
Effect of Synthetic Peptides Identified in the Bullfrog Skin on Inflammation and Oxidative Stress Control: An In Vitro Analysis

 , , , ,
, , , ,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Peptide
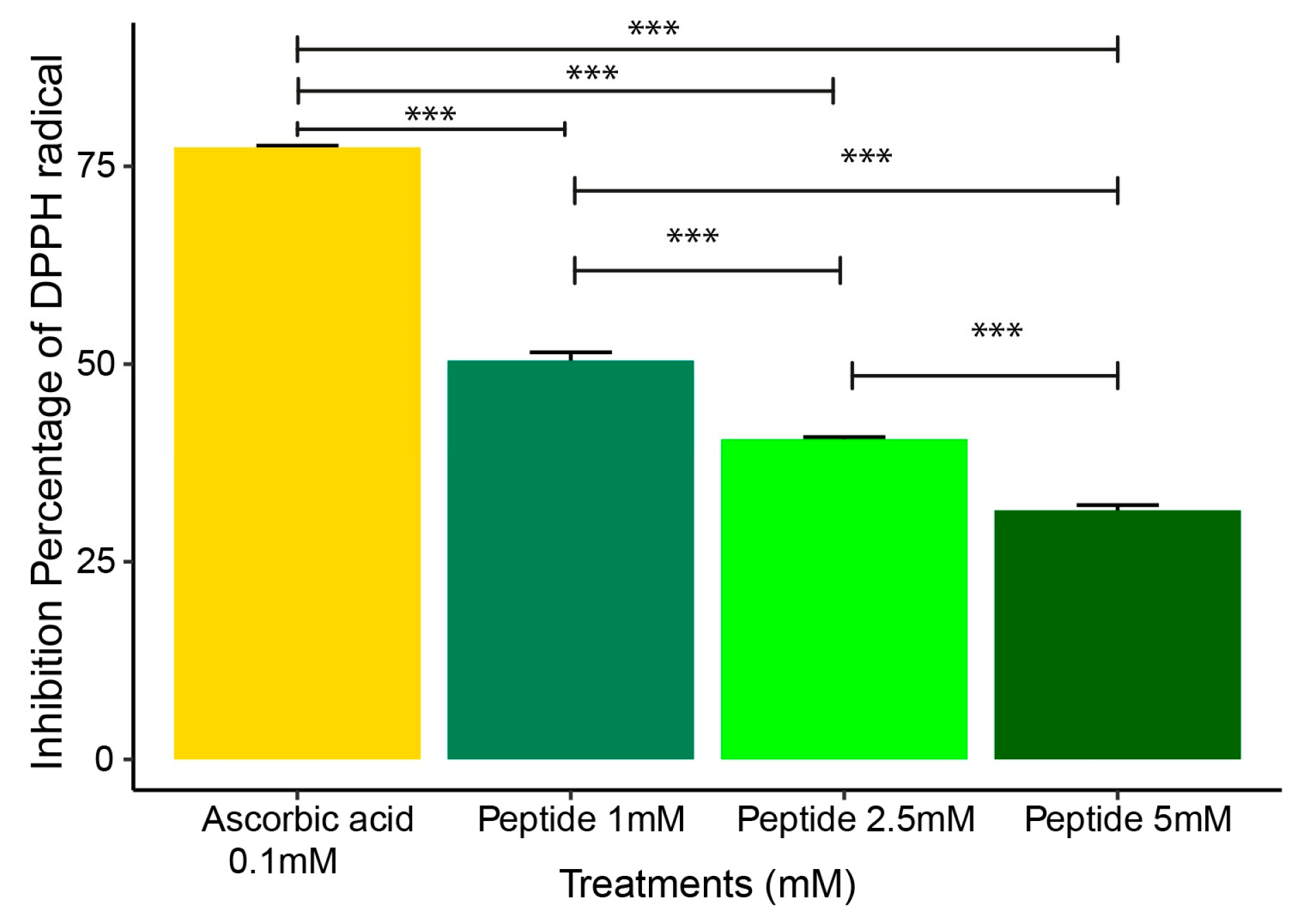
2.2. DPPH Radical Scavenging Assay
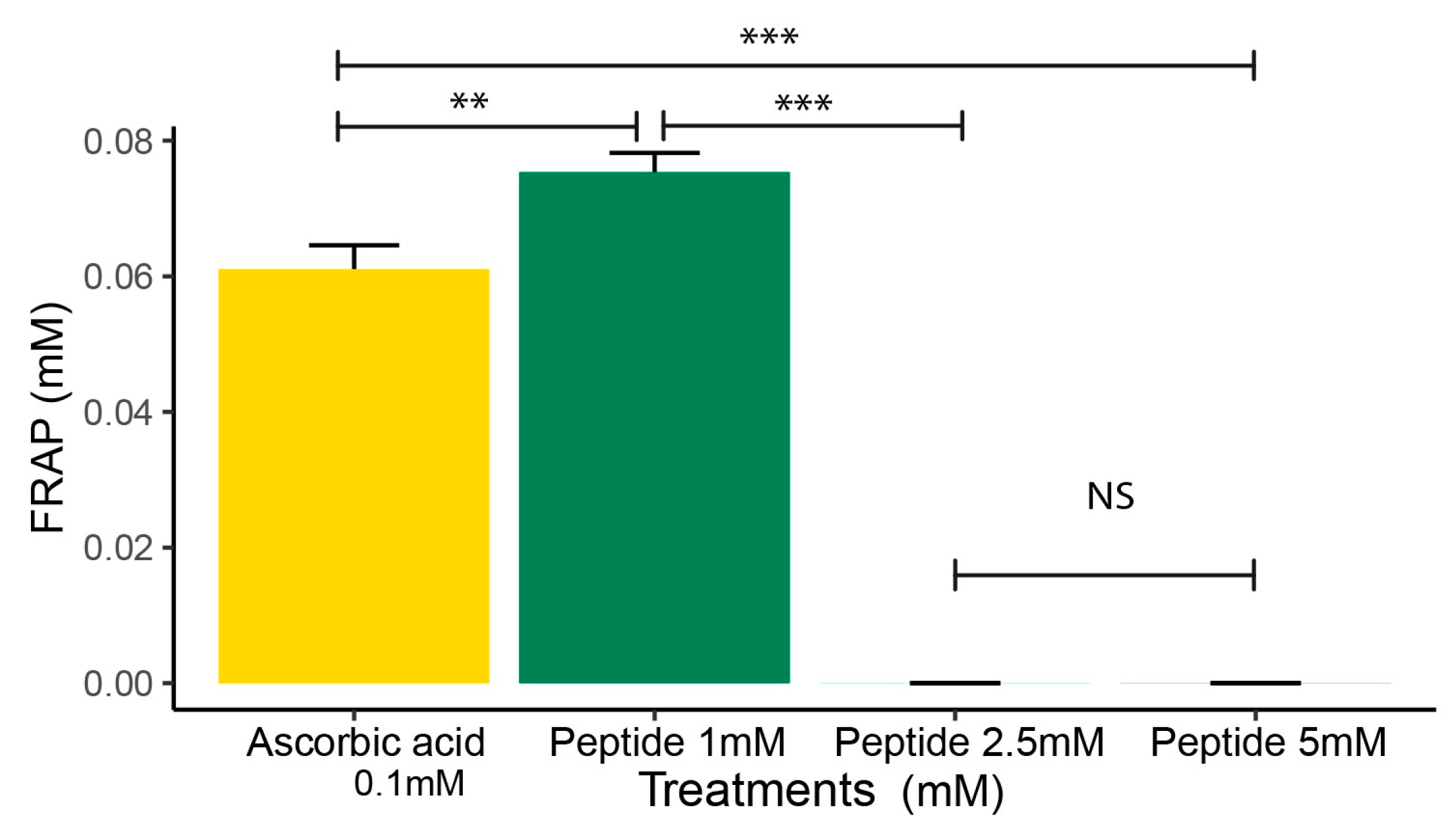
2.3. FRAP Analysis
2.4. In Vitro Analysis
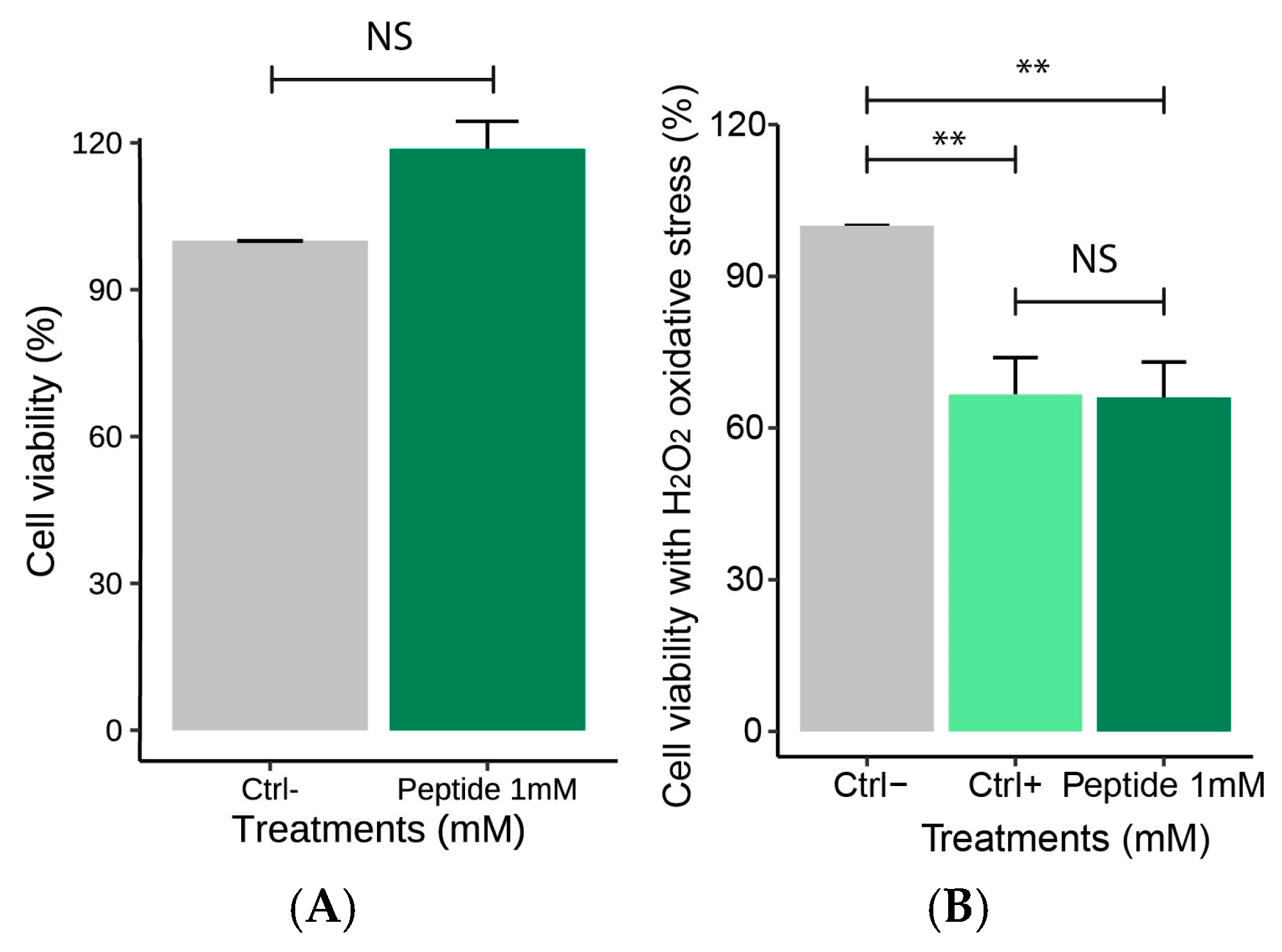
2.4.1. Effect of the Peptide on Macrophages Cell Viability
2.4.2. Effect of the Peptide on Nitric Oxide Production Following H2O2-Induced Stress
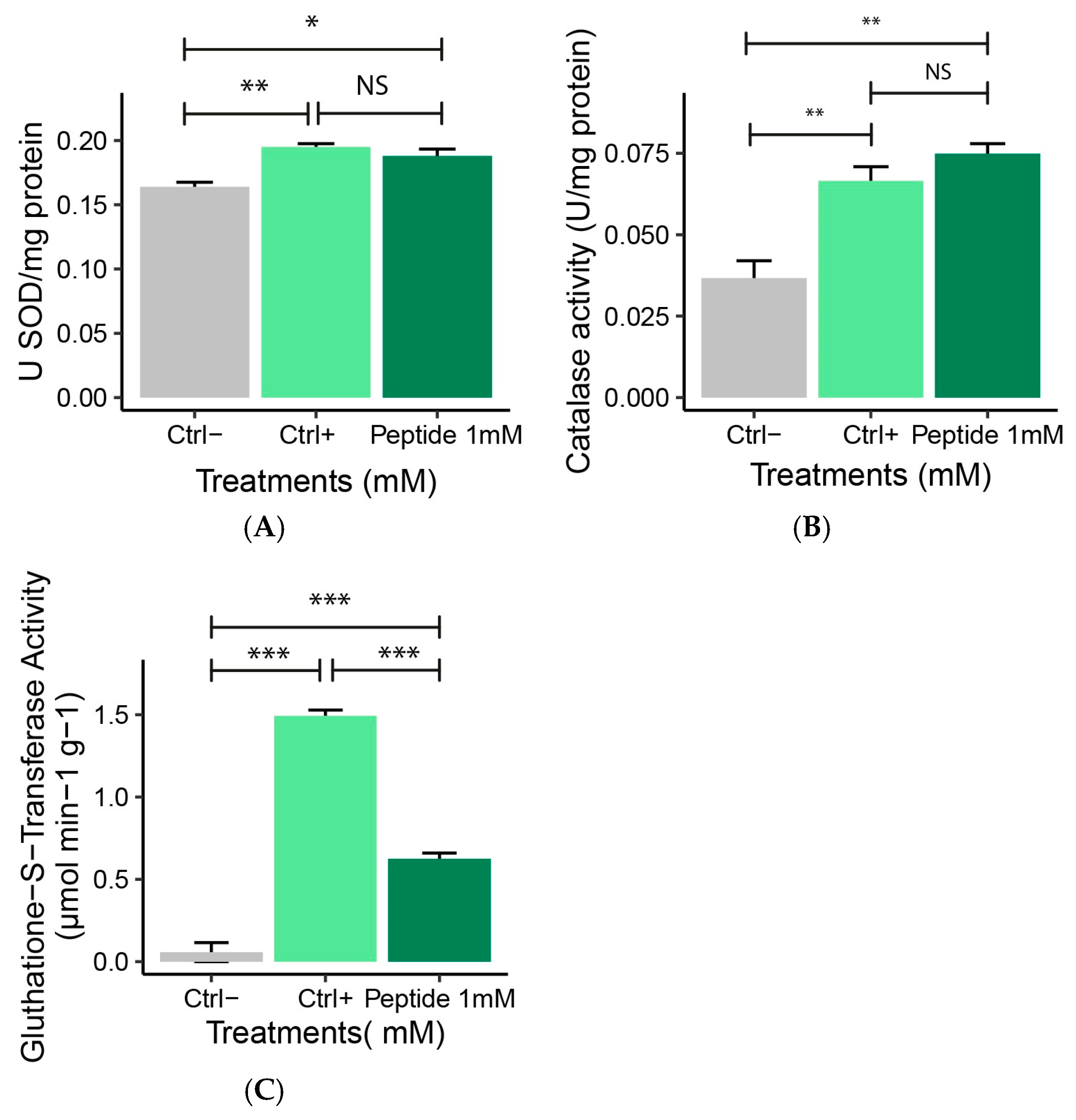
2.4.3. Antioxidant Enzyme Activity (SOD, CAT, GST)
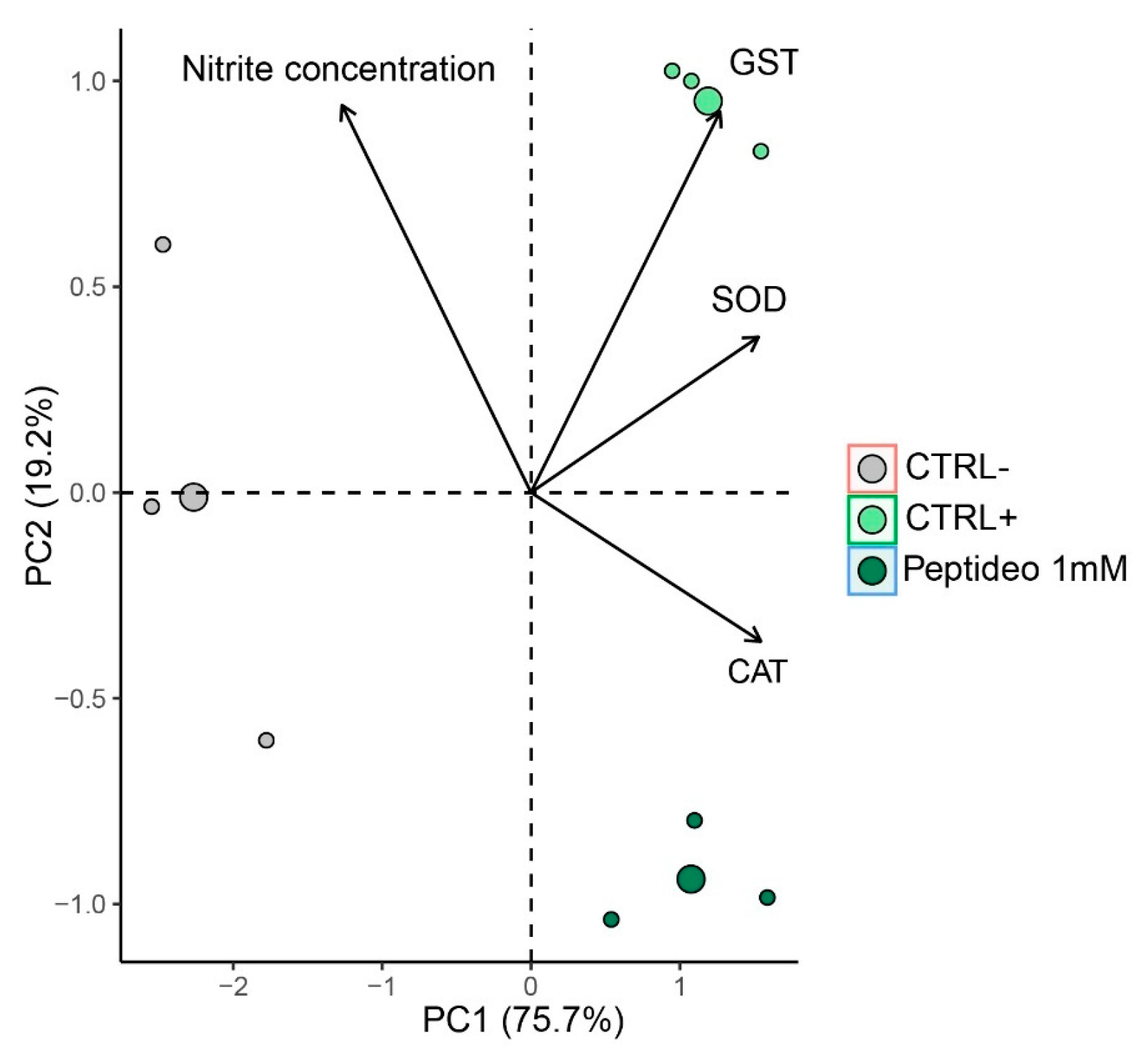
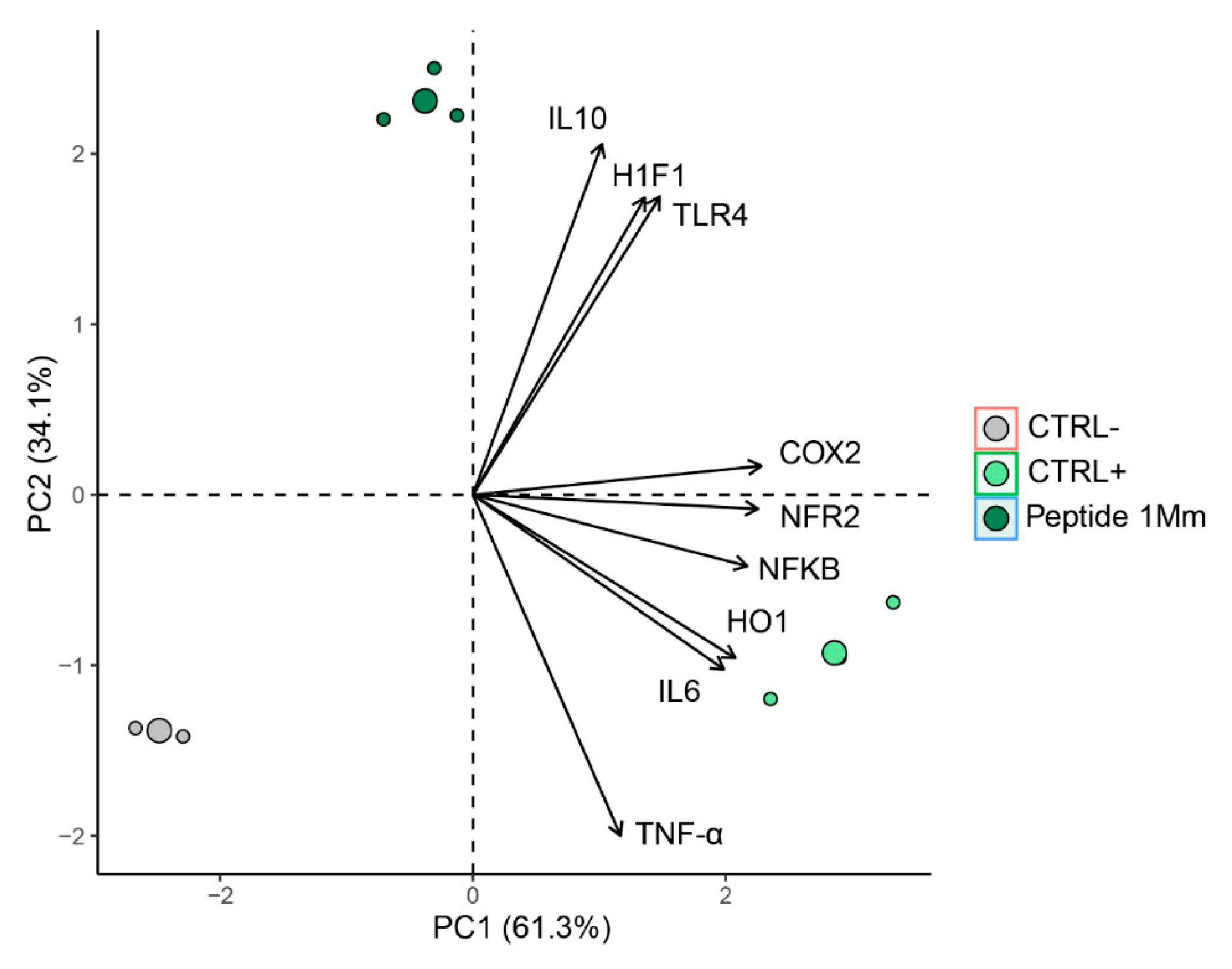
2.4.4. Multivariate Analysis
2.5. Gene Expression Analysis
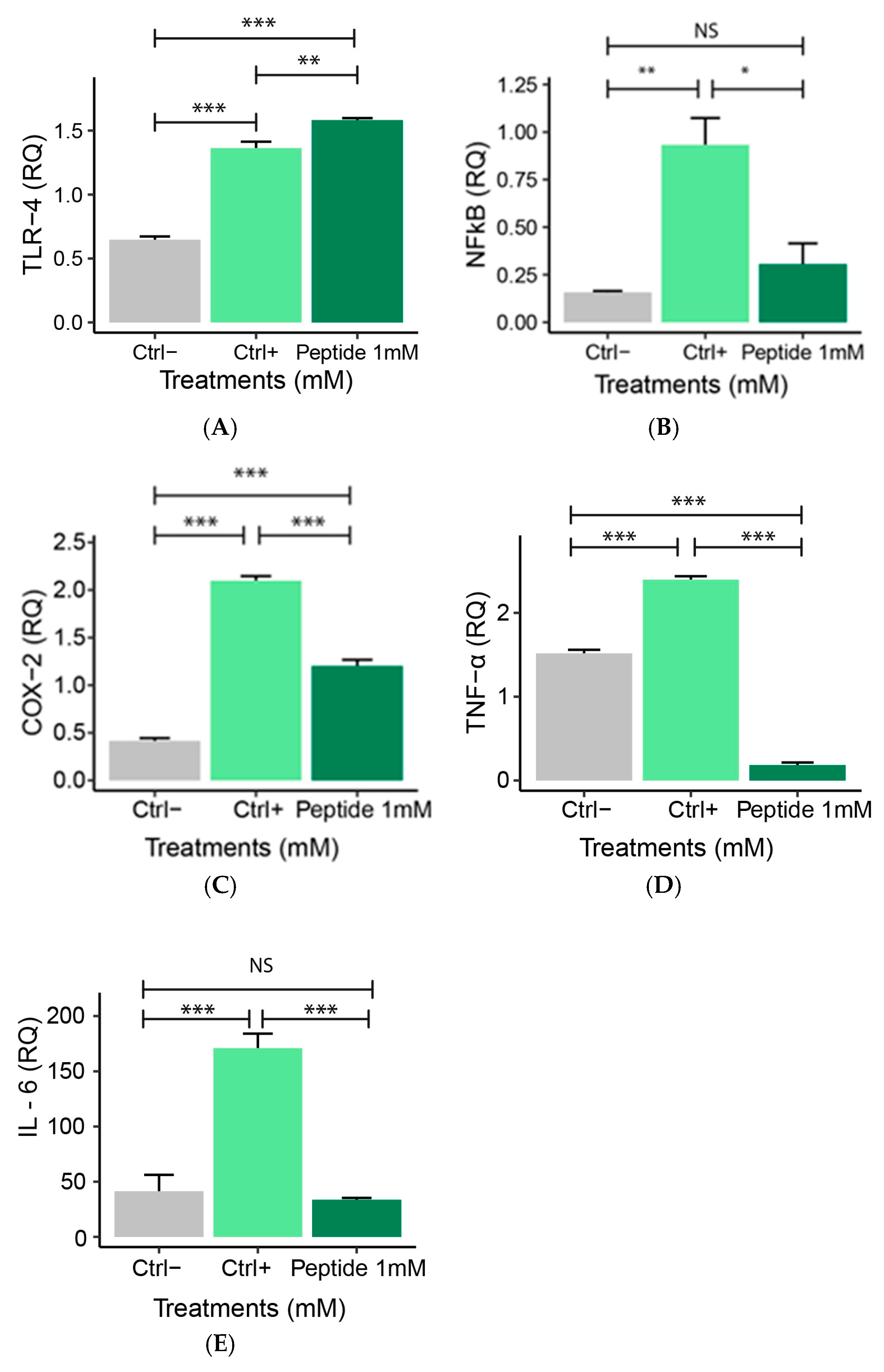
2.5.1. Expression of Pro-Inflammatory Genes
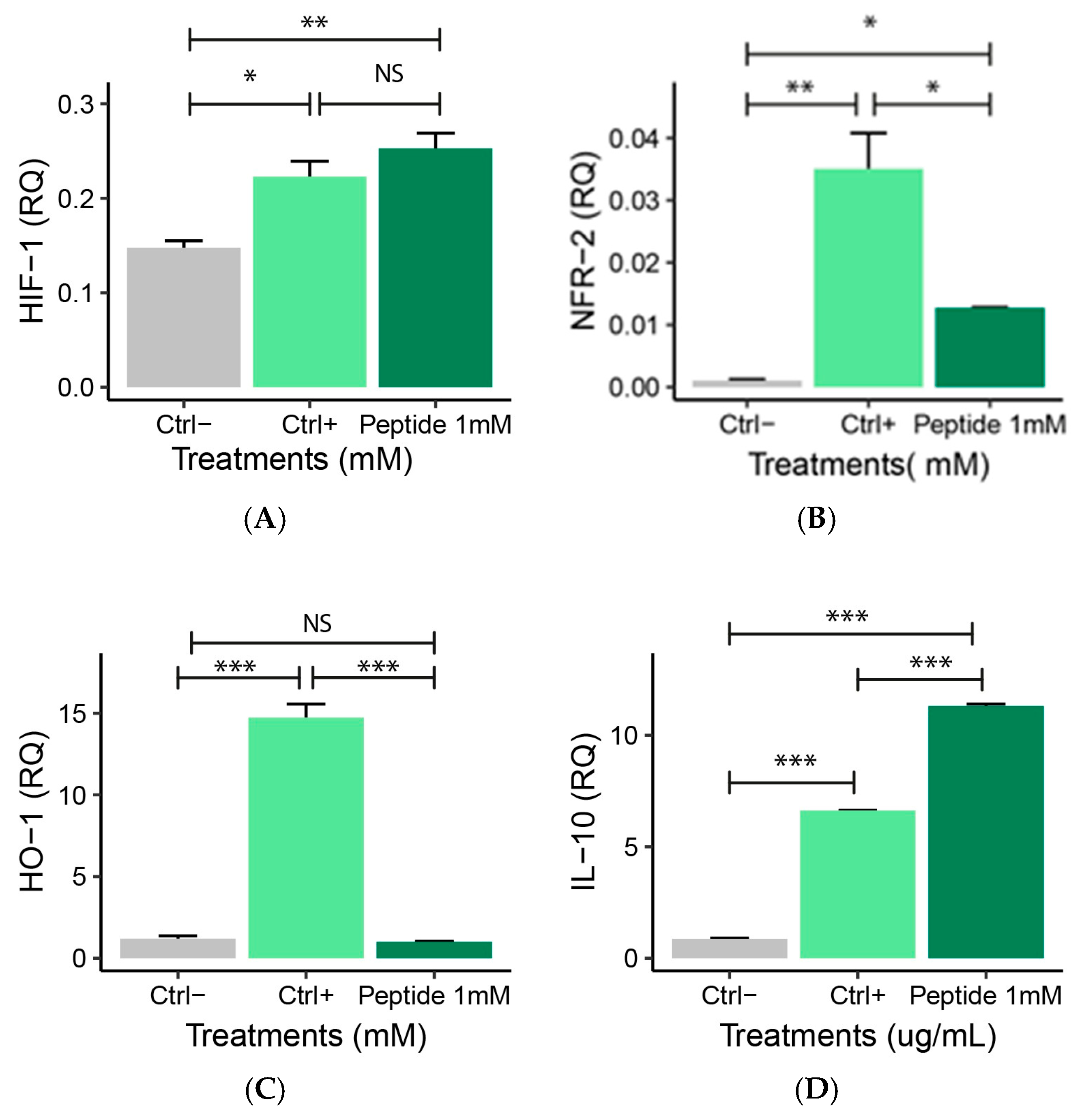
2.5.2. Expression of Anti-Inflammatory Genes
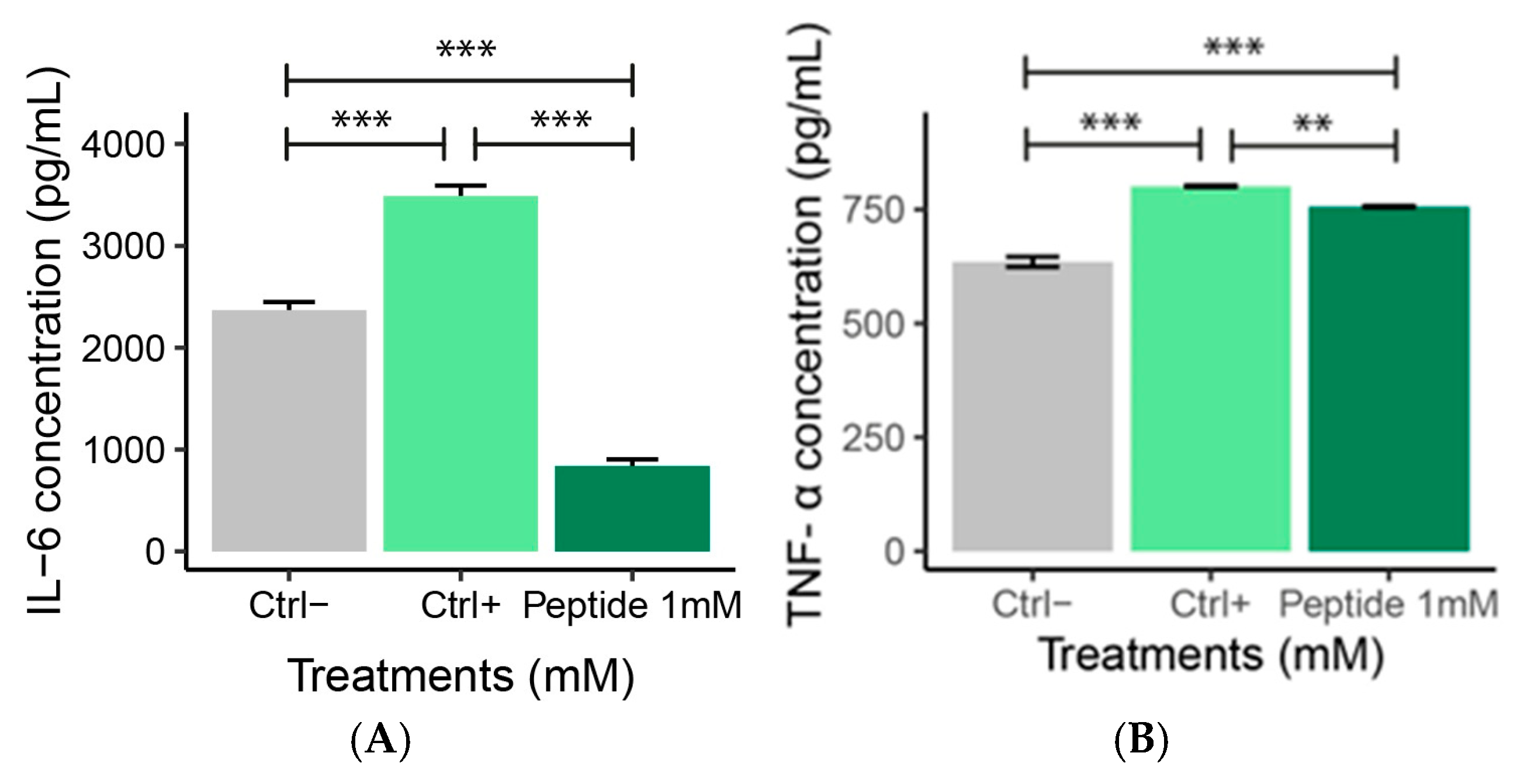
2.6. ELISA Analysis
IL-6 and TNF-α Levels
2.7. Multivariate Analysis
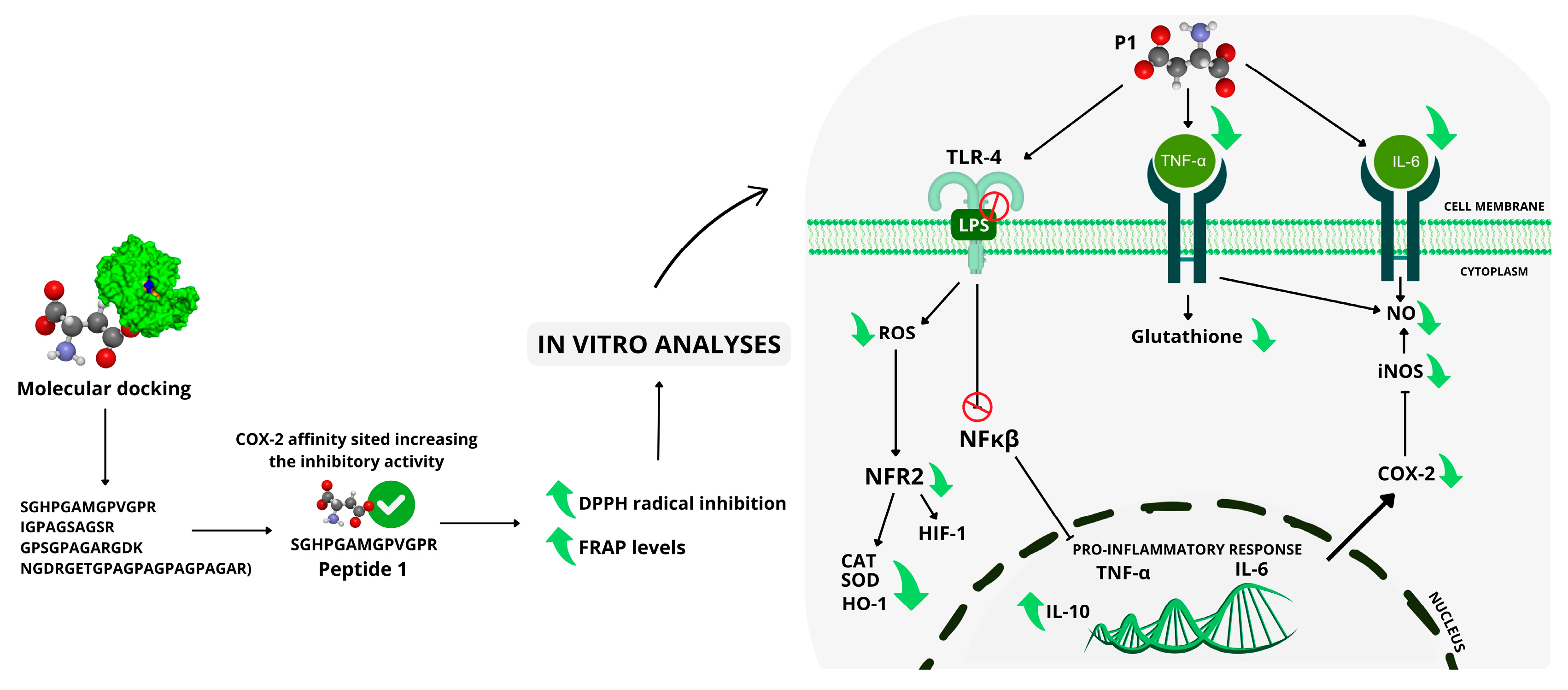
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis and Molecular Docking
4.2. DPPH Radical Scavenging Assay
4.3. FRAP Analysis
4.4. In Vitro Analyses
4.4.1. Cell Viability
4.4.2. Cell Viability After Induction of Oxidative Stress with Hydrogen Peroxide
4.4.3. Superoxide Dismutase (SOD) Analysis
4.4.4. Catalase (CAT) Activity
4.4.5. Glutathione (GST) Analysis
4.4.6. Nitric Oxide (NO) Quantification
4.5. Gene Expression Analysis
4.6. ELISA Analysis
Murine IL-6 and TNF-α Quantification
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IL-6 | Intleukin-6 |
| IL-10 | Interleukin-10 |
| TNF-alpha | Tumor Necrosis Factor-alpha |
| COX | Cyclooxygenase |
| TLR-4 | Toll-like Receptor 4 |
| NRF-2 | Nuclear Factor Erythroid-2 |
| HIF-1 | Hypoxia-Inducible Factor 1 |
| HO-1 | Heme Oxygenase 1 |
| NO | Nitric Oxide |
| SOD | Superoxide Dismutase |
| CAT | Catalase |
| GST | Glutathione S-Transferase |
| NF-κB | Nuclear Factor kappa |
| iNOS | Inducible nitric oxide synthase |
| LPS | Lipopolysaccharide |
References
- Das, D.; Sarkar, S.; Borsingh Wann, S.; Kalita, J.; Manna, P. Current perspectives on the anti-inflammatory potential of fermented soy foods. Food Res. Int. 2022, 152, 110922. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lu, H.; Chen, C.; Lyu, Y.; Cole, R.N.; Semenza, G.L. HIF-1 interacts with TRIM28 and DNA-PK to release paused RNA polymerase II and activate target gene transcription in response to hypoxia. Nat. Commun. 2022, 13, 316. [Google Scholar] [CrossRef]
