
Tailoring FXR Modulators for Intestinal Specificity: Recent Progress and Insights



Abstract

1. Introduction
1.1. FXR in the Gut
1.2. Importance of Intestinal Modulation
2. FXR Modulators
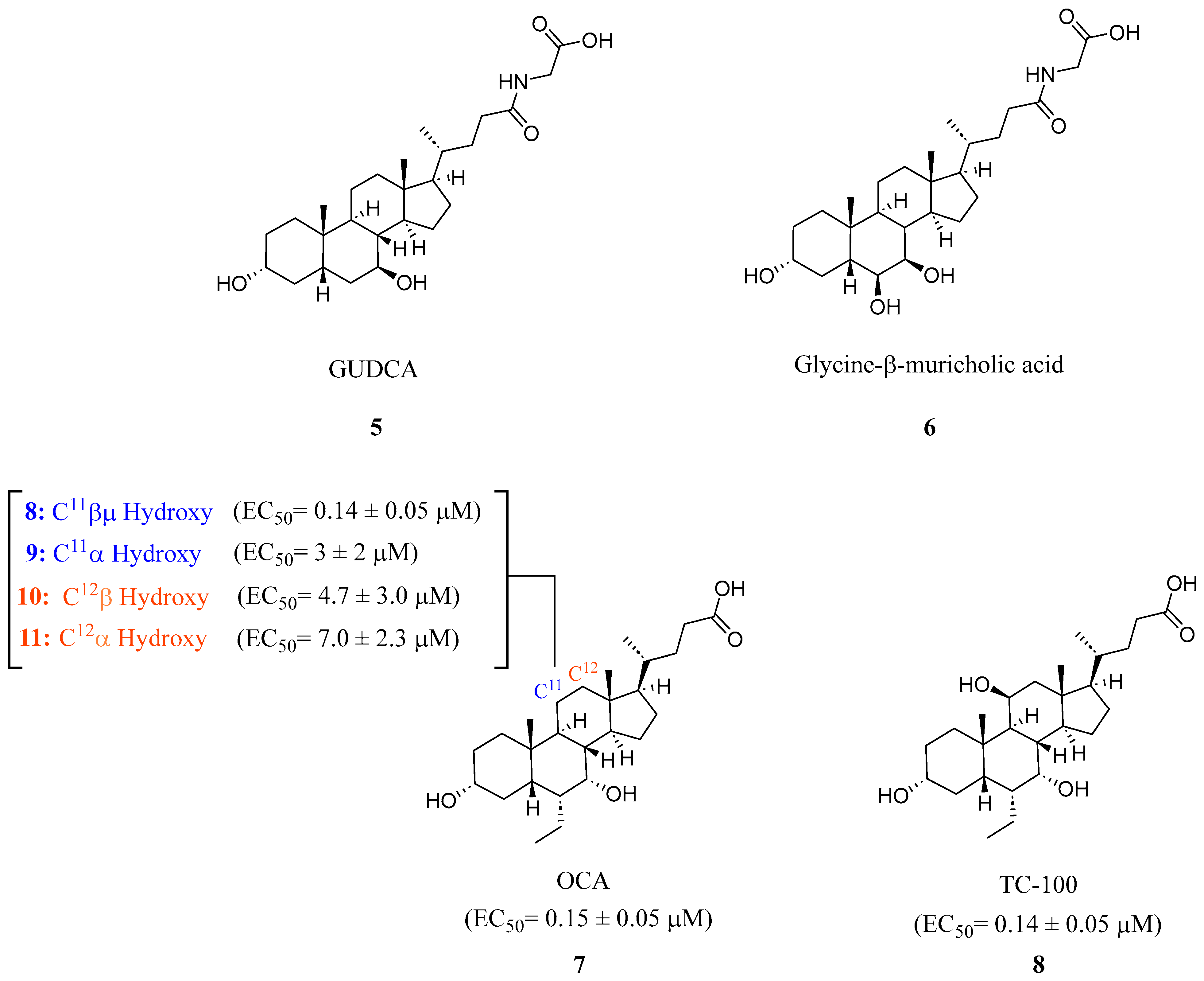
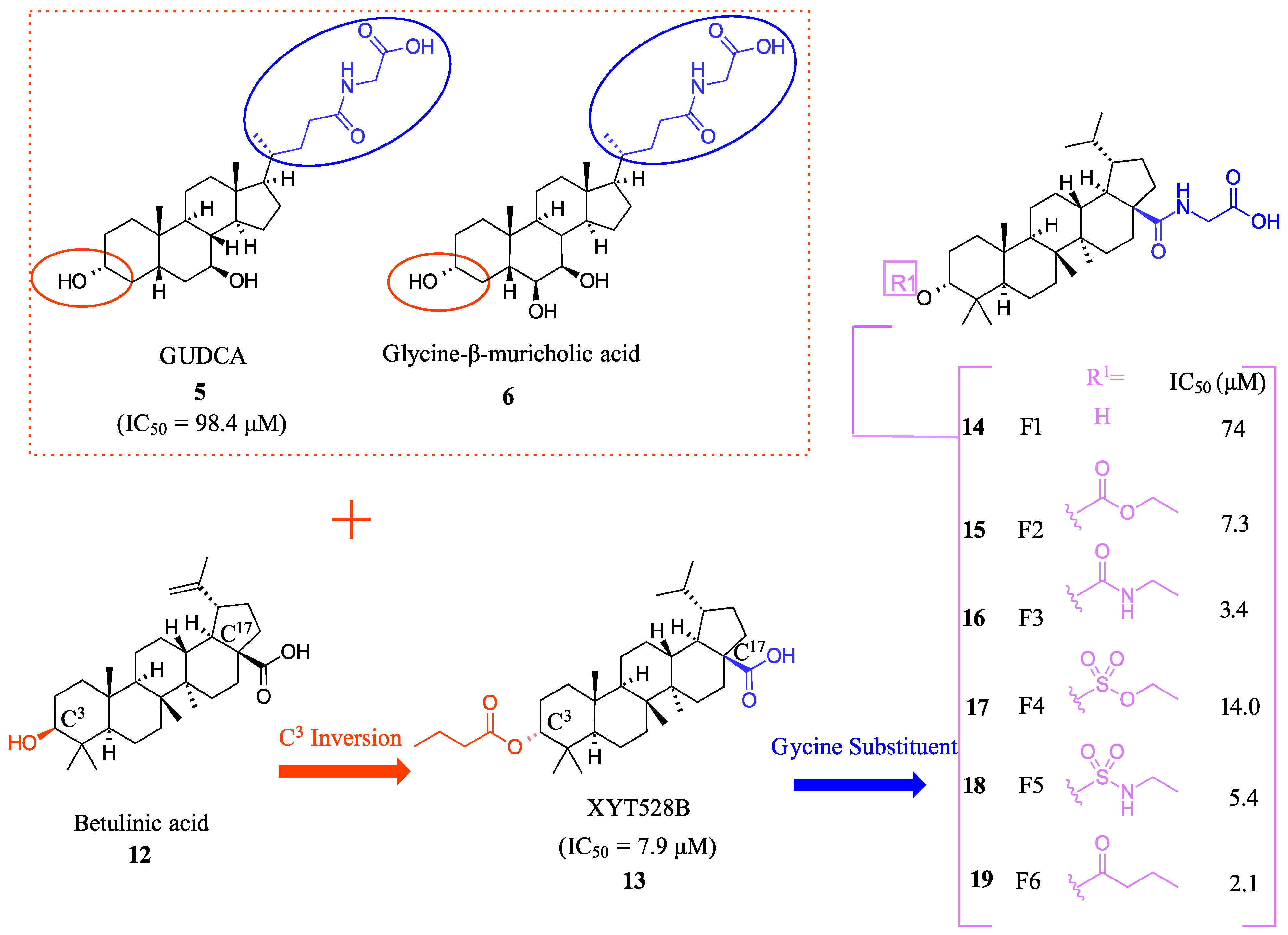
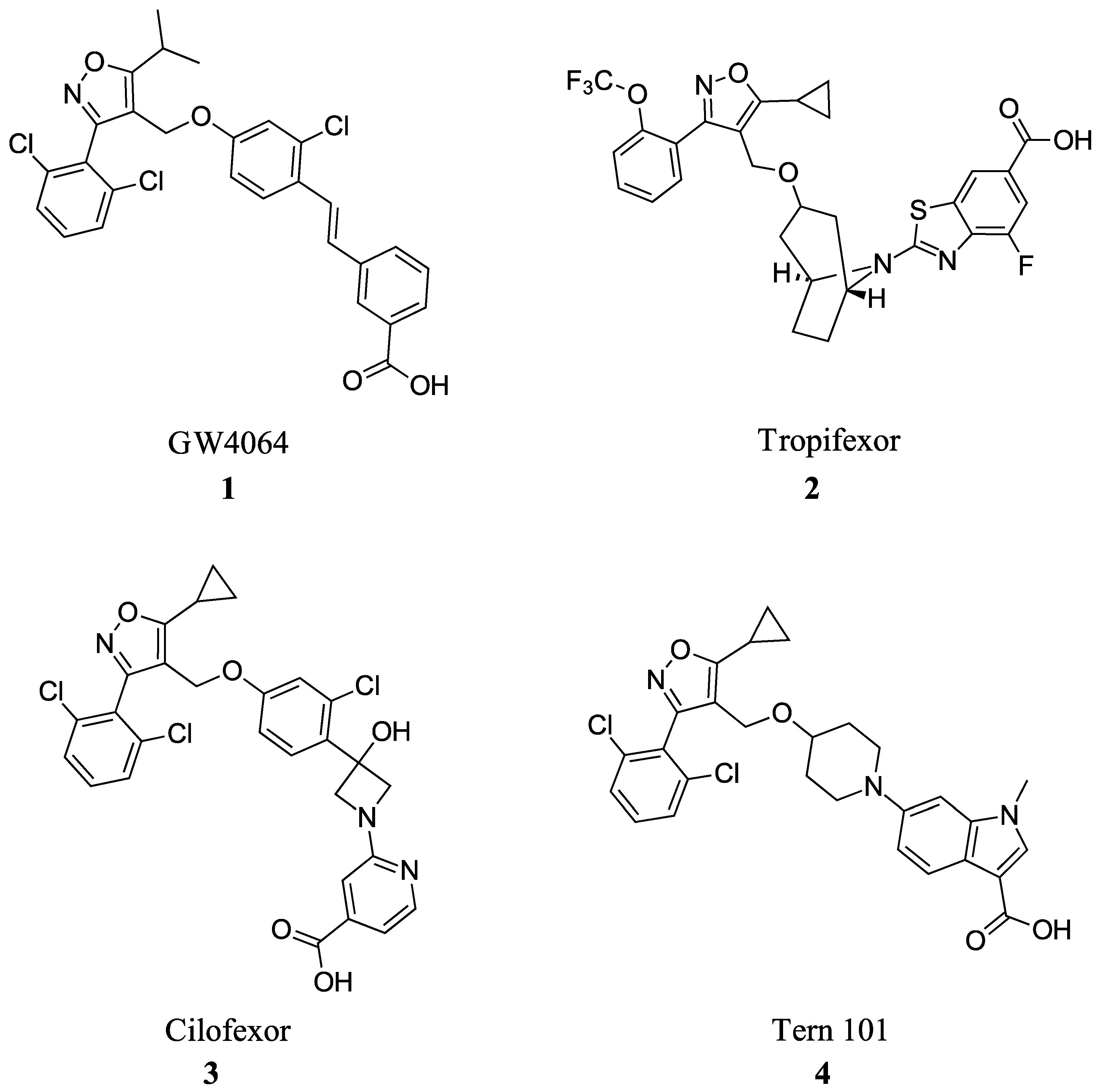
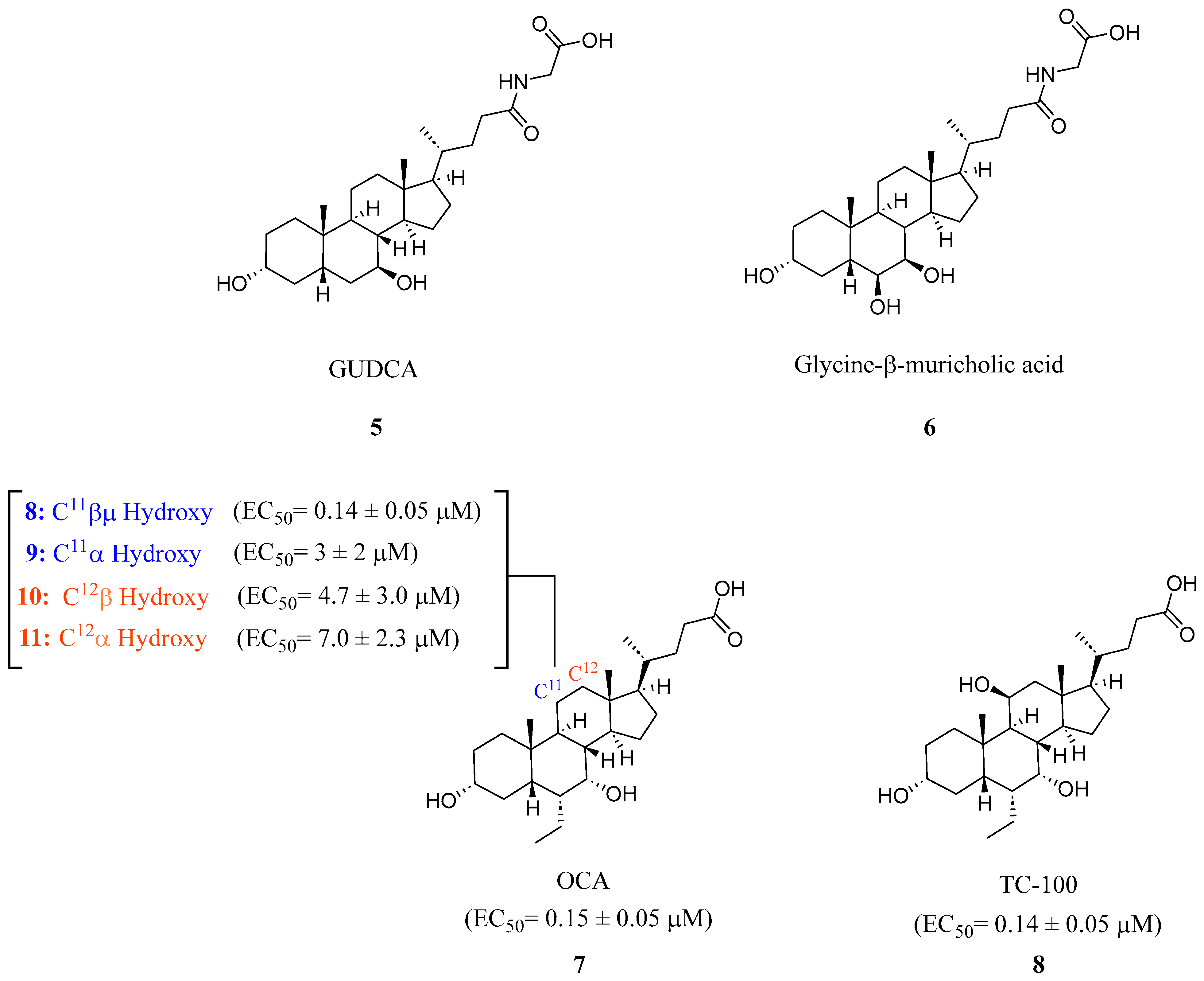
2.1. Steroidal Based Modulators
2.2. Non-Steroidal Based Modulators
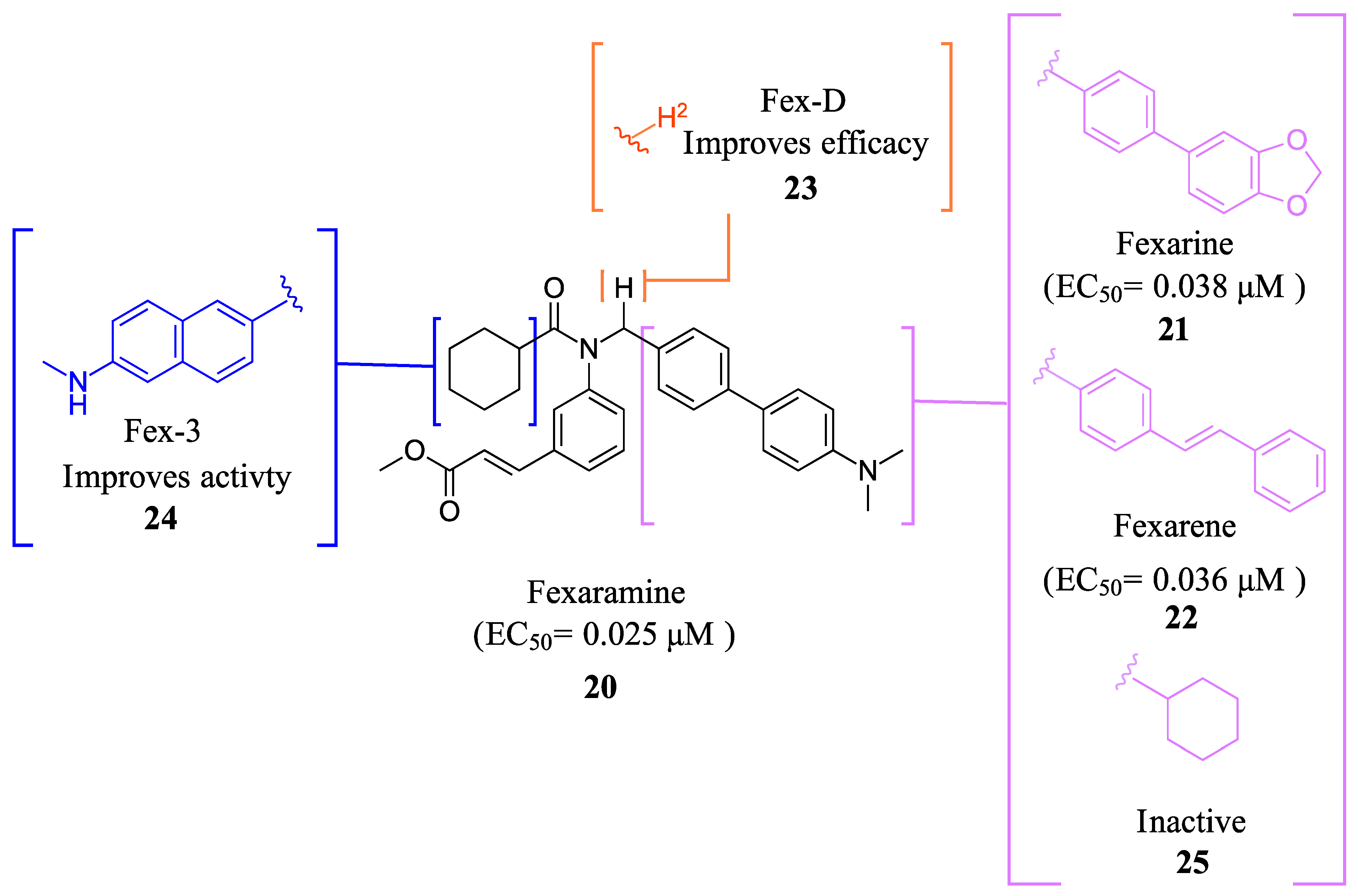
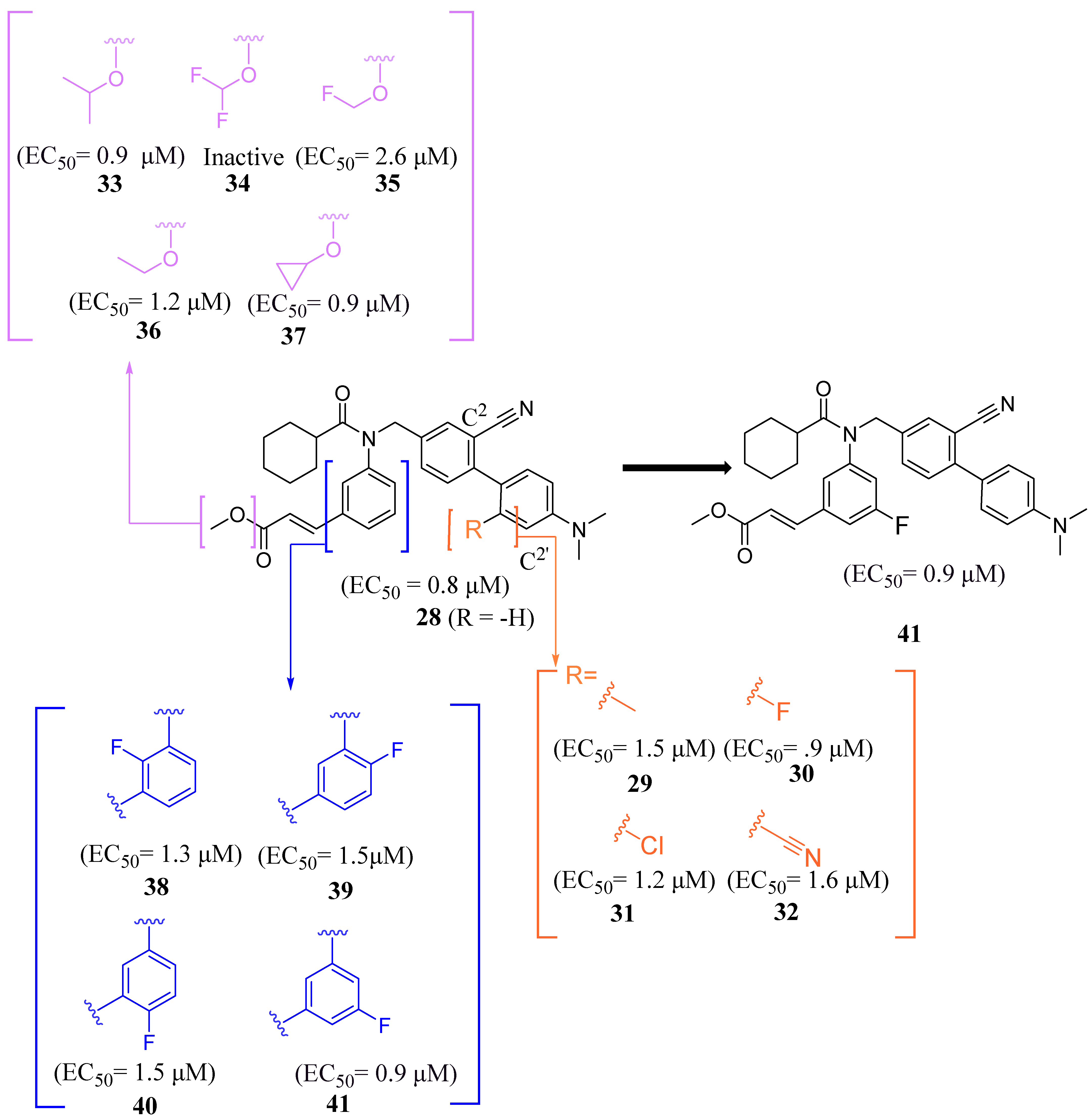
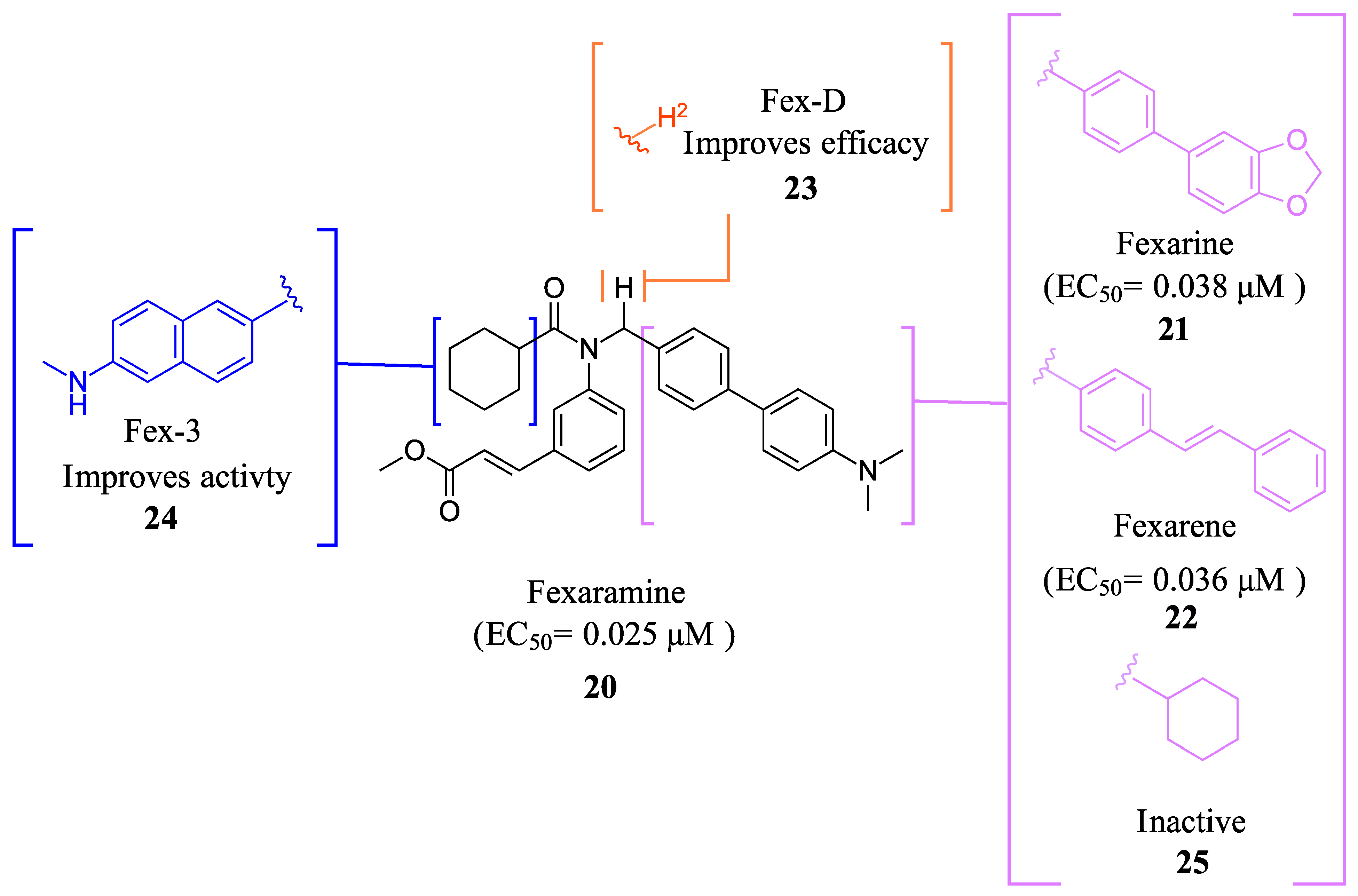
2.2.1. Fexaramine and Its Derivatives
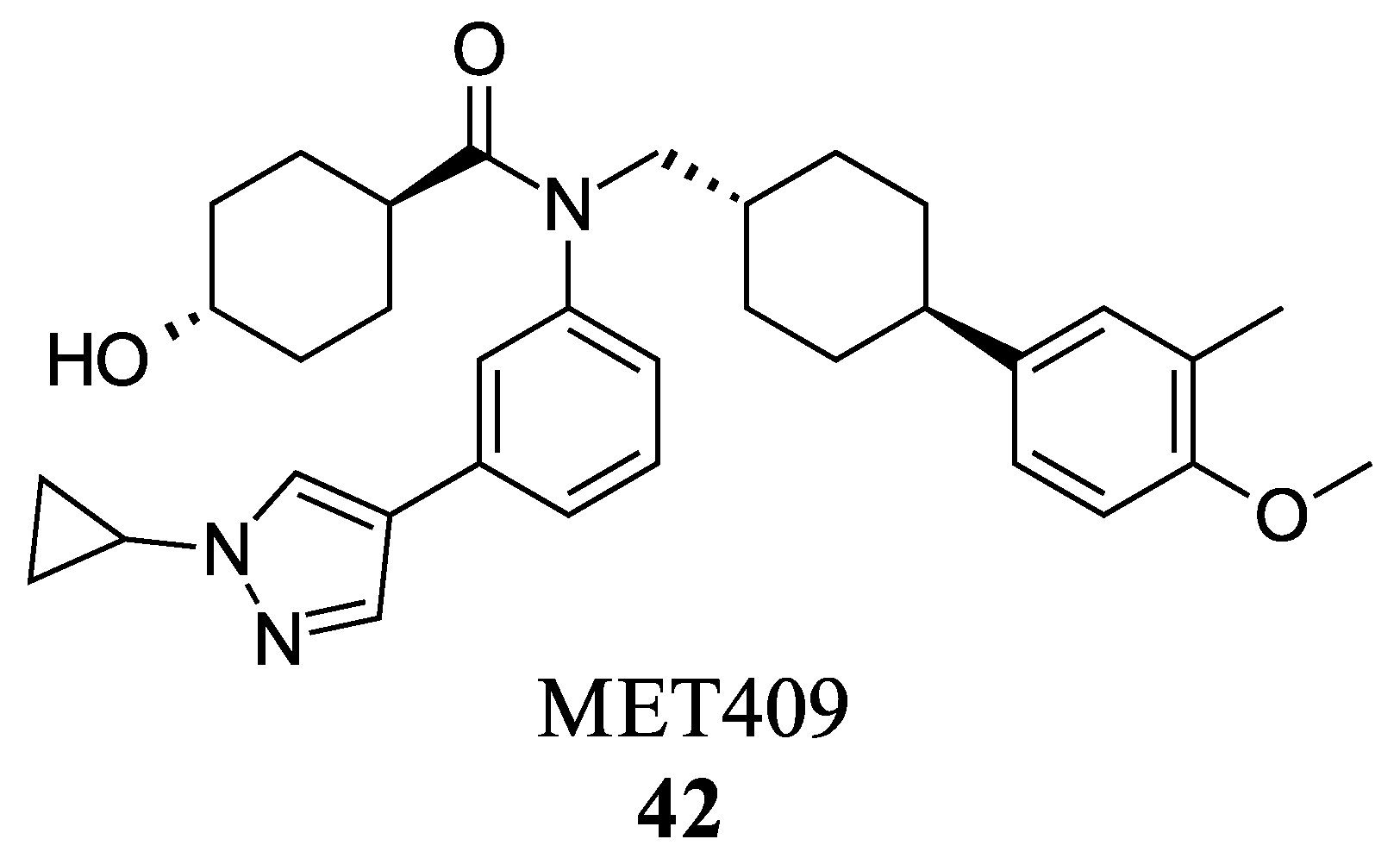
2.2.2. Miscellaneous
3. Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fang, Y.; Hegazy, L.; Finck, B.N.; Elgendy, B. Recent Advances in the Medicinal Chemistry of Farnesoid X Receptor. J. Med. Chem. 2021, 64, 17545–17571. [Google Scholar] [CrossRef] [PubMed]
- Stofan, M.; Guo, G.L. Bile Acids and FXR: Novel Targets for Liver Diseases. Front. Med. 2020, 7, 544. [Google Scholar] [CrossRef] [PubMed]
- Massafra, V.; Pellicciari, R.; Gioiello, A.; van Mil, S.W.C. Progress and Challenges of Selective Farnesoid X Receptor Modulation. Pharmacol. Ther. 2018, 191, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.Y.; Lee, H.; Hubbert, M.L.; Edwards, P.A.; Zhang, Y. FXR, a Multipurpose Nuclear Receptor. Trends Biochem. Sci. 2006, 31, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Fan, W.; Yang, W.; Samdani, A.Q.; Jackson, A.O.; Qu, S. Farnesoid X Receptor: An Important Factor in Blood Glucose Regulation. Clin. Chim. Acta 2019, 495, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Girisa, S.; Henamayee, S.; Parama, D.; Rana, V.; Dutta, U.; Kunnumakkara, A.B. Targeting Farnesoid X Receptor (FXR) for Developing Novel Therapeutics against Cancer. Mol. Biomed. 2021, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Yan, T.; Xia, Y.; Hao, H.; Wang, G.; Gonzalez, F.J. The Pathophysiological Function of Non-Gastrointestinal Farnesoid X Receptor. Pharmacol. Ther. 2021, 226, 107867. [Google Scholar] [CrossRef] [PubMed]
