
Distinction and Quantification of Noncovalent Dispersive and Hydrophobic Effects


Abstract
1. Introduction
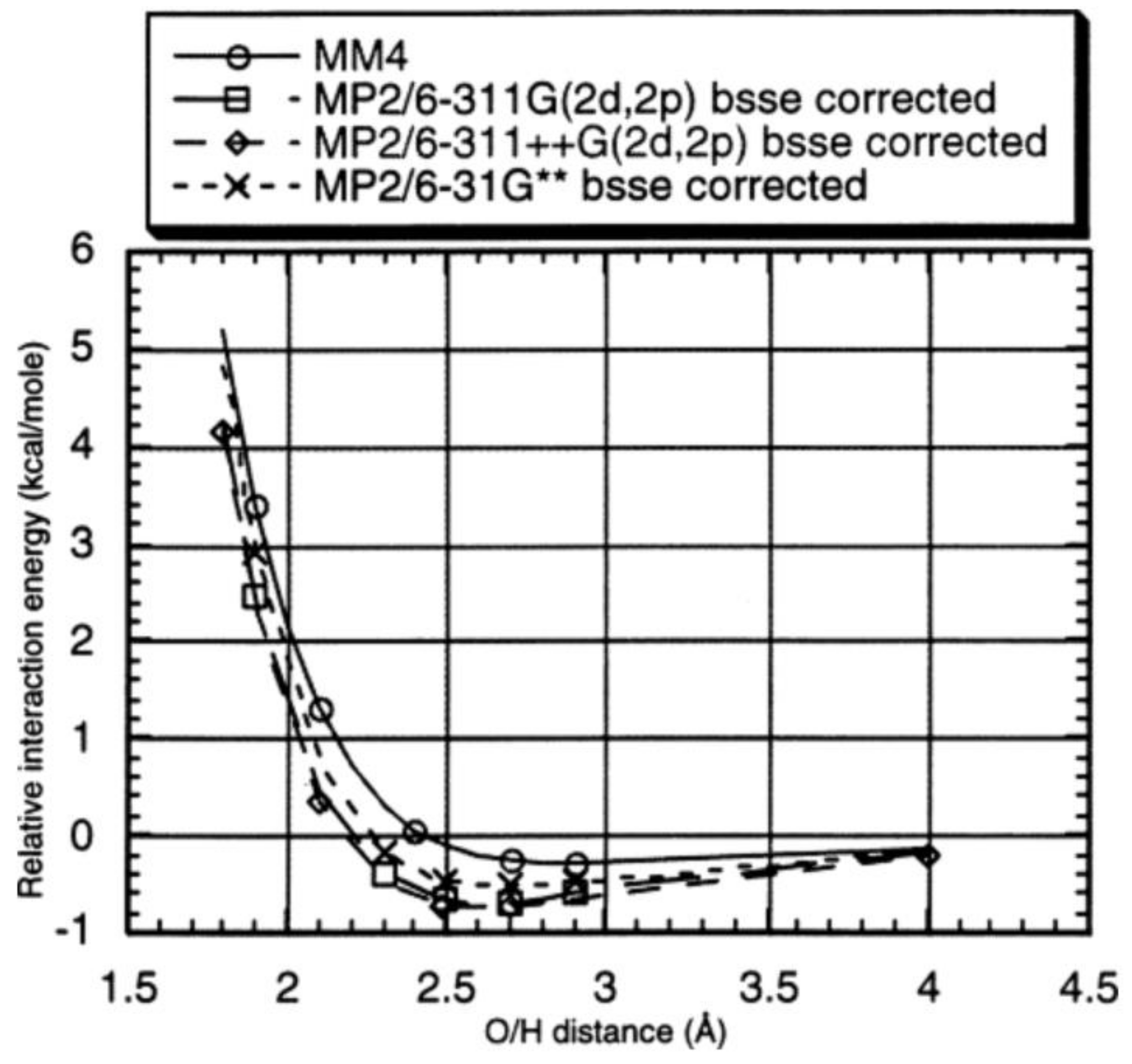
2. Evaluation of Noncovalent Interactions and Benchmarking in Different States
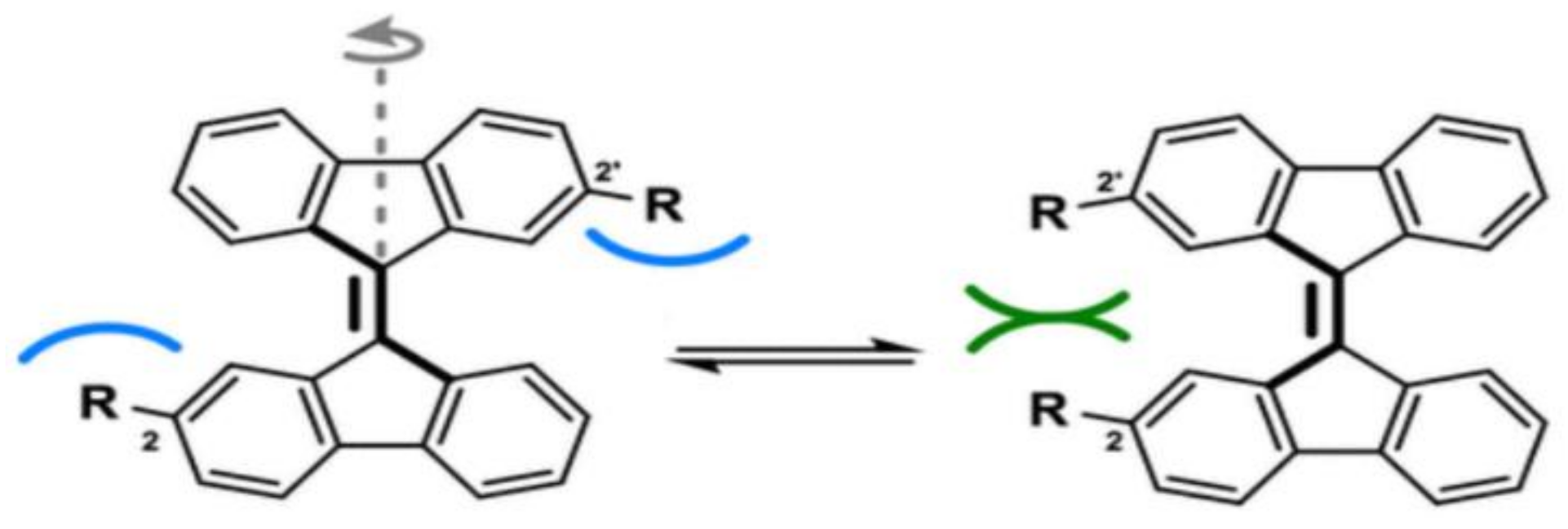
3. London Dispersive Interactions