- Leyane, T.S.; Jere, S.W.; Houreld, N.N. Oxidative Stress in Ageing and Chronic Degenerative Pathologies: Molecular Mechanisms Involved in Counteracting Oxidative Stress and Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 7273. [Google Scholar] [CrossRef]
- Hosaka, T.; Tsuji, H.; Tamaoka, A. Biomolecular Modifications Linked to Oxidative Stress in Amyotrophic Lateral Sclerosis: Determining Promising Biomarkers Related to Oxidative Stress. Processes 2021, 9, 1667. [Google Scholar] [CrossRef]
- Silveira, L.L.; Sarandy, M.M.; Novaes, R.D.; Morais-Santos, M.; Gonçalves, R.V. OxInflammation Affects Transdifferentiation to Myofibroblasts, Prolonging Wound Healing in Diabetes: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 8992. [Google Scholar] [CrossRef]
- Olejarz, W.; Łacheta, D.; Głuszko, A.; Migacz, E.; Kukwa, W.; Szczepański, M.J.; Tomaszewski, P.; Nowicka, G. RAGE and TLRs as Key Targets for Antiatherosclerotic Therapy. BioMed Res. Int. 2018, 2018, 7675286. [Google Scholar] [CrossRef]
- Yang, S.-C.; Alalaiwe, A.; Lin, Z.-C.; Lin, Y.-C.; Aljuffali, I.A.; Fang, J.-Y. Anti-Inflammatory microRNAs for Treating Inflammatory Skin Diseases. Biomolecules 2022, 12, 1072. [Google Scholar] [CrossRef]
- Valacchi, G.; Virgili, F.; Cervellati, C.; Pecorelli, A. OxInflammation: From Subclinical Condition to Pathological Biomarker. Front. Physiol. 2018, 9, 858. [Google Scholar] [CrossRef]
- Valacchi, G.; Magnani, N.; Woodby, B.; Ferreira, S.M.; Evelson, P. Particulate Matter Induces Tissue OxInflammation: From Mechanism to Damage. Antioxid. Redox Signal. 2020, 33, 308–326. [Google Scholar] [CrossRef]
- van Loo, G.; Bertrand, M.J.M. Death by TNF: A Road to Inflammation. Nat. Rev. Immunol. 2023, 23, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Derbel, H.; Elleuch, J.; Mahfoudh, W.; Michaud, P.; Fendri, I.; Abdelkafi, S. In Vitro Antioxidant and Anti-Inflammatory Activities of Bioactive Proteins and Peptides from Rhodomonas sp. Appl. Sci. 2023, 13, 3202. [Google Scholar] [CrossRef]
- Liu, H.; Guo, X.; Yi, T.; Zhu, Y.; Ren, X.; Guo, R.; Dai, Y.; Liang, S. Frog Skin Derived Peptides With Potential Protective Effects on Ultraviolet B–Induced Cutaneous Photodamage. Front. Immunol. 2021, 12, 613365. [Google Scholar] [CrossRef]
- Alves, R.S. Effects of Peptides Obtained from Animals on the Wound Healing Process in Pre-Clinical Models. Ph.D. Thesis, Federal University of Viçosa, Viçosa, Brazil, 2022; 88p. in Cellular and Structural Biology. Available online: https://locus.ufv.br/server/api/core/bitstreams/4a121ca7-7bdc-49ad-abab-77b9d2167f54/content (accessed on 16 May 2025).
- Cardoso, R.R.; Sarandy, M.M.; de Oliveira, L.L.; da Matta, S.L.P.; Novaes, R.D.; Gonçalves, R.V. Positive Effect of Peptides Obtained from Nile Tilapia (Oreochromis niloticus) on Inflammation Regulation and Wound Healing. Cosmetics 2024, 11, 133. [Google Scholar] [CrossRef]
