
Recent Advances in the Synthesis of the Marine-Derived Alkaloid Fascaplysin and Its Metabolites Homofascaplysins A–C


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Discussion
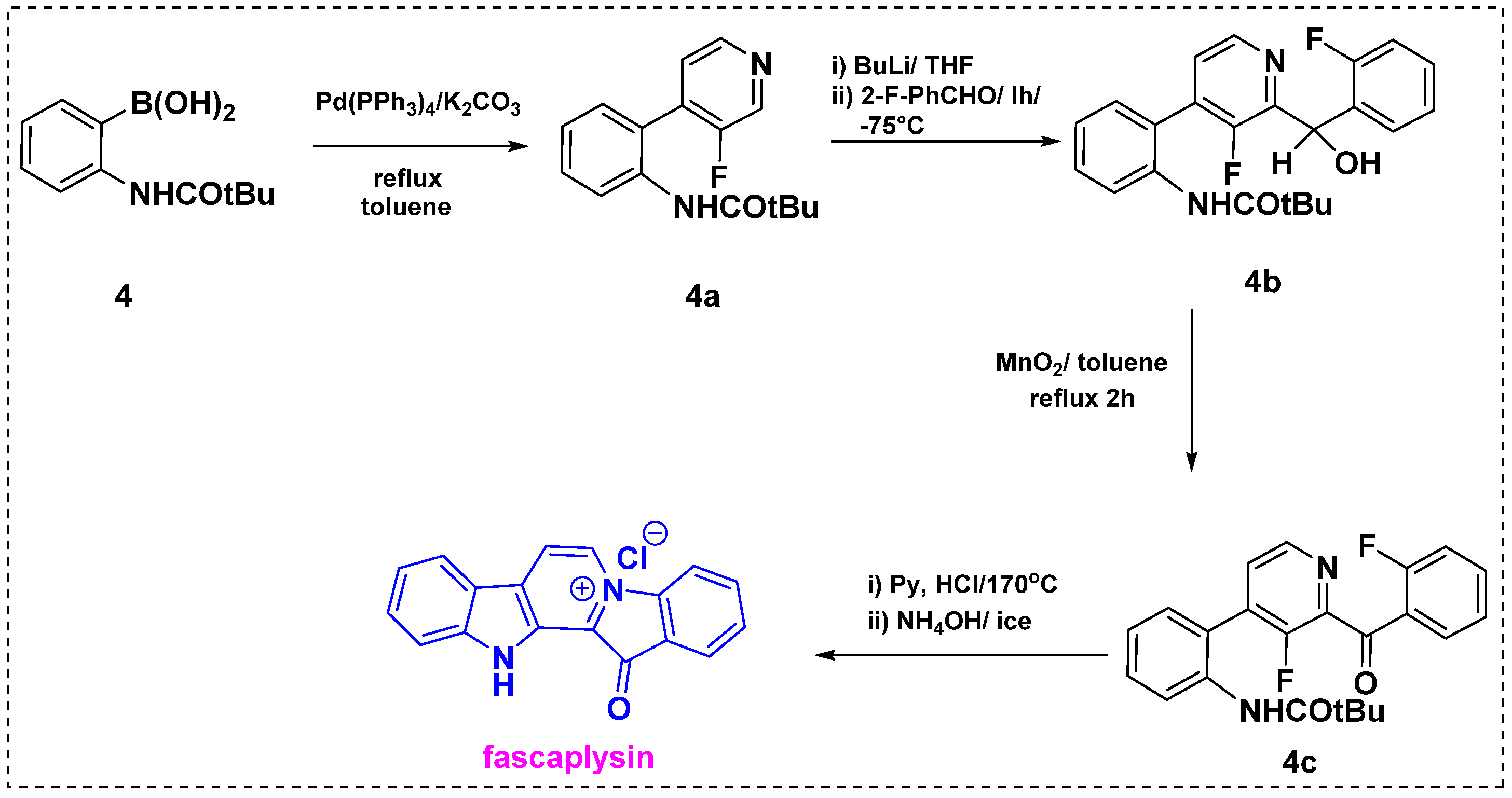
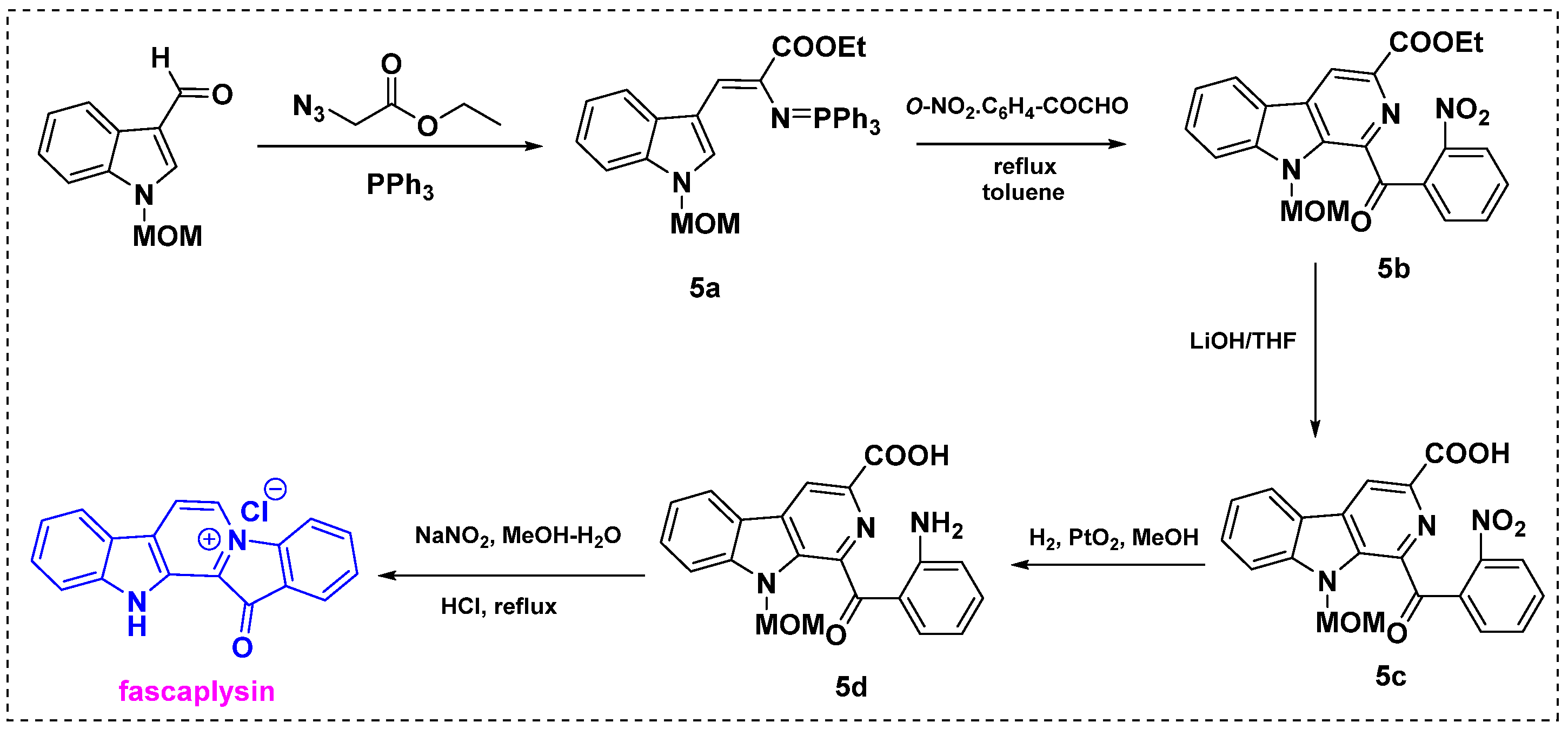
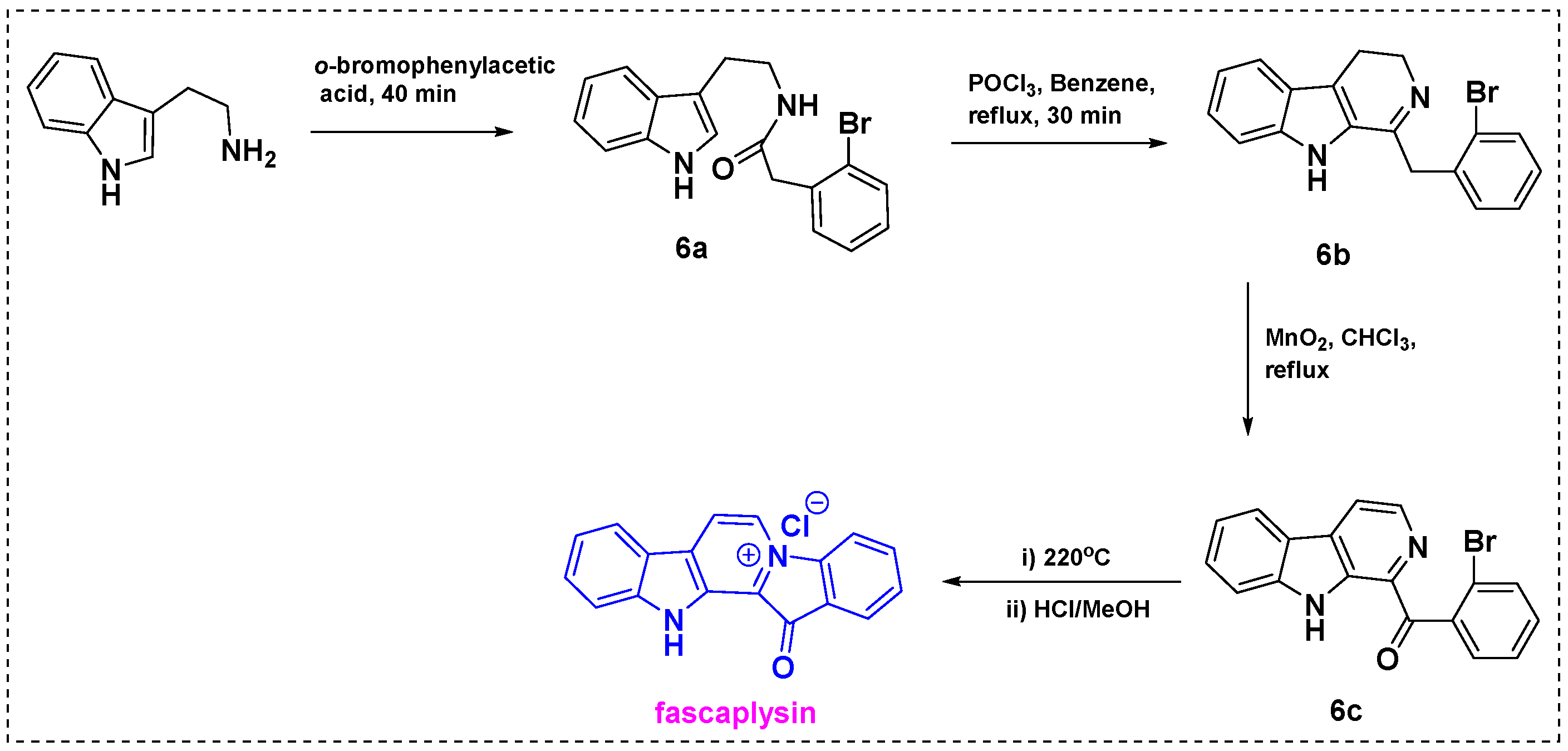
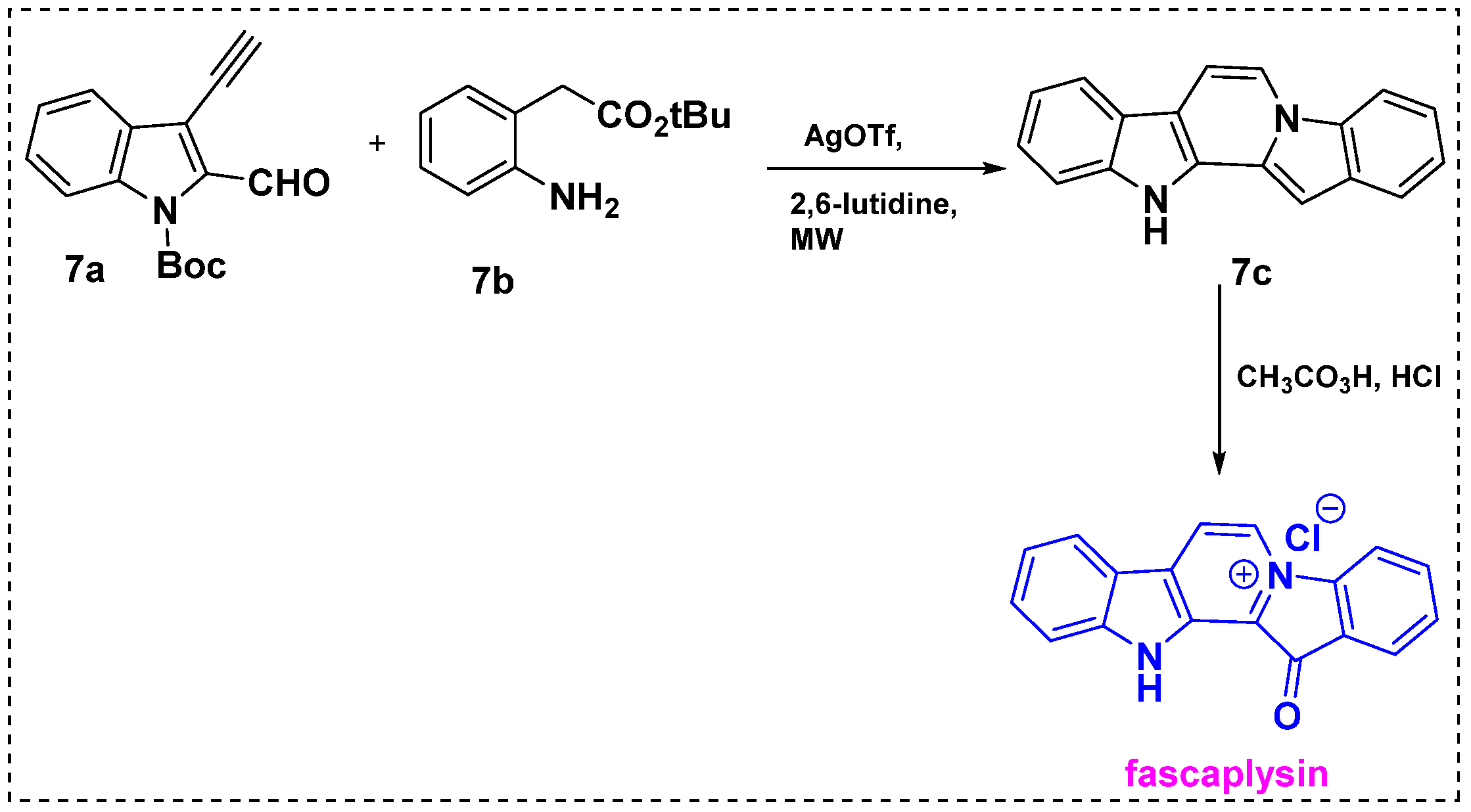
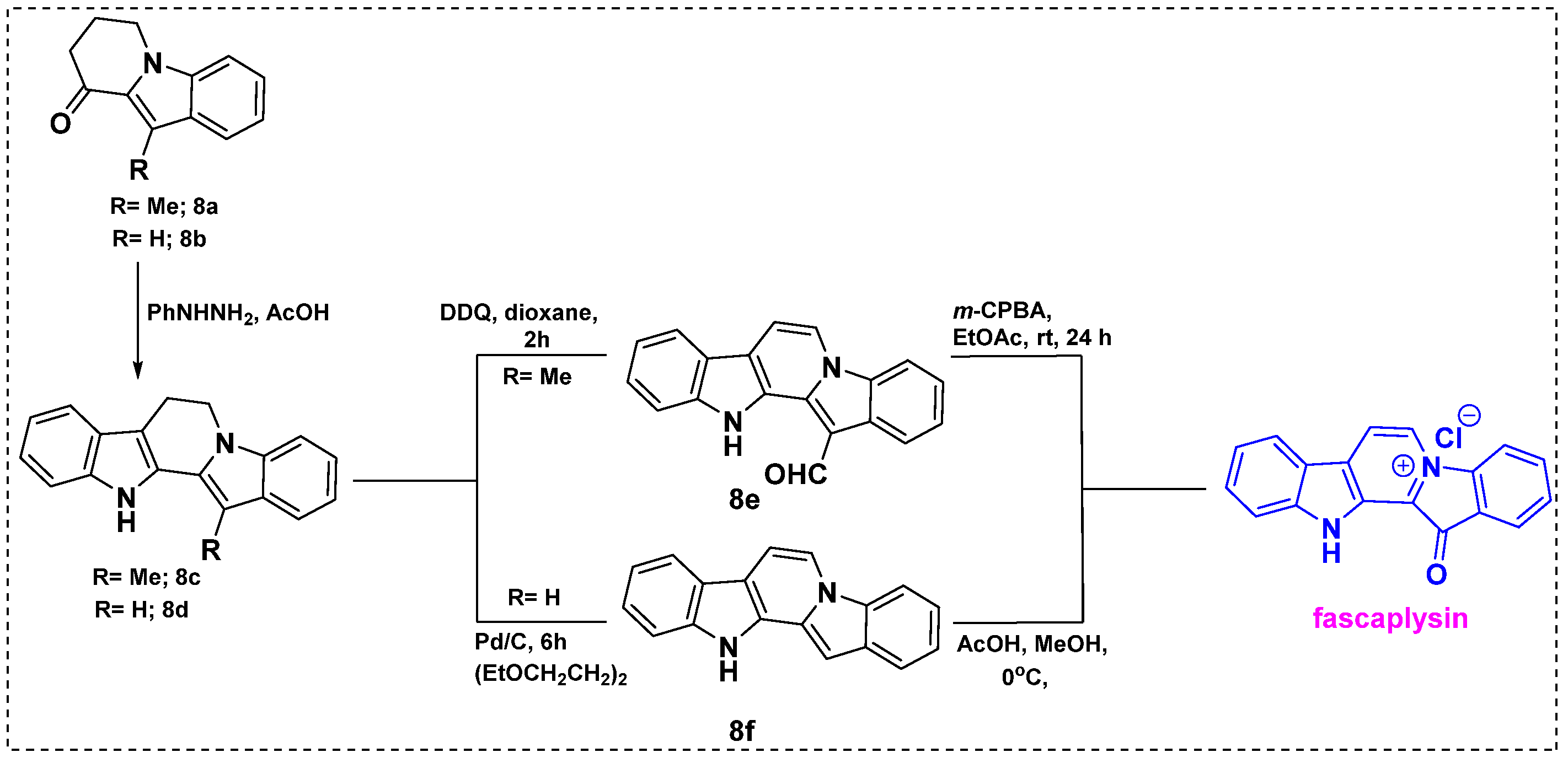
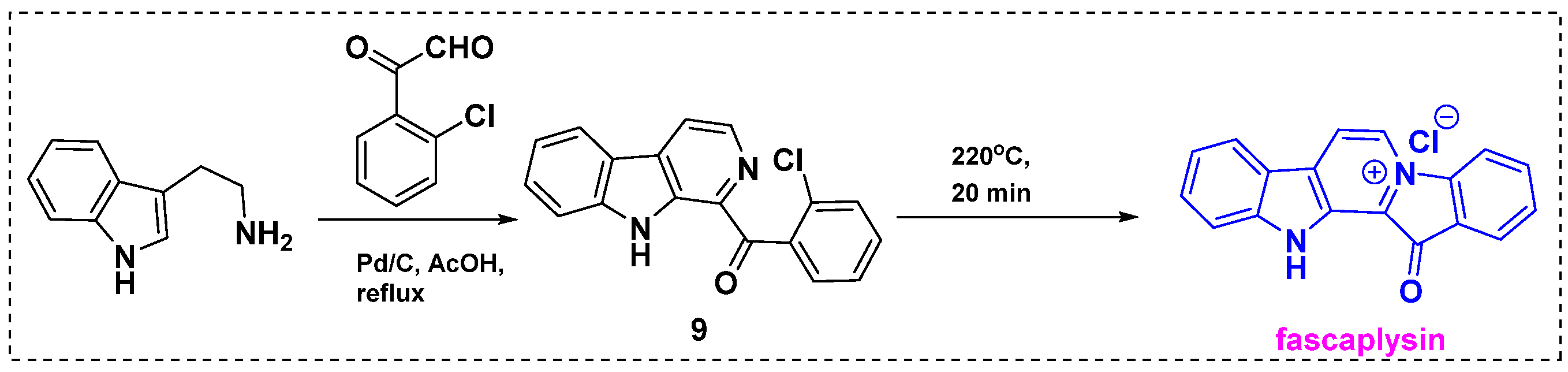
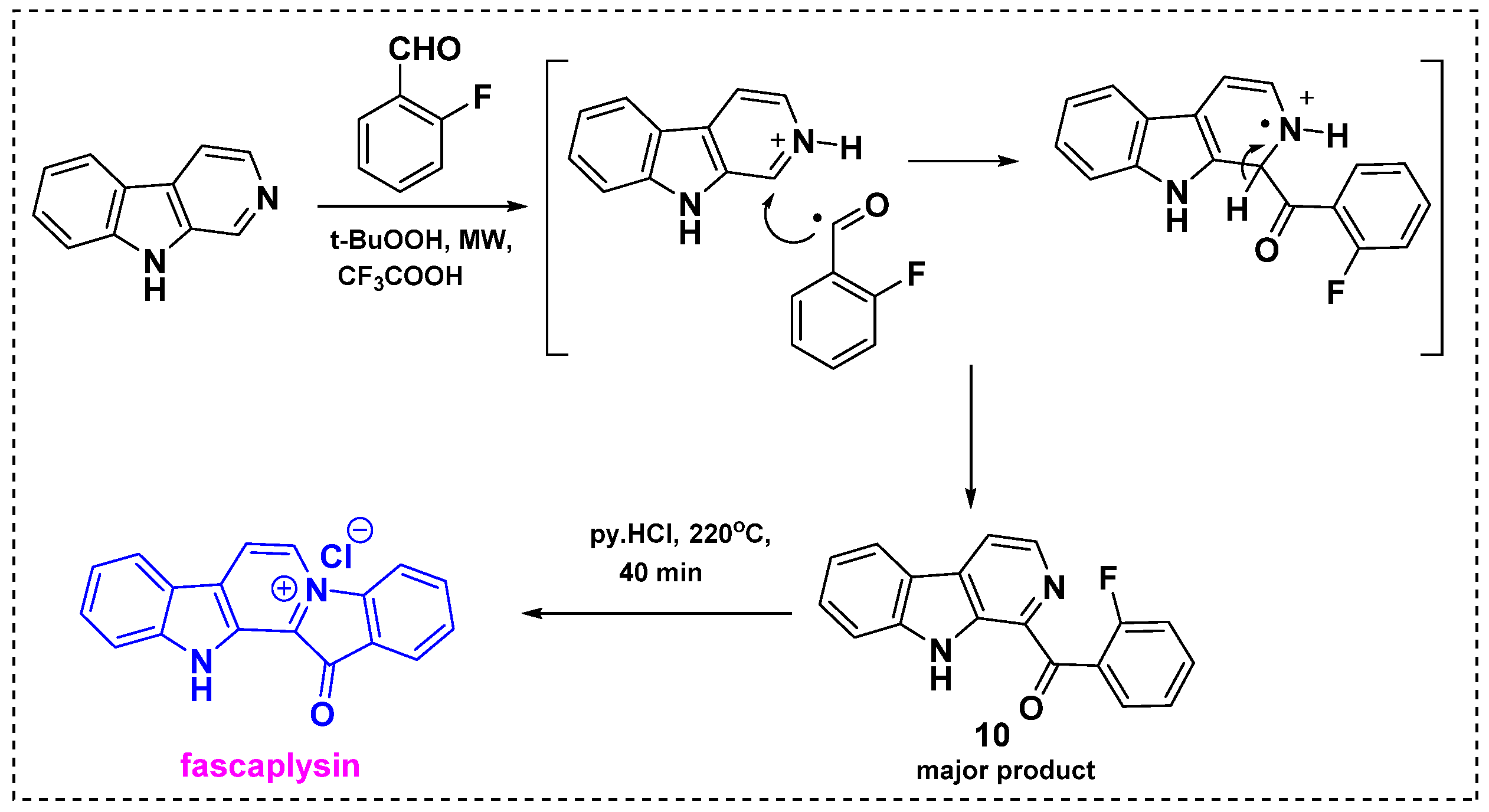
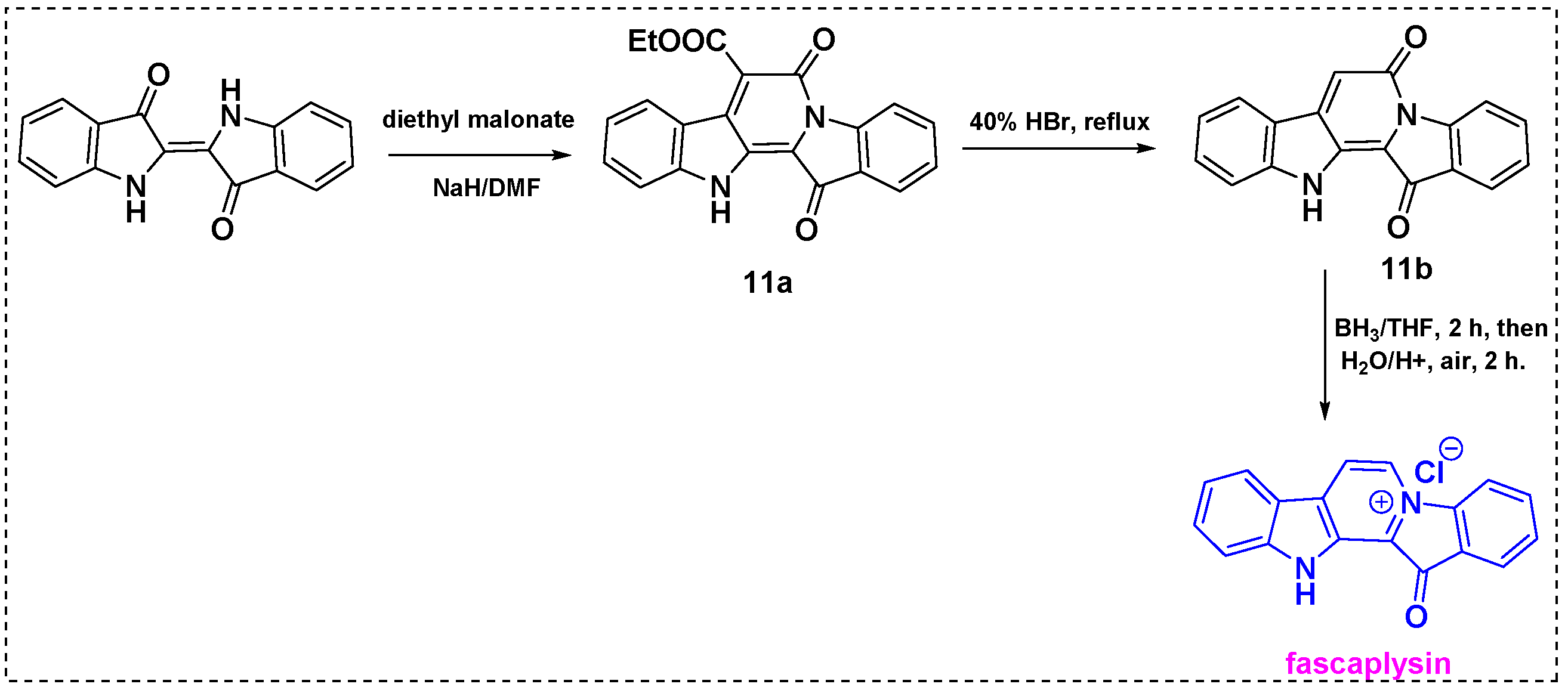
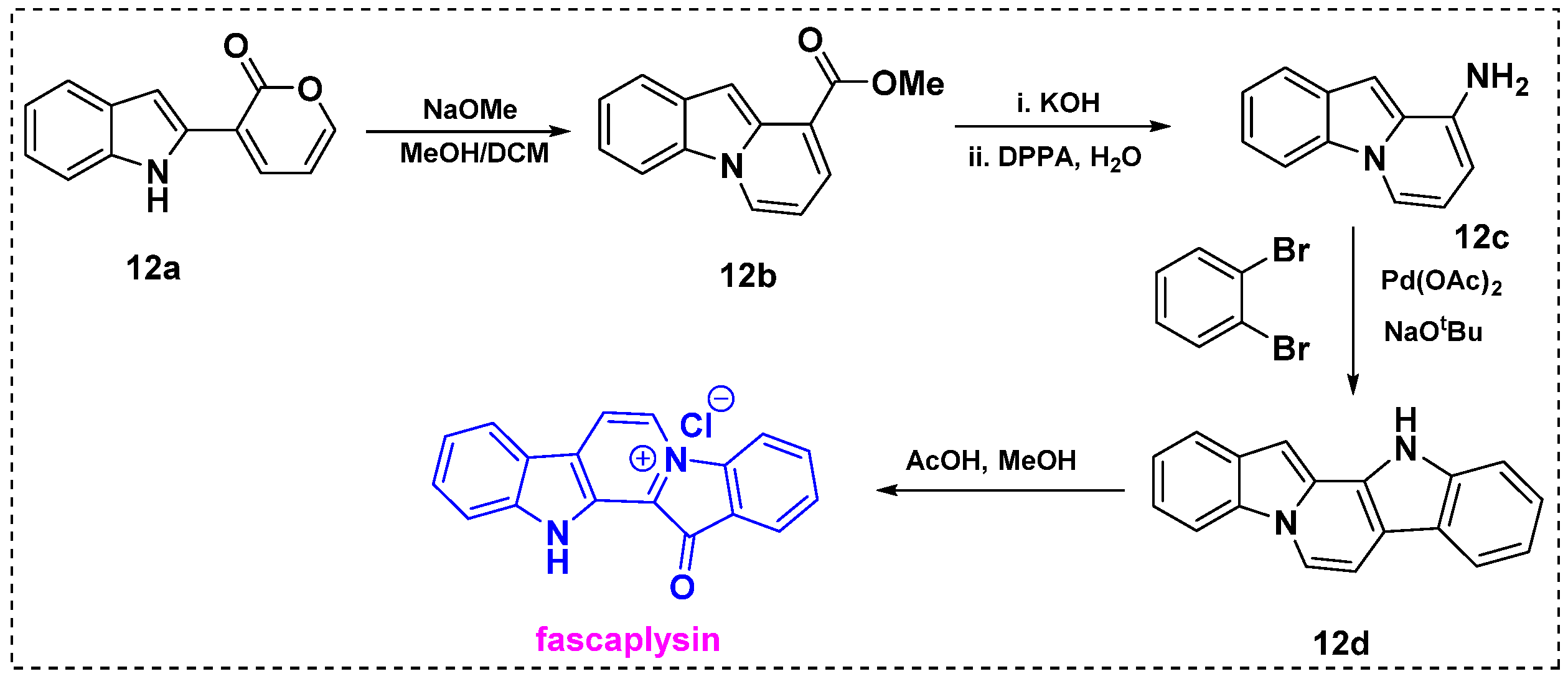
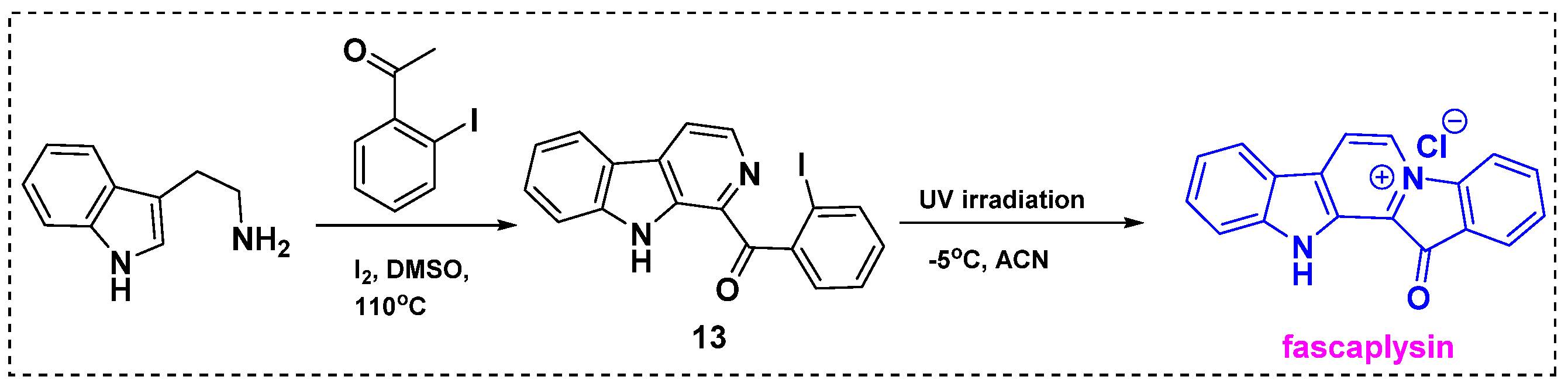
2.1. Total and Formal Synthesis of Fascaplysin
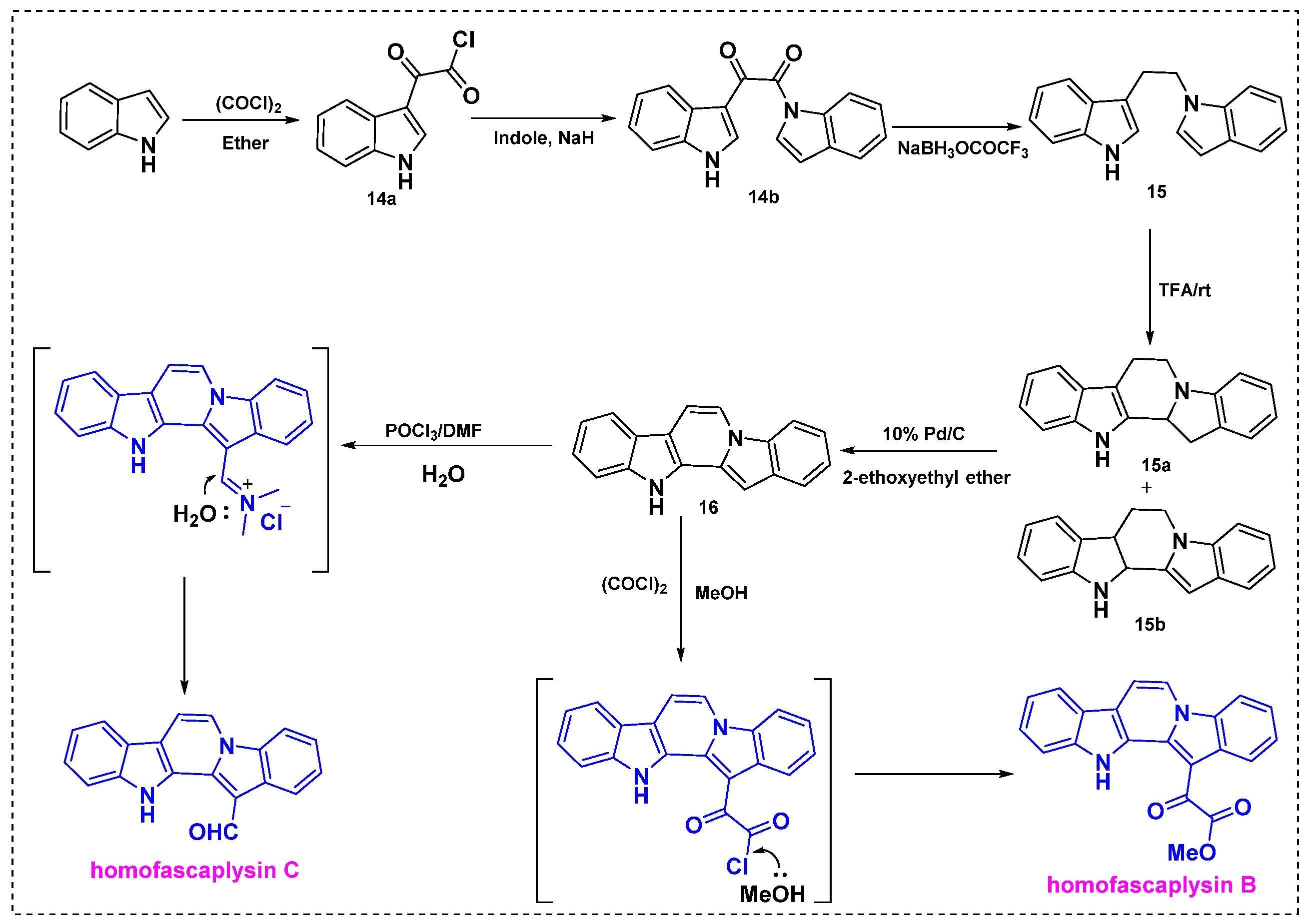
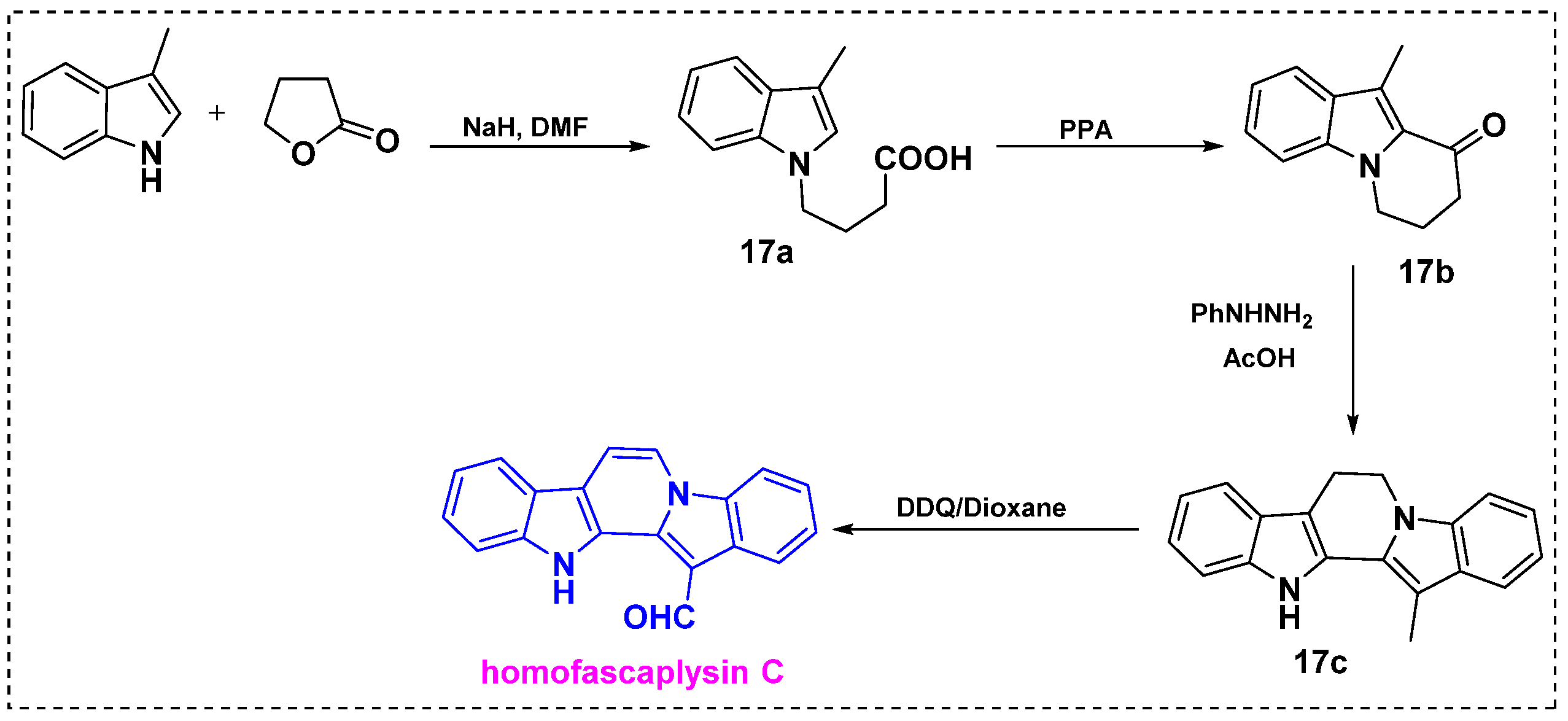
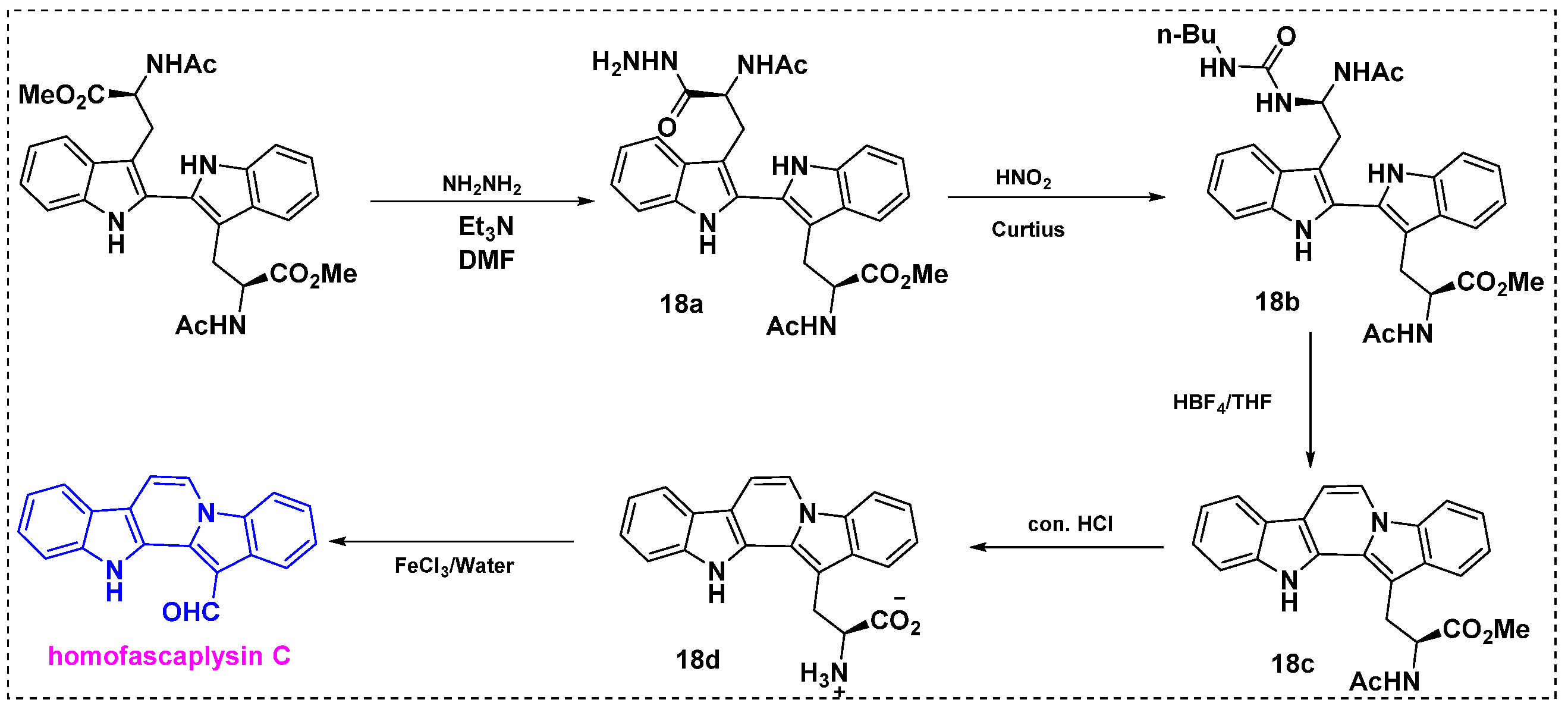
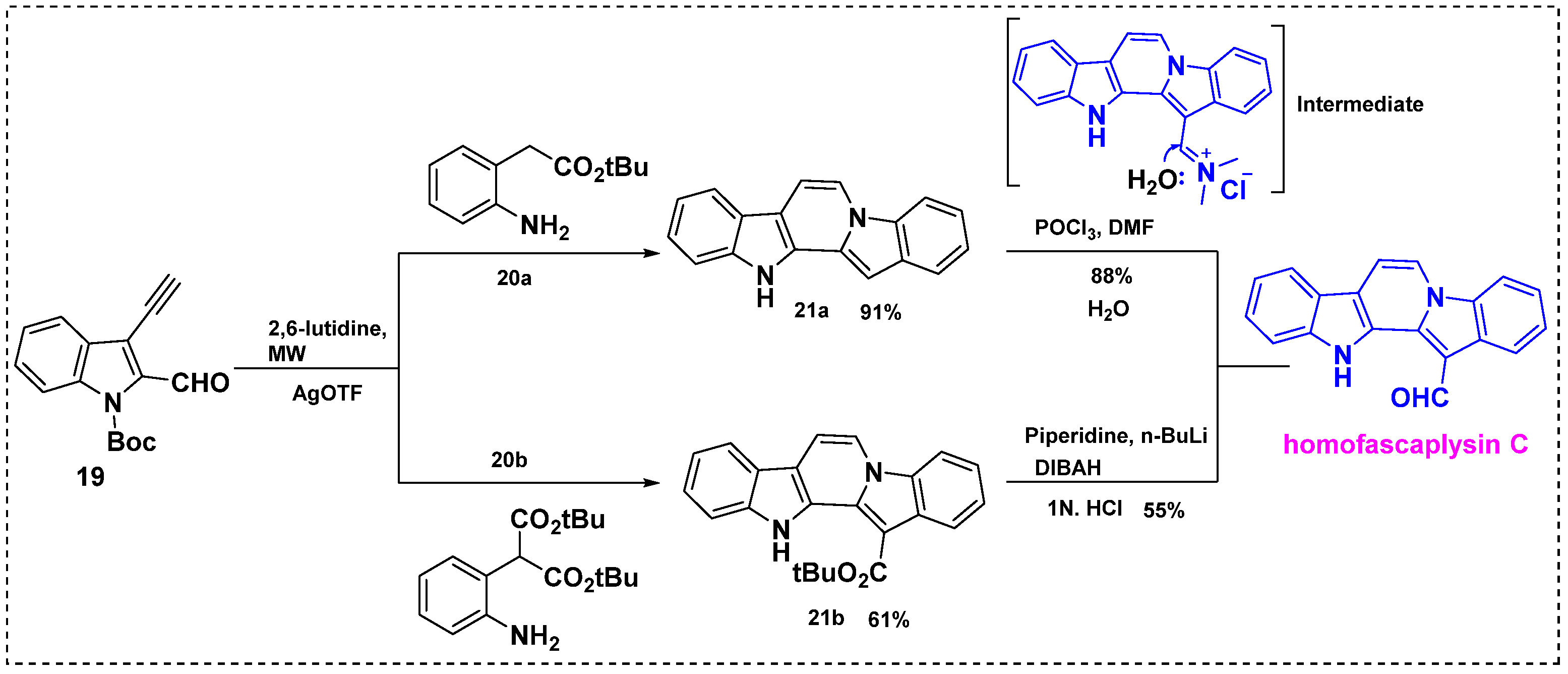
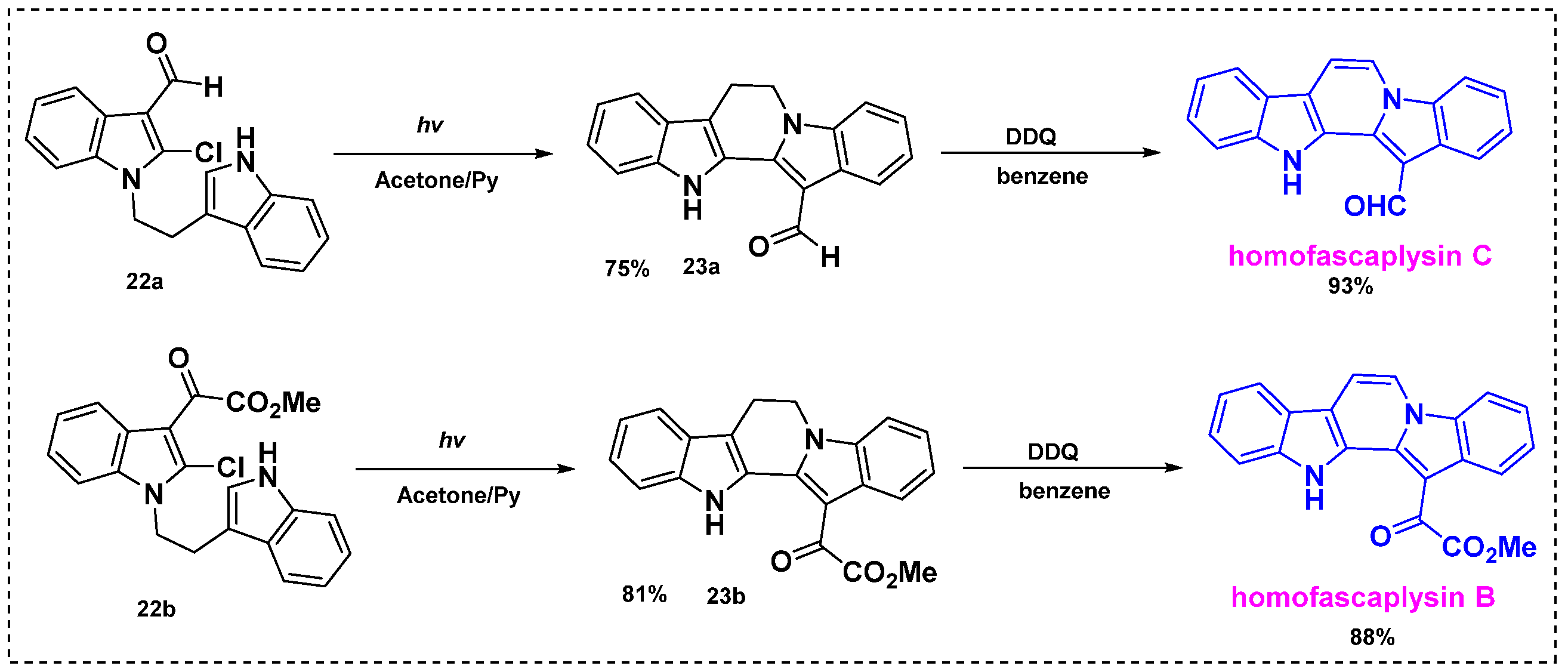
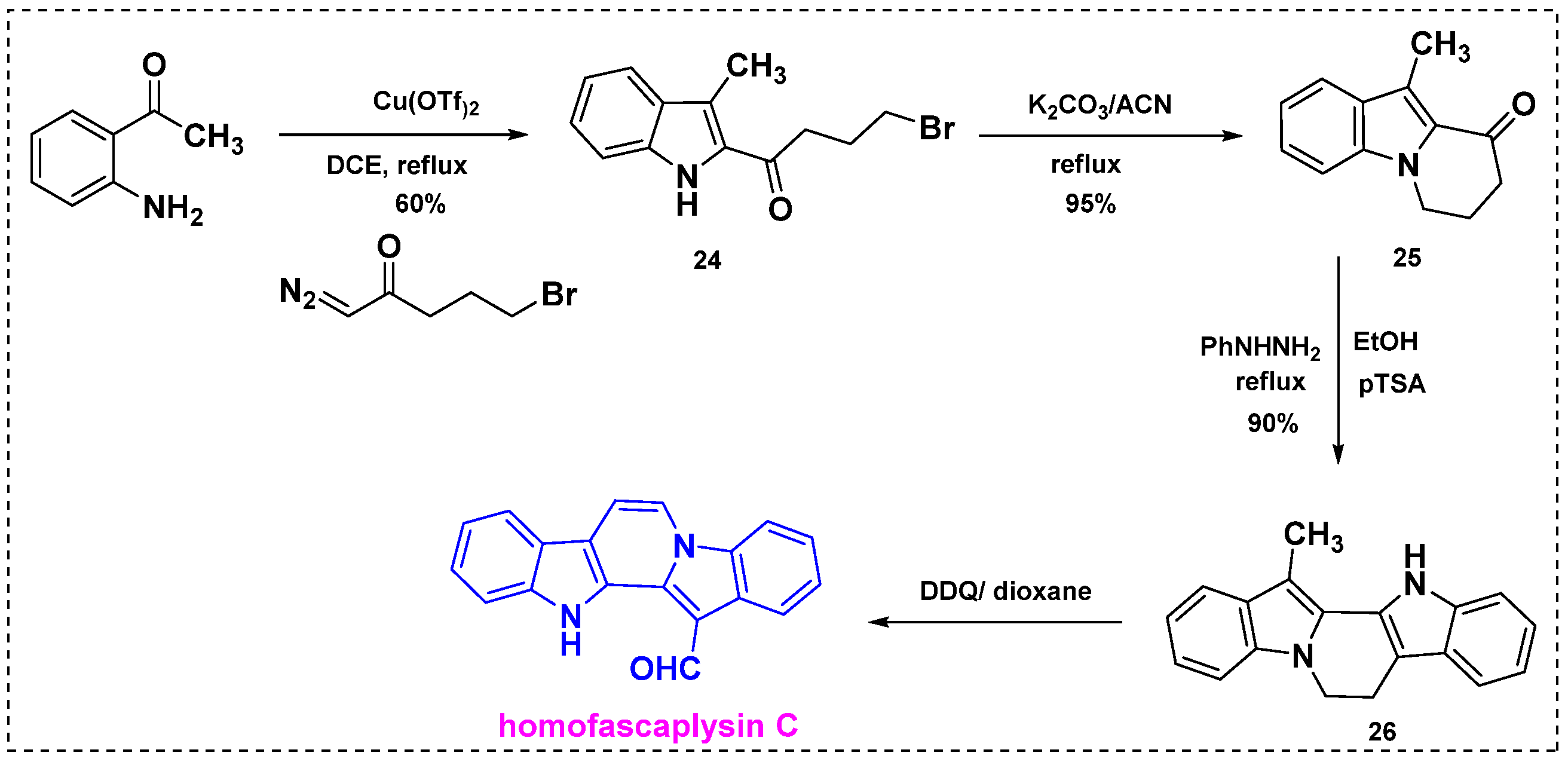
2.2. Total and Formal Synthesis of Homofascaplysins B–C
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosillo, M.; Gomez, A.G.; Dominguez, G.; Pérez-Castells, J. Chemistry of Biologically Active β-Carbolines. Targets Heterocycl. Syst. 2008, 12, 212–257. [Google Scholar]
- Lounasmaa, M.; Hanhinen, P.; Westersund, M. The Sarpagine Group of Indole Alkaloids. Alkaloids Chem. Biol. 1999, 52, 103–195. [Google Scholar]
