
Synthesis of ω-Chloroalkyl Aryl Ketones via C–C Bond Cleavage of tert-Cycloalkanols with Tetramethylammonium Hypochlorite


Abstract
1. Introduction
2. Results and Discussion
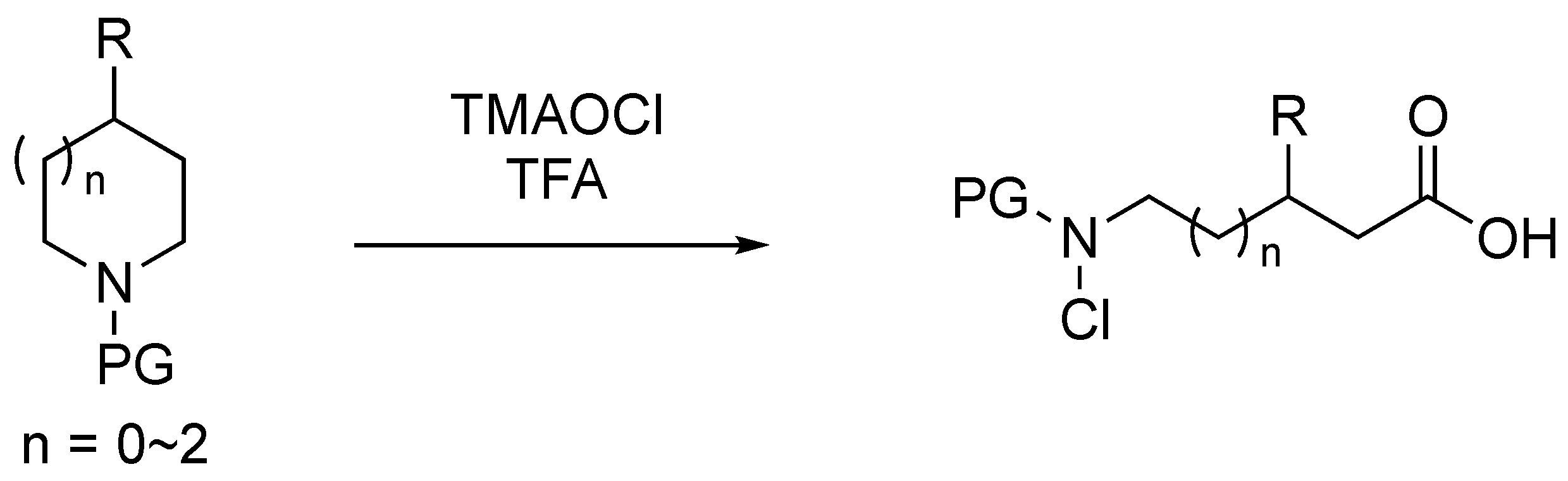
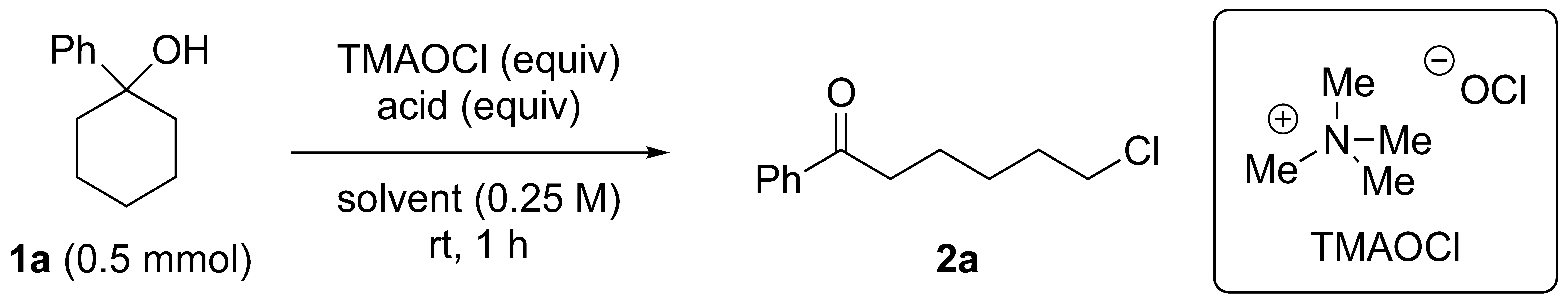
2.1. Optimization of Reaction Conditions
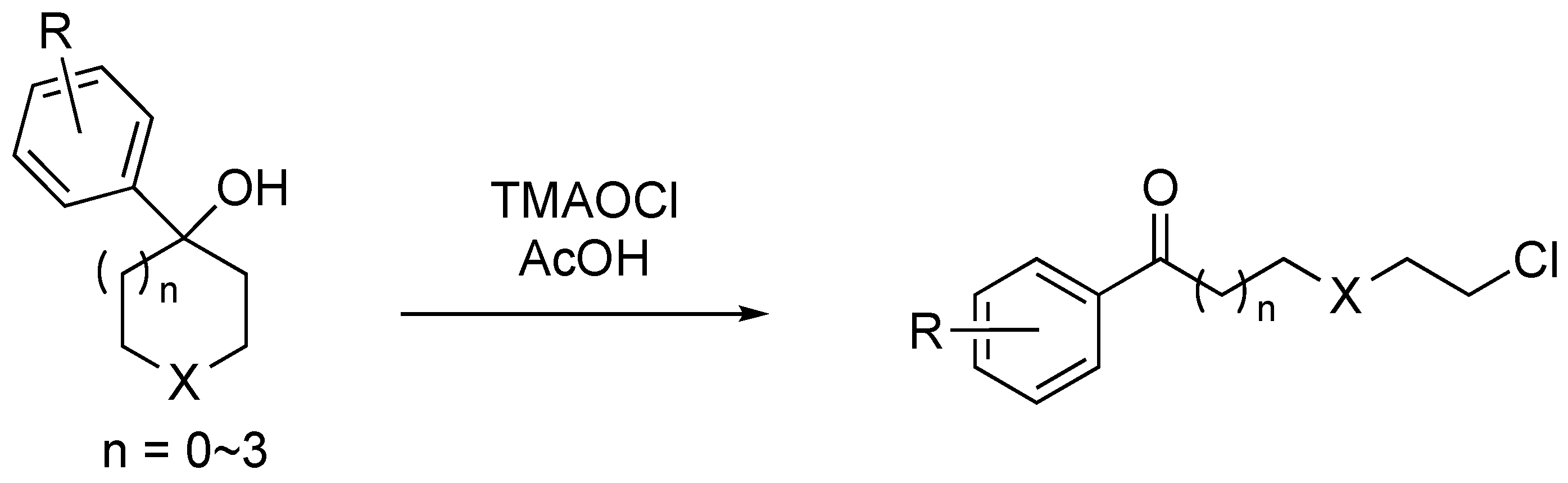
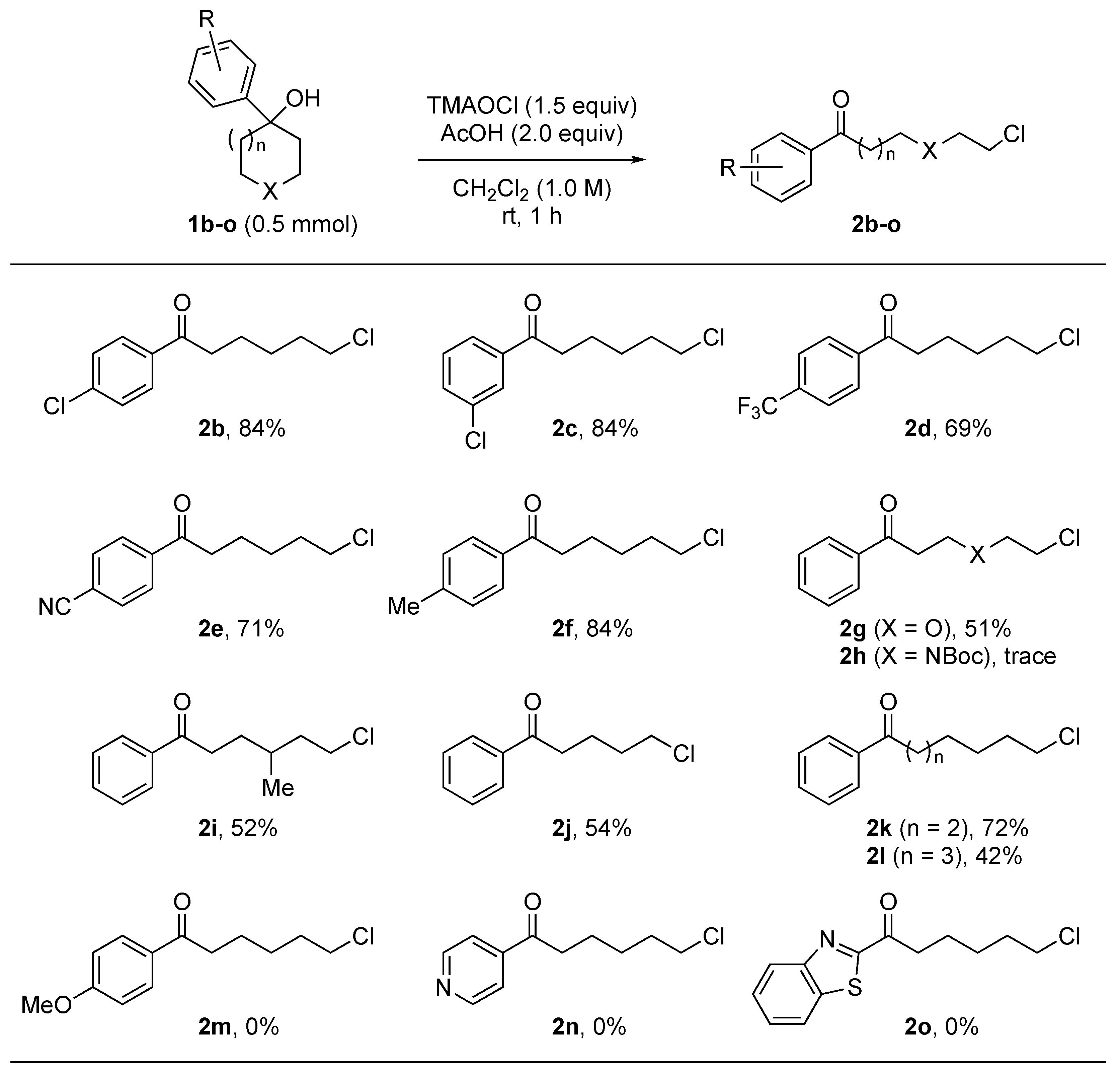
2.2. Substrate Scope
2.3. Scale-Up Experiment
2.4. Control Experiment
3. Materials and Methods
3.1. Chemicals and Instruments
3.2. General Procedure for the Oxidative C–C Bond Cleavage of tert-Cycloalkanol
3.3. Procedure for the Scale-Up Experiment
3.4. Preparation of Aqueous TMAOCl Solution
3.5. Characterization Data
- Chloro-1-phenylhexan-1-one (2a) [26]: Silica-gel column chromatography (hexane/AcOEt = 97:3) gave 89 mg of 2a (0.42 mmol, 83%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.97−7.95 (m, 2H), 7.58−7.55 (m, 1H), 7.48−7.45 (m, 2H), 3.56 (t, J = 6.6 Hz, 2H), 3.00 (t, J = 7.4 Hz, 2H), 1.86−1.76 (m, 4H), 1.56−1.52 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 200.0, 136.9, 133.0, 128.6, 128.0, 44.9, 38.3, 32.4, 26.6, 23.4; LRMS (ESI) m/z: 211 [M + H]+.