- Sagar, N.M.; McFarlane, M.; Nwokolo, C.; Bardhan, K.D.; Arasaradnam, R.P. Mechanisms of Triglyceride Metabolism in Patients with Bile Acid Diarrhea. World J. Gastroenterol. 2016, 22, 6757. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Cariello, M.; Sabbà, C.; Moschetta, A. Tissue-Specific Actions of FXR in Metabolism and Cancer. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2015, 1851, 30–39. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the Nuclear Receptor FXR Improves Hyperglycemia and Hyperlipidemia in Diabetic Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef]
- Ma, K. Farnesoid X Receptor Is Essential for Normal Glucose Homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Xu, X.; Zhang, P.; Peng, Y.; Zhao, X.; Chen, S.; Guo, F.; Xiong, Y.; Chu, X.-J.; Xu, X. Discovery of HPG1860, a Structurally Novel Nonbile Acid FXR Agonist Currently in Clinical Development for the Treatment of Nonalcoholic Steatohepatitis. J. Med. Chem. 2023, 66, 9363–9375. [Google Scholar] [CrossRef] [PubMed]
- Hollman, D.A.A.; Milona, A.; Van Erpecum, K.J.; Van Mil, S.W.C. Anti-Inflammatory and Metabolic Actions of FXR: Insights into Molecular Mechanisms. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2012, 1821, 1443–1452. [Google Scholar] [CrossRef]
- Hundt, M.; Basit, H.; John, S. Physiology, Bile Secretion. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Landrier, J.-F.; Eloranta, J.J.; Vavricka, S.R.; Kullak-Ublick, G.A. The Nuclear Receptor for Bile Acids, FXR, Transactivates Human Organic Solute Transporter-α and -β Genes. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 290, G476–G485. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Jiang, H.; Liu, W.; Zhang, X.; Chen, D.; Sun, S.; Zhou, C.; Liu, J.; Bao, S.; Wang, X.; et al. The Triterpenoid Sapogenin (2α-OH-Protopanoxadiol) Ameliorates Metabolic Syndrome via the Intestinal FXR/GLP-1 Axis through Gut Microbiota Remodelling. Cell Death Dis. 2020, 11, 770. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Liu, Y.; Gao, X.; Krausz, K.W.; Niu, B.; Gonzalez, F.J.; Xie, C. Caffeic Acid Phenethyl Ester Suppresses Intestinal FXR Signaling and Ameliorates Nonalcoholic Fatty Liver Disease by Inhibiting Bacterial Bile Salt Hydrolase Activity. Acta Pharmacol. Sin. 2023, 44, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut Microbiota and Intestinal FXR Mediate the Clinical Benefits of Metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, M.; Gu, W.; Chen, L. Intestine-Specific FXR Agonists as Potential Therapeutic Agents for Colorectal Cancer. Biochem. Pharmacol. 2021, 186, 114430. [Google Scholar] [CrossRef]
- Fang, S.; Suh, J.M.; Reilly, S.M.; Yu, E.; Osborn, O.; Lackey, D.; Yoshihara, E.; Perino, A.; Jacinto, S.; Lukasheva, Y.; et al. Intestinal FXR Agonism Promotes Adipose Tissue Browning and Reduces Obesity and Insulin Resistance. Nat. Med. 2015, 21, 159–165. [Google Scholar] [CrossRef]
- Maran, R.R.M.; Thomas, A.; Roth, M.; Sheng, Z.; Esterly, N.; Pinson, D.; Gao, X.; Zhang, Y.; Ganapathy, V.; Gonzalez, F.J.; et al. Farnesoid X Receptor Deficiency in Mice Leads to Increased Intestinal Epithelial Cell Proliferation and Tumor Development. J. Pharmacol. Exp. Ther. 2009, 328, 469–477. [Google Scholar] [CrossRef]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112.e18. [Google Scholar] [CrossRef] [PubMed]