4. Association Energy between Molecules in Solution—Entropic Contributions as a Major Problem
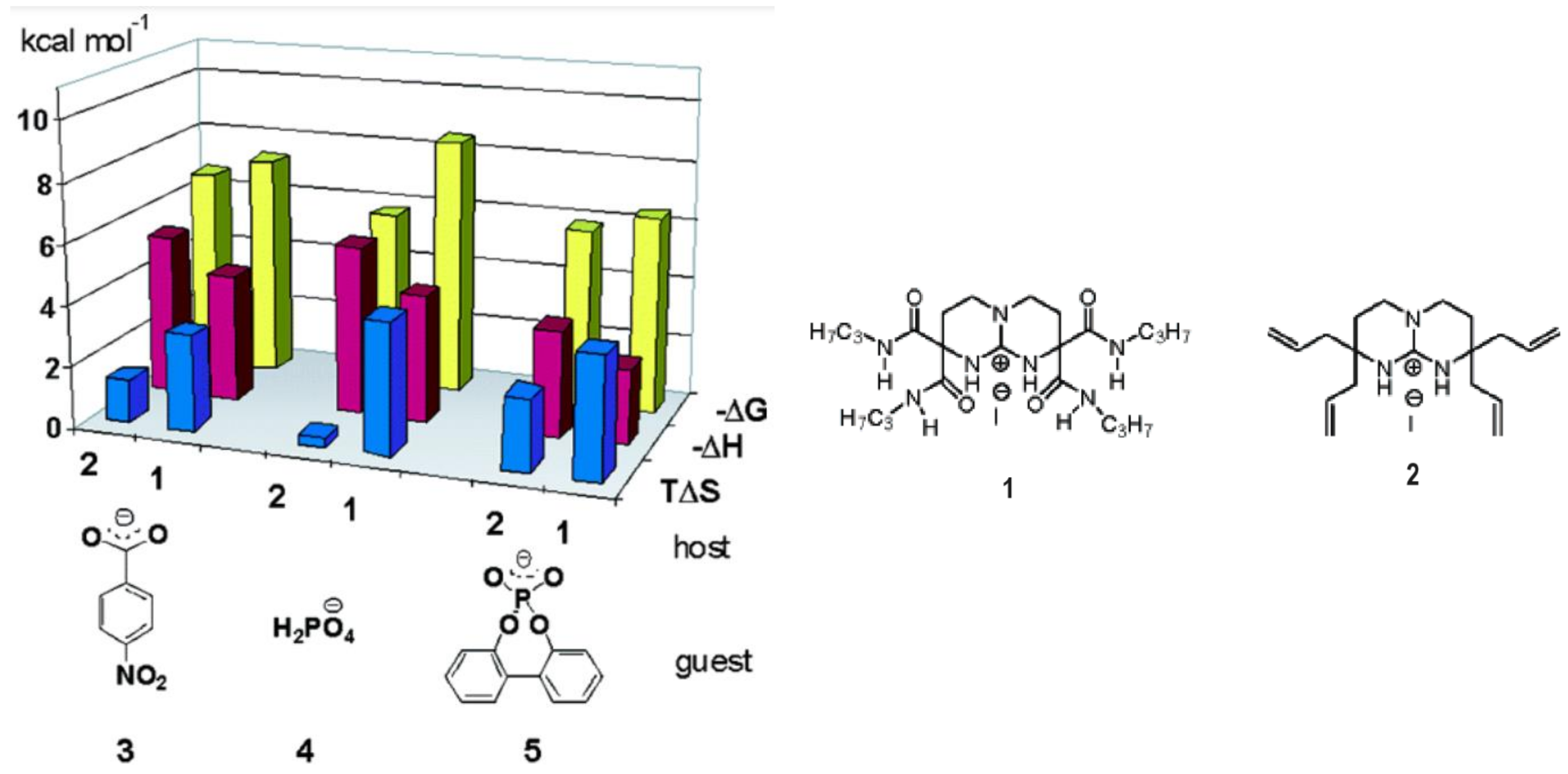
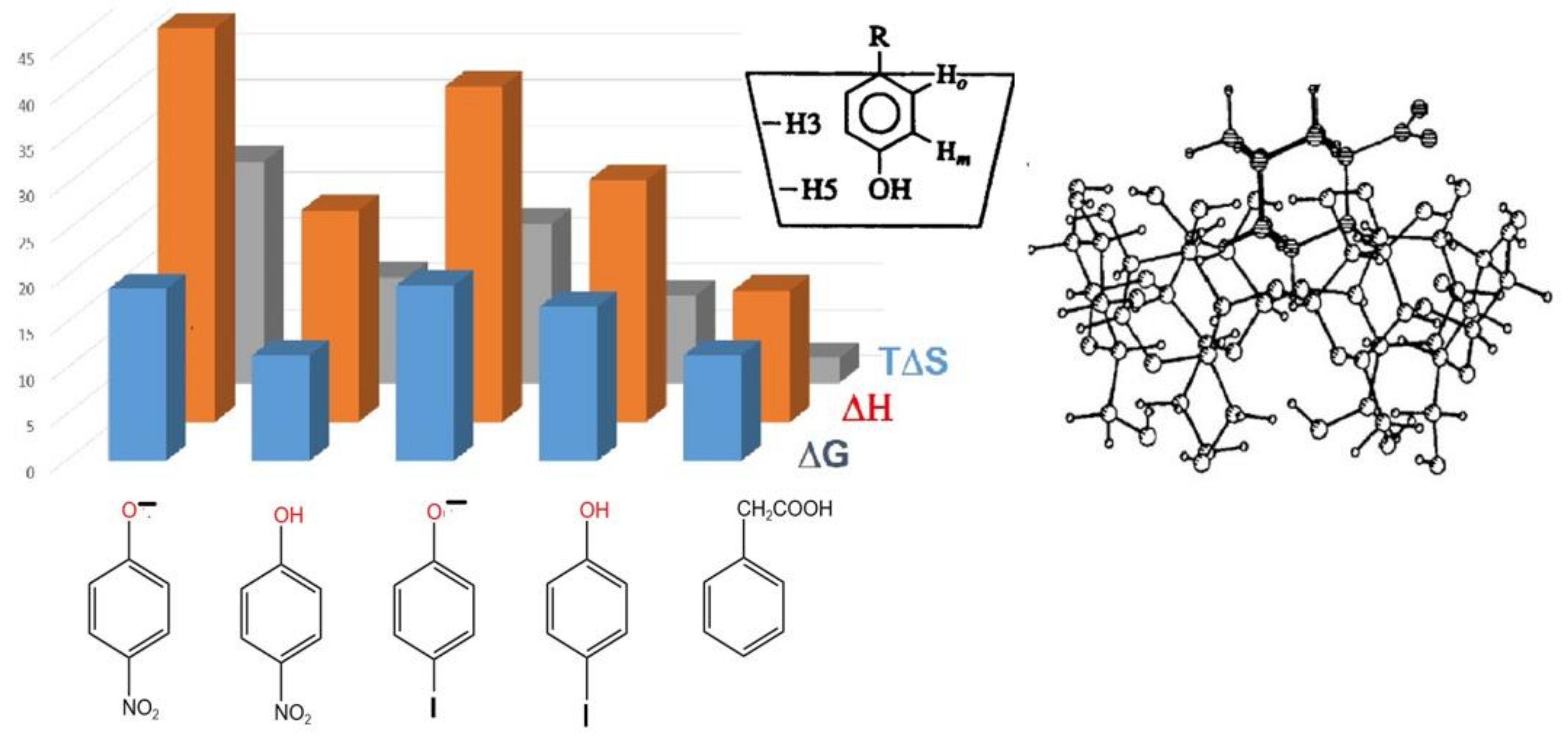
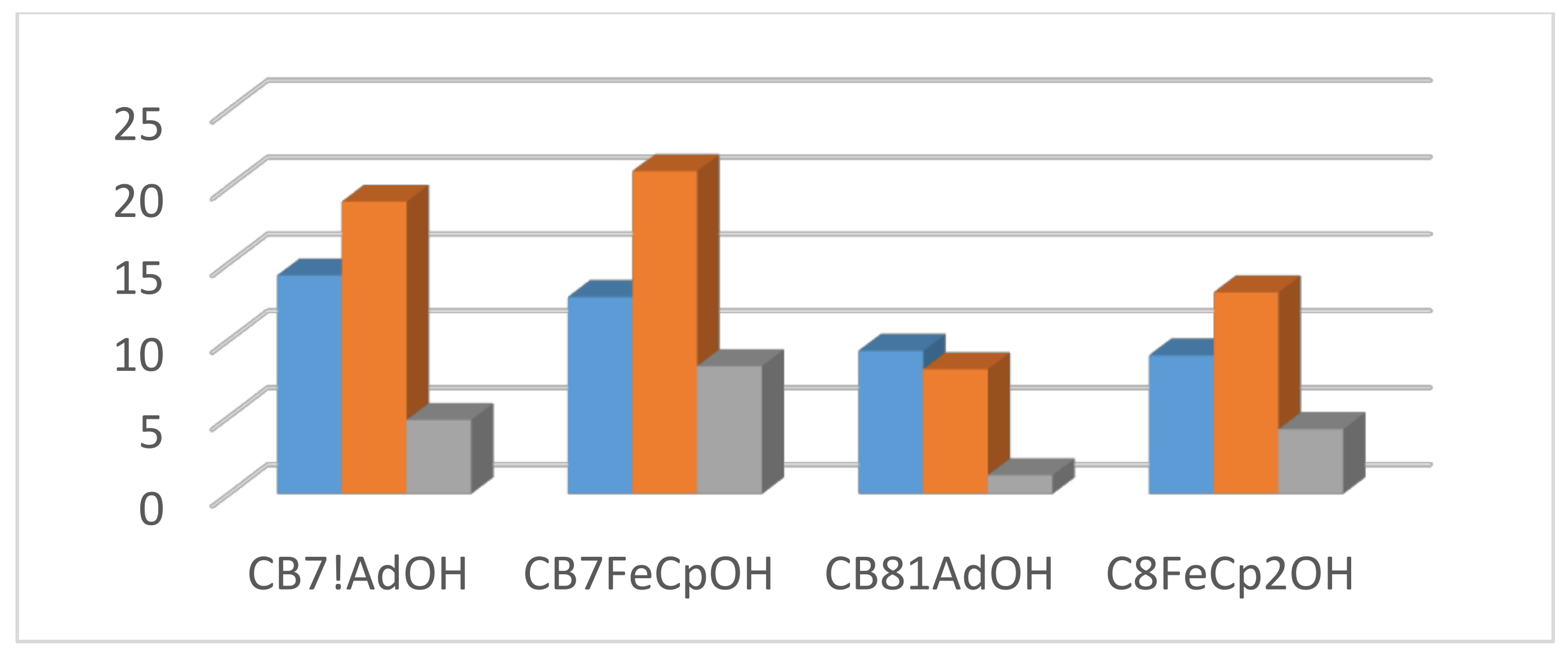
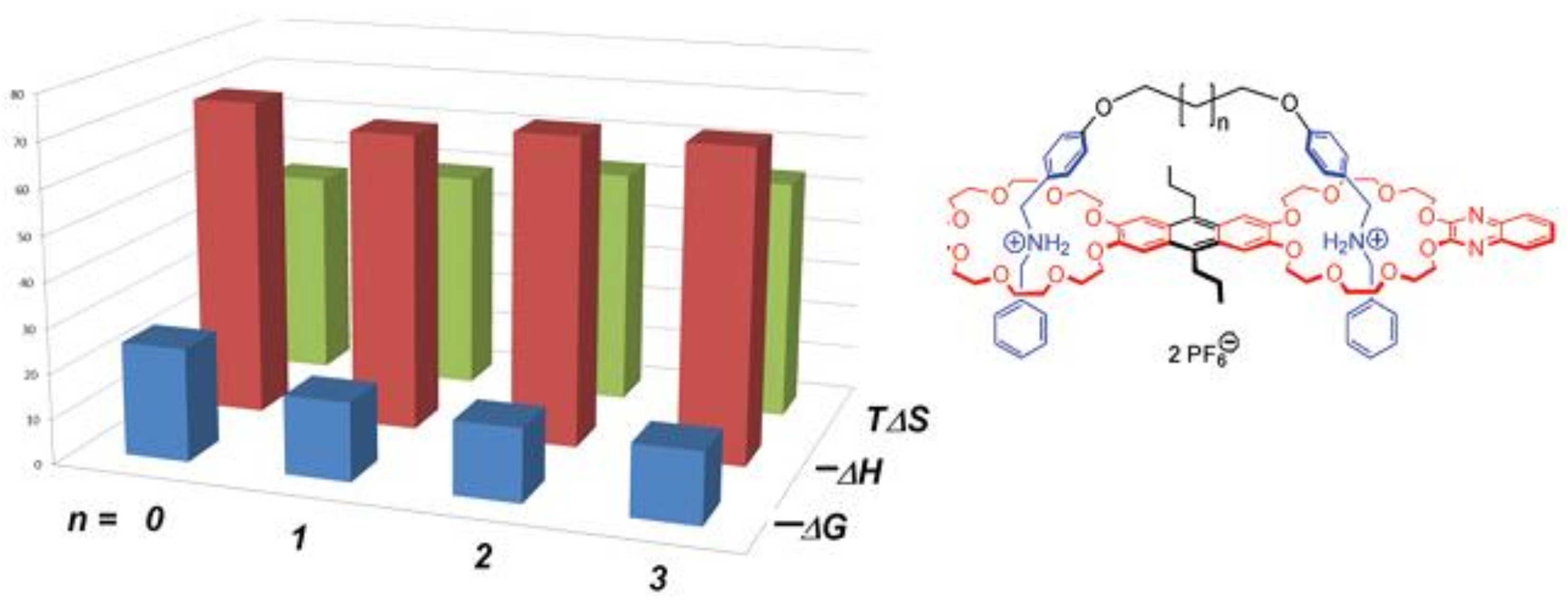
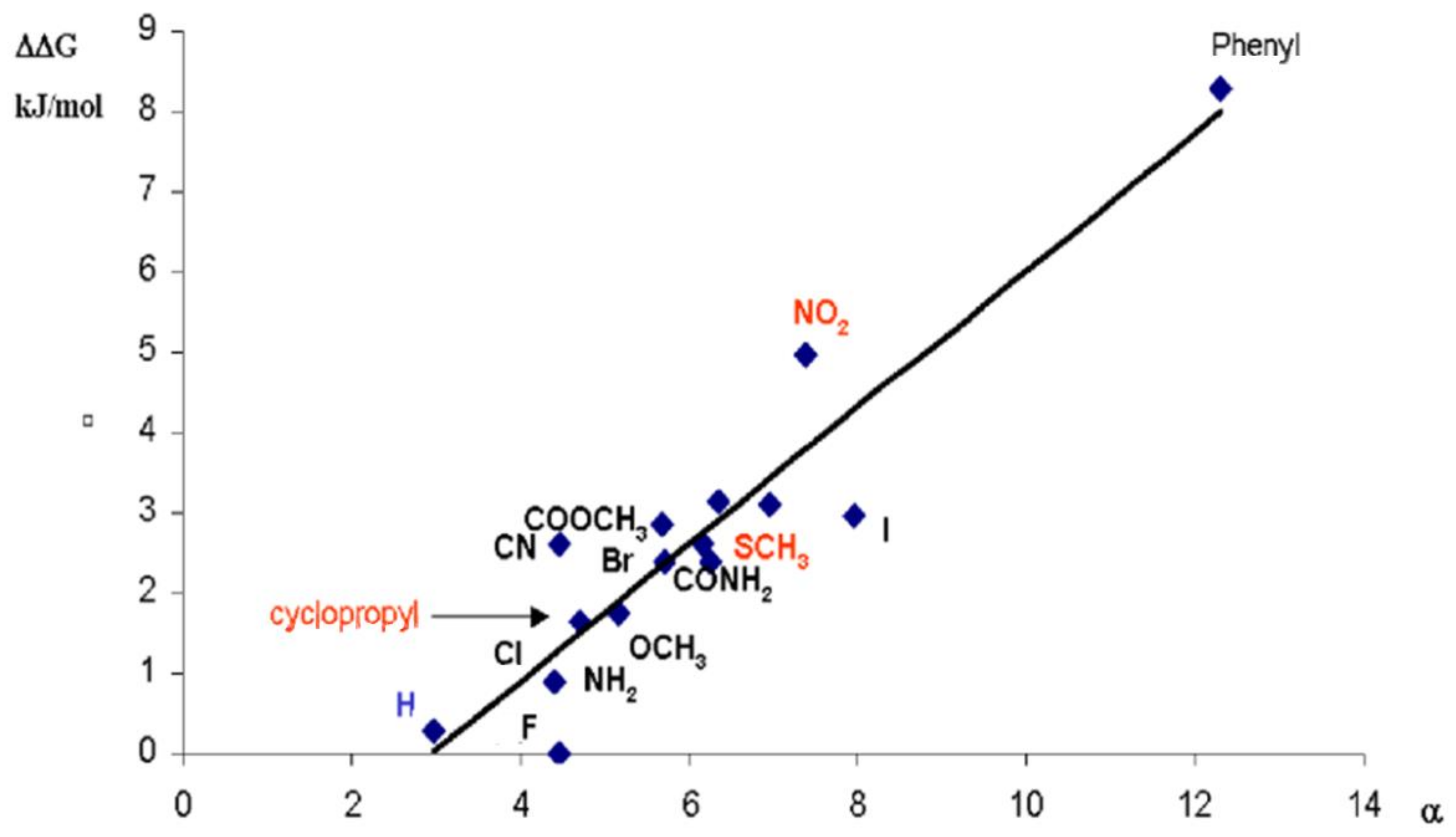
5. Consistent Experimental Free Energy Increments for Dispersive Interactions