- Hu, J.F.; Haman, R.T.; Hill, R.; Kelly, M. The manzamine alkaloids. Alkaloids Chem. Biol. 2003, 60, 207. [Google Scholar]
- Hesse, M. Alkaloids: Nature’s Curse or Blessing; Wiley–VCH: Weinheim, Germany, 2002. [Google Scholar]
- Bharate, S.B.; Manda, S.; Mupparapu, N.; Battini, N.; Vishwakarma, R.A. Chemistry and Biology of Fascaplysin, a Potent Marine-Derived CDK-4 Inhibitor. MiniRev. Med. Chem. 2012, 12, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Roll, D.M.; Ireland, C.M.; Lu, H.S.M.; Clardy, J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Kirsch, G.; Kong, G.M.; Wright, A.D.; Kaminsky, R. A new bioactive sesterterpene and antiplasmodial alkaloids from the marine sponge hyrtios cf. erecta. J. Nat. Prod. 2000, 63, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Charan, R.D.; McKee, T.C.; Gustafson, K.R.; Pannell, L.K.; Boyd, M.R. Thorectandramine, a novel β-carboline alkaloid from the marine sponge Thorectandra sp. Tetrahedron Lett. 2002, 43, 5201–5204. [Google Scholar] [CrossRef]
- Segraves, N.L.; Robinson, S.J.; Garcia, D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P.J. Comparison of fascaplysin and related alkaloids: A study of structures, cytotoxicities, and sources. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef]