- 6-Chloro-1-(4-chlorophenyl)hexan-1-one (2b) [22]: Silica-gel column chromatography (hexane/AcOEt = 97:3) gave 103 mg of 2b (0.42 mmol, 84%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.91–7.88 (m, 2H), 7.46–7.42 (m, 2H), 3.56 (t, J = 6.6 Hz, 2H), 2.97 (t, J = 6.8 Hz, 2H), 1.85–1.75 (m, 4H), 1.56–1.53 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 198.7, 139.4, 135.2, 129.4, 128.9, 44.8, 38.2, 32.4, 26.5, 23.3; LRMS (ESI) m/z: 245 [M + H]+.
- 6-Chloro-1-(3-chlorophenyl)hexan-1-one (2c): Silica-gel column chromatography (hexane/AcOEt = 97:3) gave 103 mg of 2c (0.42 mmol, 84%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.92 (s, 1H), 7.83 (d, J = 7.6 Hz, 1H), 7.55–7.51 (m, 1H), 7.43–7.39 (m, 1H), 3.56 (t, J = 6.6 Hz, 2H), 2.97 (t, J = 7.4 Hz, 2H), 1.86–1.75 (m, 4H), 1.56–1.52 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 198.5, 139.8, 132.5, 128.4, 117.9, 116.3, 44.7, 38.6, 32.3, 26.4, 23.1; IR (ATR): 2939, 2865, 1686, 1570, 1420, 1206 cm−1; HRMS (ESI) m/z: [M + H]+ calculated for C12H15Cl2O 246.1529, found 246.1530.
- 6-Chloro-1-(4-trifluoromethylphenyl)hexan-1-one (2d): Silica-gel column chromatography (hexane/AcOEt = 97:3) gave 96 mg of 2d (0.35 mmol, 69%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.06 (d, J = 8.8 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 3.57 (t, J = 6.4 Hz, 2H), 3.03 (t, J = 7.2 Hz, 2H), 1.86–1.77 (m, 4H), 1.57–1.53 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 198.9, 139.5, 134.3 (q, J = 32.4 Hz), 128.3, 125.7, 123.6 (q, J = 270.7 Hz), 44.8, 38.6, 32.4, 26.4, 23.1; 19F NMR (376 MHz, CDCl3): δ −66.3; IR (ATR): 2941, 2868, 1690, 1409, 1322, 1125, 1065 cm−1; HRMS (ESI) m/z: [M + H]+ calculated for C13H15ClF3O 279.7058, found 279.7057.
- 4-(5-Chloropentanoyl)benzonitrile (2e): Silica-gel column chromatography (hexane/AcOEt = 97:3) gave 84 mg of 2e (0.36 mmol, 71%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 8.04 (d, J = 8.8 Hz, 2H), 7.78 (d, J = 8.4 Hz, 2H), 3.56 (t, J = 6.4 Hz, 2H), 3.01 (t, J = 7.2 Hz, 2H), 1.86–1.77 (m, 4H), 1.57–1.55 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 198.5, 139.8, 132.5, 128.4, 117.9, 116.3, 44.7, 38.6, 32.3, 26.4, 23.1; IR (ATR): 2947, 2866, 2229, 1695, 1402, 1272, 1191 cm−1; HRMS (ESI) m/z: [M + H]+ calculated for C13H15ClNO 236.7173, found 236.7172.
- 6-Chloro-1-(4-methylphenyl)hexan-1-one (2f) [23]: Silica-gel column chromatography (hexane/AcOEt = 97:3) gave 94 mg of 2f (0.42 mmol, 84%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.87–7.84 (m, 2H), 7.27–7.25 (m, 2H), 3.55 (t, J = 6.8 Hz, 2H), 2.97 (t, J = 7.4 Hz, 2H), 2.41 (s, 3H), 1.85–1.77 (m, 4H), 1.56–1.53 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 199.7, 143.7, 134.5, 129.2, 128.1, 44.9, 38.2, 32.5, 26.6, 23.5, 21.6; LRMS (ESI) m/z: 225 [M + H]+.
