neighboring Particles. J. Phys. Chem. 1964, 68, 411–412. [Google Scholar] [CrossRef]
- Tan, M.; Yang, Y.; Yang, Y.; Chen, J.; Zhang, Z.; Fu, G.; Lin, J.; Wan, S.; Wang, S.; Wang, Y. Hydrogen spillover assisted by oxygenate molecules over nonreducible oxides. Nat. Commun. 2022, 13, 1457. [Google Scholar] [CrossRef]
- Mohammed, A.; Tannous, J. Catalytic Hydrodeoxygenation of Phenols and Cresols to Gasoline RangeBiofuels. Chem. Rec. 2024, 24, e202400092. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Grunwaldt, J.-D.; Jensen, P.A.; Jensen, A.D. Screening of Catalysts for Hydrodeoxygenation of Phenol as a Model Compound for Bio-oil. ACS Catal. 2013, 3, 1774–1785. [Google Scholar] [CrossRef]
- He, T.; Liu, X.; Ge, Y.; Han, D.; Li, J.; Wang, Z.; Wu, J. Gas phase hydrodeoxygenation of anisole and guaiacol to aromatics with a high selectivity over Ni-Mo/SiO2. Catal. Commun. 2017, 102, 127–130. [Google Scholar] [CrossRef]
- Yang, F.; Libretto, N.J.; Komarneni, M.R.; Zhou, W.; Miller, J.T.; Zhu, X.; Resasco, D.E. Enhancement of m-Cresol Hydrodeoxygenation Selectivity on Ni Catalysts by Surface Decoration of MoOx Species. ACS Catal. 2019, 9, 7791–7800. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J. Effects of indium on Ni/SiO2 catalytic performance in hydrodeoxygenation of anisole as model bio-oil compound: Suppression of benzene ring hydrogenation and C–C bond hydrogenolysis. Chin. J. Catal. 2017, 38, 1818–1830. [Google Scholar] [CrossRef]
- Ali, R.; Mushtag, S.; Cheng, C.; Wongsakulphasatch, S.; Haila, M.; Al-Ali, K. Hierarchically structured nanospherical fibrous silica-supported bimetalliccatalysts: An enhanced performance in methane decomposition. Int. J. Hydrogen Energy 2024, 87, 1480–1498. [Google Scholar] [CrossRef]
- Yang, F.; Liu, D.; Wang, H.; Liu, X.; Han, J.; Ge, Q.; Zhu, X. Geometric and electronic effects of bimetallic Ni–Re catalysts for selective deoxygenation of m-cresol to toluene. J. Catal. 2017, 349, 84–97. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhao, N.; Chen, J. Enhanced direct deoxygenation of anisole to benzene on SiO2-supported Ni-Ga alloy and intermetallic compound. Appl. Catal. B-Environ. 2019, 250, 280–291. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, Y.; Liang, K.; Yang, Z.; Tu, R.; Fan, X.; Cheng, S.; Yu, H.; Jiang, E.; Xu, X. Enhancing Hydrodeoxygenation of Bio-oil via Bimetallic Ni-V Catalysts Modified by Cross-Surface Migrated-Carbon from Biochar. ACS Appl. Mater. Interfaces 2021, 13, 21482–21498. [Google Scholar] [CrossRef]
- Yue, K.; Liu, S.; Yan, S.; Bi, D.; Liu, Z.; Xu, S. Analysis of the reaction mechanism of N/S co-doped carbon-based catalysts forlow-temperature NH3-SCR reduction of NOx. Sep. Purif. Technol. 2024, 344, 127302. [Google Scholar] [CrossRef]
- Sirous-Rezaei, P.; Jae, J.; Cho, K.; Ko, C.H.; Jung, S.-C.; Park, Y.-K. Insight into the effect of metal and support for mild hydrodeoxygenation of lignin-derived phenolics to BTX aromatics. Chem. Eng. J. 2019, 377, 120121. [Google Scholar] [CrossRef]