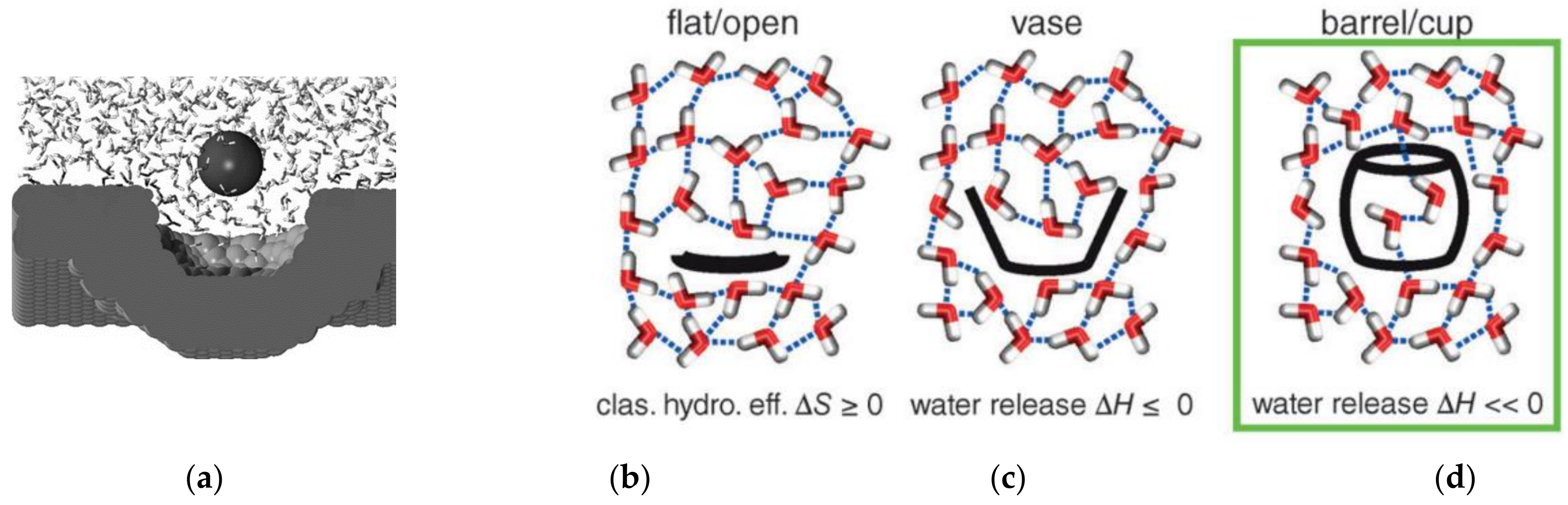
6. Hydrophobic Effects
7. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Note
- Schneider, H.-J. Dispersive interactions in solution complexes. Acc. Chem. Res. 2015, 48, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Cerny, J.; Hobza, P. Non-covalent interactions in biomacromolecules. Phys. Chem. Chem. Phys. 2007, 9, 5291–5303. [Google Scholar] [CrossRef] [PubMed]
- Gravillier, L.-A.; Cockroft, S.L. Context-Dependent Significance of London Dispersion. Acc. Chem. Res. 2023, 56, 3535–3544. [Google Scholar] [CrossRef] [PubMed]
- See Special issue “Dispersion Interactions in Chemistry” in Acc. Chem. Res. 2023, 25.