- Trabelsi, M.-S.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X Receptor Inhibits Glucagon-like Peptide-1 Production by Enteroendocrine L Cells. Nat. Commun. 2015, 6, 7629. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhang, H.; Xiao, D.; Wei, H.; Chen, Y. Farnesoid X Receptor (FXR): Structures and Ligands. Comput. Struct. Biotechnol. J. 2021, 19, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Adorini, L.; Trauner, M. FXR Agonists in NASH Treatment. J. Hepatol. 2023, 79, 1317–1331. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, C.; Lv, Y.; Li, J.; Krausz, K.W.; Shi, J.; Brocker, C.N.; Desai, D.; Amin, S.G.; Bisson, W.H.; et al. Intestine-Selective Farnesoid X Receptor Inhibition Improves Obesity-Related Metabolic Dysfunction. Nat. Commun. 2015, 6, 10166. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Passeri, D.; De Franco, F.; Mostarda, S.; Filipponi, P.; Colliva, C.; Gadaleta, R.M.; Franco, P.; Carotti, A.; Macchiarulo, A.; et al. Discovery of 3α,7α,11β-Trihydroxy-6α-Ethyl-5β-Cholan-24-Oic Acid (TC-100), a Novel Bile Acid as Potent and Highly Selective FXR Agonist for Enterohepatic Disorders. J. Med. Chem. 2016, 59, 9201–9214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, Y.; Wang, Y.; Ge, X.; Jiao, T.; Yin, J.; Wang, K.; Li, C.; Guo, S.; Xie, X.; et al. Discovery of Betulinic Acid Derivatives as Potent Intestinal Farnesoid X Receptor Antagonists to Ameliorate Nonalcoholic Steatohepatitis. J. Med. Chem. 2022, 65, 13452–13472. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Zhao, P.; Zhang, S.; Fan, S.; Yang, L.; Tong, Q.; Ji, G.; Huang, C. Betulinic Acid Alleviates Endoplasmic Reticulum Stress-mediated Nonalcoholic Fatty Liver Disease through Activation of Farnesoid X Receptors in Mice. Br. J. Pharmacol. 2019, 176, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Jiang, C.; Shi, J.; Gao, X.; Sun, D.; Sun, L.; Wang, T.; Takahashi, S.; Anitha, M.; Krausz, K.W.; et al. An Intestinal Farnesoid X Receptor–Ceramide Signaling Axis Modulates Hepatic Gluconeogenesis in Mice. Diabetes 2017, 66, 613–626. [Google Scholar] [CrossRef]