- 3-(2-Chloroethoxy)-1-phenyl-1-propanone (2g): Silica-gel column chromatography (hexane/AcOEt = 97:3) gave 54 mg of 2g (0.26 mmol, 51%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.98–7.95 (m, 2H), 7.57–7.49 (m, 1H), 7.49–7.45 (m, 2H), 3.97–3.94 (m, 2H), 3.76–3.74 (m, 2H), 3.63–3.60 (m, 2H), 3.31–3.27 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 198.0, 136.8, 133.2, 128.6, 128.0, 71.2, 66.3, 42.7, 38.6; IR (ATR): 2961, 2873, 1661, 1596, 1446, 1213, 1116 cm−1; HRMS (ESI) m/z: [M + H]+ calculated for C11H14ClO2 213.6807, found 213.6807.
- 6-Chloro-4-methyl-1-phenyl-1-hexanone (2i) [23]: Silica-gel column chromatography (hexane/toluene = 4/6) gave 58 mg of 2i (0.26 mmol, 52%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.98–7.95 (m, 2H), 7.57–7.55 (m, 1H), 7.49–7.45 (m, 2H), 3.63–3.55 (m, 2H), 3.03–2.97 (m, 2H), 1.84–1.76 (m, 3H), 1.67–1.58 (m, 2H), 0.97 (d, J = 6.8 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 200.2, 136.9, 133.0, 128.6, 128.0, 43.0, 39.5, 36.0, 30.7, 30.1, 18.9; LRMS (ESI) m/z: 225 [M + H]+.
- 5-Chloro-1-phenylpentan-1-one (2j) [26]: Silica-gel column chromatography (hexane/AcOEt = 97:3) gave 53 mg of 2j (0.27 mmol, 54%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.97–7.95 (m, 2H), 7.57–7.56 (m, 1H), 7.49–7.45 (m, 2H), 3.59 (t, J = 6.4 Hz, 2H), 3.02 (t, J = 6.8 Hz, 2H), 1.91–1.89 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3): δ 199.6, 136.8, 133.1, 128.6, 128.0, 44.7, 37.5, 32.0, 21.5; LRMS (ESI) m/z: 197 [M + H]+.
- 7-Chloro-1-phenylheptan-1-one (2k) [26]: Silica-gel column chromatography (hexane/AcOEt = 97:3) gave 81 mg of 2k (0.36 mmol, 72%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.97–7.95 (m, 2H), 7.58–7.54 (m, 1H), 7.48–7.44 (m, 2H), 3.54 (t, J = 6.8 Hz, 2H), 2.98 (t, J = 7.2 Hz, 2H), 1.82–1.75 (m, 4H), 1.50–1.42 (m, 4H); 13C{1H} NMR (100 MHz, CDCl3): δ 200.3, 137.0, 132.9, 128.6, 128.0, 45.0, 38.4, 32.4, 28.5, 26.7, 24.0; LRMS (ESI) m/z: 225 [M + H]+.
- 8-Chloro-1-phenyloctan-1-one (2l) [26]: Silica-gel column chromatography (hexane/AcOEt = 97:3) gave 50 mg of 2l (0.36 mmol, 42%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.98–7.95 (m, 2H), 7.59–7.52 (m, 1H), 7.49–7.44 (m, 2H), 3.53 (t, J = 6.8 Hz, 2H), 2.97 (t, J = 7.2 Hz, 2H), 1.80–1.73 (m, 8H), 0.93–0.82 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 200.4, 137.0, 132.9, 128.5, 128.0, 45.1, 38.5, 32.5, 29.1, 28.7, 26.7, 24.2; LRMS (ESI) m/z: 239 [M + H]+.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Wijngaarden, I.; Kruse, C.G.; van der Heyden, J.A.M.; Tulp, M.T.M. 2-Phenylpyrroles as conformationally restricted benzamide analogs. A new class of potential antipsychotics. 2. J. Med. Chem. 1988, 31, 1934–1940. [Google Scholar] [CrossRef]
- Perrone, R.; Berardi, F.; Colabufo, N.A.; Lacivita, E.; Leopoldo, M.; Tortorella, V. Synthesis and structure-affinity relationships of 1-[ω-(4-Aryl-1-piperazinyl)alkyl]-1-aryl ketones as 5-HT7 receptor ligands. J. Med. Chem. 2003, 46, 646–649. [Google Scholar] [CrossRef]
- Chen, C.-A.; Jiang, Y.; Lu, K.; Daniewska, I.; Mazza, C.G.; Negron, L.; Forray, C.; Parola, T.; Li, B.; Hegde, L.G.; et al. Synthesis and SAR investigations for novel melanin-concentrating hormone 1 receptor (MCH1) antagonists part 2: A hybrid strategy combining key fragments of HTS hits. J. Med. Chem. 2007, 50, 3883–3890. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yan, Y.-L.; Lui, B. Mild α-halogenation reactions of 1,3-dicarbonyl compounds catalyzed by Lewis acids. J. Org. Chem. 2002, 67, 7429–7431. [Google Scholar] [CrossRef]
- Frantz, R.; Hintermann, L.; Perseghini, M.; Broggini, D.; Togni, A. Titanium-catalyzed stereoselective geminal heterodihalogenation of β-ketoesters. Org. Lett. 2003, 5, 1709–1712. [Google Scholar] [CrossRef]
- Marigo, M.; Bachmann, S.; Halland, N.; Braunton, A.; Jørgensen, K.A. Highly enantioselective direct organocatalytic α-chlorination of ketones. Angew. Chem. Int. Ed. 2004, 43, 5507–5510. [Google Scholar] [CrossRef] [PubMed]