- Ghampson, I.T.; Canales, R.; Escalona, N. A study of the hydrodeoxygenation of anisole over Re-MoOx/TiO2 catalyst. Appl. Catal. A-Gen. 2018, 549, 225–236. [Google Scholar] [CrossRef]
- Ghampson, I.T.; Pecchi, G.; Fierro, J.L.G.; Videla, A.; Escalona, N. Catalytic hydrodeoxygenation of anisole over Re-MoOx/TiO2 and Re-VOx/TiO2 catalysts. Appl. Catal. B-Environ. 2017, 208, 60–74. [Google Scholar] [CrossRef]
- Huang, Y.-B.; Yan, L.; Chen, M.-Y.; Guo, Q.-X.; Fu, Y. Selective hydrogenolysis of phenols and phenyl ethers to arenes through direct C–O cleavage over ruthenium–tungsten bifunctional catalysts. Green Chem. 2015, 17, 3010–3017. [Google Scholar] [CrossRef]
- Wang, C.; Mironenko, A.V.; Raizada, A.; Chen, T.; Mao, X.; Padmanabhan, A.; Vlachos, D.G.; Gorte, R.J.; Vohs, J.M. Mechanistic Study of the Direct Hydrodeoxygenation of m-Cresol over WOx-Decorated Pt/C Catalysts. ACS Catal. 2018, 8, 7749–7759. [Google Scholar] [CrossRef]
- Meng, Q.; Yan, J.; Liu, H.; Chen, C.; Li, S.; Shen, X.; Song, J.; Zheng, L.; Han, B. Self-supported hydrogenolysis of aromatic ethers to arenes. Sci. Adv. 2019, 5, eaax6839. [Google Scholar] [CrossRef]
- Jiang, S.; Ji, N.; Diao, X.; Li, H.; Rong, Y.; Lei, Y.; Yu, Z. Vacancy Engineering in Transition Metal Sulfide and Oxide Catalysts for Hydrodeoxygenation of Lignin-Derived Oxygenates. ChemSusChem 2021, 14, 4377–4396. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Wu, K.; Zhang, K.; Jie, J.; Yang, Y. Preparation of Ni–Mo–S catalysts by hydrothermal method and their hydrodeoxygenation properties. Appl. Catal. A-Gen. 2015, 495, 8–16. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, J.; Wang, Y. Recent advances in the selective catalytic hydrodeoxygenation of lignin-derived oxygenates to arenes. Green Chem. 2020, 22, 1072–1098. [Google Scholar] [CrossRef]
- Badawi, M.; Paul, J.F.; Cristol, S.; Payen, E.; Romero, Y.; Richard, F.; Brunet, S.; Lambert, D.; Portier, X.; Popov, A.; et al. Effect of water on the stability of Mo and CoMo hydrodeoxygenation catalysts: A combined experimental and DFT study. J. Catal. 2011, 282, 155–164. [Google Scholar] [CrossRef]
- Grilc, M.; Likozar, B.; Levec, J. Hydrodeoxygenation and hydrocracking of solvolysed lignocellulosic biomass by oxide, reduced and sulphide form of NiMo, Ni, Mo and Pd catalysts. Appl. Catal. B-Environ. 2014, 150–151, 275–287. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Delichère, P.; Geantet, C. Hydrodeoxygenation of guaiacol: Part II: Support effect for CoMoS catalysts on HDO activity and selectivity. Appl. Catal. B-Environ. 2011, 101, 246–255. [Google Scholar] [CrossRef]
- Liu, G.; Robertson, A.W.; Li, M.M.-J.; Kuo, W.C.H.; Darby, M.T.; Muhieddine, M.H.; Lin, Y.-C.; Suenaga, K.; Stamatakis, M.; Warner, J.H.; et al. MoS2 monolayer catalyst doped with isolated Co atoms for the hydrodeoxygenation reaction. Nat. Chem. 2017, 9, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Zhou, S.; Hu, S.; Lai, W.; Lian, Y.; Wang, J.; Yang, W.; Wang, M.; Wang, P.; Jiang, X. Surface Engineering of CoMoS Nanosulfide for Hydrodeoxygenation of Lignin-Derived Phenols to Arenes. ACS Catal. 2019, 9, 259–268. [Google Scholar] [CrossRef]