- Downes, M.; Verdecia, M.A.; Roecker, A.J.; Hughes, R.; Hogenesch, J.B.; Kast-Woelbern, H.R.; Bowman, M.E.; Ferrer, J.-L.; Anisfeld, A.M.; Edwards, P.A.; et al. A Chemical, Genetic, and Structural Analysis of the Nuclear Bile Acid Receptor FXR. Mol. Cell 2003, 11, 1079–1092. [Google Scholar] [CrossRef]
- Hartmann, P.; Hochrath, K.; Horvath, A.; Chen, P.; Seebauer, C.T.; Llorente, C.; Wang, L.; Alnouti, Y.; Fouts, D.E.; Stärkel, P.; et al. Modulation of the Intestinal Bile Acid/Farnesoid X Receptor/Fibroblast Growth Factor 15 Axis Improves Alcoholic Liver Disease in Mice. Hepatology 2018, 67, 2150–2166. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Li, Y.; Oh, T.G.; Cayabyab, F.; He, N.; Tang, Q.; Coulter, S.; Truitt, M.; Medina, P.; He, M.; et al. FXR Mediates ILC-Intrinsic Responses to Intestinal Inflammation. Proc. Natl. Acad. Sci. USA 2022, 119, e2213041119. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Z.; Zhou, J.; Guo, Y.; Wang, G.; Hao, H.; Xu, X. A Novel Intestinal-Restricted FXR Agonist. Bioorg. Med. Chem. Lett. 2017, 27, 3386–3390. [Google Scholar] [CrossRef] [PubMed]
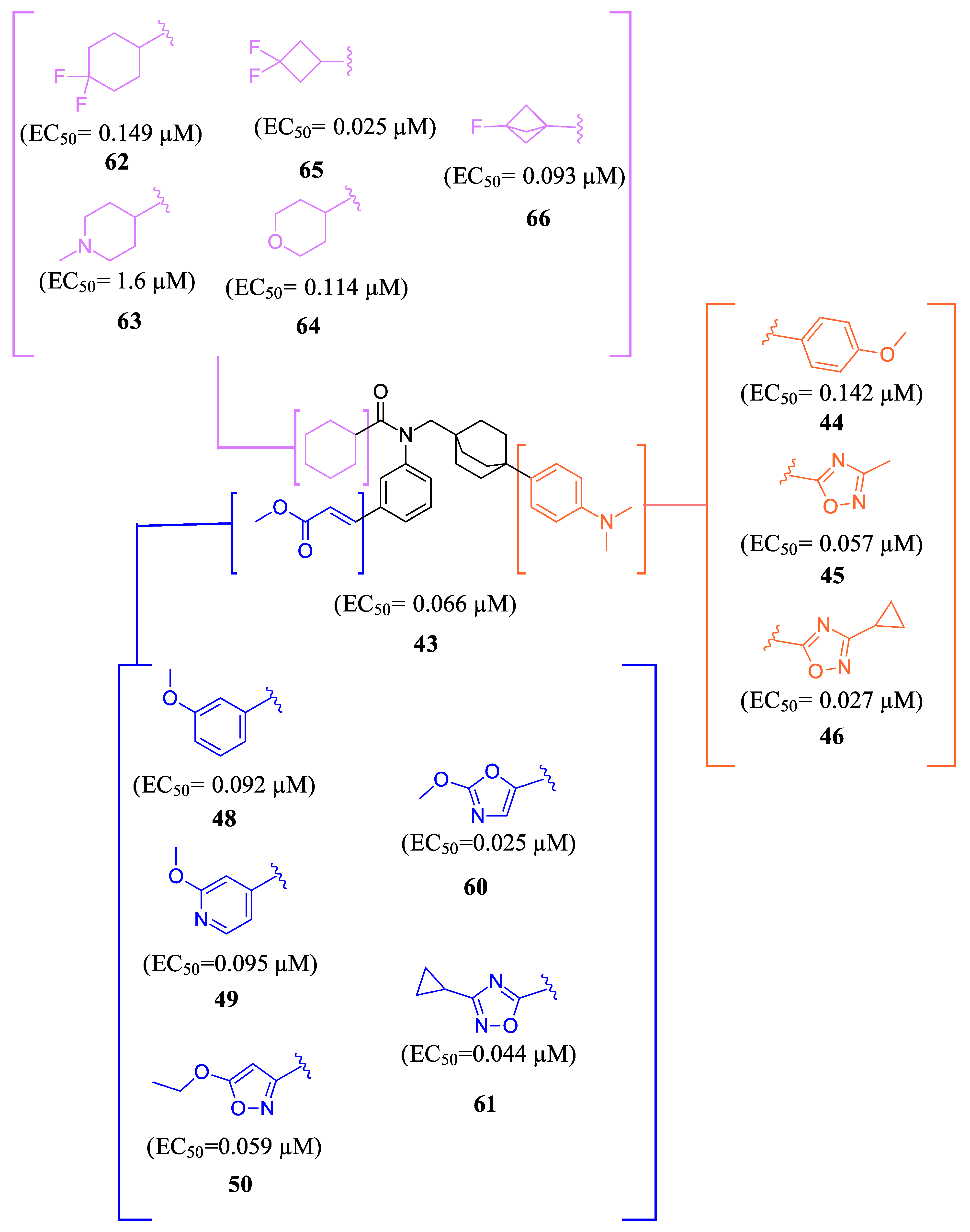
- Shim, S.; Krishnaiah, M.; Sankham, M.R.; Kim, I.; Lee, Y.; Shin, I.; Oh, A.R.; Lee, H.J.; Vu, T.N.L.; Park, J.; et al. Discovery of (E)-3-(3-((2-Cyano-4′-Dimethylaminobiphenyl-4-Ylmethyl)Cyclohexanecarbonylamino)-5-Fluorophenyl)Acrylic Acid Methyl Ester, an Intestine-Specific, FXR Partial Agonist for the Treatment of Nonalcoholic Steatohepatitis. J. Med. Chem. 2022, 65, 9974–10000. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.D.; Govek, S.P.; Nagasawa, J.Y. Farnesoid X Receptor Agonists and Uses Thereof. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018170166 (accessed on 11 February 2024).