- Assaf, K.I.; Nau, M. Dispersion Interactions in Condensed Phases and inside Molecular Containers. Acc Chem. Res. 2023, 56, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A. Quantifying Intermolecular Interactions: Guidelines for the Molecular Recognition Toolbox. Angew. Chem. Int. Ed. 2004, 43, 5310–5324. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.D. A solution to dispersion interactions. Nat. Chem. 2013, 5, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A. Van der Waals interactions in non-polar liquids. Chem. Sci. 2013, 4, 834–848. [Google Scholar] [CrossRef]
- Hwang, J.; Dial, B.E.; Li, P.; Kozik, M.E.; Smith, M.D.; Shimizu, K.D. How important are dispersion interactions to the strength of aromatic stacking interactions in solution? Chem. Sci. 2015, 6, 4358. [Google Scholar] [CrossRef] [PubMed]
- Bravin, C.; Piekos, J.A.; Licini, G.; Hunter, C.A.; Zonta, C. Dissection of the Polar and Non-Polar Contributions to Aromatic Stacking Interactions in Solution. Angew. Chem. Int. Ed. 2021, 60, 23871–23877. [Google Scholar] [CrossRef]
- Liu, H.; Shimizu, K.D. Contributions of London Dispersion Forces to Solution-Phase Association Processes. Acc. Chem. Res. 2023, 56, 3572–3580. [Google Scholar] [CrossRef]
- Schneider, H.-J. Noncovalent interactions: A brief account of a long history. J. Phys. Org. Chem. 2022, 35, e4340. [Google Scholar] [CrossRef]
- Gutmann, V. The Donor-Acceptor Approach to Molecular Interactions; Plenum: New York, NY, USA, 1978. [Google Scholar]
- Schneider, H.-J. Linear Free Energy Relations and Pairwise Interactions in Supramolecular Chemistry. Chem. Soc. Rev. 1994, 22, 227–234. [Google Scholar] [CrossRef]
- Dunitz, J.G.; Gavezzotti, A. How molecules stick together in organic crystals: Weak intermolecular interactions. Chem. Soc. Rev. 2009, 38, 2622–2633. [Google Scholar] [CrossRef] [PubMed]
- Drago, R.S. Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 1973; p. 73. [Google Scholar]
- Schneider, H.-J. Binding mechanisms in supramolecular complexes. Angew. Chem. Int. Ed. Engl. 2009, 48, 3924–3977. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, F.; Schneider, H.J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef]
- Dunitz, J.G.; Gavezzotti, A. Supramolecular Synthons: Validation and Ranking of Intermolecular Interaction Energies. Cryst. Growth Des. 2012, 12, 5873–5877. [Google Scholar] [CrossRef]
- Beran, G.J.O. Frontiers of molecular crystal structure prediction for pharmaceuticals and functional organic materials. Chem. Sci. 2023, 14, 13290–13312. [Google Scholar] [CrossRef] [PubMed]
- Beran, G.J.O.; Hartman, J.-D.; Heit, Y.N. Predicting Molecular Crystal Properties from First Principles: Finite-Temperature Thermochemistry to NMR Crystallography. Acc. Chem. Res. 2016, 49, 2501–2508. [Google Scholar] [CrossRef]
- Cervinka, C.; Beran, G.J.O. Towards reliable ab initio sublimation pressures for organic molecular crystals—Are we there yet? Phys. Chem. Chem. Phys. 2019, 21, 14799–14810. [Google Scholar] [CrossRef]
- Israelachvili, J.; Ruths, M. Brief History of Intermolecular and Intersurface Forces in Complex Fluid Systems. Langmuir 2013, 29, 9605–9619. [Google Scholar] [CrossRef]
- Holstein, B.R. The van der Waals interaction. Am. J. Phys. 2001, 69, 441–449. [Google Scholar] [CrossRef]
- Schneider, H.-J.; Yatsimirsky, A. Selectivity in supramolecular host–guest complexes. Chem. Soc. Rev. 2008, 37, 263–277. [Google Scholar] [CrossRef]
- Goldfarb, J.L. Review of Sublimation Thermodynamics of Polycyclic Aromatic Compounds and Heterocycles. Heterocycl. Chem. 2013, 50, 1243. [Google Scholar] [CrossRef]
- Chickos, J.S.; Gavezzotti, A. Sublimation Enthalpies of Organic Compounds: A Very Large Database with a Match to Crystal Structure Determinations and a Comparison with Lattice Energies. Cryst. Growth Des. 2019, 19, 6566–6576. [Google Scholar] [CrossRef]
- Acree, W.; Chickos, J.S. Phase Transition Enthalpy Measurements of Organic and Organometallic Compounds. Sublimation, Vaporization and Fusion Enthalpies from 1880 to 2010. J. Phys. Chem. Ref. Data 2010, 39, 043101. [Google Scholar] [CrossRef]
- Gavezzotti, A. Calculation of lattice energies of organic crystals: The PIXEL integration method in comparison with more traditional methods. Z. Krist.-Cryst. Mater. 2005, 220, 499–510. [Google Scholar] [CrossRef]
- Gavezzotti, A. Equilibrium structure and dynamics of organic crystals by Monte Carlo simulation: Critical assessment of force fields and comparison with static packing analysis. New J. Chem. 2013, 37, 2110–2119. [Google Scholar] [CrossRef]
- Corminboeuf, C. Minimizing Density Functional Failures for Non-Covalent Interactions Beyond van der Waals Complexes. Acc. Chem. Res. 2014, 47, 3217–3224. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.X.; Adam, C.; Nichol, G.S.; Cockroft, S.L. How much do van der Waals dispersion forces contribute to molecular recognition in solution? Nat. Chem. 2013, 5, 1006–1010. [Google Scholar] [CrossRef]
- Xu, P.; Alkan, M.; Gordon, M.S. Many-Body Dispersion. Chem. Rev. 2020, 120, 12343–12356. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-corrected mean-field electronic structure methods. Chem. Rev. 2016, 116, 5105–5154, and lit. cited therein. [Google Scholar] [CrossRef] [PubMed]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A Generally Applicable Atomic-Charge Dependent London Dispersion Correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef] [PubMed]
- Hermann, J.; DiStasio, R.A.; Tkatchenko, A. First-Principles Models for van Der Waals Interactions in Molecules and Materials: Concepts, Theory, and Applications. Chem. Rev. 2017, 117, 4714–4758. [Google Scholar] [CrossRef]