- Geng, F.; Zhong, L.; Yang, T.; Chen, J.; Yang, P.; Jiang, F.; Yan, T.; Song, B.; Yu, Z.; Yu, D.; et al. A Frog Skin-Derived Peptide Targeting SCD1 Exerts Radioprotective Effects Against Skin Injury by Inhibiting STING-Mediated Inflammation. Adv. Sci. 2024, 11, 202306253. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Quan, Z.; Xiao, P.; Duan, J.-A. New Insights into Antioxidant Peptides: An Overview of Efficient Screening, Evaluation Models, Molecular Mechanisms, and Applications. Antioxidants 2024, 13, 203. [Google Scholar] [CrossRef]
- Bazhanova, E.D.; Kozlov, A.A. Role of Apoptosis-Related Proteins P53 and Bcl-2 in the Pathogenesis of Nervous System Diseases. J. Evol. Biochem. Physiol. 2024, 60, 1475–1489. [Google Scholar] [CrossRef]
- Wang, S.; Yang, M.; Yin, S.; Zhang, Y.; Zhang, Y.; Sun, H.; Shu, L.; Liu, Y.; Kang, Z.; Liu, N.; et al. A New Peptide Originated from Amphibian Skin Alleviates the Ultraviolet B-Induced Skin Photodamage. Biomed. Pharmacother. 2022, 150, 112987. [Google Scholar] [CrossRef]
- Sun, H.; Wang, Y.; He, T.; He, D.; Hu, Y.; Fu, Z.; Wang, Y.; Sun, D.; Wang, J.; Liu, Y.; et al. Hollow Polydopamine Nanoparticles Loading with Peptide RL-QN15: A New Pro-Regenerative Therapeutic Agent for Skin Wounds. J. Nanobiotechnol. 2021, 19, 304. [Google Scholar] [CrossRef]
- Chen, X.; Liu, S.; Fang, J.; Zheng, S.; Wang, Z.; Jiao, Y.; Xia, P.; Wu, H.; Ma, Z.; Hao, L. Peptides Isolated from Amphibian Skin Secretions with Emphasis on Anti-Microbial Peptides. Toxins 2022, 14, 722. [Google Scholar] [CrossRef]
- Xie, C.; Fan, Y.; Yin, S.; Li, Y.; Liu, N.; Liu, Y.; Shu, L.; Fu, Z.; Wang, Y.; Zhang, Y.; et al. Novel Amphibian-Derived Antioxidant Peptide Protects Skin Against Ultraviolet Irradiation Damage. J. Photochem. Photobiol. B 2021, 224, 112327. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Shi, J.; Zhang, J.; Wang, Y.; Guo, Y.; Zhang, Z. Progress and Prospects of Regulatory Functions Mediated by Nitric Oxide on Immunity and Immunotherapy. Adv. Ther. 2021, 4, 2100032. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in Vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [CrossRef]
- Mendes, F.C.; de Paiva, J.C.; da Silva, E.Q.G.; Santos, M.R.; de Almeida Lima, G.D.; Moreira, G.A.; Bressan, G.C. Immunomodulatory Activity of Trifluoromethyl Arylamides Derived from the SRPK Inhibitor SRPIN340 and Their Potential Use as Vaccine Adjuvant. Life Sci. 2022, 307, 120849. [Google Scholar] [CrossRef]
- Leng, S.X.; McElhaney, J.E.; Walston, J.D.; Xie, D.; Fedarko, N.S.; Kuchel, G.A. ELISA and multiplex technologies for cytokine measurement in inflammation and aging research. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 879–884. [Google Scholar] [CrossRef]
