
The Recognition Pathway of the SARS-CoV-2 Spike Receptor-Binding Domain to Human Angiotensin-Converting Enzyme 2



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
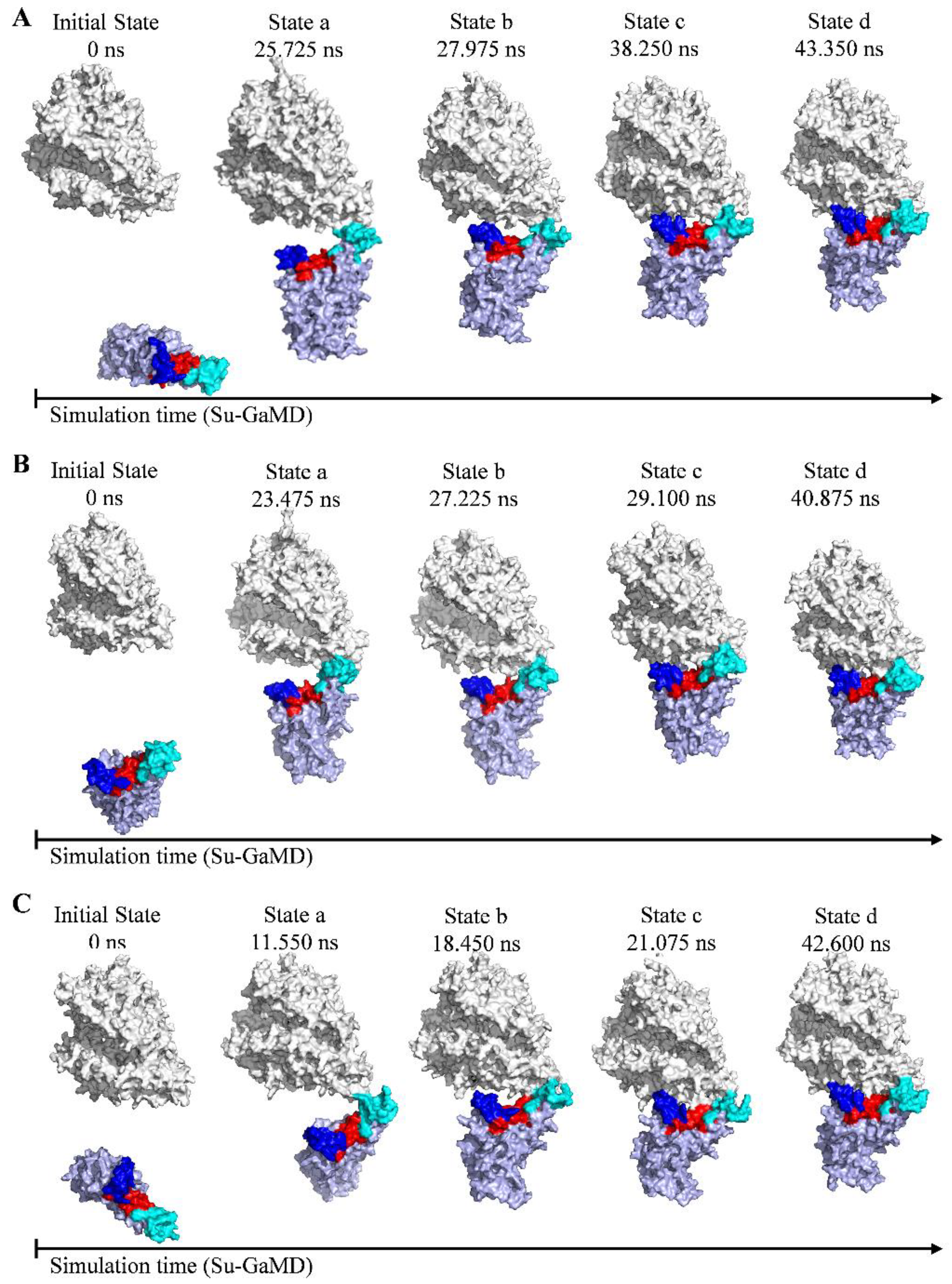
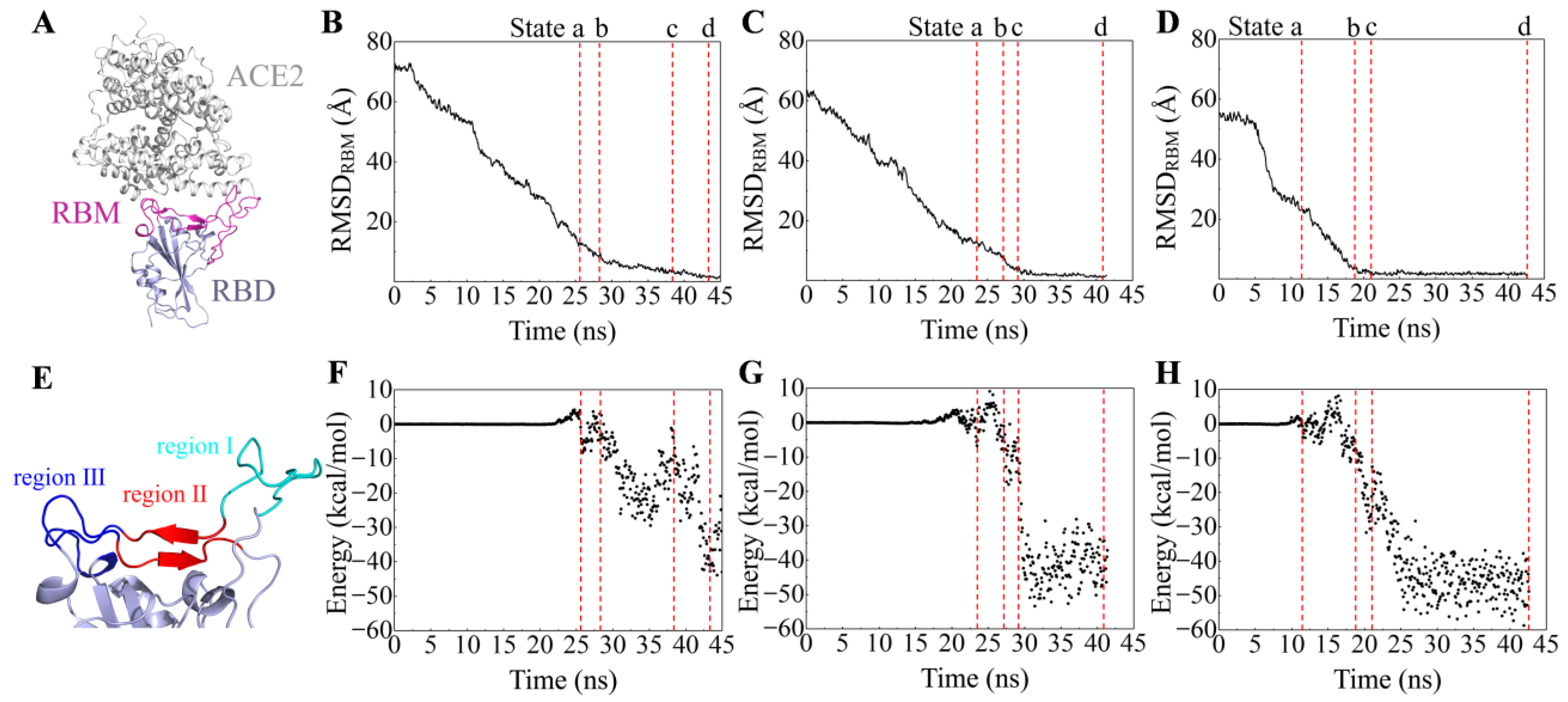
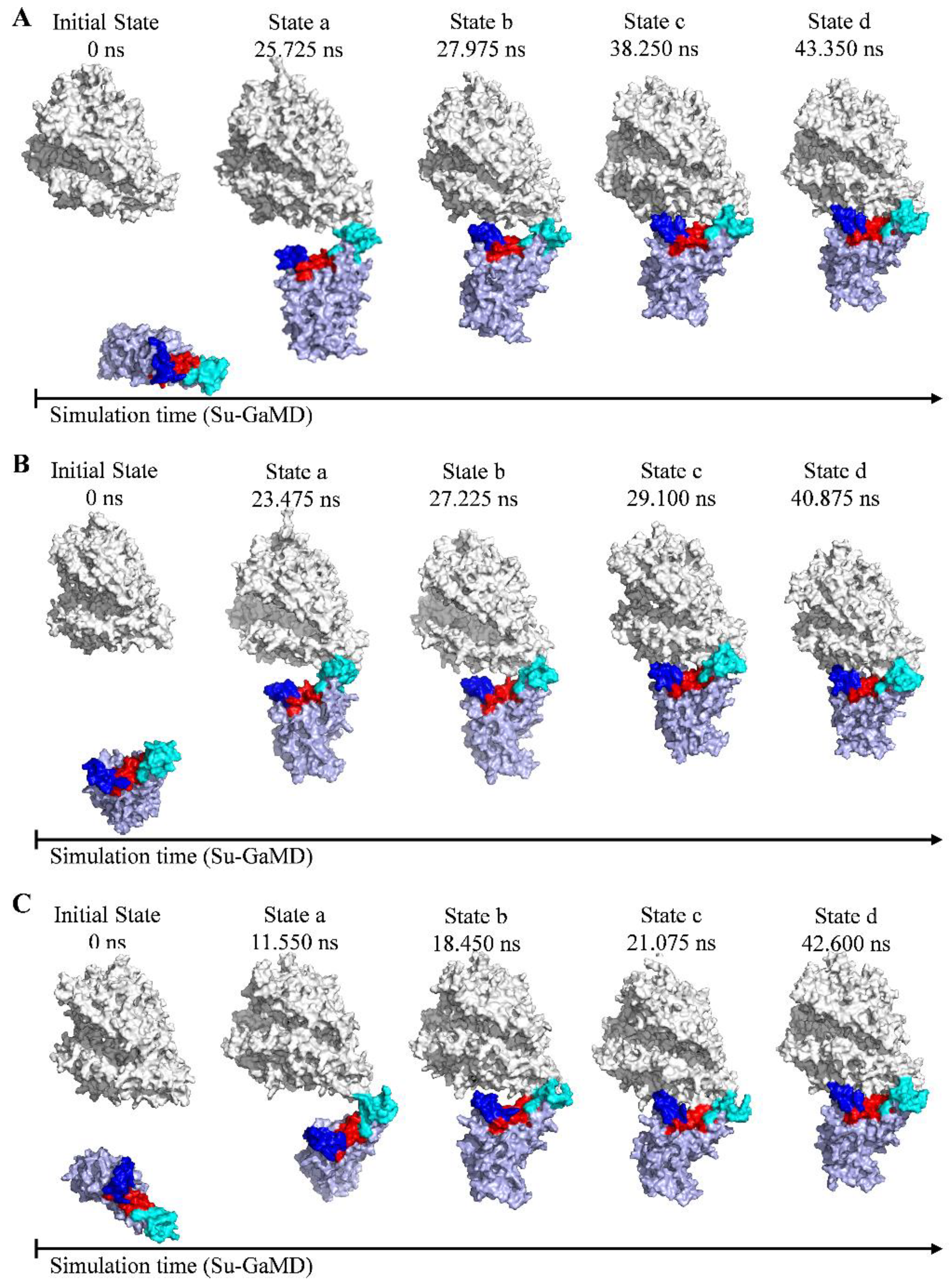
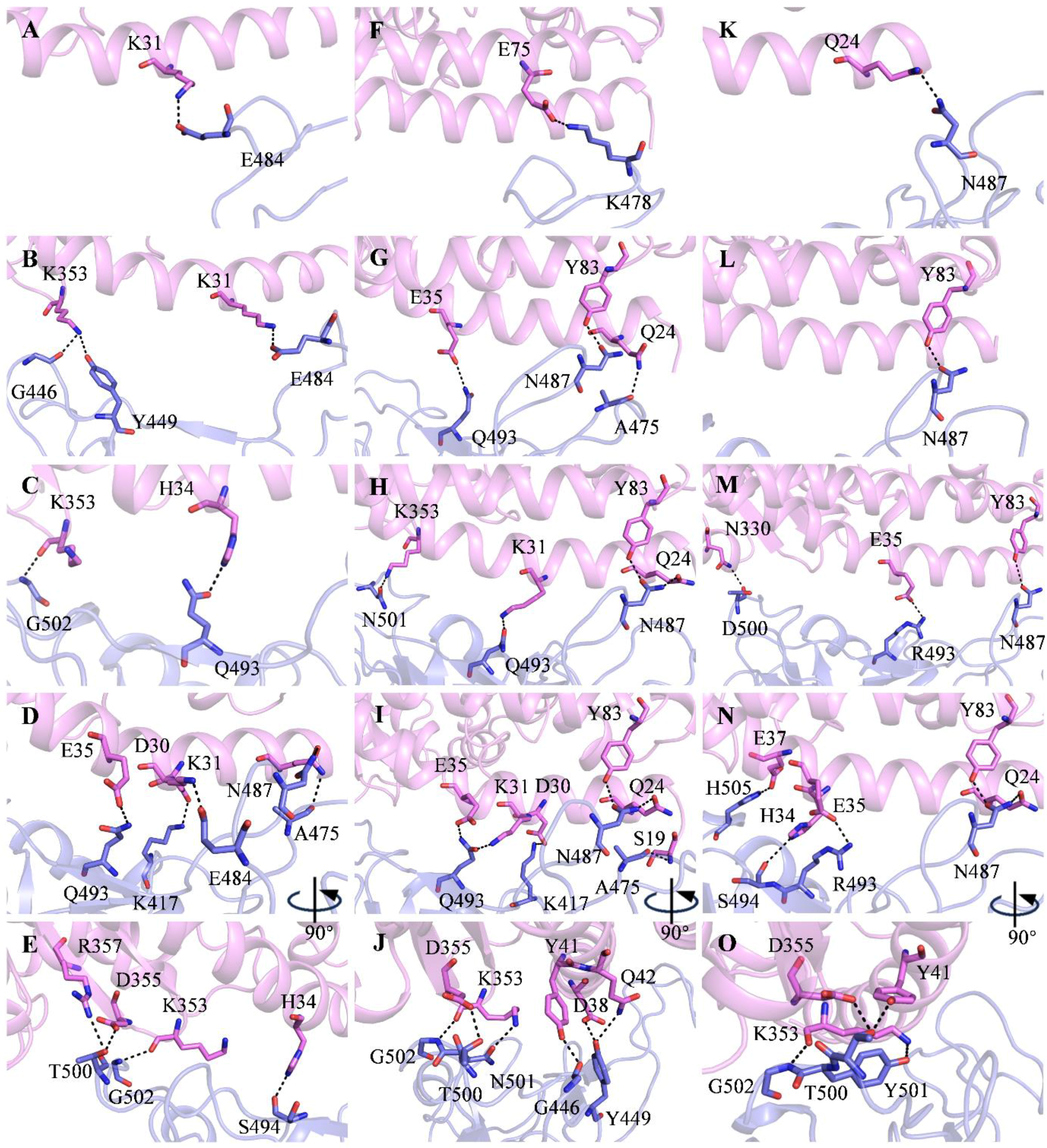
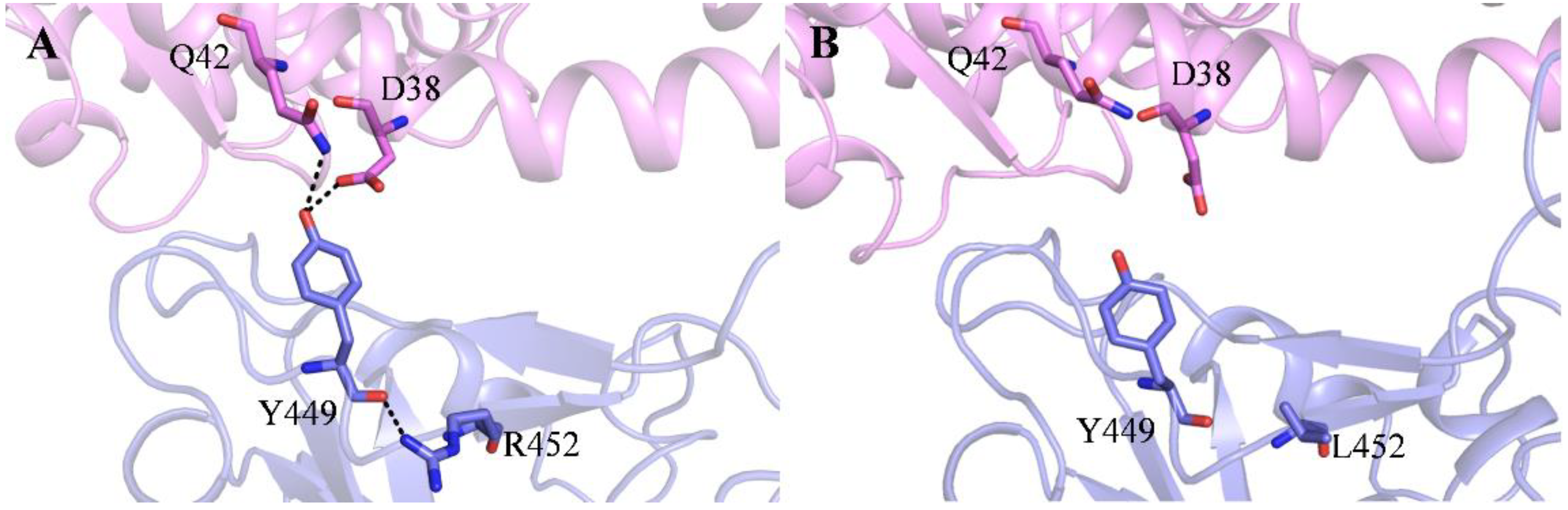