- Jimenez, C.; Crews, P. Novel marine sponge derived amino acids 13. Additional psammaplin derivatives from Psammaplysilla purpurea. Tetrahedron 1991, 47, 2097–2102. [Google Scholar] [CrossRef]
- Jiménez, C.; Quiñoá, E.; Adamczeski, M.; Hunter, L.M.; Crews, P. Novel sponge-derived amino acids. 12. Tryptophan-derived pigments and accompanying sesterterpenes from Fascaplysinopsis reticulata. J. Org. Chem. 1991, 56, 3403. [Google Scholar] [CrossRef]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Mecker, S.Z.; Fretz, H.; Chaudhuri, B. Inhibition of cyclin-dependent kinase 4 (Cdk4) by fascaplysin, a marine natural product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Soni, R.; O’Reilly, T.; Furet, P.; Muller, L.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Fabbro, D.; Chaudhuri, B. Selective In Vivo and In Vitro Effects of a Small Molecule Inhibitor of Cyclin-Dependent Kinase 4. J. Natl. Cancer Inst. 2001, 93, 436–446. [Google Scholar] [CrossRef]
- Huwe, A.; Mazitschek, R.; Giannis, A. Small Molecules as Inhibitors of Cyclin-Dependent Kinases. Angew. Chem. Int. Ed. 2003, 42, 2122–2138. [Google Scholar] [CrossRef]
