
Antitumoral Activity of the Universal Methyl Donor S-Adenosylmethionine in Glioblastoma Cells

 , ,
, , 
Abstract
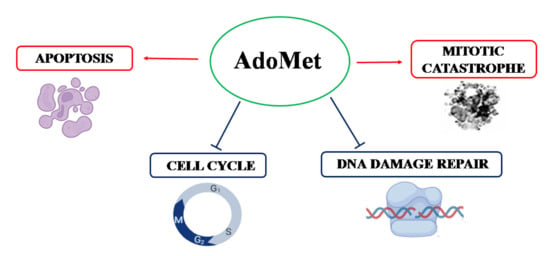
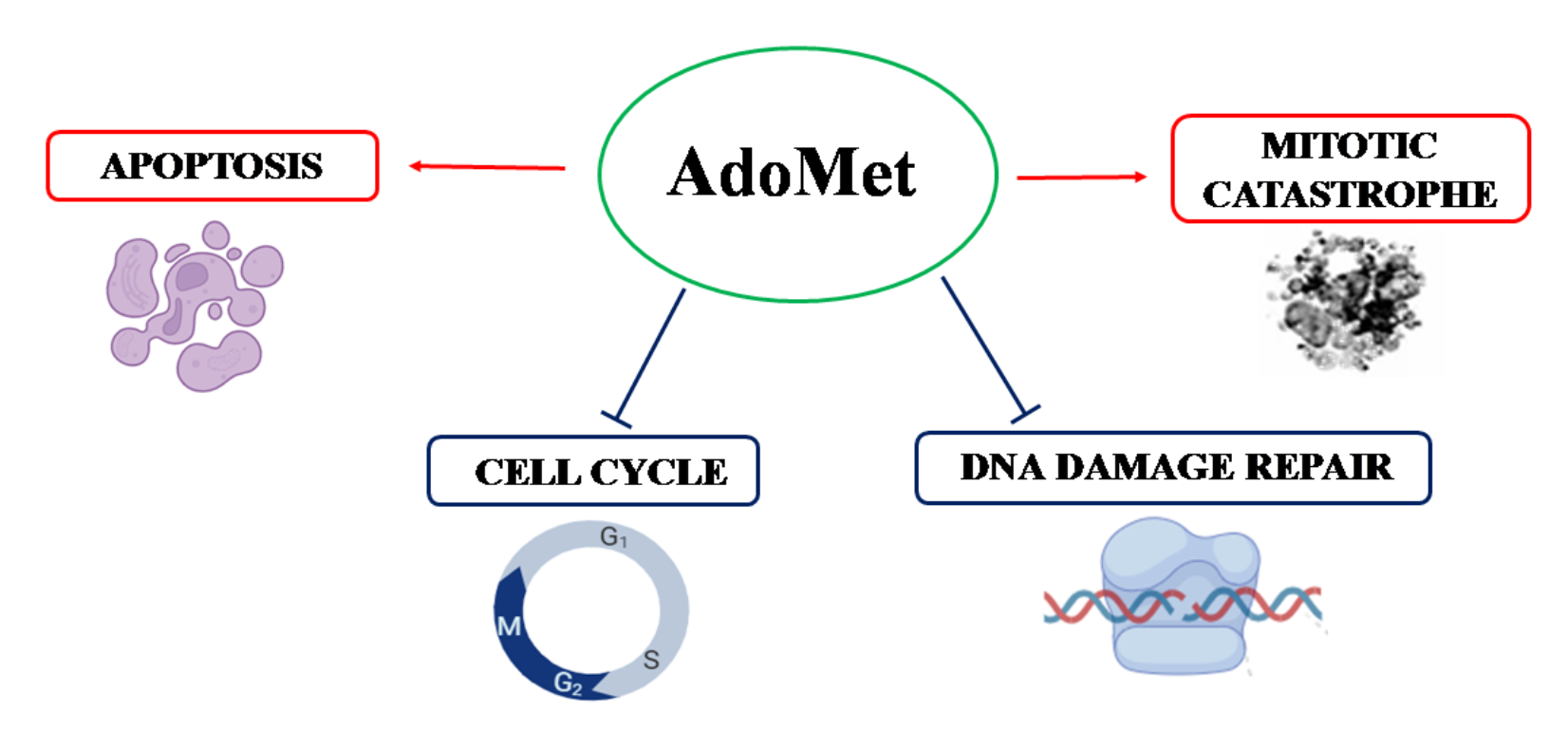{kind=link}
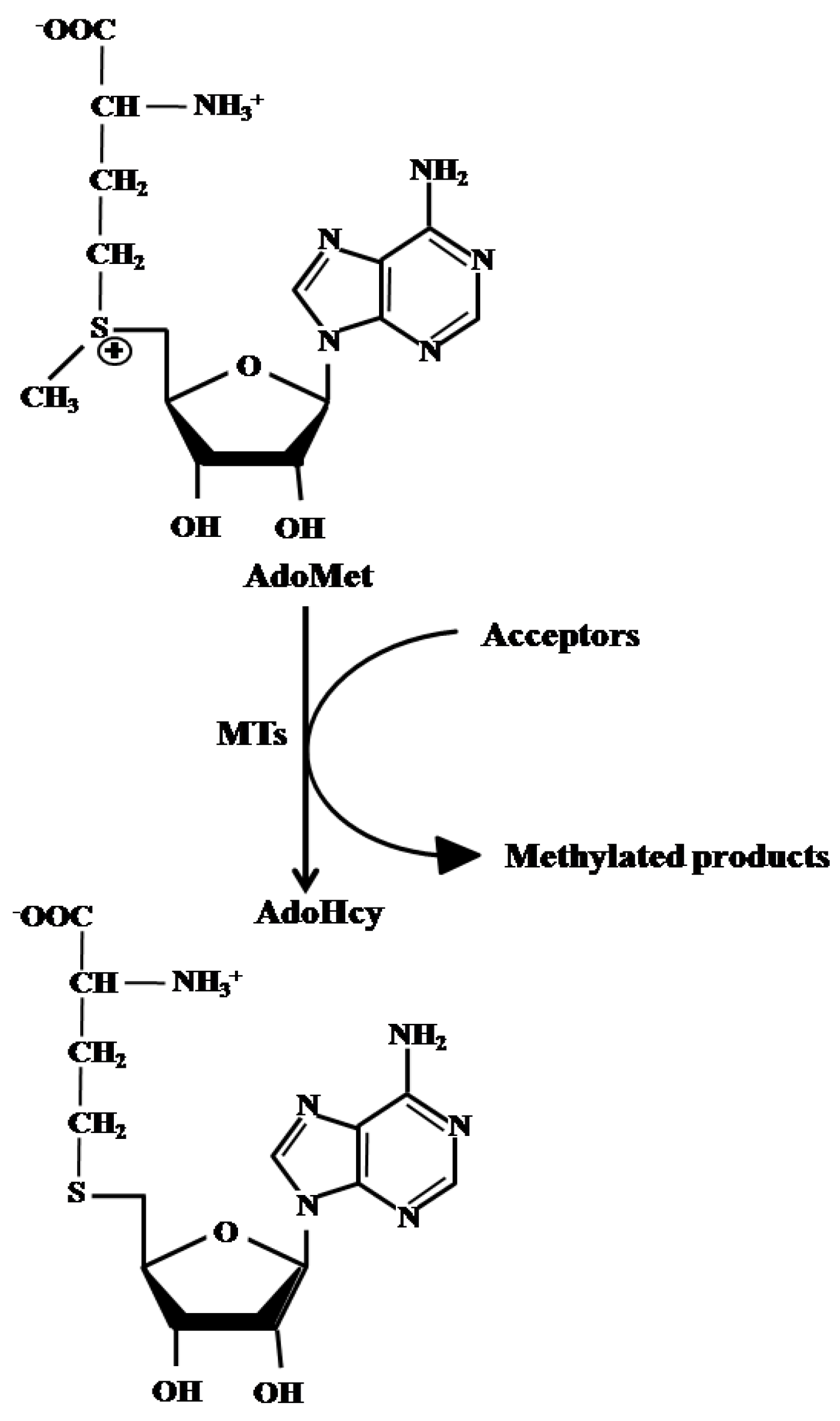{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
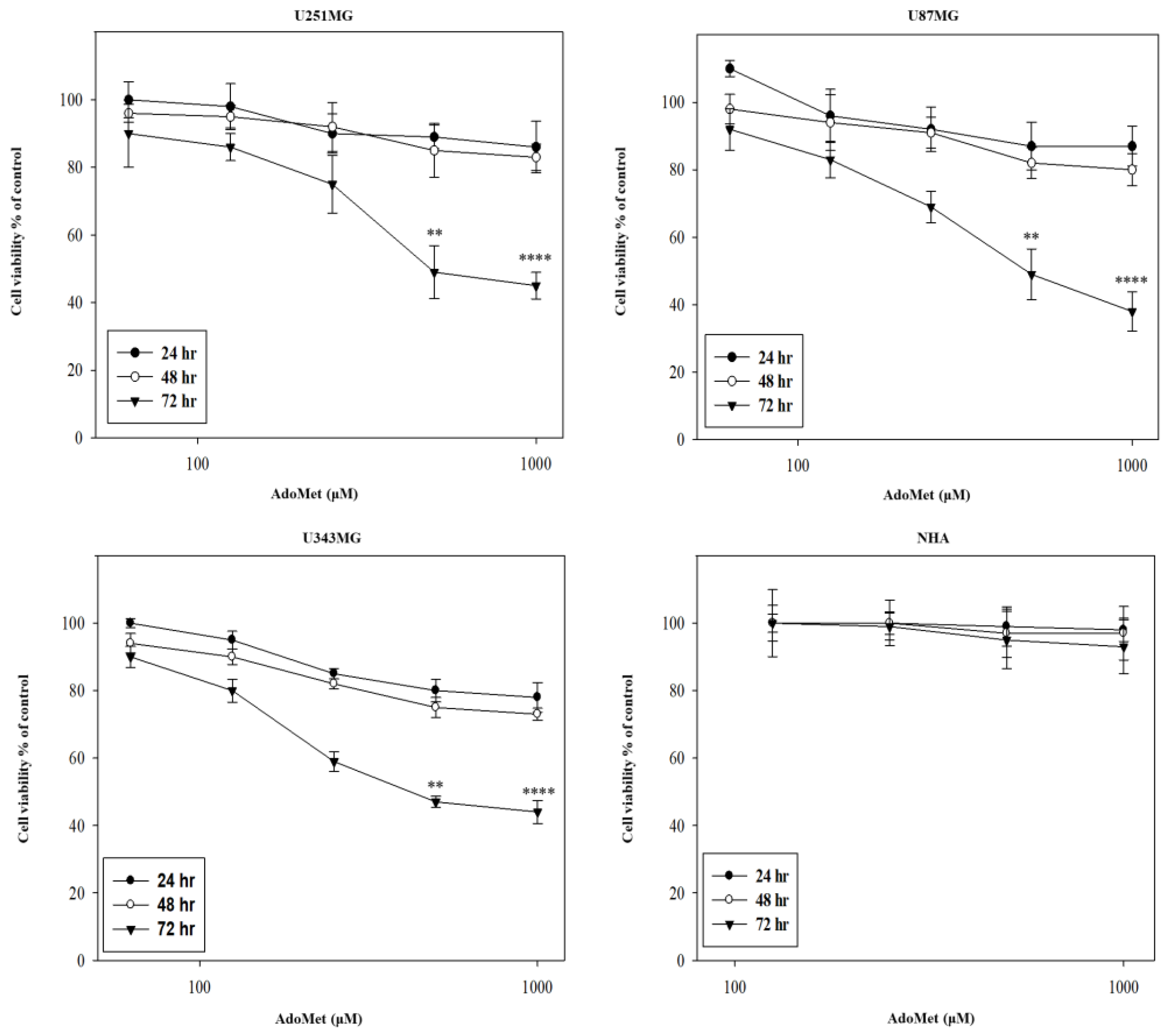
2.1. AdoMet Inhibited GBM Cell Viability
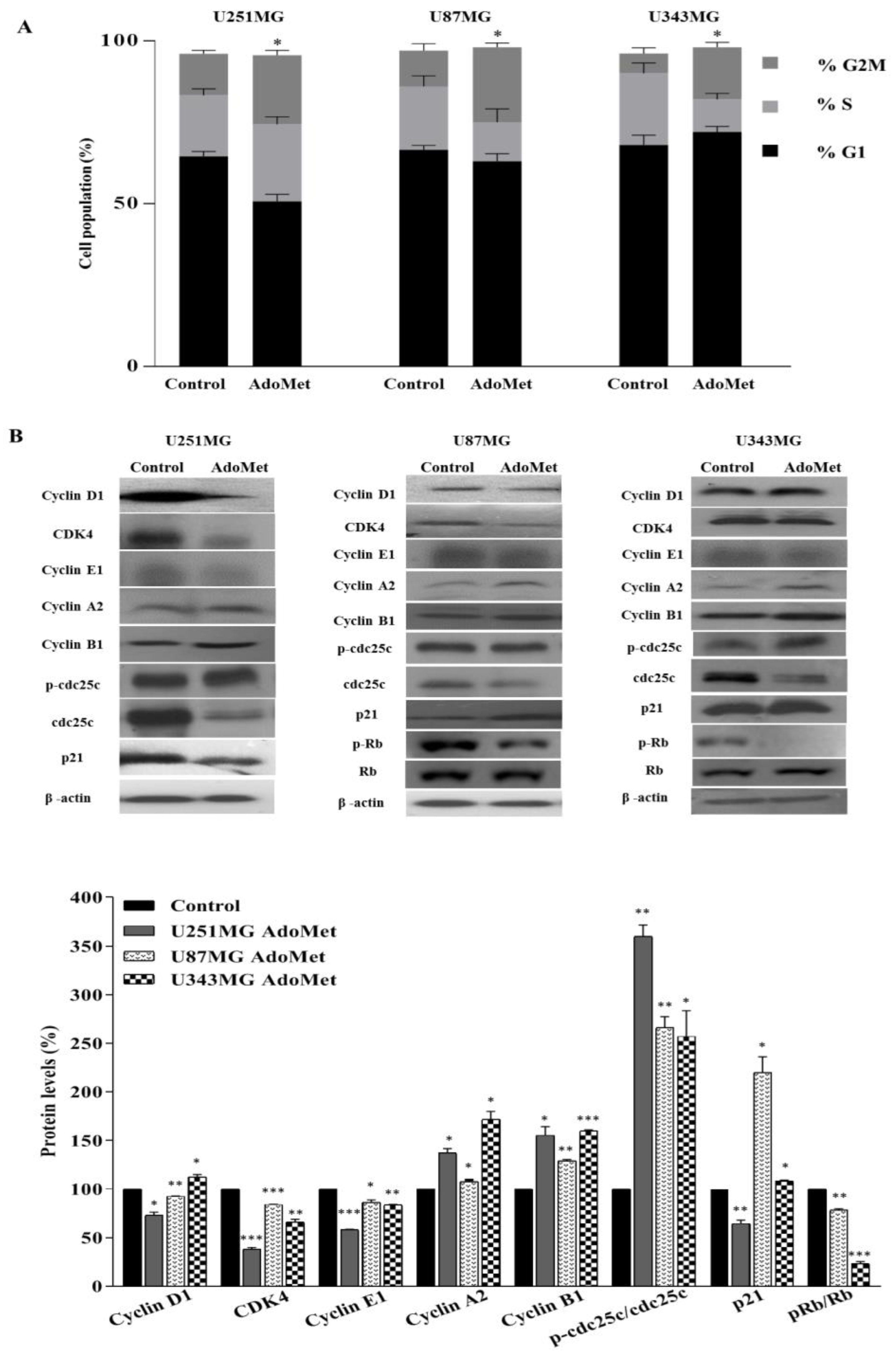
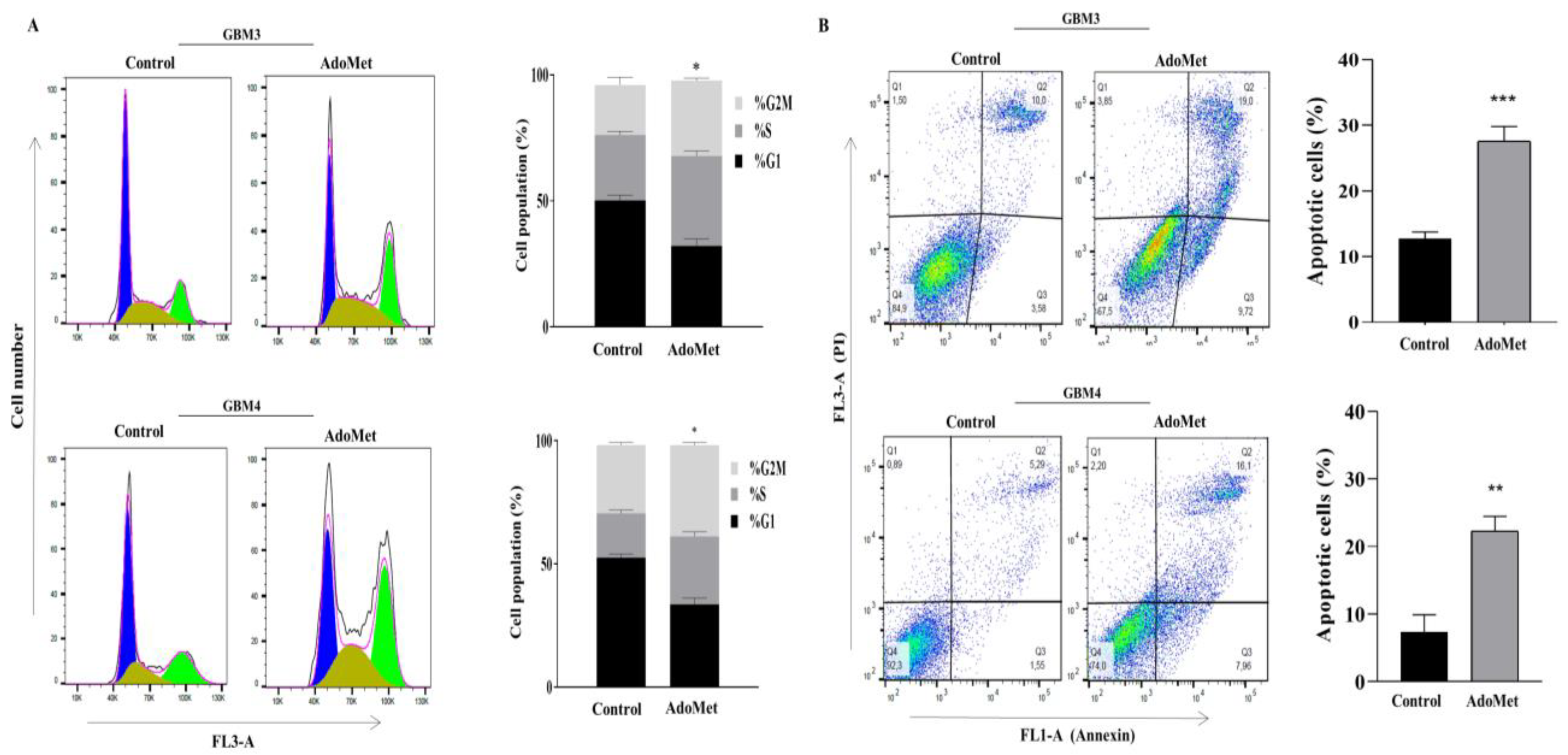
2.2. AdoMet Induced S and G2/M Cell Cycle Arrest
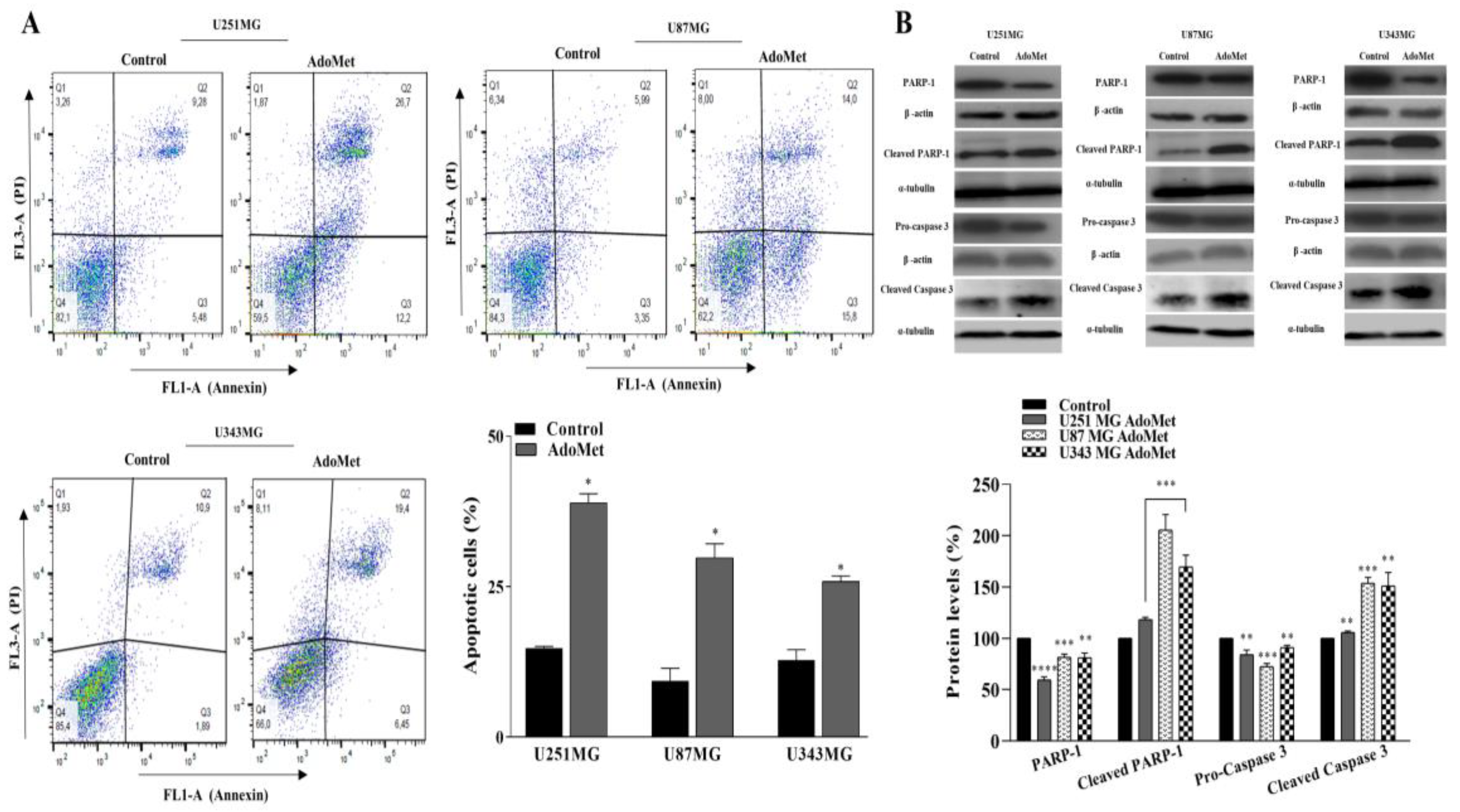
2.3. AdoMet Induced Apoptosis
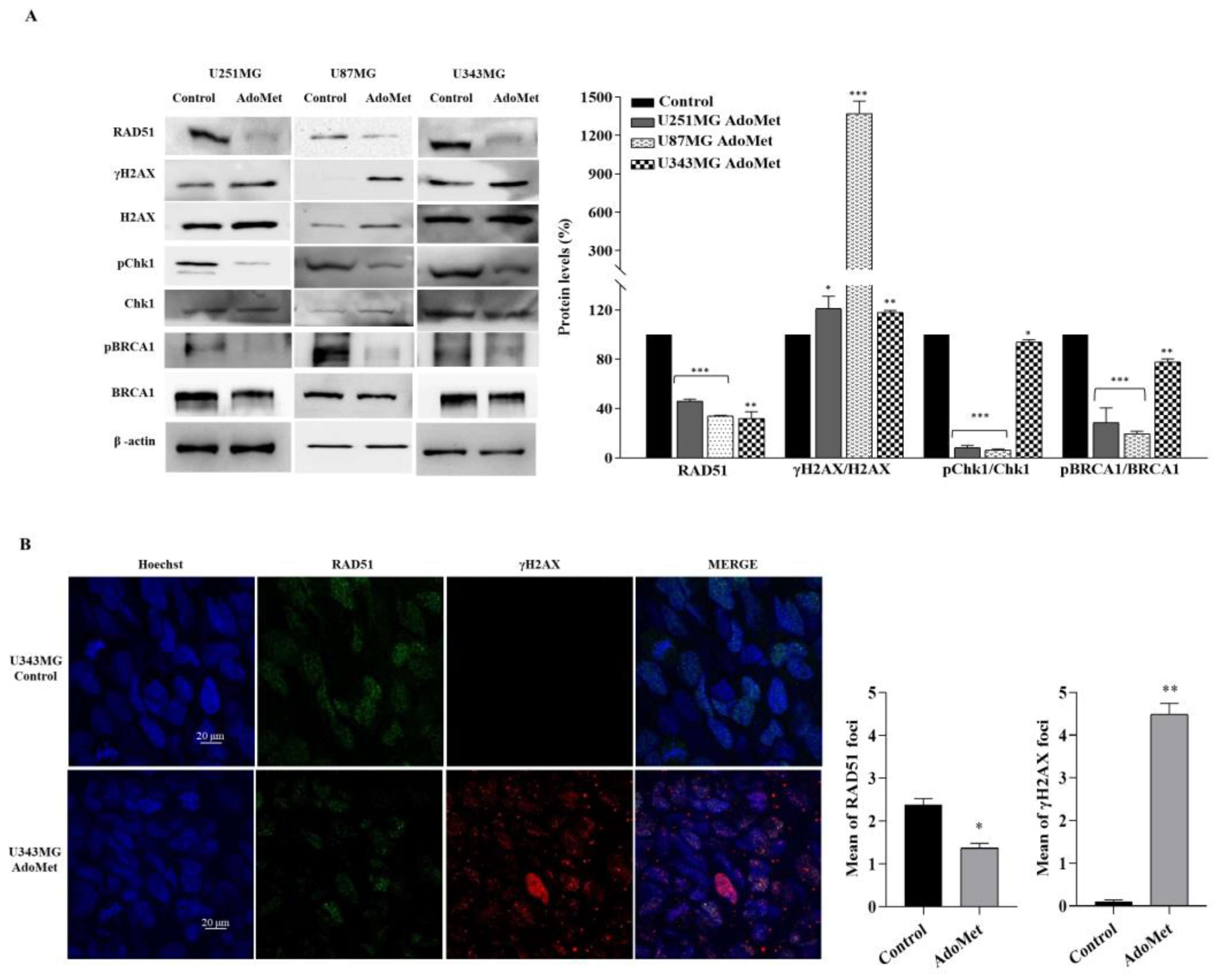
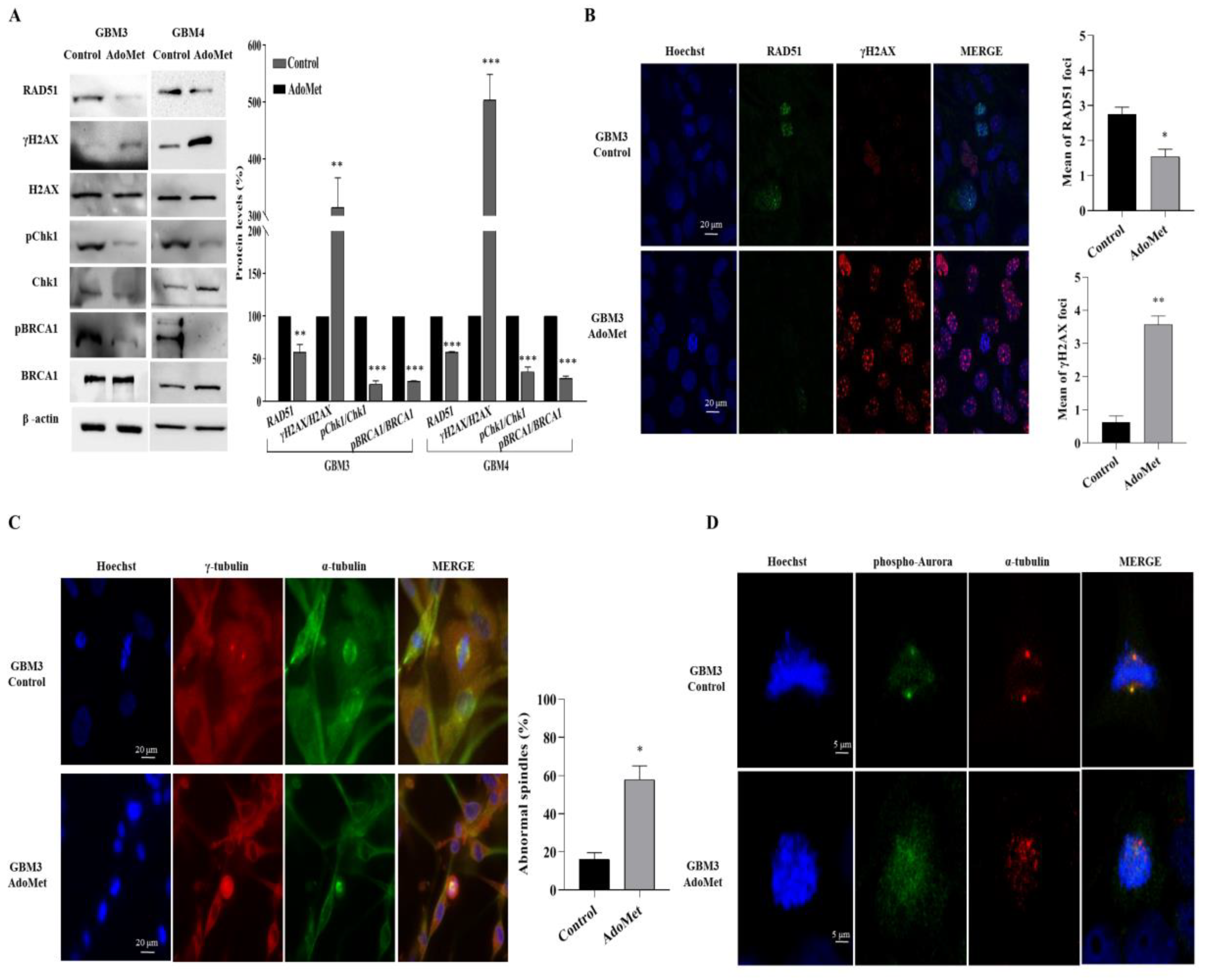
2.4. AdoMet Downregulated the Proteins Involved in Homologous Recombination Repair
2.5. AdoMet-Induced Downregulation of RAD51 Is Associated with Increased γH2AX Levels
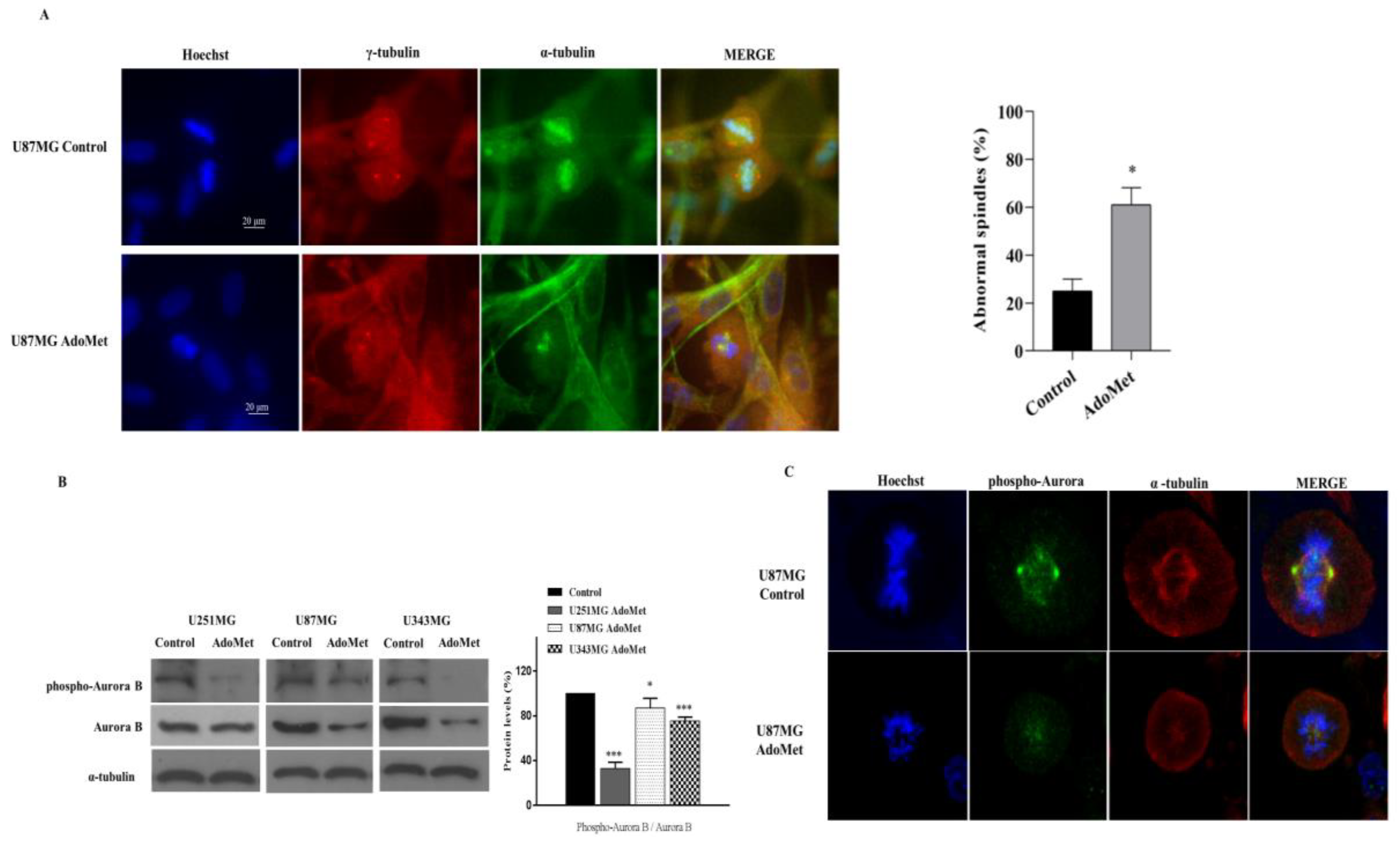
2.6. AdoMet Promoted Mitotic Catastrophe
2.7. AdoMet Targeted the Expression, Activation, and Subcellular Localization of Aurora B
2.8. AdoMet Suppressed Cell Proliferation and Survival in Patient-Derived GBM Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Cultures
4.3. Preparation of GBM Primary Cell Lines
4.4. Cell Viability Assay
4.5. Flow Cytometry Analysis of Cell Cycle
4.6. Flow Cytometry Analysis of Apoptosis
4.7. Protein Extraction and Western Blot Analysis
4.8. Immunofluorescence Analysis
4.9. Confocal Laser Scanning Microscopy
4.10. Statistical Analysis
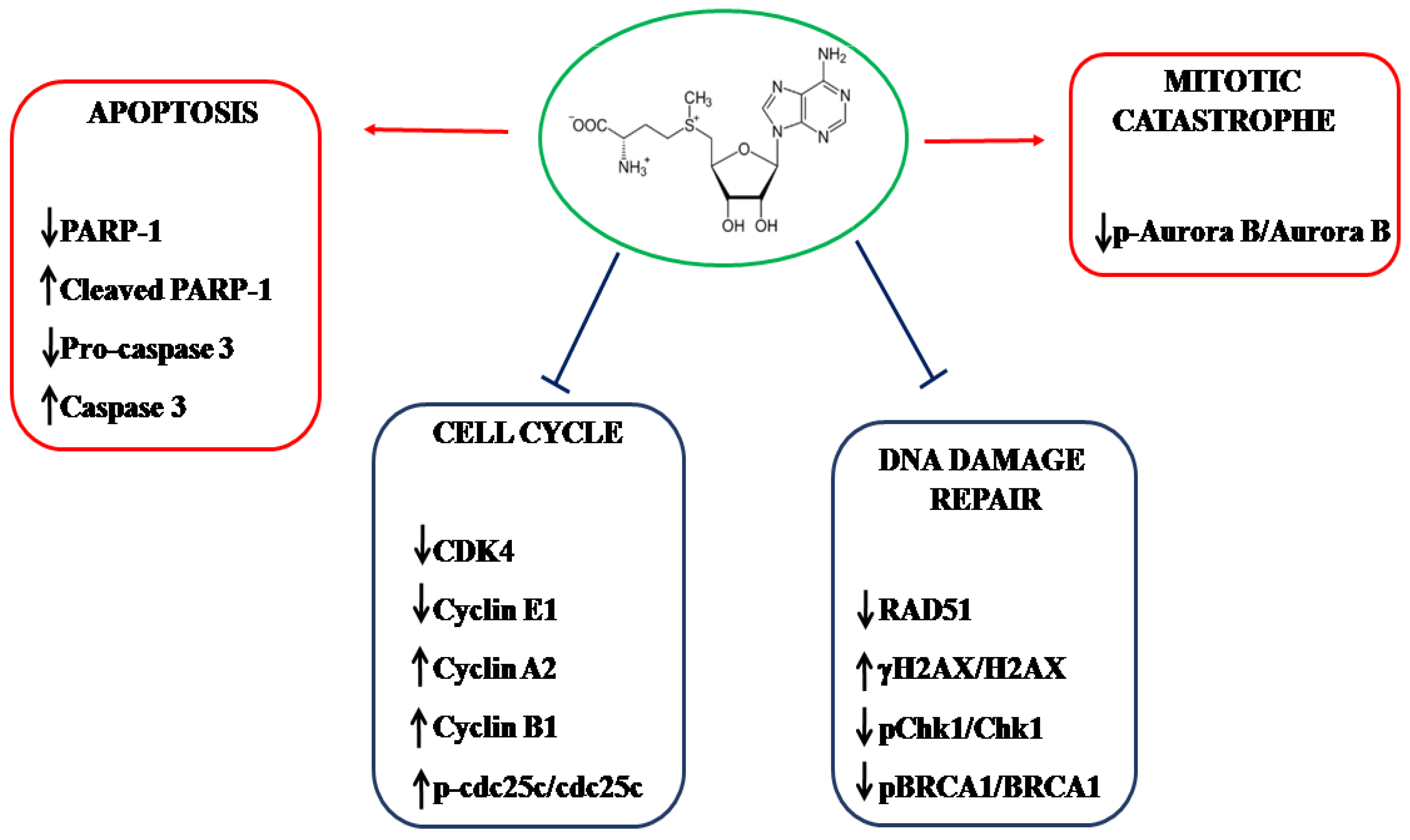
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Muir, M.; Gopakumar, S.; Traylor, J.; Lee, S.; Rao, G. Glioblastoma multiforme: Novel therapeutic targets. Expert Opin. Ther. Targets 2020, 24, 605–614. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. 2014, 9, 1–25. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer. Prev. 2017, 18, 3–9. [Google Scholar]
- Ferri, A.; Stagni, V.; Barilà, D. Targeting the DNA Damage Response to Overcome Cancer Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 4910. [Google Scholar] [CrossRef]
- Majd, N.K.; Yap, T.A.; Koul, D.; Balasubramaniyan, V.; Li, X.; Khan, S.; Gandy, K.S.; Yung, W.; de Groot, J.F. The promise of DNA damage response inhibitors for the treatment of glioblastoma. Neuro Oncol. Adv. 2021, 3, vdab015. [Google Scholar] [CrossRef]