- Frey, J.A.; Holzer, C.; Klopper, W.; Leutwyler, S. Experimental and Theoretical Determination of Dissociation Energies of Dispersion-Dominated Aromatic Molecular Complexes. Chem. Rev. 2016, 116, 5614–5641. [Google Scholar] [CrossRef] [PubMed]
- Lii, J.H.; Allinger, N.L. Molecular Mechanics. The MM3 Force Field for Hydrocarbons. 2. Vibrational Frequencies and Thermodynamics. J. Am. Chem. Soc. 1989, 111, 8576–8582, and lit. cited therein. [Google Scholar] [CrossRef]
- Allinger, N.L.; Durkin, K.A. Van der Waals Effects between Hydrogen and First-Row Atoms in Molecular Mechanics (MM3/MM4). J. Comput. Chem. 2000, 21, 1229–1242, and lit. cited therein. [Google Scholar] [CrossRef]
- Pollice, R.; Chen, P. A Universal Quantitative Descriptor of the Dispersion Interaction Potential. Angew. Chem. Int. Ed. 2019, 58, 9758–9769. [Google Scholar] [CrossRef] [PubMed]
- Via-Nadal, M.; Rodríguez-Mayorga, M.; Matito, E. Salient signature of van der Waals interactions. Phys. Rev. A 2017, 96, 050501. [Google Scholar] [CrossRef]
- Yang, L.; Sun, L.; Deng, W.Q. Van der Waals Function for Molecular Mechanics. J. Phys. Chem. A 2020, 124, 2102–2107. [Google Scholar] [CrossRef]
- Kraus, P.; Obenchain, D.A.; Frank, I. Benchmark-Quality Semiexperimental Structural Parameters of van der Waals Complexes. J. Phys. Chem. A 2018, 122, 1077–1087. [Google Scholar] [CrossRef]
- Kraus, P.; Frank, I. Density Functional Theory for Microwave Spectroscopy of Noncovalent Complexes: A Benchmark Study. J. Phys. Chem. A 2018, 122, 4894–4901. [Google Scholar] [CrossRef]
- Leko, K.; Hanzevacki, M.; Brkljaca, Z.; Piculjan, K.; Ribic, R.; Pozar, J. Solvophobically Driven Complexation of Adamantyl Mannoside with β-Cyclodextrin in Water and Structured Organic Solvents. Chem. Eur. J. 2020, 26, 5208–5219. [Google Scholar] [CrossRef] [PubMed]
- Jencks, W.P. Binding energy, specificity, and enzymic catalysis: The circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 43, 219–410. [Google Scholar] [PubMed]
- Zhou, H.-X.; Gilson, M.K. Theory of Free Energy and Entropy in Noncovalent Binding. Chem. Rev. 2009, 109, 4092–4107. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Nowosinski, L.; Löw, N.L.; Dzyuba, E.V.; Klautzsch, F.; Schäfer, A.; Huuskonen, J.; Rissanen, K.; Schalley, C.A. Chelate Cooperativity and Spacer Length Effects on the Assembly Thermodynamics and Kinetics of Divalent Pseudorotaxanes. J. Am. Chem. Soc. 2012, 134, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, B.; Grimme, S. Quantum Chemistry of FLPs and Their Activation of Small Molecules: Methodological Aspects. Top. Curr. Chem. 2013, 332, 213–230. [Google Scholar] [PubMed]
- Groslambert, L.; Cornaton, Y.; Ditte, M.; Aubert, E.; Pale, P.; Tkatchenko, A.; Djukic, J.P.; Mamane, V. Affinity of Telluronium Chalcogen Bond Donors for Lewis Bases in Solution: A Critical Experimental-Theoretical Joint Study. Eur. J. 2024, 30, e20230293. [Google Scholar] [CrossRef] [PubMed]
- Arnaud-Neu, F.; Delgado, R.; Chaves, S. Critical evaluation of stability constants and thermodynamic functions of metal complexes of crown ethers. Pure Appl. Chem. 2003, 75, 71–102. [Google Scholar] [CrossRef]
- Rekharsky, M.V.; Inoue, Y. Complexation Thermodynamics of Cyclodextrins. Chem. Rev. 1998, 98, 1875–1918. [Google Scholar] [CrossRef]
- Walter, S.M.; Kniep, F.; Rout, L.; Schmidtchen, F.P.; Herdtweck, E.; Huber, S.M. Isothermal Calorimetric Titrations on Charge-Assisted Halogen Bonds: Role of Entropy, Counterions, Solvent, and Temperature. J. Am. Chem. Soc. 2012, 134, 8507–8512, and lit. cited therein. [Google Scholar] [CrossRef]
- Jadhav, V.D.; Schmidtchen, F.P. Surprises in the Design of Anion Receptors: Calorimetry Prevents False Reasoning. Org. Lett. 2005, 7, 3311–3314. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, V.; Eliseev, A.; Simova, S.; Schneider, H.J.; Blandamer, M.J.; Cullis, P.M.; Meyer, A.J. Conformational, calorimetric and NMR spectroscopic studies on inclusion complexes of cyclodextrins with substituted phenyl and adamantane derivatives. J. Chem. Soc. Perkin Trans. 2 1996, 2119–2123. [Google Scholar] [CrossRef]
- Biedermann, F.; Uzunova, V.D.; Scherman, O.A.; Nau, W.M.; De Simone, A. Release of High-Energy Water as an Essential Driving Force for the High-Affinity Binding of Cucurbit[n]urils. J. Am. Chem. Soc. 2012, 134, 15318–15323. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.K. Classical vs. nonclassical hydrophobic interactions underlying various interaction processes: Application of isothermal titration calorimetry. Chem. Physics Impact 2022, 5, 100104. [Google Scholar] [CrossRef]
- Ghai, R.; Falconer, R.J.; Collins, B.M. Applications of isothermal titration calorimetry in pure and applied research—Survey of the literature from 2010. J. Mol. Recognit. 2012, 25, 32–52. [Google Scholar] [CrossRef] [PubMed]
- Nandy, A.; Shekhar, S.; Paul, B.K.; Mukherjee, S. Exploring the nucleobase specific hydrophobic interaction of cryptolepine hydrate with RNA and its subsequent sequestration. Langmuir 2021, 37, 11176–11187. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, M.S.; Shimizu, S.; Chan, H.S. Temperature Dependence of Three-Body Hydrophobic Interactions: Potential of Mean Force, Enthalpy, Entropy, Heat Capacity, and Nonadditivity. J. Am. Chem. Soc. 2005, 127, 303–331. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Fu, Y.; Wang, W. Temperature effects on hydrophobic interactions: Implications for protein unfolding. Chem. Phys. 2022, 559, 111550. [Google Scholar] [CrossRef]