- Liu, X.; Hou, X.; Zhang, Y.; Yuan, H.; Hong, X.; Liu, G. In Situ Formation of CoMoS Interfaces for Selective Hydrodeoxygenation of p-Cresol to Toluene. Ind. Eng. Chem. Res. 2020, 59, 15921–15928. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Xia, Q.; Jia, H.; Hong, X.; Liu, G. Tailoring of Surface Acidic Sites in Co–MoS2 Catalysts for Hydrodeoxygenation Reaction. J. Phys. Chem. Lett. 2021, 12, 5668–5674. [Google Scholar] [CrossRef]
- Diao, X.; Ji, N.; Li, X.; Rong, Y.; Zhao, Y.; Lu, X.; Song, C.; Liu, C.; Chen, G.; Ma, L.; et al. Fabricating high temperature stable Mo-Co9S8/Al2O3 catalyst for selective hydrodeoxygenation of lignin to arenes. Appl. Catal. B-Environ. 2022, 305, 121067. [Google Scholar] [CrossRef]
- Wu, K.; Wang, W.; Guo, H.; Yang, Y.; Huang, Y.; Li, W.; Li, C. Engineering Co Nanoparticles Supported on Defect MoS2–x for Mild Deoxygenation of Lignin-Derived Phenols to Arenes. ACS Energy Lett. 2020, 5, 1330–1336. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Tan, S.; Wu, K.; Zhu, G.; Liu, Y.; Xu, Y.; Yang, Y. Preparation of NiS2//MoS2 catalysts by two-step hydrothermal method and their enhanced activity for hydrodeoxygenation of p-cresol. Fuel 2016, 179, 1–9. [Google Scholar] [CrossRef]
- Yoosuk, B.; Tumnantong, D.; Prasassarakich, P. Amorphous unsupported Ni–Mo sulfide prepared by one step hydrothermal method for phenol hydrodeoxygenation. Fuel 2012, 91, 246–252. [Google Scholar] [CrossRef]
- Wang, W.; Wu, K.; Li, L.; Tan, S.; Zhu, G.; Li, W.; He, Z.; Yang, Y. Microwave-assisted hydrothermal synthesis of NiSx and their promotional effect for the hydrodeoxygenation of p-cresol on MoS2. Catal. Commun. 2016, 74, 60–64. [Google Scholar] [CrossRef]
- Laskar, D.D.; Tucker, M.P.; Chen, X.; Helms, G.L.; Yang, B. Noble-metal catalyzed hydrodeoxygenation of biomass-derived lignin to aromatic hydrocarbons. Green Chem. 2014, 16, 897–910. [Google Scholar] [CrossRef]
- Whiffen, V.M.L.; Smith, K.J. Hydrodeoxygenation of 4-Methylphenol over Unsupported MoP, MoS2, and MoOx Catalysts. Energy Fuels 2010, 24, 4728–4737. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, Q.; Deng, W.; Zhang, Q.; Wang, Y. Chapter One—Catalytic valorization of biomass and bioplatforms to chemicals through deoxygenation. Adv. Catal. 2020, 66, 1–108. [Google Scholar]
- Rensel, D.J.; Rouvimov, S.; Gin, M.E.; Hicks, J.C. Highly selective bimetallic FeMoP catalyst for C–O bond cleavage of aryl ethers. J. Catal. 2013, 305, 256–263. [Google Scholar] [CrossRef]
- Rensel, D.J.; Kim, J.; Bonita, Y.; Hicks, J.C. Investigating the multifunctional nature of bimetallic FeMoP catalysts using dehydration and hydrogenolysis reactions. Appl. Catal. A-Gen. 2016, 524, 85–93. [Google Scholar] [CrossRef]