- Shibatomi, K.; Soga, Y.; Narayama, A.; Fujisawa, I.; Iwasa, S. Highly enantioselective chlorination of β-ketoesters and subsequent SN2 displacement of tertiary chlorides: A flexible method for the construction of quaternary stereogenic centers. J. Am. Chem. Soc. 2012, 134, 9836–9839. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Hasegawa, M.; Tomisawa, K.; Mitsukuchi, M. Synthesis and Structure–Activity Relationships of 5-Phenylthiophenecarboxylic Acid Derivatives as Antirheumatic Agents. Bioorg. Med. Chem. 2003, 11, 4729–4742. [Google Scholar] [CrossRef] [PubMed]
- Close, W.J. An Improved Synthesis of Cyclopropyl Phenyl Ketone and Related Substances. J. Am. Chem. Soc. 1957, 79, 1455–1458. [Google Scholar] [CrossRef]
- Lukin, K.; Hsu, M.C.; Chambournier, G.; Kotecki, B.; Venkatramani, C.J.; Leanna, M.R. Development of a Large Scale Asymmetric Synthesis of Vanilloid Receptor (TRPV1) Antagonist ABT-102. Org. Process Res. Dev. 2007, 11, 578–584. [Google Scholar] [CrossRef]
- Majetich, G.; Wheless, K. Remote intramolecular free radical functionalizations: An update. Tetrahedron 1995, 51, 7095–7129. [Google Scholar] [CrossRef]
- Čeković, Ź. Reactions of δ-carbon radicals generated by 1,5-hydrogen transfer to alkoxy radicals. Tetrahedron 2003, 59, 8073–8090. [Google Scholar] [CrossRef]
- Chiba, S.; Chen, H. sp3 C−H oxidation by remote H-radical shift with oxygen- and nitrogen-radicals: A recent update. Org. Biomol. Chem. 2014, 12, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ishida, N. β-Scission of Alkoxy Radicals in Synthetic Transformations. Chem. Lett. 2017, 46, 1692–1700. [Google Scholar] [CrossRef]
- Kapustina, N.I.; Sokova, L.L.; Nikishin, G.I. Electrochemical oxidative ring opening of 1-methylcyclobutanol. Russ. Chem. Bull. 1996, 45, 1246–1248. [Google Scholar] [CrossRef]
- Allen, B.D.W.; Hareram, M.D.; Seastram, A.C.; McBride, T.; Wirth, T.; Browne, D.L.; Morrill, L.C. Manganese-Catalyzed Electrochemical Deconstructive Chlorination of Cycloalkanols via Alkoxy Radicals. Org. Lett. 2019, 21, 9241–9246. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yang, D.; Zhang, J.; Tan, C.; Li, J.; Wang, S.; Zhang, H.; Huang, Z.; Lei, A. Electrophotochemical Ce-Catalyzed Ring-Opening Functionalization of Cycloalkanols under Redox-Neutral Conditions: Scope and Mechanism. J. Am. Chem. Soc. 2022, 144, 13895–13902. [Google Scholar] [CrossRef] [PubMed]
- Huan, L.; Zhu, C. Manganese-catalyzed ring-opening chlorination of cyclobutanols: Regiospecific synthesis of γ-chloroketones. Org. Chem. Front. 2016, 3, 1467–1471. [Google Scholar] [CrossRef]
- Kapustina, B.I.; Spektor, S.S.; Nikishin, G.I. Oxidative cleavage of tertiary cycloalkanols by the lead tetraacetate-metal halide system. Russ. Chem. Bull. 1983, 32, 1394–1398. [Google Scholar] [CrossRef]
- Yamamoto, K.; Toguchi, H.; Kuriyama, M.; Watanabe, S.; Iwasaki, F.; Onomura, O. Electrophotochemical Ring-Opening Bromination of tert-Cycloalkanols. J. Org. Chem. 2021, 86, 16177–16186. [Google Scholar] [CrossRef] [PubMed]
- Han, W.-J.; Zhan, J.-L.; Yang, F.-L.; Liu, L. Ring-Opening Functionalization/Cyclization Reactions of Cycloalkanols under Transition-Metal-Free Conditions. Eur. J. Org. Chem. 2024, 27, e202301215. [Google Scholar] [CrossRef]
- Huang, F.-Q.; Xie, J.; Sun, J.-G.; Wang, Y.-W.; Dong, X.; Qi, L.-W.; Zhang, B. Regioselective Synthesis of Carbonyl-Containing Alkyl Chlorides via Silver-Catalyzed Ring-Opening Chlorination of Cycloalkanols. Org. Lett. 2016, 18, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhao, H.; Yu, J.; Bao, X.; Zhu, C. Regiospecific synthesis of distally chlorinated ketones via C−C bond cleavage of cycloalkanols. Org. Chem. Front. 2016, 3, 227–232. [Google Scholar] [CrossRef]