- Möller, B.; Villiger, P.M. Inhibition of IL-1, IL-6, and TNF-α in immune-mediated inflammatory diseases *. In Springer Seminars in Immunopathology; Springer: Berlin, Germany, 2006; Volume 27, pp. 391–408. [Google Scholar]
- Bhat, Z.F.; Kumar, S.; Bhat, H.F. Bioactive Peptides of Animal Origin: A Review. J. Food Sci. Technol. 2015, 52, 5377–5392. [Google Scholar] [CrossRef]
- Bashir, K.M.I.; Sohn, J.H.; Kim, J.-S.; Choi, J.-S. Identification and Characterization of Novel Antioxidant Peptides from Mackerel (Scomber japonicus) Muscle Protein Hydrolysates. Food Chem. 2020, 323, 126809. [Google Scholar] [CrossRef]
- Tao, J.; Zhao, Y.-Q.; Chi, C.-F.; Wang, B. Bioactive Peptides from Cartilage Protein Hydrolysate of Spotless Smoothhound and Their Antioxidant Activity In Vitro. Mar. Drugs 2018, 16, 100. [Google Scholar] [CrossRef]
- Kim, N.Y.; Jung, H.Y.; Kim, J.K. Identification and Characterisation of a Novel Heptapeptide Mackerel By-Product Hydrolysate, and Its Potential as a Functional Fertiliser Component. J. Chromatogr. B 2021, 1180, 122881. [Google Scholar] [CrossRef]
- Yin, S.; Wang, Y.; Yang, X. Amphibian-Derived Wound Healing Peptides: Chemical Molecular Treasure Trove for Skin Wound Treatment. Front. Pharmacol. 2023, 14, 1120228. [Google Scholar] [CrossRef]
- Tian, M.; Liu, J.; Chai, J.; Wu, J.; Xu, X. Antimicrobial and Anti-Inflammatory Effects of a Novel Peptide From the Skin of Frog Microhyla pulchra. Front. Pharmacol. 2021, 12, 783108. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O. Interrelation of Oxidative Stress and Inflammation in Neurodegenerative Disease: Role of TNF. Oxidative Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-Induced DNA Damage, Mutations and Cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef]
- Udenigwe, C.C.; Aluko, R.E. Chemometric Analysis of the Amino Acid Requirements of Antioxidant Food Protein Hydrolysates. Int. J. Mol. Sci. 2011, 12, 3148–3161. [Google Scholar] [CrossRef]
- Ketnawa, S.; Wickramathilaka, M.; Liceaga, A.M. Changes on Antioxidant Activity of Microwave—Treated Protein Hydrolysates after Simulated Gastrointestinal Digestion: Purification and Identification. Food Chem. 2018, 254, 36–46. [Google Scholar] [CrossRef]
- Torres-Fuentes, C.; Contreras, M.D.M.; Recio, I.; Alaiz, M.; Vioque, J. Identification and Characterization of Antioxidant Peptides from Chickpea Protein Hydrolysates. Food Chem. 2015, 180, 194–202. [Google Scholar] [CrossRef]