- Jain, V.; Bonita, Y.; Brown, A.; Taconi, A.; Hicks, J.C.; Rai, N. Mechanistic insights into hydrodeoxygenation of phenol on bimetallic phosphide catalysts. Catal. Sci. Technol. 2018, 8, 4083–4096. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Li, D.; Bui, P.; Oyama, S.T. Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing catalysts. Appl. Catal. A-Gen. 2011, 391, 305–310. [Google Scholar] [CrossRef]
- García-Rollán, M.; Bertran-Llorens, S.; Palazzolo, M.; Deuss, P.; Heeres, H.; Ruiz-Rosas, R.; Rosas, J.; Rodriguez-Mirasol, J.; Cordero, T. Lignin hydrotreatment to aromatics products on metallic phosphides carbon-based catalysts produced from lignin. Fuel. 2025, 390, 134622. [Google Scholar] [CrossRef]
- Ge, F.; Li, H.; Wu, B.; Yang, X.; Jiang, J.; Zhang, Y.; Zhou, M. MOFs-derived carbon-covered nickel phosphide for catalytic transfer hydrodeoxygenation of lignin-derived vanillin. Chem. Eng. J. 2024, 489, 151367. [Google Scholar] [CrossRef]
- Moon, J.-S.; Kim, E.-G.; Lee, Y.-K. Active sites of Ni2P/SiO2 catalyst for hydrodeoxygenation of guaiacol: A joint XAFS and DFT study. J. Catal. 2014, 311, 144–152. [Google Scholar] [CrossRef]
- Li, Y.; Fu, J.; Chen, B. Highly selective hydrodeoxygenation of anisole, phenol and guaiacol to benzene over nickel phosphide. RSC Adv. 2017, 7, 15272–15277. [Google Scholar] [CrossRef]
- Pitakjakpipop, P.; Song, C. Effects of CeO2, TiO2, and TiO2-CeO2 Supports on Catalytic Performance of Ni2P in Hydrodeoxygenation of Anisole. Energy Fuels. 2023, 37, 8311–8323. [Google Scholar] [CrossRef]
- Gonçalves, V.O.O.; de Souza, P.M.; da Silva, V.T.; Noronha, F.B.; Richard, F. Kinetics of the hydrodeoxygenation of cresol isomers over Ni2P/SiO2: Proposals of nature of deoxygenation active sites based on an experimental study. Appl. Catal. B-Environ. 2017, 205, 357–367. [Google Scholar] [CrossRef]
- Prins, R.; Bussell, M.E. Metal Phosphides: Preparation, Characterization and Catalytic Reactivity. Catal. Lett. 2012, 142, 1413–1436. [Google Scholar] [CrossRef]
- Lan, X.; Hensen, E.J.M.; Weber, T. Hydrodeoxygenation of guaiacol over Ni2P/SiO2–reaction mechanism and catalyst deactivation. Appl. Catal. A-Gen. 2018, 550, 57–66. [Google Scholar] [CrossRef]
- Wang, S.; Xu, D.; Chen, Y.; Zhou, S.; Zhu, D.; Wen, X.; Yang, Y.; Li, Y. Hydrodeoxygenation of anisole to benzene over an Fe2P catalyst by a direct deoxygenation pathway. Catal. Sci. Technol. 2020, 10, 3015–3023. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Hu, C.; Bao, X.; Yuan, P. Advances in enhancing hydrodeoxygenation selectivity of lignin-derived oxygenates: From synthetic strategies to fundamental techniques. Green Energy Environ. 2025, 10, 292–321. [Google Scholar] [CrossRef]
- Sharma, D.; Choudhary, P.; Kumar, S.; Krishnan, K. Interfacial nanoarchitectonics of nickel phosphide supported on activated carbon for transfer hydrogenation of nitroarenes under mild conditions. J. Colloid Interface Sci. 2024, 657, 449–462. [Google Scholar] [CrossRef]
