
Acridine–Isoxazole and Acridine–Azirine Hybrids: Synthesis, Photochemical Transformations in the UV/Visible Radiation Boundary Region, and Anticancer Activity

 , and
, and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
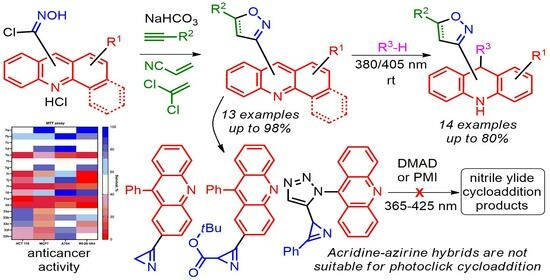
2. Results and Discussion
3. Materials and Methods
3.1. General Instrumentation
3.2. General Experimental Procedures
3.2.1. General Procedure A (GP-A) for the Preparation of Chlorooxime Hydrochlorides 2-HCl
3.2.2. General Procedure B (GP-B) for the Preparation of Isoxazoles 7
3.2.3. General Procedure C (GP-C) for the Reaction of Isoxazoles 7a–e,l and 10 with Hydrogen Donor Solvents
3.2.4. Specific Procedures and Characterization
- 2-Methyl-9-phenylacridine (4e). Compound 4e was prepared according to the published procedure [54] from 4-methyl-N-phenylaniline (1.83 g, 10 mmol), benzoic acid (2.44 g, 20 mmol), and anhydrous ZnCl2 (2.72 g, 20 mmol) at 220 °C overnight to give a pure product of 1.62 g (60% yield), after column chromatography on silica (light petroleum/ethyl acetate, 5:1–1:1, (v/v)) as a light yellow solid: mp 110–111 °C (ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.26 (d, 1H, J = 8.8 Hz), 8.18 (d, 1H, J = 8.9 Hz), 7.73 (ddd, 1H, J = 8.5, 6.5, 1.3 Hz), 7.66 (dd, 1H, J = 8.8, 1.3 Hz), 7.67–7.58 (m, 4H), 7.45–7.38 (m, 4H), 2.46 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 148.3 (C), 147.8 (C), 146.0 (C), 136.2 (C), 135.5 (C), 132.9 (CH), 130.5 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 128.5 (CH), 128.2 (CH), 126.8 (CH), 125.5 (CH), 125.3 (C), 125.2 (C), 124.8 (CH), 22.0 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C20H16N+ 270.1277, found 270.1280.
- 2-Nitroacridine-9-carbaldehyde (5c). Compound 5c was prepared according to the published procedure [55] from 9-methyl-2-nitroacridine 4c (325 mg, 1.35 mmol) and SeO2 (160 mg, 1.43 mmol) in 1,4-dioxane (5 mL) at 115 °C (bath temperature) to give a pure product of 214 mg (62% yield), after column chromatography on silica (light petroleum/ethyl acetate, 1:1–0:1, (v/v)) as a brown-orange solid: mp 182–184 °C (ethyl acetate); 1H NMR (400 MHz, DMSO-d6) δ 11.52 (s, 1H), 9.78 (d, 1H, J = 2.6 Hz), 8.94 (d, 1H, J = 8.8 Hz), 8.55 (dd, 1H, J = 9.4, 2.7 Hz), 8.44 (d, 1H, J = 9.6 Hz), 8.34 (d, 1H, J = 8.7 Hz), 8.09–8.06 (m, 1H), 7.92–7.88 (m, 1H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 195.1 (CH), 150.9 (C), 148.9 (C), 146.3 (C), 136.0 (C), 132.6 (CH), 131.8 (CH), 130.1 (CH), 129.7 (CH), 124.4 (C), 124.1 (CH), 123.2 (CH), 122.9 (CH), 120.4 (C); HRMS (ESI) m/z [M + H]+ calcd for C14H9N2O3+ 253.0608, found 253.0599.
- 9-Methylbenzo[c]acridine-7-carbaldehyde (5d). Compound 5d was prepared according to the published procedure [55] from 7,9-dimethylbenzo[c]acridine 4d (1.5 g, 7.24 mmol) and SeO2 (843 mg, 7.6 mmol) in 1,4-dioxane (50 mL) at 115 °C (bath temperature) to give a pure product of 1.32 g (82% yield), after column chromatography on silica (light petroleum/ethyl acetate, 1:1–0:1, (v/v)) as an orange solid: mp 165–166 °C (ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 11.44 (d, 1H, J = 6.7 Hz), 9.50–9.47 (m, 1H), 8.49 (d, 1H, J = 4.5 Hz), 8.45–8.41 (m, 1H), 8.33 (dd, 1H, J = 8.7, 5.0 Hz), 7.88–7.73 (m, 4H), 7.69 (dt, 1H, J = 9.3, 2.2 Hz), 2.64 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 193.9 (CH), 146.9 (C), 146.4 (C), 138.8 (C), 132.7 (C), 132.2 (CH), 131.4 (C), 131.0 (C), 130.8 (CH), 130.3 (CH), 129.2 (CH), 127.79 (CH), 127.75 (CH), 125.2 (CH), 123.6 (C), 123.1 (C), 121.8 (CH), 120.3 (CH), 22.4 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C19H14NO+ 272.1070, found 272.1065.