- Hormann, A.; Chaudhuri, B.; Fretz, H. DNA binding properties of the marine sponge pigment fascaplysin. Bioorg. Med. Chem. 2001, 9, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; Chapter 17. [Google Scholar]
- Wang, F.; Chen, H.; Yan, X.; Zheng, Y. Fascaplysin Sensitizes Cells to TRAIL-Induced Apoptosis through Upregulating DR5 Expression. Chin. J. Oceanol. Limnol. 2013, 31, 560–569. [Google Scholar] [CrossRef]
- Oh, T.I.; Lee, Y.M.; Nam, T.J.; Ko, Y.S.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.H.; Kim, Y.J.; Hong, S.; et al. Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 29, 2074. [Google Scholar] [CrossRef]
- Hamilton, G. Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines. Mar. Drugs. 2014, 12, 1377–1389. [Google Scholar] [CrossRef]
- Manda, S.; Sharma, S.; Wani, A.; Joshi, P.; Kumar, V.; Guru, S.K.; Bharate, S.S.; Bhushan, S.; Vishwakarma, R.A.; Kumar, A. Discovery of a Marine-Derived Bis-Indole Alkaloid Fascaplysin, as a New Class of Potent P-Glycoprotein Inducer and Establishment of Its Structure–Activity Relationship. Eur. J. Med. Chem. 2016, 107, 1–11. [Google Scholar] [CrossRef]
- Wang, C.; Wang, S.; Li, H.; Hou, Y.; Cao, H.; Hua, H.H.; Li, D. Marine-Derived Lead Fascaplysin: Pharmacological Activity, Total Synthesis, and Structural Modification. Mar. Drugs. 2023, 21, 226. [Google Scholar] [CrossRef]