- Rominiyi, O.; Collis, S.J. DDRugging glioblastoma: Understanding and targeting the DNA damage response to improve future therapies. Mol. Oncol. 2022, 16, 11–41. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009, 100, 2235–2241. [Google Scholar] [CrossRef]
- Romani, M.; Pistillo, M.P.; Banelli, B. Epigenetic Targeting of Glioblastoma. Front. Oncol. 2018, 8, 448–456. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma-A comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef]
- Shen, D.; Liu, T.; Lin, Q.; Lu, X.; Wang, Q.; Lin, F.; Mao, W. MGMT promoter methylation correlates with an overall survival benefit in Chinese high-grade glioblastoma patients treated with radiotherapy and alkylating agent-based chemotherapy: A single-institution study. PLoS ONE 2014, 9, e107558–e107563. [Google Scholar] [CrossRef]
- Chiang, P.K.; Gordon, R.K.; Tal, J.; Zeng, G.C.; Doctor, B.P.; Pardhasaradhi, K.; McCann, P.P. S-Adenosylmethionine and methylation. FASEB J. 1996, 10, 471–480. [Google Scholar] [CrossRef]
- Mato, J.M.; Martínez-Chantar, M.L.; Lu, S.C. S-Adenosylmethionine metabolism and liver disease. Ann. Hepatol. 2013, 12, 183–189. [Google Scholar] [CrossRef]
- Pascale, R.M.; Simile, M.M.; Calvisi, D.F.; Feo, C.F.; Feo, F. S-Adenosylmethionine: From the Discovery of Its Inhibition of Tumorigenesis to Its Use as a Therapeutic Agent. Cells 2022, 11, 409. [Google Scholar] [CrossRef]
- Hao, X.; Zhou, M.; Li, H.; Angres, I.A. Novel immunoassays to detect methionine adenosyl transferase activity and quantify S-adenosylmethionine. FEBS Lett. 2017, 591, 1114–1125. [Google Scholar] [CrossRef]
- Serefidou, M.; Venkatasubramani, A.V.; Imhof, A. The Impact of One Carbon Metabolism on Histone Methylation. Front. Genet. 2019, 10, 764–770. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Y.G. SAM/SAH Analogs as Versatile Tools for SAM-Dependent methyltransferases. ACS Chem. Biol. 2016, 11, 583–597. [Google Scholar] [CrossRef]
- Hermes, M.; Geisler, H.; Osswald, H.; Riehle, R.; Kloor, D. Alterations in S-adenosylhomocysteine metabolism decrease O6-methylguanine DNA methyltransferase gene expression without affecting promoter methylation. Biochem. Pharmacol. 2008, 75, 2100–2111. [Google Scholar] [CrossRef]
- Mosca, L.; Vitiello, F.; Pagano, M.; Coppola, A.; Veglia Tranchese, R.; Grillo, R.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine, a Promising Antitumor Agent in Oral and Laryngeal Cancer. Appl. Sci. 2022, 12, 1746. [Google Scholar] [CrossRef]