- Yayla, H.G.; Wang, H.; Tarantino, K.T.; Orbe, H.S.; Knowles, R.R. Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O–H Bonds. J. Am. Chem. Soc. 2016, 138, 10794–10797. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Ma, S. Halogen-Substituted Allenyl Ketones through Ring Opening of Nonstrained Cycloalkanols. Org. Lett. 2021, 23, 2533–2537. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Bai, M.; Xu, P.-F.; Sun, Q.-X.; Duan, X.-H.; Guo, L.-N. Copper-catalyzed radical ring-opening halogenation with HX. Chem. Commun. 2021, 57, 8652–8655. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Ma, Y.; Huang, R.; Li, X.; Yu, S. N-Haloimide-enabled halogenation via halogen-bond-assisted C–C activation of alkanols. Green Chem. 2023, 25, 221–228. [Google Scholar] [CrossRef]
- Wilt, J.W.; Hill, J.W. On the Rearrangement of t-Cycloalkyl Hypochlorites. J. Org. Chem. 1961, 26, 3523–3525. [Google Scholar] [CrossRef]
- Stevens, R.V.; Chapman, K.T.; Stubbs, C.A.; Tam, W.W.; Albizati, K.F. Further Studies on the Utility of Sodium Hypochlorite in Organic Synthesis. Selective Oxidation of Diols and Direct Conversion of Aldehydes to Esters. Tetrahedron Lett. 1982, 23, 4647–4650. [Google Scholar] [CrossRef]
- McDonald, C.E.; Nice, L.E.; Shaw, A.W.; Nestor, N.B. Calcium hypochlorite-mediated oxidation of primary alcohols to methyl esters. Tetrahedron Lett. 1993, 34, 2741–2744. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Cabaj, J.E.; Havens, J.L. Use of a Δ1-Piperidine for the Synthesis of a Differentially Protected Diamine Intermediate for the Preparation of the Iron Siderophore Desferrioxamine. J. Org. Chem. 1994, 59, 6470–6471. [Google Scholar] [CrossRef]
- Kirihara, M.; Okada, T.; Sugiyama, Y.; Akiyoshi, M.; Matsunaga, T.; Kimura, Y. Sodium Hypochlorite Pentahydrate Crystals (NaOCl·5H2O): Convenient and Environmentally Benign Oxidant for Organic Synthesis. Org. Process Res. Dev. 2017, 21, 1925–1937. [Google Scholar] [CrossRef]
- Lee, G.A.; Freedman, H.H. Phase transfer catalyzed oxidations of alcohols and amines by aqueous hypochlorite. Tetrahedron Lett. 1976, 17, 1641–1644. [Google Scholar] [CrossRef]
- Ishii, F.; Kishi, K.-i. Oxidation of Hydroquinone and Catechols with Aqueous Sodium Hypochlorite under Phase-Transfer Catalysis. Synthesis 1980, 1980, 706–708. [Google Scholar] [CrossRef]
- Shimoda, A.; Kikkawa, Y.; Sato, T.; Negishi, T. Quaternary Alkylammonium Hypochlorite Solution, Manufacturing Method Thereof, and Semiconductor Wafer Processing Method. JP Patent JP 2021-90051 A, 10 June 2021. [Google Scholar]
- Murata, A.; Umezu, N.; Tono, S.; Taira, H. Process for Producing Tetraalkylammonium Salt, and Process for Producing Tetraalkylammonium Hydroxide Using Same as Raw Material. PCT Int. Appl. WO 2012/090699 A1, 5 July 2012. [Google Scholar]
- Kaieda, Y.; Yamamoto, K.; Toguchi, H.; Hanazawa, N.; Kuriyama, M.; Onomura, O. Oxidative C–N Bond Cleavage of Cyclic Amines with Ammonium Hypochlorite. Synthesis 2023, eFirst. [Google Scholar] [CrossRef]
- Reger, D.L.; Habib, M.M.; Fauth, D.J. Use of phase-transfer reaction conditions for the hydrogenation of conjugated dienes and α,β-unsaturated ketones with a homogeneous metal hydride catalyst. J. Org. Chem. 1980, 45, 3860–3865. [Google Scholar] [CrossRef]
- Sasson, Y.; Negussie, S.; Royz, M.; Mushkin, N. Tetramethylammonium chloride as a selective and robust phase transfer catalyst in a solid–liquid halex reaction: The role of water. Chem. Commun. 1996, 297–298. [Google Scholar] [CrossRef]
- Dailey, J.I.; Hays, R.S.; Lee, H.; Mitchell, R.M.; Ries, J.J.; Landolt, R.G.; Husmann, H.H.; Lockridge, J.B.; Hendrickson, W.H. β-Scission of Tertiary Alkyl Hypochlorites Promoted by Phase-Transfer Catalysis. J. Org. Chem. 2000, 65, 2568–2571. [Google Scholar] [CrossRef] [PubMed]
- The reaction of 1-(2-chlorophenyl)cyclohexanol afforded an inseparable mixture of the desired product (22% NMR yield) and unknown compounds. 2024; Unpublished work.