- Stohr, M.; van Voorish, T.; Tkatchenko, A. Theory and practice of modeling van der Waals interactions in electronic-structure calculations. Chem. Soc. Rev. 2019, 48, 4118–4154, and lit. cited therein. [Google Scholar] [CrossRef]
- Stahn, M.; Ehlert, S.; Grimme, S. Extended Conductor-like Polarizable Continuum Solvation Model (CPCM-X) for Semiempirical Methods Marcel Stahn. J. Phys. Chem. A 2023, 127, 7036–7043, and lit. cited therein. [Google Scholar] [CrossRef]
- Schneider, H.-J. Comment on “HYDROPHOBE Challenge: A Joint Experimental and Computational Study on the Host-Guest Binding of Hydrocarbons to Cucurbiturils, Allowing Explicit Evaluation of Guest Hydration Free-Energy Contributions”. J. Phys. Chem. B 2018, 122, 2825–2826. [Google Scholar] [CrossRef] [PubMed]
- Elmi, A.; Cockroft, S.L. Quantifying Interactions and Solvent Effects using Molecular Balances and Model Complexes. Acc. Chem. Res. 2021, 54, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Cockroft, S.L.; Hunter, C.A. Chemical double-mutant cycles: Dissecting non-covalent interactions. Chem. Soc. Rev. 2007, 36, 172–188. [Google Scholar] [CrossRef]
- Liptrot, D.; Power, P. London dispersion forces in sterically crowded inorganic and organometallic molecules. Nat. Chem. Rev. 2017, 1, 0004. [Google Scholar] [CrossRef]
- Wilming, F.M.; Marazzi, B.; Debes, P.P.; Becker, J.; Schreiner, P.R. Probing the Size Limit of Dispersion Energy Donors with a Bifluorenylidene Balance: Magic Cyclohexyl. J. Org. Chem. 2023, 88, 1024–1035. [Google Scholar] [CrossRef]
- Wagner, J.P.; Schreiner, P.R. London Dispersion in Molecular Chemistry-Reconsidering Steric Effects. Angew. Chem. Int. Ed. 2015, 54, 12274–12296, and lit. cited therein. [Google Scholar] [CrossRef] [PubMed]
- Leibiger, B.; Stapf, M.; Mazik, M. Cycloalkyl Groups as Building Blocks of Artificial Carbohydrate Receptors. Molecules 2022, 27, 7630. [Google Scholar] [CrossRef]
- Giese, M.; Albrecht, M. Alkyl-Alkyl Interactions in the Periphery of Supramolecular Entities: From the Evaluation of Weak Forces to Applications. PlusChem 2020, 85, 715. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L. Energetics of Protein Folding. J. Mol. Biol. 2007, 371, 283–301. [Google Scholar] [CrossRef]
- Sun, S.-S. Peptide folding driven by Van der Waals interactions. Protein Sci. 2015, 24, 1383–1388. [Google Scholar]
- Gupta, S.K.; Banerjee, S.; Prabhakaran, E.N. Van der Waals interactions to control amide cis–trans isomerism. New J. Chem. 2022, 46, 12470. [Google Scholar] [CrossRef]
- Frank, H.S.; Evans, M.W. Free volume and entropy in condensed systems III. Entropy in binary liquid mixtures; partial molal entropy in dilute solutions; structure and thermodynamics in aqueous electrolytes. J. Chem. Phys. 1945, 13, 507–532. [Google Scholar] [CrossRef]
- Kauzmann, W. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 1959, 14, 1–63. [Google Scholar]
- Sun, Q. The Hydrophobic Effects: Our Current Understanding. Molecules 2022, 27, 7009. [Google Scholar] [CrossRef] [PubMed]
- Ben-Amotz, D. Water-Mediated Hydrophobic Interactions. Annu. Rev. Phys. Chem. 2016, 67, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Kellogg, G.E. Hydrophobicity—Shake Flasks, Protein Folding and Drug Discovery. Curr. Top. Med. Chem. 2010, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef]
- Hummer, G. Molecular binding under water’s influence. Nat. Chem. 2010, 2, 906–907. [Google Scholar] [CrossRef]
- Hillyer, M.B.; Gibb, B.C. Molecular Shape and the Hydrophobic Effect. Annu. Rev. Phys. Chem. 2016, 67, 307–329. [Google Scholar] [CrossRef]
- Rego, N.B.; Patel, A.J. Understanding Hydrophobic Effects: Insights from Water Density Fluctuations. Annu. Rev. Condens. Matter Phys. 2022, 13, 303–324. [Google Scholar] [CrossRef]
- Franzese, G.; Bianco, V.; Iskrov, S. Water at Interface with Proteins. Food Biophys. 2011, 6, 186–198. [Google Scholar] [CrossRef]
- Syme, N.R.; Dennis, C.; Phillips, S.E.V.; Homans, S.W. Origin of heat capacity changes in a “Nonclassical” hydrophobic interaction. ChemBioChem 2007, 8, 1509–1511. [Google Scholar] [CrossRef]
- Cooper, A. Heat capacity effects in protein folding and ligand binding: A re-evaluation of the role of water in biomolecular thermodynamics. Biophys. Chem. 2005, 115, 89–97. [Google Scholar] [CrossRef]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A View of the Hydrophobic Effect. J. Phys. Chem. B 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Oshovsky, G.V.; Reinhoudt, D.N.; Verboom, W. Supramolecular Chemistry in Water. Angew. Chem. Int. Ed. 2007, 46, 2366–2393. [Google Scholar] [CrossRef]
- Escobar, L.; Ballester, P. Molecular Recognition in Water Using Macrocyclic Synthetic Receptors. Chem. Rev. 2021, 121, 2445–2514. [Google Scholar] [CrossRef]
- Kubik, S. When Molecules Meet in Water-Recent Contributions of Supramolecular Chemistry to the Understanding of Molecular Recognition Processes in Water. ChemistryOpen 2022, 11, e202200028. [Google Scholar] [CrossRef]
- Davis, A.P.; Kubik, S.; Dalla Cort, A. Supramolecular chemistry in water. Org. Biomol. Chem. 2015, 13, 2499–2500, web collection of 50 papers. [Google Scholar] [CrossRef]
- Schneider, H.-J.; Yatsimirsky, A. Principles and Methods in Supramolecular Chemistry; Wiley: Chichester, UK; New York, NY, USA; Weinheim, Germany; Brisbane, Australia; Singapore; Toronto, ON, Canada, 2000. [Google Scholar]
- Schneider, H.-J. Quantification of noncovalent interactions–promises and problems. New J. Chem. 2019, 43, 15498–15512. [Google Scholar] [CrossRef]