- Souza Macedo, L.; Oliveira, R.R.; van Haasterecht, T.; Teixeira da Silva, V.; Bitter, H. Influence of synthesis method on molybdenum carbide crystal structure and catalytic performance in stearic acid hydrodeoxygenation. Appl. Catal. B-Environ. 2019, 241, 81–88. [Google Scholar] [CrossRef]
- Batool, M.; Hameed, A.; Nadeem, M. Recent developments on iron and nickel-based transition metal nitrides foroverall water splitting: A critical review. Coord. Chem. Rev. 2023, 480, 215029. [Google Scholar] [CrossRef]
- Liu, S.; Wang, H.; Putra, R.D.D.; Kim, C.S.; Smith, K.J. Impact of Carbon Properties on Mo2C/Carbon Catalysts for the Hydrodeoxygenation of 4-Methylphenol. Energy Fuels 2019, 33, 4506–4514. [Google Scholar] [CrossRef]
- Lee, W.-S.; Wang, Z.; Wu, R.J.; Bhan, A. Selective vapor-phase hydrodeoxygenation of anisole to benzene on molybdenum carbide catalysts. J. Catal. 2014, 319, 44–53. [Google Scholar] [CrossRef]
- Chen, C.-J.; Lee, W.-S.; Bhan, A. Mo2C catalyzed vapor phase hydrodeoxygenation of lignin-derived phenolic compound mixtures to aromatics under ambient pressure. Appl. Catal. A-Gen. 2016, 510, 42–48. [Google Scholar] [CrossRef]
- Murugappan, K.; Anderson, E.M.; Teschner, D.; Jones, T.E.; Skorupska, K.; Román-Leshkov, Y. Operando NAP-XPS unveils differences in MoO3 and Mo2C during hydrodeoxygenation. Nat. Catal. 2018, 1, 960–967. [Google Scholar] [CrossRef]
- Wang, H.; Liu, S.; Smith, K.J. Synthesis and Hydrodeoxygenation Activity of Carbon Supported Molybdenum Carbide and Oxycarbide Catalysts. Energy Fuels 2016, 30, 6039–6049. [Google Scholar] [CrossRef]
- Lu, Q.; Chen, C.-J.; Luc, W.; Chen, J.G.; Bhan, A.; Jiao, F. Ordered Mesoporous Metal Carbides with Enhanced Anisole Hydrodeoxygenation Selectivity. ACS Catal. 2016, 6, 3506–3514. [Google Scholar] [CrossRef]
- Iida, T.; Shetty, M.; Murugappan, K.; Wang, Z.; Ohara, K.; Wakihara, T.; Román-Leshkov, Y. Encapsulation of Molybdenum Carbide Nanoclusters inside Zeolite Micropores Enables Synergistic Bifunctional Catalysis for Anisole Hydrodeoxygenation. ACS Catal. 2017, 7, 8147–8151. [Google Scholar] [CrossRef]
- Smirnov, A.A.; Geng, Z.; Khromova, S.A.; Zavarukhin, S.G.; Bulavchenko, O.A.; Saraev, A.A.; Kaichev, V.V.; Ermakov, D.Y.; Yakovlev, V.A. Nickel molybdenum carbides: Synthesis, characterization, and catalytic activity in hydrodeoxygenation of anisole and ethyl caprate. J. Catal. 2017, 354, 61–77. [Google Scholar] [CrossRef]
- Tran, C.-C.; Han, Y.; Garcia-Perez, M.; Kaliaguine, S. Synergistic effect of Mo–W carbides on selective hydrodeoxygenation of guaiacol to oxygen-free aromatic hydrocarbons. Catal. Sci. Technol. 2019, 9, 1387–1397. [Google Scholar] [CrossRef]
- Sullivan, M.M.; Bhan, A. Acetone Hydrodeoxygenation over Bifunctional Metallic–Acidic Molybdenum Carbide Catalysts. ACS Catal. 2016, 6, 1145–1152. [Google Scholar] [CrossRef]
- Sepúlveda, C.; Leiva, K.; García, R.; Radovic, L.R.; Ghampson, I.T.; DeSisto, W.J.; Fierro, J.L.G.; Escalona, N. Hydrodeoxygenation of 2-methoxyphenol over Mo2N catalysts supported on activated carbons. Catal. Today 2011, 172, 232–239. [Google Scholar] [CrossRef]