- Greene, F.D.; Savitz, M.L.; Osterholtz, F.D.; Lau, H.H.; Smith, W.N.; Zanet, P.M. Decomposition of tertiary alkyl hypochlorites. J. Org. Chem. 1963, 28, 55–64. [Google Scholar] [CrossRef]
- Fonouni, H.E.; Krishnan, S.; Kuhn, D.G.; Hamilton, G.A. Mechanisms of epoxidations and chlorinations of hydrocarbons by inorganic hypochlorite in the presence of a phase-transfer catalyst. J. Am. Chem. Soc. 1983, 105, 7672–7676. [Google Scholar] [CrossRef]
- Kurouchi, H.; Andujar-De Sanctis, I.L.; Singleton, D.A. Controlling Selectivity by Controlling Energy Partitioning in a Thermal Reaction in Solution. J. Am. Chem. Soc. 2016, 138, 14534–14537. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.-Y.; Li, L.-G.; Liu, Q.-R.; Pan, C.-X.; Su, G.-F.; Mo, D.-L. Synthesis of β-Acetoxy Alcohols by PhI(OAc)2-Mediated Metal-Free Diastereoselective β-Acetoxylation of Alcohols. Org. Biomol. Chem. 2016, 14, 6795–6803. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.P.; Reddy, K.R.; Reddy, A.P.; Krupadanam, G.L.D.; Mukkera, V.; Sudhakar, N.; Subrahmanyam, L.V.L. Preparation of Novel Substituted Betulinic Amide Derivatives as HIV Inhibitors. PCT Int. Appl. WO 2017017630 Al, 2 February 2017. [Google Scholar]
- Chang, M.-Y.; Chen, Y.-C.; Chan, C.-K. N-Bromosuccinimide-mediated reaction of cyclic styrenes with chloramine-T. Tetrahedron Lett. 2014, 55, 4767–4770. [Google Scholar] [CrossRef]
- Deng, W.; Ye, C.; Li, Y.; Li, D.; Bao, H. Iron-Catalyzed Oxyalkylation of Terminal Alkynes with Alkyl Iodides. Org. Lett. 2019, 21, 261–265. [Google Scholar] [CrossRef] [PubMed]
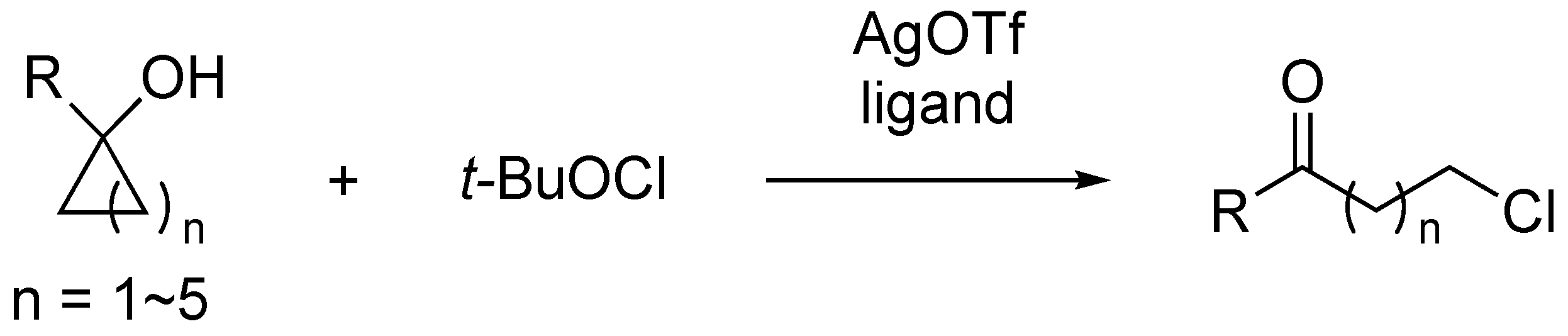
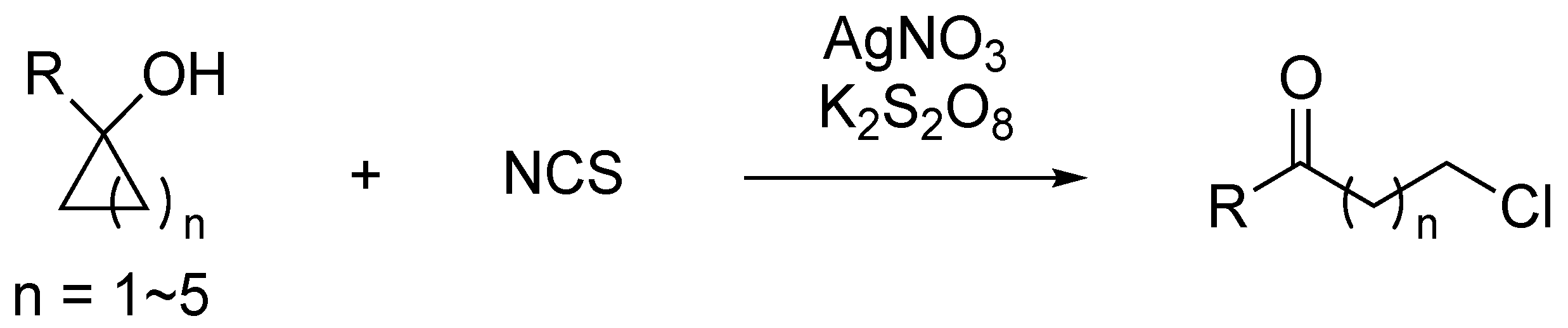
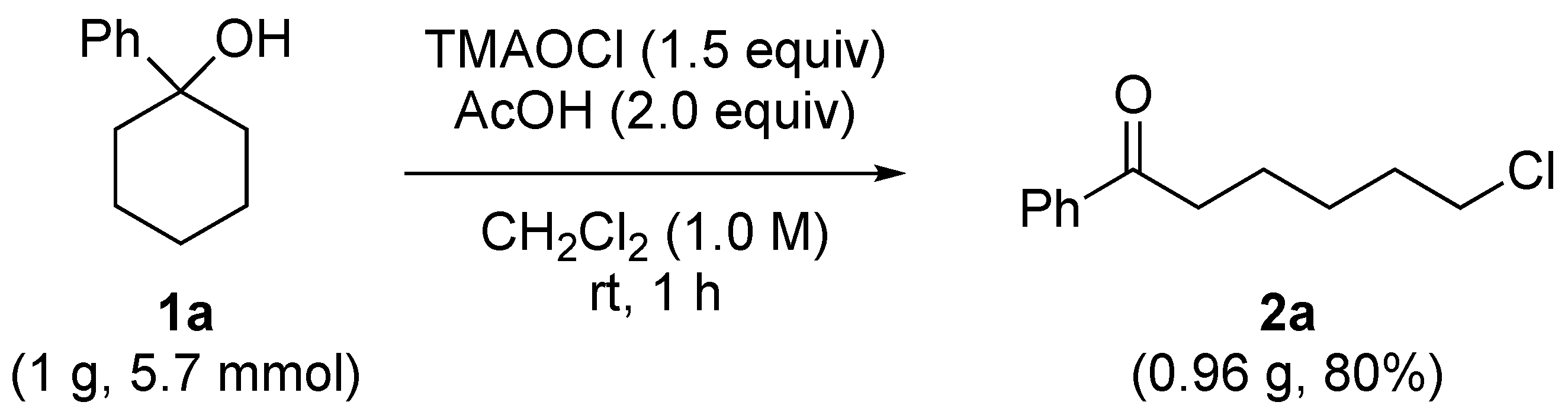

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | TMAOCl (Equiv) | Acid (Equiv) | Solvent | Yield (%) a |
| 1 | 1.5 | 35% HCl (1.5) | CH2Cl2 | 47 |