- Mosca, L.; Pagano, M.; Borzacchiello, L.; Mele, L.; Russo, A.; Russo, G.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine increases the sensitivity of human colorectal cancer cells to 5-fluorouracil by inhibiting P-glycoprotein expression and NF-kB activation. Int. J. Mol. Sci. 2021, 22, 9286. [Google Scholar] [CrossRef]
- Ouyang, Y.; Wu, Q.; Li, J.; Sun, S.; Sun, S. S-adenosylmethionine: A metabolite critical to the regulation of autophagy. Cell Prolif. 2020, 53, e12891–e12903. [Google Scholar] [CrossRef]
- Mahmood, N.; Cheishvili, D.; Arakelian, A.; Tanvir, I.; Khan, H.A.; Pépin, A.S.; Szyf, M.; Rabbani, S.A. Methyl donor S-adenosylmethionine (SAM) supplementation attenuates breast cancer growth, invasion and metastasis in vivo; therapeutic and chemopreventive applications. Oncotarget 2018, 9, 5169–5183. [Google Scholar] [CrossRef]
- Yan, L.; Liang, X.; Huang, H.; Zhang, G.; Liu, T.; Zhang, J.; Chen, Z.; Zhang, Z.; Chen, Y. S-Adenosylmethionine Affects Cell Cycle Pathways and Suppresses Proliferation in Liver Cells. J. Cancer 2019, 10, 4368–4379. [Google Scholar] [CrossRef]
- Mahmood, N.; Arakelian, A.; Cheishvili, D.; Szyf, M.; Rabbani, S.A. S-adenosylmethionine in combination with decitabine shows enhanced anti-cancer effects in repressing breast cancer growth and metastasis. J. Cell. Mol. Med. 2020, 24, 10322–10337. [Google Scholar] [CrossRef]
- Mosca, L.; Pagano, M.; Ilisso, C.P.; Delle Cave, D.; Desiderio, V.; Mele, L.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. AdoMet triggers apoptosis in head and neck squamous cancer by inducing ER-stress and potentiates cell sensitivity to cisplatin. J. Cell. Physiol. 2019, 234, 13277–13291. [Google Scholar] [CrossRef]
- Cave, D.D.; Desiderio, V.; Mosca, L.; Ilisso, C.P.; Mele, L.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine-mediated apoptosis is potentiated by autophagy inhibition induced by chloroquine in human breast cancer cells. J. Cell. Physiol. 2017, 233, 1370–1383. [Google Scholar] [CrossRef]
- Mosca, L.; Vitiello, F.; Coppola, A.; Borzacchiello, L.; Ilisso, C.P.; Pagano, M.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. Therapeutic Potential of the Natural Compound S-Adenosylmethionine as a Chemoprotective Synergistic Agent in Breast, and Head and Neck Cancer Treatment: Current Status of Research. Int. J. Mol. Sci. 2020, 21, 8547. [Google Scholar] [CrossRef]