- Schneider, H.-J.; Kramer, R.; Simova, S.; Schneider, U. Solvent and Salt Effects on Binding Constants of Organic Substrates in Macrocyclic Host Compounds. J. Am. Chem. Soc. 1988, 110, 6442. [Google Scholar] [CrossRef]
- Schneider, H.-J.; Wang, M. Ligand-Porphyrin Complexes: Quantitative Evaluation of Stacking and Ionic Contributions. J. Org. Chem. 1994, 59, 7464–7472. [Google Scholar] [CrossRef]
- Kinoshita, M.; Hayashi, T. Unified elucidation of the entropy-driven and -opposed hydrophobic effects. Phys. Chem. Chem. Phys. 2017, 19, 25891. [Google Scholar] [CrossRef]
- Ferguson, S.B.; Seward, E.M.; Diederich, F.; Sanford, E.M.; Chou, A.; Inocencio-Szweda, P.; Knobler, C.B. Cyclophane-Arene Inclusion Complexation in Protic Solvents: Solvent Effects versus Electron Donor-Acceptor Interactions. J. Org. Chem. 1988, 53, 5593–5595. [Google Scholar] [CrossRef]
- Smithrud, D.B.; Wyman, T.B.; Diederich, F. Enthalpically Driven Cyclophane-Arene Inclusion Complexation: Solvent-Dependent Calorimetric Studies. J. Am. Chem. Soc. 1991, 113, 5420–5426. [Google Scholar] [CrossRef]
- Biedermann, F.; Nau, W.M.; Schneider, H.-J. The Hydrophobic Effect Revisited—Studies with Supramolecular Complexes Imply High-Energy Water as a Noncovalent Driving Force. Angew. Chem. Int. Ed. 2014, 53, 11158–11171. [Google Scholar] [CrossRef]
- Grimm, L.M.; Spicher, D.; Tkatchenko, A.; Schreiner, P.R.; Grimme, S.; Biedermann, F. The Role of Packing, Dispersion, Electrostatics, and Solvation in High-Affinity Complexes of Cucurbit[n]urils with Uncharged Polar Guests. Chem. Eur. J. 2022, 28, e20220052. [Google Scholar] [CrossRef]
- Baron, R.; Setny, P.; McCammon, J.A. How Can Hydrophobic Association Be Enthalpy Driven? J. Am. Chem. Soc. 2010, 132, 12091–12097. [Google Scholar] [CrossRef]
- Clark, G.N.I.; Cappa, C.D.; Smith, J.D.; Saykally, R.J.; Head-Gordon, T. The structure of ambient water. Mol. Phys. 2010, 108, 1415–1433. [Google Scholar] [CrossRef]
- Nilsson, A.; Pettersson, L.G.M. Perspective on the structure of liquid water. Chem. Phys. 2011, 389, 1–34. [Google Scholar] [CrossRef]
- Komiyama, M.; Bender, M.L. Importance of Apolar Binding in Complex Formation of Cyclodextrins with Adamantanecarboxylate. J. Am. Chem. Soc. 1978, 100, 2259–2260. [Google Scholar] [CrossRef]
- Schneider, H.J. Mechanisms of Molecular Recognition: Investigations of Organic Host–Guest Complexes. Angew. Chem. Int. Ed. Engl. 1991, 30, 1417–1436. [Google Scholar] [CrossRef]
- Assaf, K.I.; Nau, W.M. Cucurbiturils: From synthesis to high-affinity binding and catalysis. Chem. Soc. Rev. 2015, 44, 394–418. [Google Scholar] [CrossRef]
- Assaf, K.I.; Florea, M.; Antony, J.; Henriksen, N.M.; Yin, J.; Hansen, A.; Qu, Z.-w.; Sure, R.; Klapstein, D.; Gilson, M.K.; et al. HYDROPHOBE Challenge: A Joint Experimental and Computational Study on the Host–Guest Binding of Hydrocarbons to Cucurbiturils, Allowing Explicit Evaluation of Guest Hydration Free-Energy Contributions. J. Phys. Chem. B 2017, 121, 11144–11162. [Google Scholar] [CrossRef]
- Barrow, S.J.; Assaf, K.I.; Palma, A.; Nau, W.M.; Scherman, O.A. Preferential binding of unsaturated hydrocarbons in aryl-bisimidazolium·cucurbit [8]uril complexes furbishes evidence for small-molecule interactions. Chem. Sci. 2019, 10, 10240–10246. [Google Scholar] [CrossRef]
- He, S.; Biedermann, F.; Vankova, N.; Zhechkov, L.; Heine, T.; Hoffman, R.E.; De Simone, A.; Duignan, T.T.; Nau, W.M. Cavitation energies can outperform dispersion interactions. Nat. Chem. 2018, 10, 1252–1257, and lit. cited therein. [Google Scholar] [CrossRef]
- Leo, A.J. Hydrophobic parameter—Measurement and calculation. Methods Enzymol. 1991, 202, 544–591. [Google Scholar]
- Hua, L.; Zangi, R.; Berne, B.J. Hydrophobic interactions and dewetting between plates with hydrophobic and hydrophilic domains. J. Phys. Chem. C 2009, 113, 5244–5525. [Google Scholar] [CrossRef]
- Huang, X.; Margulis, C.J.; Berne, B.J. Dewetting-induced collapse of hydrophobic particles. Proc. Natl. Acad. Sci. USA 2003, 100, 11953–11958. [Google Scholar] [CrossRef]
- Caporale, A.; Adorinni, S.; Lamba, D.; Saviano, M. Peptide–Protein Interactions: From Drug Design to Supramolecular Biomaterials. Molecules 2021, 2 6, 121. [Google Scholar] [CrossRef]
- Kapon, R.; Nevo, R.; Reich, Z. Protein energy landscape roughness. Biochem. Soc. Trans. 2008, 36, 1404–1408. [Google Scholar] [CrossRef]
- Paul, B.K.; Ghosh, N.; Mukherjee, S. Interaction of bile salts with β-cyclodextrins reveals nonclassical hydrophobic effect and enthalpy–entropy compensation. J. Phys. Chem. B 2016, 120, 3963–3968. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MM2 | MM3 | Exp. * | |
|---|---|---|---|
| C6H6 | 11.18 | 10.32 | 10.42 |
| C6Me6 | 24.81 | 16.83 | 17.86 |
| n-hexane | 19.31 | 11.59 | 9.76 |
| n-dodecane | 38.77 | 24.52 | 23.78 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, H.-J. Distinction and Quantification of Noncovalent Dispersive and Hydrophobic Effects. Molecules 2024, 29, 1591. https://doi.org/10.3390/molecules29071591
Schneider H-J. Distinction and Quantification of Noncovalent Dispersive and Hydrophobic Effects. Molecules. 2024; 29(7):1591. https://doi.org/10.3390/molecules29071591
Chicago/Turabian StyleSchneider, Hans-Jörg. 2024. "Distinction and Quantification of Noncovalent Dispersive and Hydrophobic Effects" Molecules 29, no. 7: 1591. https://doi.org/10.3390/molecules29071591
APA StyleSchneider, H.-J. (2024). Distinction and Quantification of Noncovalent Dispersive and Hydrophobic Effects. Molecules, 29(7), 1591. https://doi.org/10.3390/molecules29071591