- Gul, W.; Hamann, M. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef]
- Pelcman, B.; Gribble, G.W. Total synthesis of the marine sponge pigment fascaplysin. Tetrahedron Lett. 1990, 31, 2381. [Google Scholar] [CrossRef]
- Rocca, P.; Marsais, F.; Godard, A.; Qukguiner, G. A short synthesis of the antimicrobial marine sponge pigment fascaplysin. Tetrahedron Lett. 1993, 34, 7917–7918. [Google Scholar] [CrossRef]
- Molina, P.; Fresneda, P.M.; Zafra, S.G.; Almendros, P. Iminophosphorane-mediated syntheses of the fascaplysin alkaloid of marine origin and nitramarine. Tetrahedron Lett. 1994, 35, 8851–8854. [Google Scholar] [CrossRef]
- Radchenko, O.S.L.; Novikov, V.L.; Elyakov, G.B. A simple and practical approach to the synthesis of the marine sponge pigment fascaplysin and related compounds. Tetrahedron Lett. 1997, 38, 5339. [Google Scholar] [CrossRef]
- Waldmann, H.; Eberhardt, L.; Wittsteinab, K.; Kumar, K. Silver catalyzed cascade synthesis of alkaloid ring systems: Concise total synthesis of fascaplysin, homofascaplysin C and analogues. Chem. Commun. 2010, 46, 4622–4624. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Baranova, O.V.; Kravchenko, N.S.; Dubovitskii, S.V. A new method for the synthesis of the marine alkaloid fascaplysin. Tetrahedron Lett. 2010, 51, 6498–6499. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Joshi, P.; Singh, B.; Vishwakarma, R. Total synthesis and anti-cholinesterase activity of marine-derived bis-indole alkaloid fascaplysin. Med. Chem. Commun. 2012, 3, 1098–1103. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kaminskii, V.A. A new method for the synthesis of the marine alkaloid fascaplysin based on the microwave-assisted Minisci reaction. Tetrahedron Lett. 2013, 54, 3530–3532. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kantemirov, A.V.; Koisevnikov, A.V.; Andin, A.N.; Kuzmich, A.S. Syntheses of the marine alkaloids 6-oxofascaplysin, fascaplysin and their derivatives. Tetrahedron Lett. 2018, 59, 708–711. [Google Scholar] [CrossRef]
- Palani, V.; Perea, M.A.; Gardner, K.E.; Sarpong, R. A pyrone remodeling strategy to access diverse heterocycles: Application to the synthesis of fascaplysin natural products. Chem. Sci. 2021, 12, 1528–1534. [Google Scholar] [CrossRef]
- Tryapkin, O.A.; Kantemirov, A.V.; Dyshlovoy, S.A.; Prassolov, V.S.; Spirin, P.V.; Amsberg, G.V.; Sidorova, M.A.; Zhidkov, M.E. A New Mild Method for Synthesis of Marine Alkaloid Fascaplysin and Its Therapeutically Promising Derivatives. Mar. Drugs 2023, 21, 424. [Google Scholar] [CrossRef]