- Ilisso, C.P.; Cave, D.D.; Mosca, L.; Pagano, M.; Coppola, A.; Mele, L.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. Adenosylmethionine regulates apoptosis and autophagy in MCF-7 breast cancer cells through the modulation of specific microRNAs. Cancer Cell Int. 2018, 18, 197–209. [Google Scholar] [CrossRef]
- Uddin, M.S.; Mamun, A.A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of glioblastoma multiforme: From molecular mechanisms to therapeutic approaches. Semin. Cancer Biol. 2022, 83, 100–120. [Google Scholar] [CrossRef]
- Li, P.; Wu, M. Epigenetic Mechanisms of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, QLD, Australia, 2017. [Google Scholar]
- Wang, Y.; Sun, Z.; Szyf, M. S-adenosyl-methionine (SAM) alters the transcriptome and methylome and specifically blocks growth and invasiveness of liver cancer cells. Oncotarget 2017, 8, 111866–111881. [Google Scholar] [CrossRef]
- Luo, J.; Li, Y.N.; Wang, F.; Zhang, W.M.; Geng, X. S-adenosylmethionine inhibits the growth of cancer cells by reversing the hypomethylation status of c-myc and H-ras in human gastric cancer and colon cancer. Int. J. Biol. Sci. 2010, 6, 784–795. [Google Scholar] [CrossRef]
- Coppola, A.; Ilisso, C.P.; Stellavato, A.; Schiraldi, C.; Caraglia, M.; Mosca, L.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine Inhibits Cell Growth and Migration of Triple Negative Breast Cancer Cells through Upregulating MiRNA-34c and MiRNA-449a. Int. J. Mol. Sci. 2020, 22, 286. [Google Scholar] [CrossRef]
- Pagano, M.; Mosca, L.; Vitiello, F.; Ilisso, C.P.; Coppola, A.; Borzacchiello, L.; Mele, L.; Caruso, F.P.; Ceccarelli, M.; Caraglia, M.; et al. Mi-RNA-888-5p is involved in S-adenosylmethionine antitumor effects in laryngeal squamous cancer cells. Cancers 2020, 12, 3665. [Google Scholar] [CrossRef]
- Mosca, L.; Vitiello, F.; Borzacchiello, L.; Coppola, A.; Veglia Tranchese, R.; Pagano, M.; Caraglia, M.; Cacciapuoti, G.; Porcelli, M. Mutual Correlation between Non-Coding RNA and S-Adenosylmethionine in Human Cancer: Roles and Therapeutic Opportunities. Cancers 2021, 13, 3264. [Google Scholar] [CrossRef]
- Borzacchiello, L.; Veglia Tranchese, R.; Grillo, R.; Arpino, R.; Mosca, L.; Cacciapuoti, G.; Porcelli, M. S-Adenosylmethionine Inhibits Colorectal Cancer Cell Migration through Mirna-Mediated Targeting of Notch Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 7673. [Google Scholar] [CrossRef]