- Ghampson, I.T.; Sepúlveda, C.; Garcia, R.; Radovic, L.R.; Fierro, J.L.G.; DeSisto, W.J.; Escalona, N. Hydrodeoxygenation of guaiacol over carbon-supported molybdenum nitride catalysts: Effects of nitriding methods and support properties. Appl. Catal. A-Gen. 2012, 439–440, 111–124. [Google Scholar] [CrossRef]
- Ghampson, I.T.; Sepúlveda, C.; Garcia, R.; Frederick, B.G.; Wheeler, M.C.; Escalona, N.; DeSisto, W.J. Guaiacol transformation over unsupported molybdenum-based nitride catalysts. Appl. Catal. A-Gen. 2012, 413–414, 78–84. [Google Scholar] [CrossRef]
- Boullosa-Eiras, S.; Lødeng, R.; Bergem, H.; Stöcker, M.; Hannevold, L.; Blekkan, E.A. Catalytic hydrodeoxygenation (HDO) of phenol over supported molybdenum carbide, nitride, phosphide and oxide catalysts. Catal. Today 2014, 223, 44–53. [Google Scholar] [CrossRef]
- de Souza, P.M.; Rabelo-Neto, R.C.; Borges, L.E.P.; Jacobs, G.; Davis, B.H.; Sooknoi, T.; Resasco, D.E.; Noronha, F.B. Role of Keto Intermediates in the Hydrodeoxygenation of Phenol over Pd on Oxophilic Supports. ACS Catal. 2015, 5, 1318–1329. [Google Scholar] [CrossRef]
- de Souza, P.M.; Rabelo-Neto, R.C.; Borges, L.E.P.; Jacobs, G.; Davis, B.H.; Graham, U.M.; Resasco, D.E.; Noronha, F.B. Effect of Zirconia Morphology on Hydrodeoxygenation of Phenol over Pd/ZrO2. ACS Catal. 2015, 5, 7385–7398. [Google Scholar] [CrossRef]
- Lai, Q.; Zhang, C.; Holles, J.H. Hydrodeoxygenation of guaiacol over Ni@Pd and Ni@Pt bimetallic overlayer catalysts. Appl. Catal. A-Gen. 2016, 528, 1–13. [Google Scholar] [CrossRef]
- Phan, T.N.; Park, Y.-K.; Lee, I.-G.; Ko, C.H. Enhancement of CO bond cleavage to afford aromatics in the hydrodeoxygenation of anisole over ruthenium-supporting mesoporous metal oxides. Appl. Catal. A-Gen. 2017, 544, 84–93. [Google Scholar] [CrossRef]
- Zheng, Z.; Luo, Z.; Zhao, C. Morphologically Cross-Shaped Ru/HZSM-5 Catalyzes Tandem Hydrogenolysis of Guaiacol to Benzene in Water. ChemCatChem 2018, 10, 1376–1384. [Google Scholar] [CrossRef]
- Li, B.; Li, L.; Zhao, C. A highly stable Ru/LaCO3OH catalyst consisting of support-coated Ru nanoparticles in aqueous-phase hydrogenolysis reactions. Green Chem. 2017, 19, 5412–5421. [Google Scholar] [CrossRef]
- Chang, J.; Danuthai, T.; Dewiyanti, S.; Wang, C.; Borgna, A. Hydrodeoxygenation of Guaiacol over Carbon-Supported Metal Catalysts. ChemCatChem 2013, 5, 3041–3049. [Google Scholar] [CrossRef]
- Nie, L.; Resasco, D.E. Kinetics and mechanism of m-cresol hydrodeoxygenation on a Pt/SiO2 catalyst. J. Catal. 2014, 317, 22–29. [Google Scholar] [CrossRef]
- Phan, D.-P.; Le, V.N.; Kim, J.; Lee, E.Y. Controlled hydrodeoxygenation of lignin-derived anisole over supported Pt on UiO-66 based-catalysts through defect engineering approach. Fuel Process. Technol. 2021, 224, 107001. [Google Scholar] [CrossRef]
- Li, N.; Zhang, X.; Zhang, Q.; Chen, L.; Li, Y.; Wang, C.; Ma, L. Mo-based catalyst for chemical looping deoxygenation of phenolic compounds to aromatic hydrocarbons. Fuel Process. Technol. 2021, 221, 106936. [Google Scholar] [CrossRef]