2.1. Recognition Process of Spike RBD of Different Variants to ACE2
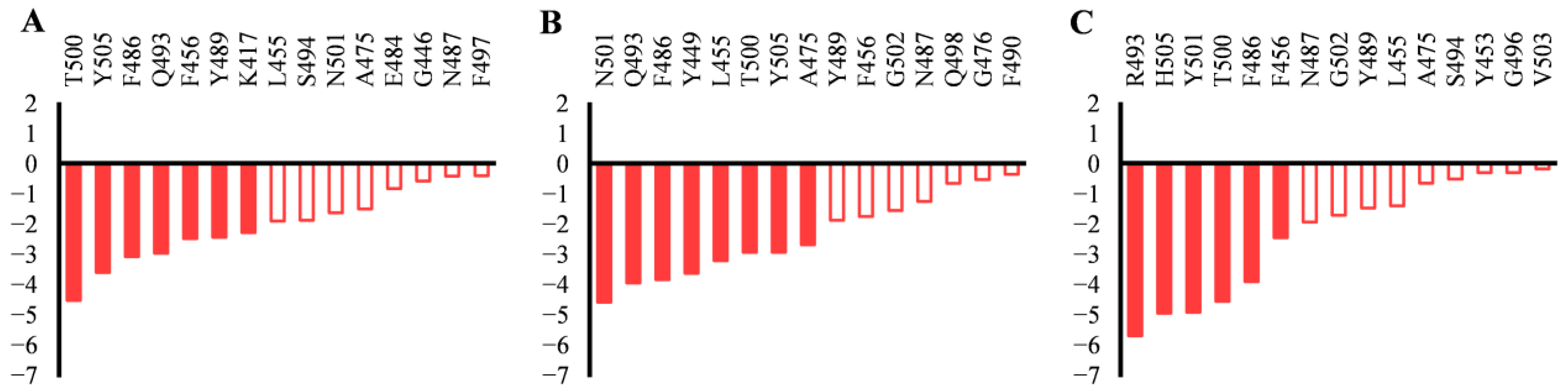
2.2. Effect of the Mutations on Spike RBD to the RBDDelta-ACE2 Recognition