- Topacio, B.R.; Zatulovskiy, E.; Cristea, S.; Xie, S.; Tambo, C.S.; Rubin, S.M.; Sage, J.; Kõivomägi, M.; Skotheim, J.M. Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix. Mol. Cell 2019, 74, 758–770. [Google Scholar] [CrossRef]
- Godoy, P.R.; Mello, S.S.; Magalhães, D.A.; Donaires, F.S.; Nicolucci, P.; Donadi, E.A.; Passos, G.A.; Sakamoto-Hojo, E.T. Ionizing radiation-induced gene expression changes in TP53 proficient and deficient glioblastoma cell lines. Mutat. Res. 2013, 756, 46–55. [Google Scholar] [CrossRef]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213–228. [Google Scholar] [CrossRef]
- Bonm, A.; Kesari, S. DNA Damage Response in Glioblastoma: Mechanism for Treatment Resistance and Emerging Therapeutic Strategies. Cancer J. 2021, 27, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Fehrmann, R.S.; De Vries, E.G.; Van Vugt, M.A. Regulators of homologous recombination repair as novel targets for cancer treatment. Front. Genet. 2015, 6, 96–110. [Google Scholar] [CrossRef]
- Peng, G.; Lin, S.Y. Exploiting the homologous recombination DNA repair network for targeted cancer therapy. World J. Clin. Oncol. 2011, 2, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jia, R.; Wang, L.; Yang, Q.; Hu, X.; Fu, Q.; Zhang, X.; Li, W.; Ren, Y. The Emerging Roles of Rad51 in Cancer and Its Potential as a Therapeutic Target. Front. Oncol. 2022, 12, 935593–935599. [Google Scholar] [CrossRef] [PubMed]
- Christou, C.M.; Kyriacou, K. BRCA1 and Its Network of Interacting Partners. Biology 2013, 2, 40–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer. 2014, 134, 1013–1023. [Google Scholar] [CrossRef]
- Collins, P.L.; Purman, C.; Porter, S.I.; Nganga, V.; Saini, A.; Hayer, K.E.; Gurewitz, G.L.; Sleckman, B.P.; Bednarski, J.J.; Bassing, C.H.; et al. DNA double-strand breaks induce H2AX phosphorylation domains in a contact-dependent manner. Nat. Commun. 2020, 11, 3158–3166. [Google Scholar] [CrossRef]
- Ji, J.; Zhang, Y.; Redon, C.E.; Reinhold, W.C.; Chen, A.P.; Fogli, L.K.; Holbeck, S.L.; Parchment, R.E.; Hollingshead, M.; Tomaszewski, J.E.; et al. Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay. PLoS ONE. 2017, 12, e0171582–e0171599. [Google Scholar] [CrossRef]
- Sazonova, E.V.; Petrichuk, S.V.; Kopeina, G.S.; Zhivotovsky, B. A link between mitotic defects and mitotic catastrophe: Detection and cell fate. Biol. Direct. 2021, 16, 25. [Google Scholar] [CrossRef]