- Wagner, B.; Brigham, D.; Lee, K.-J.; Ortiz, A.; Lu, N.; Douglas, K.; Govek, S.; Feigh, M.; Veidal, S.; Vrang, N.; et al. M480, a Novel Non-Bile Acid FXR Agonist, Shows Improvement across Multiple Parameters in a Diet-Induced Obese Mouse Model with Biopsy-Confirmed NASH. J. Hepatol. 2017, 66, S433. [Google Scholar] [CrossRef]
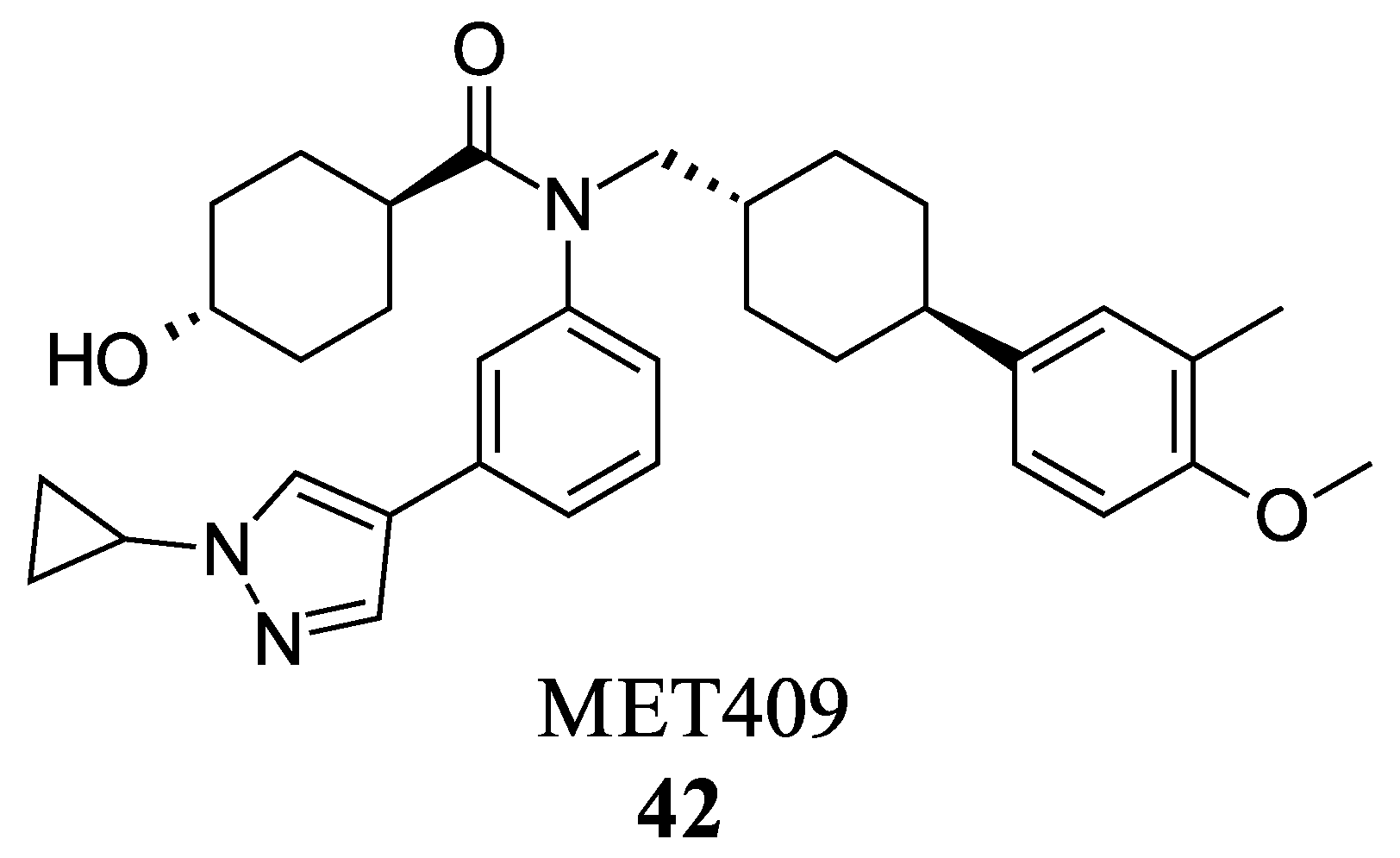
- Harrison, S.A.; Bashir, M.R.; Lee, K.-J.; Shim-Lopez, J.; Lee, J.; Wagner, B.; Smith, N.D.; Chen, H.C.; Lawitz, E.J. A Structurally Optimized FXR Agonist, MET409, Reduced Liver Fat Content over 12 Weeks in Patients with Non-Alcoholic Steatohepatitis. J. Hepatol. 2021, 75, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Nara, S.J.; Jogi, S.; Cheruku, S.; Kandhasamy, S.; Jaipuri, F.; Kathi, P.K.; Reddy, S.; Sarodaya, S.; Cook, E.M.; Wang, T.; et al. Discovery of BMS-986339, a Pharmacologically Differentiated Farnesoid X Receptor Agonist for the Treatment of Nonalcoholic Steatohepatitis. J. Med. Chem. 2022, 65, 8948–8960. [Google Scholar] [CrossRef] [PubMed]
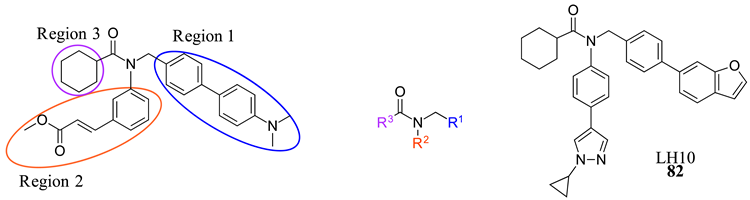
- Huang, W.; Cao, Z.; Wang, W.; Yang, Z.; Jiao, S.; Chen, Y.; Chen, S.; Zhang, L.; Li, Z. Discovery of LH10, a Novel Fexaramine-Based FXR Agonist for the Treatment of Liver Disease. Bioorganic Chem. 2024, 143, 107071. [Google Scholar] [CrossRef]
- Ren, Q.; Chen, Y.; Zhou, Z.; Cai, Z.; Jiao, S.; Huang, W.; Wang, B.; Chen, S.; Wang, W.; Cao, Z.; et al. Discovery of the First-in-Class Intestinal Restricted FXR and FABP1 Dual Modulator ZLY28 for the Treatment of Nonalcoholic Fatty Liver Disease. J. Med. Chem. 2023, 66, 6082–6104. [Google Scholar] [CrossRef]
- Huang, H.; McIntosh, A.L.; Martin, G.G.; Landrock, D.; Chung, S.; Landrock, K.K.; Dangott, L.J.; Li, S.; Kier, A.B.; Schroeder, F. FABP1: A Novel Hepatic Endocannabinoid and Cannabinoid Binding Protein. Biochemistry 2016, 55, 5243–5255. [Google Scholar] [CrossRef]
- Teno, N.; Yamashita, Y.; Iguchi, Y.; Fujimori, K.; Une, M.; Nishimaki-Mogami, T.; Hiramoto, T.; Gohda, K. Nonacidic Chemotype Possessing N-Acylated Piperidine Moiety as Potent Farnesoid X Receptor (FXR) Antagonists. ACS Med. Chem. Lett. 2018, 9, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Teno, N.; Yamashita, Y.; Masuda, A.; Iguchi, Y.; Oda, K.; Fujimori, K.; Hiramoto, T.; Nishimaki-Mogami, T.; Une, M.; Gohda, K. Identification of Potent Farnesoid X Receptor (FXR) Antagonist Showing Favorable PK Profile and Distribution toward Target Tissues: Comprehensive Understanding of Structure-Activity Relationship of FXR Antagonists. Bioorg. Med. Chem. 2019, 27, 2220–2227. [Google Scholar] [CrossRef] [PubMed]
- Teno, N.; Iguchi, Y.; Oda, K.; Yamashita, Y.; Masuda, A.; Fujimori, K.; Une, M.; Gohda, K. Discovery of Orally Active and Nonsteroidal Farnesoid X Receptor (FXR) Antagonist with Propensity for Accumulation and Responsiveness in Ileum. ACS Med. Chem. Lett. 2021, 12, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.S.; Obach, R.S.; Rotter, C.; Miller, H.R.; Chang, G.; Steyn, S.J.; El-Kattan, A.; Troutman, M.D. Physicochemical Space for Optimum Oral Bioavailability: Contribution of Human Intestinal Absorption and First-Pass Elimination. J. Med. Chem. 2010, 53, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Dorel, R.; Wong, A.R.; Crawford, J.J. Trust Your Gut: Strategies and Tactics for Intestinally Restricted Drugs. ACS Med. Chem. Lett. 2023, 14, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Small-Molecule Drug Discovery Suite. QikProp; Version 4.2; Schrödinger, LLC: New York, NY, USA, 2014.