- Popov, A.M.; Stonik, V.A. Physiological activity of fascaplysine, an unusual pigment from tropic sea sponges. Antibiot. Khimioter. 1991, 36, 12–14. [Google Scholar]
- Gribble, G.W.; Pelcman, B. Total syntheses of the marine sponge pigments fascaplysin and homofascaplysin B and C. J. Org. Chem. 1992, 57, 3636–3642. [Google Scholar] [CrossRef]
- Dubovitskii, S.V. Method for synthesis of 12H-pyrido[1,2-a:3,4-b′]diindoles. Total synthesis of homofascaplysin C. Tetrahedron Lett. 1996, 37, 5207–5208. [Google Scholar] [CrossRef]
- Carter, D.S.; Van Vranken, D.L. Synthesis of Homofascaplysin C and Indolo[2,3-a] carbazole from Ditryptophans. J. Org. Chem. 1999, 64, 8537–8545. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, W.; Wang, K.; Wang, W.; Zhang, W. Synthesis of homofascaplysin B, C, and analogues by the photocyclization of 3-acyl-2-chloro-1-[2-(indol-3-yl) ethyl] indoles. Tetrahedron 2013, 69, 1912–1918. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Reddy, M.R.; Rao, Y.G.; Yadav, J.S.; Sridhar, B. Cu(OTf)2-Catalyzed Synthesis of 2,3-Disubstituted Indoles and 2,4,5-Trisubstituted Pyrroles from α-Diazoketones. Org. Lett. 2013, 15, 464–467. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Sidorova, M.A.; Lyakhova, I.A. One-step transformation of the marine alkaloid fascaplysin into homofascaplysins B and B-1. The first syntheses of 3-bromohomofascaplysin B and 3–bromohomofascaplysin B-1. Tetrahedron Lett. 2018, 59, 1417–1420. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mittapalli, R.R.; Kumari, H. Recent Advances in the Synthesis of the Marine-Derived Alkaloid Fascaplysin and Its Metabolites Homofascaplysins A–C. Molecules 2024, 29, 1590. https://doi.org/10.3390/molecules29071590
Mittapalli RR, Kumari H. Recent Advances in the Synthesis of the Marine-Derived Alkaloid Fascaplysin and Its Metabolites Homofascaplysins A–C. Molecules. 2024; 29(7):1590. https://doi.org/10.3390/molecules29071590
Chicago/Turabian StyleMittapalli, Ramana Reddy, and Harshita Kumari. 2024. "Recent Advances in the Synthesis of the Marine-Derived Alkaloid Fascaplysin and Its Metabolites Homofascaplysins A–C" Molecules 29, no. 7: 1590. https://doi.org/10.3390/molecules29071590
APA StyleMittapalli, R. R., & Kumari, H. (2024). Recent Advances in the Synthesis of the Marine-Derived Alkaloid Fascaplysin and Its Metabolites Homofascaplysins A–C. Molecules, 29(7), 1590. https://doi.org/10.3390/molecules29071590