- Mc Gee, M.M. Targeting the Mitotic Catastrophe Signaling Pathway in Cancer. Mediat. Inflamm. 2015, 8, 146282. [Google Scholar] [CrossRef] [PubMed]
- Vitovcova, B.; Skarkova, V.; Rudolf, K.; Rudolf, E. Biology of Glioblastoma Multiforme-Exploration of Mitotic Catastrophe as a Potential Treatment Modality. Int. J. Mol. Sci. 2020, 21, 5324. [Google Scholar] [CrossRef] [PubMed]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef]
- Kitagawa, M.; Lee, S.H. The chromosomal passenger complex (CPC) as a key orchestrator of orderly mitotic exit and cytokinesis. Front. Cell Dev. Biol. 2015, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef]
- Pagano, C.; Navarra, G.; Pastorino, O.; Avilia, G.; Coppola, L.; Della Monica, R.; Chiariotti, L.; Florio, T.; Corsaro, A.; Torelli, G.; et al. N6-Isopentenyladenosine Hinders the Vasculogenic Mimicry in Human Glioblastoma Cells through Src-120 Catenin Pathway Modulation and RhoA Activity Inhibition. Int. J. Mol. Sci. 2021, 22, 10530. [Google Scholar] [CrossRef]
- Shen, B. A New Golden Age of Natural Products Drug Discovery. Cell 2015, 163, 1297–1300. [Google Scholar] [CrossRef]
- Zhai, K.; Siddiqui, M.; Abdellatif, B.; Liskova, A.; Kubatka, P.; Büsselberg, D. Natural Compounds in Glioblastoma Therapy: Preclinical Insights, Mechanistic Pathways, and Outlook. Cancers 2021, 13, 2317. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Feng, Y.; Gao, S.; Mu, Q.; Liu, C. Nanotherapeutics Overcoming the Blood-Brain Barrier for Glioblastoma Treatment. Front. Pharmacol. 2021, 12, 786700. [Google Scholar] [CrossRef]
- Sharma, A.; Gerbarg, P.; Bottiglieri, T.; Massoumi, L.; Carpenter, L.L.; Lavretsky, H.; Muskin, P.R.; Brown, R.P.; Mischoulon, D.; Work Group of the American Psychiatric Association Council on Research. S-Adenosylmethionine (SAMe) for neuropsychiatric disorders: A clinician-oriented review of research. J. Clin. Psychiatry 2017, 78, e656–e667. [Google Scholar] [CrossRef]
- Cavallaro, R.A.; Fuso, A.; d’Erme, M.; Miraglia, N.; Martire, S. Role of S-adenosylmethionine in the Modulation of Oxidative Stress-Related Neurodegeneration. Int. J. Clin. Nutr. Diet. 2016, 2, 1–5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chishty, M.; Reichel, A.; Abbott, N.J.; Begley, D.J. S-Adenosylmethionine is substrate for carrier mediated transport at the blood-brain barrier in vitro. Brain Res. 2002, 942, 46–50. [Google Scholar] [CrossRef]