- Yin, S.; Wang, Y.; Liu, N.; Yang, M.; Hu, Y.; Li, X.; Fu, Y.; Luo, M.; Sun, J.; Yang, X. Potential Skin Protective Effects After UVB Irradiation Afforded by an Antioxidant Peptide from Odorrana andersonii. Biomed. Pharmacother. 2019, 120, 109535. [Google Scholar] [CrossRef]
- Yin, S.; Li, S.; Bian, W.; Yang, M.; Liu, N.; Hu, Y.; Li, X.; Wang, Y.; Li, Z.; Sun, J.; et al. Antioxidant Peptide AOP-P1 Derived from Odorous Frog Showed Protective Effects Against UVB-Induced Skin Damages. Int. J. Pept. Res. Ther. 2020, 26, 557–565. [Google Scholar] [CrossRef]
- Zhang, B.; Qin, Y.; Wang, Y. A Nitric Oxide-Eluting and REDV Peptide-Conjugated Coating Promotes Vascular Healing. Biomaterials 2022, 284, 121478. [Google Scholar] [CrossRef]
- Doyle, J.; Llewellyn, L.E.; Brinkworth, C.S.; Bowie, J.H.; Wegener, K.L.; Rozek, T.; Wabnitz, P.A.; Wallace, J.C.; Tyler, M.J. Amphibian Peptides That Inhibit Neuronal Nitric Oxide Synthase. Eur. J. Biochem. 2002, 269, 100–109. [Google Scholar] [CrossRef]
- Vieira, N.F.; Barbosa, T.P.; Cárnio, E.C. Performance of Nitric Oxide in Sepsis: A Scoping Review. Acta Paul. de Enferm. 2024, 37, eAPE00512. [Google Scholar] [CrossRef]
- Trist, B.G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angew. Chem. Int. Ed. 2021, 60, 9215–9246. [Google Scholar] [CrossRef]
- Coulibaly, K.; Thauvin, M.; Melenbacher, A.; Testard, C.; Trigoni, E.; Vincent, A.; Stillman, M.J.; Vriz, S.; Policar, C.; Delsuc, N. A Di-Copper Peptidyl Complex Mimics the Activity of Catalase, a Key Antioxidant Metalloenzyme. Inorg. Chem. 2021, 60, 9309–9319. [Google Scholar] [CrossRef]
- Jomova, K.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Several Lines of Antioxidant Defense Against Oxidative Stress: Antioxidant Enzymes, Nanomaterials with Multiple Enzyme-Mimicking Activities, and Low-Molecular-Weight Antioxidants. Arch. Toxicol. 2024, 98, 1323–1367. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of Glutathione Synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Liu, R.-M.; Gao, L.; Choi, J.; Forman, H.J. γ-Glutamylcysteine Synthetase: mRNA Stabilization and Independent Subunit Transcription by 4-Hydroxy-2-Nonenal. Am. J. Physiol. Lung Cell. Mol. Physiol. 1998, 275, L861–L869. [Google Scholar] [CrossRef]
- Kondo, T.; Higashiyama, Y.; Goto, S.; Iida, T.; Cho, S.; Iwanaga, M.; Mori, K.; Tani, M.; Urata, Y. Regulation of γ-glutamycysteine synthetase expression in response to oxidative stress. Free Radic. Res. 1999, 31, 325–334. [Google Scholar] [CrossRef]
- Jang, D.; Lee, A.-H.; Shin, H.-Y.; Song, H.-R.; Park, J.-H.; Kang, T.-B.; Lee, S.-R.; Yang, S.-H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef] [PubMed]