2.3. Effect of the Mutations on Spike RBD to RBDOmicronBA.2-ACE2 Recognition
3. Materials and Methods
3.1. General
3.2. System Setup
3.3. System Equilibration
3.4. Su-GaMD Simulation
3.5. Binding Free Energy Calculations
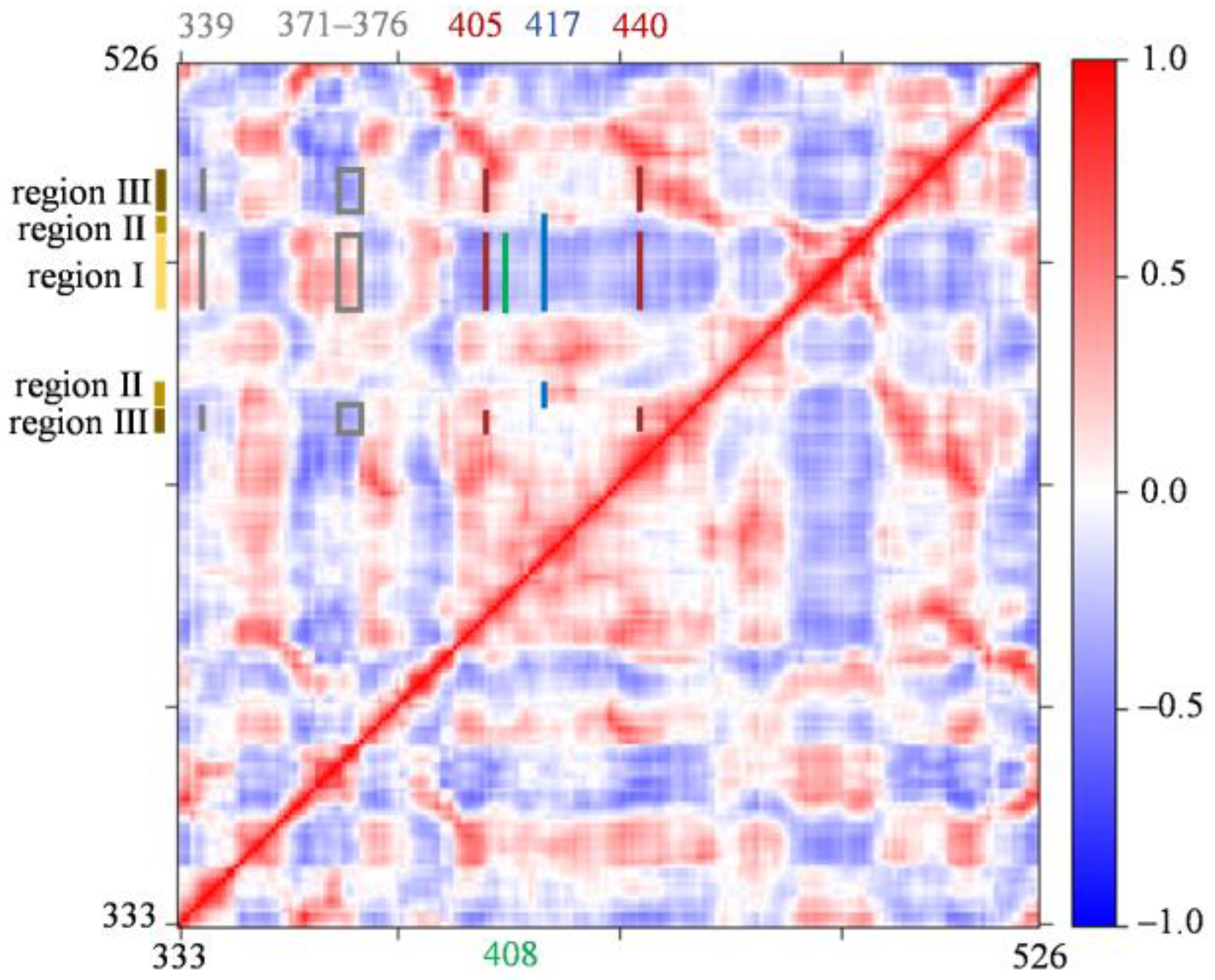
3.6. Dynamic Cross-Correlation Matrix (DCCM) Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Coronavirus (COVID-19) Dashboard; World Health Organization: Geneva, Switzerland, 2020; Available online: https://covid19.who.int/ (accessed on 1 December 2023).