| 2 | 1.5 | TFA (1.5) | CH2Cl2 | 77 |
| 3 | 1.5 | AcOH (1.5) | CH2Cl2 | 79 |
| 4 | 1.5 | H3PO4 (0.75) | CH2Cl2 | 76 |
| 5 | 1.5 | NaH2PO4 (1.5) | CH2Cl2 | 75 |
| 6 | 1.5 | AcOH (2.0) | CH2Cl2 | 85 |
| 7 | 1.5 | AcOH (2.25) | CH2Cl2 | 83 |
| 8 | 2.0 | AcOH (2.0) | CH2Cl2 | 70 |
| 9 b | 1.5 | AcOH (2.0) | CH2Cl2 | 85 |
| 10 c | 1.5 | AcOH (2.0) | CH2Cl2 | 85 (83) |
| 11 c,d | 1.5 | AcOH (2.0) | CH2Cl2 | 84 |
| 12 c | 1.5 | AcOH (2.0) | ClPh | 70 |
| 13 c | 1.5 | AcOH (2.0) | AcOEt | 67 |
| 14 c | 1.5 | AcOH (2.0) | MeCN | 62 |
| 15 c,e | - | AcOH (2.0) | CH2Cl2 | 56 |
| 16 c,f | - | AcOH (2.0) | CH2Cl2 | 74 |
| 17 c,g | - | AcOH (2.0) | CH2Cl2 | 79 |
| 18 | 1.5 | - | CH2Cl2 | trace |
| 19 h | 1.5 | AcOH (2.0) | CH2Cl2 | 77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanazawa, N.; Kuriyama, M.; Yamamoto, K.; Onomura, O. Synthesis of ω-Chloroalkyl Aryl Ketones via C–C Bond Cleavage of tert-Cycloalkanols with Tetramethylammonium Hypochlorite. Molecules 2024, 29, 1874. https://doi.org/10.3390/molecules29081874
Hanazawa N, Kuriyama M, Yamamoto K, Onomura O. Synthesis of ω-Chloroalkyl Aryl Ketones via C–C Bond Cleavage of tert-Cycloalkanols with Tetramethylammonium Hypochlorite. Molecules. 2024; 29(8):1874. https://doi.org/10.3390/molecules29081874
Chicago/Turabian StyleHanazawa, Natsumi, Masami Kuriyama, Kosuke Yamamoto, and Osamu Onomura. 2024. "Synthesis of ω-Chloroalkyl Aryl Ketones via C–C Bond Cleavage of tert-Cycloalkanols with Tetramethylammonium Hypochlorite" Molecules 29, no. 8: 1874. https://doi.org/10.3390/molecules29081874
APA StyleHanazawa, N., Kuriyama, M., Yamamoto, K., & Onomura, O. (2024). Synthesis of ω-Chloroalkyl Aryl Ketones via C–C Bond Cleavage of tert-Cycloalkanols with Tetramethylammonium Hypochlorite. Molecules, 29(8), 1874. https://doi.org/10.3390/molecules29081874