- Shin, E.-J.; Keane, M.A. Gas-Phase Hydrogenation/Hydrogenolysis of Phenol over Supported Nickel Catalysts. Ind. Eng. Chem. Res. 2000, 39, 883–892. [Google Scholar] [CrossRef]
- Do, P.T.M.; Foster, A.J.; Chen, J.; Lobo, R.F. Bimetallic effects in the hydrodeoxygenation of meta-cresol on γ-Al2O3 supported Pt–Ni and Pt–Co catalysts. Green Chem. 2012, 14, 1388–1397. [Google Scholar] [CrossRef]
- Hong, Y.; Wang, Y. Elucidation of reaction mechanism for m-cresol hydrodeoxygenation over Fe based catalysts: A kinetic study. Catal. Commun. 2017, 100, 43–47. [Google Scholar] [CrossRef]
- Yang, Y.; Tan, M.; Garcia, A.; Zhang, Z.; Lin, J.; Wan, S.; McEwen, J.-S.; Wang, S.; Wang, Y. Controlling the Oxidation State of Fe-Based Catalysts through Nitrogen Doping toward the Hydrodeoxygenation of m-Cresol. ACS Catal. 2020, 10, 7884–7893. [Google Scholar] [CrossRef]
- Tan, Q.; Wang, G.; Long, A.; Dinse, A.; Buda, C.; Shabaker, J.; Resasco, D.E. Mechanistic analysis of the role of metal oxophilicity in the hydrodeoxygenation of anisole. J. Catal. 2017, 347, 102–115. [Google Scholar] [CrossRef]
- Olcese, R.; Bettahar, M.M.; Malaman, B.; Ghanbaja, J.; Tibavizco, L.; Petitjean, D.; Dufour, A. Gas-phase hydrodeoxygenation of guaiacol over iron-based catalysts. Effect of gases composition, iron load and supports (silica and activated carbon). Appl. Catal. B-Environ. 2013, 129, 528–538. [Google Scholar] [CrossRef]
- Yohe, S.L.; Choudhari, H.J.; Mehta, D.D.; Dietrich, P.J.; Detwiler, M.D.; Akatay, C.M.; Stach, E.A.; Miller, J.T.; Delgass, W.N.; Agrawal, R.; et al. High-pressure vapor-phase hydrodeoxygenation of lignin-derived oxygenates to hydrocarbons by a PtMo bimetallic catalyst: Product selectivity, reaction pathway, and structural characterization. J. Catal. 2016, 344, 535–552. [Google Scholar] [CrossRef]
- González-Borja, M.Á.; Resasco, D.E. Anisole and Guaiacol Hydrodeoxygenation over Monolithic Pt–Sn Catalysts. Energy Fuels 2011, 25, 4155–4162. [Google Scholar] [CrossRef]
- Bui, V.N.; Laurenti, D.; Afanasiev, P.; Geantet, C. Hydrodeoxygenation of guaiacol with CoMo catalysts. Part I: Promoting effect of cobalt on HDO selectivity and activity. Appl. Catal. B-Environ. 2011, 101, 239–245. [Google Scholar] [CrossRef]
- Gonçalves, V.O.O.; Brunet, S.; Richard, F. Hydrodeoxygenation of Cresols Over Mo/Al2O3 and CoMo/Al2O3 Sulfided Catalysts. Catal. Lett. 2016, 146, 1562–1573. [Google Scholar] [CrossRef]
- Viljava, T.R.; Komulainen, R.S.; Krause, A.O.I. Effect of H2S on the stability of CoMo/Al2O3 catalysts during hydrodeoxygenation. Catal. Today 2000, 60, 83–92. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Wu, K.; Zhu, G.; Tan, S.; Liu, Y.; Yang, Y. Highly selective catalytic conversion of phenols to aromatic hydrocarbons on CoS2/MoS2 synthesized using a two step hydrothermal method. RSC Adv. 2016, 6, 31265–31271. [Google Scholar] [CrossRef]