- Kumar, V. Understanding the complexities of SARS-CoV2 infection and its immunology: A road to immune-based therapeutics. Int. Immunopharmacol. 2020, 88, 106980. [Google Scholar] [CrossRef] [PubMed]
- Shiehzadegan, S.; Alaghemand, N.; Fox, M.; Venketaraman, V. Analysis of the Delta variant B.1.617.2 COVID-19. Clin. Pract. 2021, 11, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Roohani, J.; Keikha, M. Global challenge with the SARS-CoV-2 omicron BA.2 (B.1.1.529.2) subvariant: Should we be concerned? World J. Virol. 2022, 11, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Yamasoba, D.; Kimura, I.; Nasser, H.; Morioka, Y.; Nao, N.; Ito, J.; Uriu, K.; Tsuda, M.; Zahradnik, J.; Shirakawa, K.; et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell 2022, 185, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 variant: A new chapter in the COVID-19 pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef] [PubMed]
- Desingu, P.A.; Nagarajan, K.; Dhama, K. Emergence of Omicron third lineage BA.3 and its importance. J. Med. Virol. 2022, 94, 1808–1810. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: What we know about the BA.4 and BA.5 omicron variants. Br. Med. J. 2022, 378, o1969. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.W.; Yu, J.F.; Shan, S.S.; Zhou, H.; Fan, S.L.; Zhang, Q.; Shi, X.L.; Wang, Q.S.; Zhang, L.Q.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.-f.; Xu, W.; Liu, S.-w. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Nutalai, R.; Zhou, D.; Tuekprakhon, A.; Ginn, H.M.; Supasa, P.; Liu, C.; Huo, J.; Mentzer, A.J.; Duyvesteyn, H.M.E.; Dijokaite-Guraliuc, A.; et al. Potent cross-reactive antibodies following Omicron breakthrough in vaccinees. Cell 2022, 185, 2116–2131. [Google Scholar] [CrossRef]
- Xu, Z.; Kang, X.; Han, P.; Du, P.; Li, L.; Zheng, A.; Deng, C.; Qi, J.; Zhao, X.; Wang, Q.; et al. Binding and structural basis of equine ACE2 to RBDs from SARS-CoV, SARS-CoV-2 and related coronaviruses. Nat. Commun. 2022, 13, 3547. [Google Scholar] [CrossRef] [PubMed]
- Magazine, N.; Zhang, T.; Wu, Y.; McGee, M.C.; Veggiani, G.; Huang, W. Mutations and Evolution of the SARS-CoV-2 Spike Protein. Viruses 2022, 14, 640. [Google Scholar] [CrossRef]
- Han, P.; Su, C.; Zhang, Y.; Bai, C.; Zheng, A.; Qiao, C.; Wang, Q.; Niu, S.; Chen, Q.; Zhang, Y.; et al. Molecular insights into receptor binding of recent emerging SARS-CoV-2 variants. Nat. Commun. 2021, 12, 6103. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural basis for SARS-CoV-2 Delta variant recognition of ACE2 receptor and broadly neutralizing antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Kodchakorn, K.; Kongtawelert, P. Molecular dynamics study on the strengthening behavior of Delta and Omicron SARS-CoV-2 spike RBD improved receptor-binding affinity. PLoS ONE 2022, 17, e0277745. [Google Scholar] [CrossRef] [PubMed]
- Pitsillou, E.; Liang, J.J.; Beh, R.C.; Hung, A.; Karagiannis, T.C. Molecular dynamics simulations highlight the altered binding landscape at the spike-ACE2 interface between the Delta and Omicron variants compared to the SARS-CoV-2 original strain. Comput. Biol. Med. 2022, 149, 106035. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Feng, Y.; Ni, H.; Zhi, F.; Miao, Y.; Fang, B.; Zhang, L.; Zhang, J.Z.H. Anchor-locker binding mechanism of the coronavirus spike protein to human ACE2: Insights from computational analysis. J. Chem. Inf. Model. 2021, 61, 3529–3542. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Liu, Y.; Lei, Z.; Dicker, J.; Cao, Y.; Zhang, X.F.; Im, W. Differential interactions between human ACE2 and spike RBD of SARS-CoV-2 variants of concern. J. Theor. Comput. Chem. 2021, 17, 7972–7979. [Google Scholar] [CrossRef] [PubMed]