- Bai, J.; Li, Y.; Zhang, G. Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 2017, 14, 348–362. [Google Scholar]
- Donzelli, M.; Draetta, G.F. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003, 4, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Kabakci, Z.; Käppeli, S.; Cantù, C.; Jensen, L.D.; König, C.; Toggweiler, J.; Gentili, C.; Ribaudo, G.; Zagotto, G.; Basler, K.; et al. Pharmacophore-guided discovery of CDC25 inhibitors causing cell cycle arrest and tumor regression. Sci. Rep. 2019, 9, 1335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dube, C.; Gibert, M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef]
- Bonilla, B.; Hengel, S.R.; Grundy, M.K.; Bernstein, K.A. RAD51 Gene Family Structure and Function. Annu. Rev. Genet. 2020, 54, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Wassing, I.E.; Graham, E.; Saayman, X.; Rampazzo, L.; Ralf, C.; Bassett, A.; Esashi, F. The RAD51 recombinase protects mitotic chromatin in human cells. Nat. Commun. 2021, 12, 5380–5396. [Google Scholar] [CrossRef]
- Morrison, C.; Weterings, E.; Mahadevan, D.; Sanan, A.; Weinand, M.; Stea, B. Expression Levels of RAD51 Inversely Correlate with Survival of Glioblastoma Patients. Cancers 2021, 13, 5358. [Google Scholar] [CrossRef]
- Zhang, J.; Willers, H.; Feng, Z.; Ghosh, J.C.; Kim, S.; Weaver, D.T.; Chung, J.H.; Powell, S.N.; Xia, F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol. Cell. Biol. 2004, 24, 708–718. [Google Scholar] [CrossRef]
- Orhan, E.; Velazquez, C.; Tabet, I.; Sardet, C.; Theillet, C. Regulation of RAD51 at the Transcriptional and Functional Levels: What Prospects for Cancer Therapy? Cancers 2021, 13, 2930. [Google Scholar] [CrossRef] [PubMed]
- Bahassi, E.M.; Ovesen, J.L.; Riesenberg, A.L.; Bernstein, W.Z.; Hasty, P.E.; Stambrook, P.J. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 2008, 27, 3977–3985. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu, H.; Olsson, M.; Zhivotovsky, B. Death through a tragedy: Mitotic catastrophe. Cell Death. Differ. 2008, 15, 1153–1162. [Google Scholar] [CrossRef]
- Huang, X.; Tran, T.; Zhang, L.; Hatcher, R.; Zhang, P. DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proc. Natl. Acad. Sci. USA 2005, 102, 1065–1070. [Google Scholar] [CrossRef]