- Azman, M.; Sabri, A.H.; Anjani, Q.K.; Mustaffa, M.F.; Hamid, K.A. Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery. Pharmaceuticals 2022, 15, 975. [Google Scholar] [CrossRef]
- Winiwarter, S.; Ax, F.; Lennernäs, H.; Hallberg, A.; Pettersson, C.; Karlén, A. Hydrogen Bonding Descriptors in the Prediction of Human in Vivo Intestinal Permeability. J. Mol. Graph. Model. 2003, 21, 273–287. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Comp. | R1 | R2 | R3 | EC50 (µM) |
| 67 | 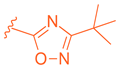 | 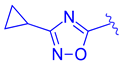 |  | 0.70 |
| 68 | 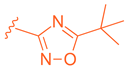 | | | 0.029 |
| 69 | | 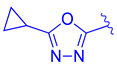 | | 0.045 |
| 70 | | 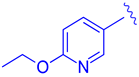 | | 0.104 |
| 71 | |  | | 0.041 |
| 72 | | |  | 0.111 |
| 73 | 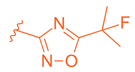 |  | | 0.025 |
| 74 | | |  | 0.034 |
 | ||||
| Comp. | R1 | R2 | R3 | EC50 (µM) |
| 75 | 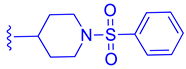 |  |  | 4.98 |
| 76 | 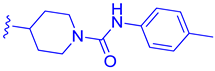 | | | 3.81 |
| 77 | 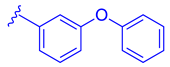 | | | 1.07 |
| 78 |  | 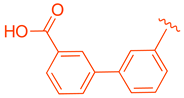 | | >10 |
| 79 | |  | | 3.87 |
| 80 | |  | | 9.58 |
| 81 | | 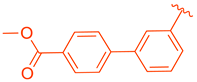 | | >10 |
| 82 | 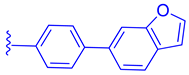 | 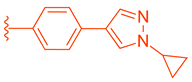 | | 0.14 |
| 83 | 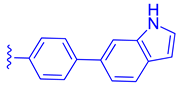 | | | 0.28 |
| 84 | | 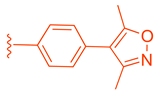 | | 0.57 |
| 85 | 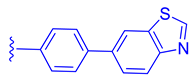 | 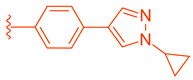 | | 2.81 |
| 86 | | | 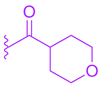 | 0.45 |
| Comp. | Systemic Effects | Gene Effects | EC50 μM | IC50 μM | CLogp | pKa (Acidic) | MW |
|---|---|---|---|---|---|---|---|
| GUDCA (5) | Increased metabolic rate and GLP1 production | Decreased FGF19 and SHP | - | 77.2 | 2.61 | 3.77 | 449.31 |
| Gly-MCA (6) | Reduced ceramide levels | Decreased SHP and FGF15 | - | N/A | 4.2 | 3,1 | 465.62 |
| F6 (19) | Reduced weight gain Improved insulin sensitivity, glucose tolerance, and reduced ceramide levels | Increased FGF15, OSTα, and SHP Lowered ALT, AST, and TG | - | 2.1 | 7.47 | 4.11 | 585.87 |
| TC-100 (8) | N/A | Increased CYP7A1, SHP, OSTα, FGF15, ANG1, and BSEP | 0.14 | - | 3.12 | 4.48 | 436.63 |
| Fex (20) | Increased energy expenditure and weight loss Lowered glucose, insulin levels, Improved ethanol induced hepatic inflammation | SHP, MRP-2, BSEP; PLTP, intestinal Nr0b2, and FGF15 | 0.025 | - | 7.21 | 4.82 | 496.65 |
| Fex-D (23) | Decreased ILC3, total intestinal serum BA | SHP, FGF15, OSTα, cytokine genes, and fibrotic genes | - | - | 7.21 | 4.82 | 496.65 |
| Fex-3 (24) | Increased intestinal selectivity | BSEP, CYP7A1, FGF15, and SHP | - | - | 7.41 | 4.96 | 569.71 |