- Philip, A.M.; Ahmed, W.S.; Biswas, K.H. Reversal of the unique Q493R mutation increases the affinity of Omicron S1-RBD for ACE2. Comput. Struct. Biotechnol. J. 2023, 21, 1966–1977. [Google Scholar] [CrossRef] [PubMed]
- Abeywardhana, S.; Bandaranayake, U.; Perera, D.; Premathilaka, M.; Peiris, D.C. In silico study of SARS-CoV-2 spike protein RBD and human ACE-2 affinity dynamics across variants and Omicron subvariants. J. Med. Virol. 2022, 95, e28406. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kang, Y.; Duan, M.; Hou, T. Regulation mechanism for the binding between the SARS-CoV-2 spike protein and host angiotensin-converting enzyme II. J. Phys. Chem. Lett. 2021, 12, 6252–6261. [Google Scholar] [CrossRef]
- Deganutti, G.; Prischi, F.; Reynolds, C.A. Supervised molecular dynamics for exploring the druggability of the SARS-CoV-2 spike protein. J. Comput. Aided Mol. Des. 2021, 35, 195–207. [Google Scholar] [CrossRef]
- Pipitò, L.; Reynolds, C.A.; Mobarec, J.C.; Vickery, O.; Deganutti, G. A pathway model to understand the evolution of spike protein binding to ACE2 in SARS-CoV-2 variants. Biomolecules 2022, 12, 1607. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, J.; Li, D.; Lin, J. The full activation mechanism of the adenosine A1 receptor revealed by GaMD and Su-GaMD simulations. Proc. Natl. Acad. Sci. USA 2022, 119, e2203702119. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; McCammon, J.A. Gaussian accelerated molecular dynamics: Theory, implementation, and applications. Annu. Rep. Comput. Chem. 2017, 13, 231–278. [Google Scholar] [PubMed]
- Chakraborty, S.; Saha, A.; Saha, C.; Ghosh, S.; Mondal, T. Decoding the effects of spike receptor binding domain mutations on antibody escape abilities of omicron variants. Biochem. Biophys. Res. Commun. 2022, 627, 168–175. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. Amber20; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 2006, 65, 1409–1419. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Dalby, P.A. Chapter Two—A beginner’s guide to molecular dynamics simulations and the identification of cross-correlation networks for enzyme engineering. In Methods in Enzymology; Tawfik, D.S., Ed.; Academic Press: Amsterdam, The Netherlands, 2020; Volume 643, pp. 15–49. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, C.; Lv, X.; Zhang, Z.; Lin, J.; Li, D. The Recognition Pathway of the SARS-CoV-2 Spike Receptor-Binding Domain to Human Angiotensin-Converting Enzyme 2. Molecules 2024, 29, 1875. https://doi.org/10.3390/molecules29081875
Peng C, Lv X, Zhang Z, Lin J, Li D. The Recognition Pathway of the SARS-CoV-2 Spike Receptor-Binding Domain to Human Angiotensin-Converting Enzyme 2. Molecules. 2024; 29(8):1875. https://doi.org/10.3390/molecules29081875
Chicago/Turabian StylePeng, Can, Xinyue Lv, Zhiqiang Zhang, Jianping Lin, and Dongmei Li. 2024. "The Recognition Pathway of the SARS-CoV-2 Spike Receptor-Binding Domain to Human Angiotensin-Converting Enzyme 2" Molecules 29, no. 8: 1875. https://doi.org/10.3390/molecules29081875
APA StylePeng, C., Lv, X., Zhang, Z., Lin, J., & Li, D. (2024). The Recognition Pathway of the SARS-CoV-2 Spike Receptor-Binding Domain to Human Angiotensin-Converting Enzyme 2. Molecules, 29(8), 1875. https://doi.org/10.3390/molecules29081875