- Uciechowski, P.; Dempke, W.C.M. Interleukin-6: A Masterplayer in the Cytokine Network. Oncology 2020, 98, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Calvano, S.E.; Lowry, S.F. Inflammatory Cytokines and Cell Response in Surgery. Surgery 2000, 127, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Su, W.-W.; Wang, S.; Li, P.-B. Naringin inhibits chemokine production in an LPS-induced RAW 264.7 macrophage cell line. Mol. Med. Rep. 2012, 6, 1343–1350. [Google Scholar] [CrossRef]
- Smith, J.K. IL-6 and the dysregulation of immune, bone, muscle, and metabolic homeostasis during spaceflight. NPJ Microgravity 2018, 4, 24. [Google Scholar] [CrossRef]
- Wang, L.; Walia, B.; Evans, J.; Gewirtz, A.T.; Merlin, D.; Sitaraman, S.V. IL-6 Induces NF-κB Activation in the Intestinal Epithelia. J. Immunol. 2003, 171, 3194–3201. [Google Scholar] [CrossRef]
- Tomolonis, J.A.; Xu, X.; Dholakia, K.H.; Zhang, C.; Guo, L.; Courtney, A.N.; Wang, S.; Balzeau, J.; Barragn, G.A.; Tian, G.; et al. Interaction between tumor cell TNFR2 and monocyte membrane-bound TNF-α triggers tumorigenic inflammation in neuroblastoma. J. Immunother. Cancer 2023, 11, e005478. [Google Scholar] [CrossRef]
- Fu, Y.; Li, C.; Li, X.; Zeng, L.; Wang, Y.; Fu, Z.; Shu, L.; Liu, Y.; Liu, N.; Yang, Y.; et al. Amphibian-Derived Peptide Homodimer Promotes Regeneration of Skin Wounds. Biomed. Pharmacother. 2022, 146, 112539. [Google Scholar] [CrossRef]
- Park, Y.R.; Park, C.-I.; Soh, Y. Antioxidant and Anti-Inflammatory Effects of NCW Peptide from Clam Worm (Marphysa sanguinea). J. Microbiol. Biotechnol. 2020, 30, 1387–1394. [Google Scholar] [CrossRef]
- Awasthi, S.; Kumar, G.; Ramani, V.; Awasthi, V.; Rodgers, K.K.; Xie, J.; Beierle, J.; Kyere-Davies, G.; Singh, B.; Rahman, N.; et al. Mechanism of Anti-Inflammatory Activity of TLR4-Interacting SPA4 Peptide. Immunohorizons 2021, 5, 659–674. [Google Scholar] [CrossRef]
- Li, C.; Xiong, Y.; Fu, Z.; Ji, Y.; Yan, J.; Kong, Y.; Peng, Y.; Ru, Z.; Huang, Y.; Li, Y.; et al. The Direct Binding of Bioactive Peptide Andersonin-W1 to TLR4 Expedites the Healing of Diabetic Skin Wounds. Cell. Mol. Biol. Lett. 2024, 29, 24. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Shin, H.-J.; Shah, M.; Cho, H.-Y.; Anwar, M.A.; Achek, A.; Kwon, H.-K.; Lee, B.; Yoo, T.H.; Choi, S. TLR4/MD2 Specific Peptides Stalled In Vivo LPS-Induced Immune Exacerbation. Biomaterials 2017, 126, 49–60. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Jin, X.; Gong, L.; Peng, Y.; Li, L.; Liu, G. Enhancer-Bound Nrf2 Licenses HIF-1α Transcription under Hypoxia to Promote Cisplatin Resistance in Hepatocellular Carcinoma Cells. Aging 2021, 13, 364–375. [Google Scholar] [CrossRef]