| 41 | Reduced liver fibrosis and dyslipidemia effects | Increased IBABP, FGF15, SHP, and OSTα | 0.9 | - | 7.21 | 4.60 | 539.65 |
| Met409 (42) | Lowered liver fat content, liver fibrosis, and HDL in NASH models | Improved Col1a1 and Liver galectin-3 levels Decreased ALT | 0.016 | - | 7.0 | 16.1 | 541.732 |
| BMS-986318 (74) | Lower liver fibrosis | Increased FGF15/19, IBABP | 0.034 | - | 6.55 | 11.23 | 643.72 |
| LH10 (82) | Improved steatosis, ballooning, and inflammatory in NASH models | Improved ALT, AST, ALP, LDH, and TBA Decreased TBIL | 0.14 | - | 7.7 | 2.3 (basic) | 515.657 |
| ZLY28 (94) | Reduced liver body weight ratio Lowered hepatic lipid levels Improved hepatic steatosis, lobular inflammation, and ballooning | SHP, FGF15, and OSTβ | 0.143 | (FABP1) 2.7 | 7.4 | 3.4 | 520.41 |
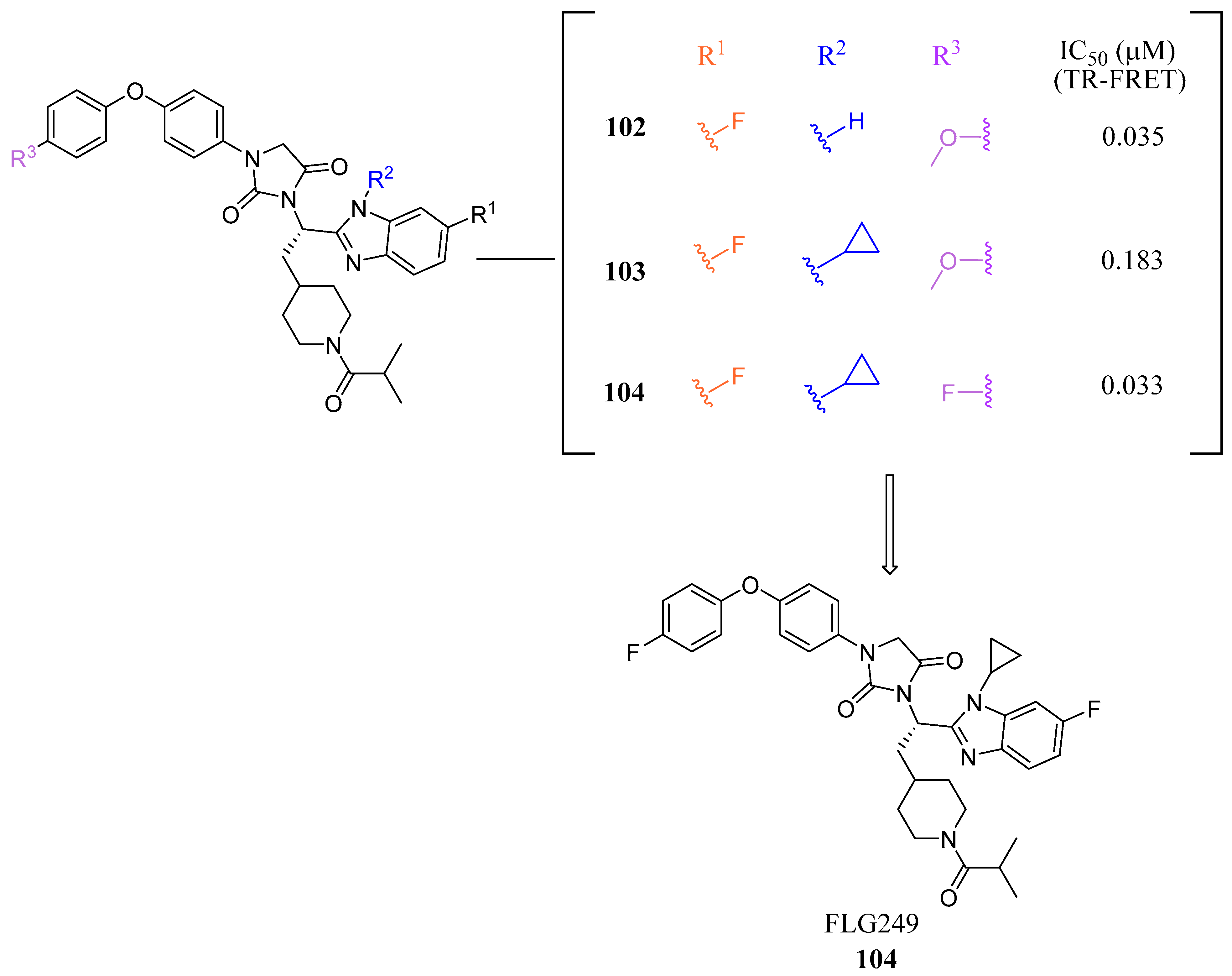
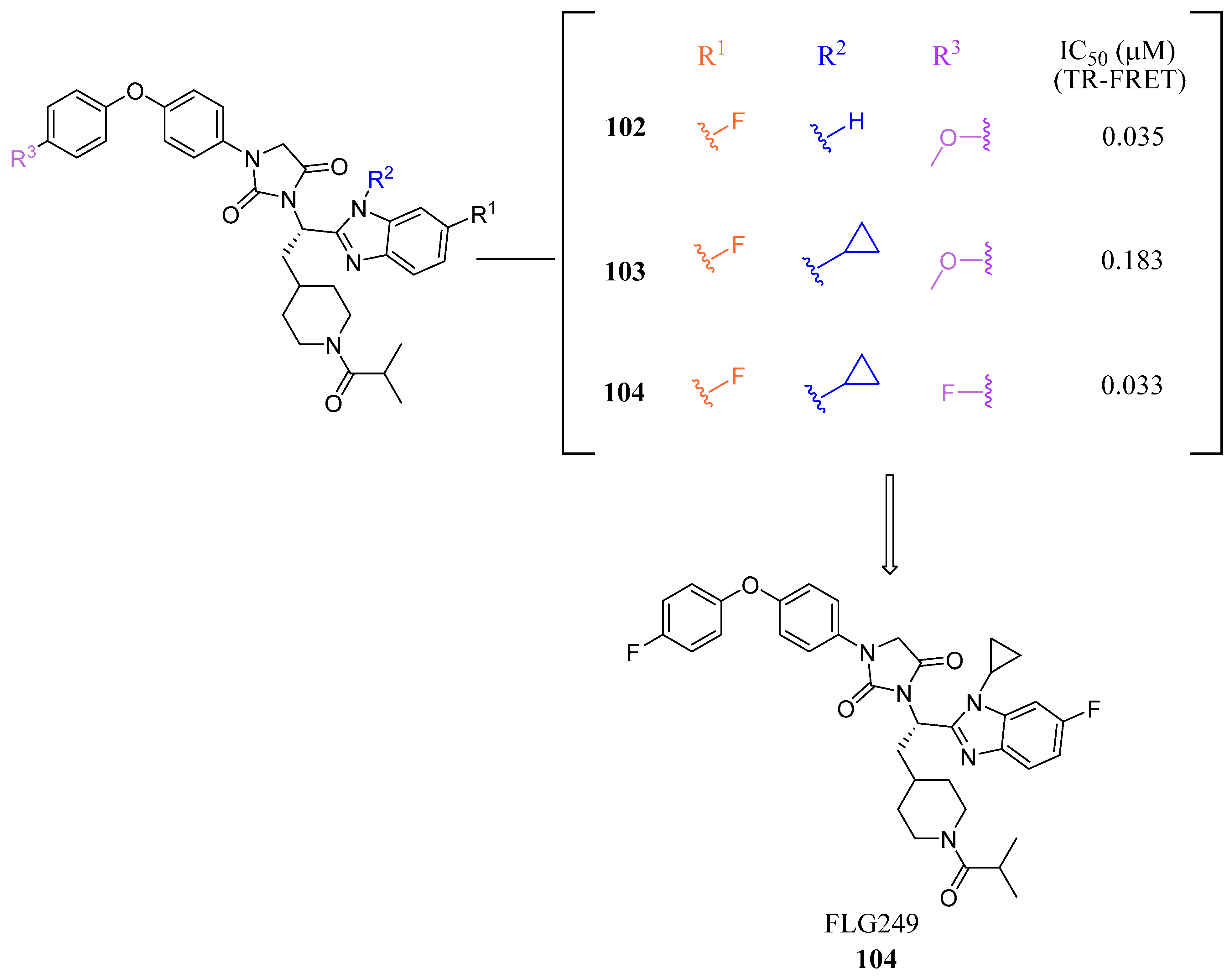
| FLG249 (104) | N/A | Decreased FGF15 and SHP Increased ASBT | N/A | 0.033 | 5.75 | 12.33 | 641.72 |
| Comp. | SASA | HBA | HBD | QPlogPo/w | QPlogS | Human Oral Absorption | % Human Oral Absorption | Rule of 5 Violations |
|---|---|---|---|---|---|---|---|---|
| GUDCA (5) | 746.058 | 9 | 4 | 2.392 | −4.085 | 2 | 62.258 | 0 |
| Gly-MCA (6) | 749.774 | 7 | 3 | 3.192 | −4.884 | 2 | 56.576 | 0 |
| F6 (19) | 923.429 | 6 | 1 | 7.246 | −9.174 | 1 | 75.705 | 2 |
| TC-100 (8) | 674.678 | 7 | 4 | 3.593 | −4.480 | 3 | 77.265 | 0 |
| Fex (20) | 925.627 | 6 | 0 | 7.120 | −9.151 | 1 | 100 | 1 |
| Fex-D (23) | 924.731 | 6 | 0 | 7.125 | −9.134 | 1 | 100 | 1 |
| Fex-3 (24) | 1019.959 | 7 | 1 | 7.823 | −10.537 | 1 | 100 | 2 |
| 41 | 909.912 | 7 | 0 | 6.472 | −9.109 | 1 | 89.651 | 2 |
| Met409 (42) | 974.504 | 6 | 1 | 7.304 | −10.380 | 1 | 100 | 2 |
| BMS-986318 (74) | 1001.255 | 7 | 2 | 7.966 | −10.923 | 1 | 95.677 | 2 |
| LH10 (82) | 896.199 | 5 | 0 | 7.97 | −9.768 | 1 | 100 | 2 |
| ZLY28 (94) | 825.370 | 4 | 1 | 7.474 | −9.362 | 1 | 83.381 | 2 |
| FLG249 | 1003.014 | 8 | 0 | 14.075 | −9.226 | 1 | 100 | 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morrison, A.; Elgendy, B. Tailoring FXR Modulators for Intestinal Specificity: Recent Progress and Insights. Molecules 2024, 29, 2022. https://doi.org/10.3390/molecules29092022
Morrison A, Elgendy B. Tailoring FXR Modulators for Intestinal Specificity: Recent Progress and Insights. Molecules. 2024; 29(9):2022. https://doi.org/10.3390/molecules29092022
Chicago/Turabian StyleMorrison, Amanda, and Bahaa Elgendy. 2024. "Tailoring FXR Modulators for Intestinal Specificity: Recent Progress and Insights" Molecules 29, no. 9: 2022. https://doi.org/10.3390/molecules29092022
APA StyleMorrison, A., & Elgendy, B. (2024). Tailoring FXR Modulators for Intestinal Specificity: Recent Progress and Insights. Molecules, 29(9), 2022. https://doi.org/10.3390/molecules29092022