- Wang, C.; Wu, Z.; Tang, C.; Li, L.; Wang, D. The effect of nickel content on the hydrodeoxygenation of 4-methylphenol over unsupported NiMoW sulfide catalysts. Catal. Commun. 2013, 32, 76–80. [Google Scholar] [CrossRef]
- Whiffen, V.M.L.; Smith, K.J.; Straus, S.K. The influence of citric acid on the synthesis and activity of high surface area MoP for the hydrodeoxygenation of 4-methylphenol. Appl. Catal. A-Gen. 2012, 419–420, 111–125. [Google Scholar] [CrossRef]
- Wu, S.-K.; Lai, P.-C.; Lin, Y.-C.; Wan, H.-P.; Lee, H.-T.; Chang, Y.-H. Atmospheric Hydrodeoxygenation of Guaiacol over Alumina-, Zirconia-, and Silica-Supported Nickel Phosphide Catalysts. ACS Sustain. Chem. Eng. 2013, 1, 349–358. [Google Scholar] [CrossRef]
- Baddour, F.G.; Witte, V.A.; Nash, C.P.; Griffin, M.B.; Ruddy, D.A.; Schaidle, J.A. Late-Transition-Metal-Modified β-Mo2C Catalysts for Enhanced Hydrogenation during Guaiacol Deoxygenation. ACS Sustain. Chem. Eng. 2017, 5, 11433–11439. [Google Scholar] [CrossRef]
- Chen, C.-J.; Bhan, A. Mo2C Modification by CO2, H2O, and O2: Effects of Oxygen Content and Oxygen Source on Rates and Selectivity of m-Cresol Hydrodeoxygenation. ACS Catal. 2017, 7, 1113–1122. [Google Scholar] [CrossRef]
- Jongerius, A.L.; Gosselink, R.W.; Dijkstra, J.; Bitter, J.H.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Carbon Nanofiber Supported Transition-Metal Carbide Catalysts for the Hydrodeoxygenation of Guaiacol. ChemCatChem 2013, 5, 2964–2972. [Google Scholar] [CrossRef]
- Liu, J.; Li, C.; Yang, Y.; Chen, Y.; Chen, H.; Wang, X.; Yang, H. Hydrodeoxygenation of lignin-derived oxygenated aromatic compounds catalyzed by nano-floral hydrotalcite for the preparation of liquid-phase organic hydrogen carriers. Appl. Catal. B-Environ. 2025, 371, 125285. [Google Scholar] [CrossRef]
- Pang, T.; Xue, Z.; Wang, G.; Li, J.; Sui, W.; Si, C. Non-Carbonized Pd Single-Atom Catalyst Supported on Lignin-Functionalized Phenolic Resin for Potent Catalytic Transfer Hydrogenation of Lignin-Derived Aldehydes. Angew. Chem. Int. Ed. 2025, e202503195. [Google Scholar] [CrossRef]
- Pang, T.; Wang, G.; Sui, W.; Xu, T.; Wang, D.; Si, C. Lignin-based support for metal catalysts: Synthetic strategies, performance boost, and application advances. Coord. Chem. Rev. 2025, 528, 216426. [Google Scholar] [CrossRef]
- Yu, Y.; Li, Y.; Lou, Y.; Chen, M.; Liu, Y.; Yu, H. Tunable Transfer-Hydrodeoxygenated Upgrading of Lignin-Derived Propylphenols to Versatile Value-Added Alkane-Based Chemicals. Adv. Sci. 2025, 12, 2500687. [Google Scholar] [CrossRef]
- Erasmo, B.; Perepichka, I.; Su, H.; Vaccaro, L.; Li, C. Accessing Arenes via the Hydrodeoxygenation of Phenolic Derivatives Enabled by Hydrazine. ACS Catal. 2025, 15, 3367–3376. [Google Scholar] [CrossRef]
- Yangcheng, R.; Li, J.; He, J.; Zheng, Y.; Yu, H.; Chen, C.; Wang, J. Carboxyl-Decorated UiO-66 Supporting Pd Nanoparticles for Efficient Room-Temperature Hydrodeoxygenation of Lignin Derivatives. Small 2024, 20, 2309821. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).