- Toth, R.; Warfel, N. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants 2017, 6, 27. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, B.; Shi, Q.; Wang, X.; Wang, D.; Zhu, L. Brusatol Inhibits HIF-1 Signaling Pathway and Suppresses Glucose Uptake under Hypoxic Conditions in HCT116 Cells. Sci. Rep. 2016, 6, 39123. [Google Scholar] [CrossRef]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 Blockade Suppresses Colon Tumor Angiogenesis by Inhibiting Hypoxia-Induced Activation of HIF-1α. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef]
- Yang, X.; Lee, W.-H.; Zhang, Y. Extremely Abundant Antimicrobial Peptides Existed in the Skins of Nine Kinds of Chinese Odorous Frogs. J. Proteome Res. 2012, 11, 306–319. [Google Scholar] [CrossRef]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a Free Radical Method to Evaluate Antioxidant Activity. LWT-Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Musa, K.H.; Abdullah, A.; Kuswandi, B.; Hidayat, M.A. A Novel High Throughput Method Based on the DPPH Dry Reagent Array for Determination of Antioxidant Activity. Food Chem. 2013, 141, 4102–4106. [Google Scholar] [CrossRef]
- Benzie, I.F.; Devaki, M. The ferric reducing/antioxidant power (FRAP) assay for non-enzymatic antioxidant capacity: Concepts, procedures, limitations and applications. In Measurement of Antioxidant Activity & Capacity: Recent Trends and Applications; Wiley: Hoboken, NJ, USA, 2018; pp. 77–106. [Google Scholar] [CrossRef]
- Tyszka-Czochara, M.; Paśko, P.; Reczyński, W.; Szlósarczyk, M.; Bystrowska, B.; Opoka, W. Zinc and Propolis Reduces Cytotoxicity and Proliferation in Skin Fibroblast Cell Culture: Total Polyphenol Content and Antioxidant Capacity of Propolis. Biol. Trace Elem. Res. 2014, 160, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Ewing, J.F.; Janero, D.R. Microplate superoxide dismutase assay employing a nonenzymatic superoxide generator. Anal. Biochem. 1995, 232, 243–248. [Google Scholar] [CrossRef]
- Dieterich, S.; Bieligk, U.; Beulich, S.; Hasenfuss, G.; Prestle, J. Gene Expression of Antioxidative Enzymes in the Human Heart: Increased Expression of Catalase in the End-Stage Failing Heart. Circulation 2000, 101, 33–39. [Google Scholar] [CrossRef]
- Shangari, N.; O’Brien, P.J. Catalase activity assays. Curr. Protoc. Toxicol. 2006, 27, 7.7.1–7.7.6. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Hearne, J.L.; Colman, R.F. Delineation of xenobiotic substrate sites in rat glutathione S-transferase M1-1. Protein Sci. 2005, 14, 2526–2536. [Google Scholar] [CrossRef]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [CrossRef]
- Yucel, A.A.; Gulen, S.; Dincer, S.; Yucel, A.E.; Yetkin, G.I. Comparison of two different applications of the Griess method for nitric oxide measurement. J. Exp. Integr. Med. 2012, 2, 167. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. The Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction: Twenty-Something Years On. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef]