- De Almeida Magalhães, T.; de Sousa, G.R.; Alencastro Veiga Cruzeiro, G.; Tone, L.G.; Valera, E.T.; Borges, K.S. The therapeutic potential of Aurora kinases targeting in glioblastoma: From preclinical research to translational oncology. J. Mol. Med. 2020, 98, 495–512. [Google Scholar] [CrossRef] [PubMed]
- Borah, N.A.; Reddy, M.M. Aurora Kinase B Inhibition: A Potential Therapeutic Strategy for Cancer. Molecules 2021, 26, 1981. [Google Scholar] [CrossRef] [PubMed]
- Mele, L.; Del Vecchio, V.; Marampon, F.; Regad, T.; Wagner, S.; Mosca, L.; Bimonte, S.; Giudice, A.; Liccardo, D.; Prisco, C.; et al. 2-AR blockade potentiates MEK1/2 inhibitor effect on HNSCC by regulating the Nrf2-mediated defense mechanism. Cell Death Dis. 2020, 11, 850–863. [Google Scholar] [CrossRef]
- Squillaci, G.; Vitiello, F.; Mosca, L.; La Cara, F.; Cacciapuoti, G.; Porcelli, M.; Morana, A. Polyphenol Extract from “Greco” Grape Canes: Characterization, Antioxidant Capacity, and Antitumor Effects on Cal-33 and JHU-SCC-011 Head and Neck Squamous Cell Carcinoma. Molecules 2022, 27, 2576. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosca, L.; Pagano, C.; Tranchese, R.V.; Grillo, R.; Cadoni, F.; Navarra, G.; Coppola, L.; Pagano, M.; Mele, L.; Cacciapuoti, G.; et al. Antitumoral Activity of the Universal Methyl Donor S-Adenosylmethionine in Glioblastoma Cells. Molecules 2024, 29, 1708. https://doi.org/10.3390/molecules29081708
Mosca L, Pagano C, Tranchese RV, Grillo R, Cadoni F, Navarra G, Coppola L, Pagano M, Mele L, Cacciapuoti G, et al. Antitumoral Activity of the Universal Methyl Donor S-Adenosylmethionine in Glioblastoma Cells. Molecules. 2024; 29(8):1708. https://doi.org/10.3390/molecules29081708
Chicago/Turabian StyleMosca, Laura, Cristina Pagano, Roberta Veglia Tranchese, Roberta Grillo, Francesca Cadoni, Giovanna Navarra, Laura Coppola, Martina Pagano, Luigi Mele, Giovanna Cacciapuoti, and et al. 2024. "Antitumoral Activity of the Universal Methyl Donor S-Adenosylmethionine in Glioblastoma Cells" Molecules 29, no. 8: 1708. https://doi.org/10.3390/molecules29081708
APA StyleMosca, L., Pagano, C., Tranchese, R. V., Grillo, R., Cadoni, F., Navarra, G., Coppola, L., Pagano, M., Mele, L., Cacciapuoti, G., Laezza, C., & Porcelli, M. (2024). Antitumoral Activity of the Universal Methyl Donor S-Adenosylmethionine in Glioblastoma Cells. Molecules, 29(8), 1708. https://doi.org/10.3390/molecules29081708