- Beutler, B. TLRs and innate immunity. Nat. Rev. Immunol. 2009, 9, 393–404. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024; Available online: https://www.r-project.org/ (accessed on 15 May 2025).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Origin Protein | COX-2 Binding Energy | COX-2 Inhibition Active Site |
|---|---|---|---|
| SGHPGAMGPVGPR | Collagen α-2 (I) * | −9.5 (kcal/mol) | 3 |
| Gene | Forward | Reverse |
|---|---|---|
| NF-κβ | 5′-GCT GCC AAA GAA GGA CAC GAC A-3′ | 5′-GGC AGG CTA TTG CTC ATC ACA G-3′ |
| COX-2 | 5′-TGC ACT ATG GTT ACA AAA GCT GG-3′ | 5′-TCA GGA AGC TCC TTA TTT CCC TT-3′ |
| IL-6 | 5′-TCC TTC CTA CCC CAA TTT CC-3′ | 5′-GCC ACT CCT TCT GTG ACT CC-3′ |
| TNF-α | 5′-TAT GGC TCA GGG TCC AAC TC-3′ | 5′-CCC ATT TGA GTC CTT GAT GG-3′ |
| TLR | 5′-CAG GTG GAA TTG TAT CGC CT-3′ | 5′-CGA GGC TTT TCC ATC CAA TA-3′ |
| HIF-1 | 5′-CGA AGT TAC AG CTT TCC GAC CAG-3′ | 5′-GTT TGT GTC GGT CAG CAC CAC T-3′ |
| IL-10 | 5′-TTA ATA AGC TCC AAG ACC AAG G-3′ | 5′-GATO GATO GTA TGC TTC TAT GCA G-3′ |
| Nfr2 | 5′-CTG AAC TCC TGG ACG GGA CTA-3′ | 5′-CGG TGG GTC TCC GTA AAT GG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelinsari, S.M.; Mattosinhos, P.d.S.; Dias, M.M.d.S.; de Paula, R.A.; Novaes, R.D.; Vilela, E.F.; Valacchi, G.; Gonçalves, R.V. Effect of Synthetic Peptides Identified in the Bullfrog Skin on Inflammation and Oxidative Stress Control: An In Vitro Analysis. Molecules 2025, 30, 2223. https://doi.org/10.3390/molecules30102223
Pelinsari SM, Mattosinhos PdS, Dias MMdS, de Paula RA, Novaes RD, Vilela EF, Valacchi G, Gonçalves RV. Effect of Synthetic Peptides Identified in the Bullfrog Skin on Inflammation and Oxidative Stress Control: An In Vitro Analysis. Molecules. 2025; 30(10):2223. https://doi.org/10.3390/molecules30102223
Chicago/Turabian StylePelinsari, Silvânia Mól, Patricia da Silva Mattosinhos, Manoela Maciel dos Santos Dias, Rosinéa Aparecida de Paula, Romulo Dias Novaes, Emerson Ferreira Vilela, Giuseppe Valacchi, and Reggiani Vilela Gonçalves. 2025. "Effect of Synthetic Peptides Identified in the Bullfrog Skin on Inflammation and Oxidative Stress Control: An In Vitro Analysis" Molecules 30, no. 10: 2223. https://doi.org/10.3390/molecules30102223
APA StylePelinsari, S. M., Mattosinhos, P. d. S., Dias, M. M. d. S., de Paula, R. A., Novaes, R. D., Vilela, E. F., Valacchi, G., & Gonçalves, R. V. (2025). Effect of Synthetic Peptides Identified in the Bullfrog Skin on Inflammation and Oxidative Stress Control: An In Vitro Analysis. Molecules, 30(10), 2223. https://doi.org/10.3390/molecules30102223