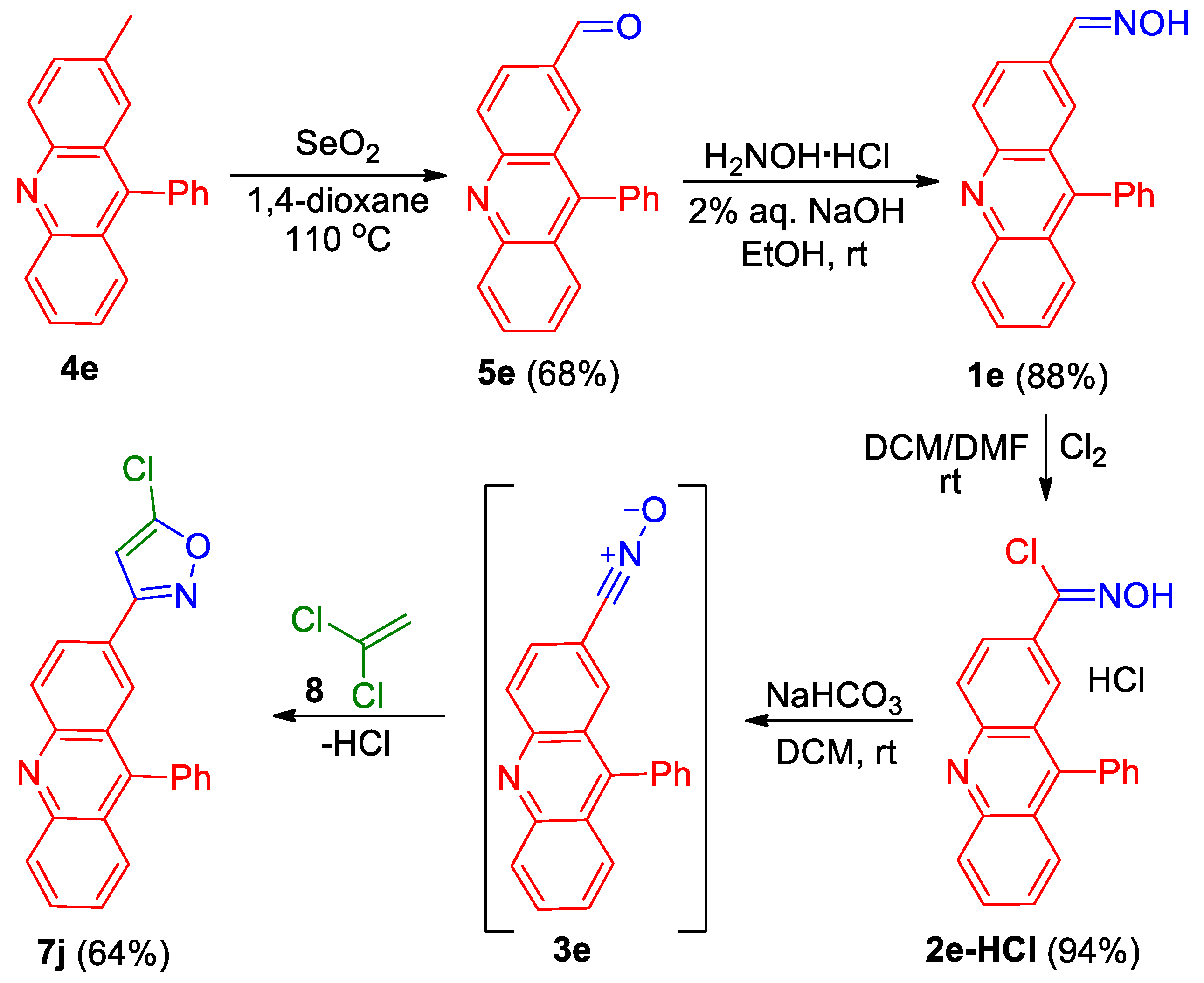
- 9-Phenylacridine-2-carbaldehyde (5e). Compound 5e was prepared according to the published procedure [55] from 2-methyl-9-phenylacridine 4e (1.0 g, 3.7 mmol) and SeO2 (4.3 g, 38.7 mmol, 6 portions every 12 h) 1,4-dioxane (35 mL) at 115 °C (bath temperature) to give a pure product of 714 mg (68% yield), after column chromatography on silica (light petroleum/ethyl acetate, 1:1–0:1, (v/v)) as a light yellow solid: mp 185–186 °C (ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 10.02 (d, 1H, J = 0.8 Hz), 8.32 (td, 2H, J = 9.6, 0.8 Hz), 8.23–8.20 (m, 2H), 7.77–7.75 (m, 1H), 7.68–7.64 (m, 3H), 7.52–7.47 (, 3H); 13C{1H} NMR (CDCl3, 100 MHz) δ 191.5 (CH), 150.5 (C), 150.4 (C), 149.9 (C), 135.4 (CH), 134.9 (C), 133.7 (C), 131.5 (CH), 131.1 (CH), 130.4 (CH), 129.8 (CH), 129.0 (CH), 128.7 (CH), 127.2 (CH), 126.5 (CH), 125.7 (CH), 125.5 (C), 124.3 (C); HRMS (ESI) m/z [M + H]+ calcd for C20H14NO+ 284.1070, found 284.1062.
- 2-Methylacridine-9-carbaldehyde oxime (1b). Compound 1b was prepared according to the published procedure [29] from acridinecarbaldehyde 5b (1.0 g, 4.5 mmol) and H2NOH·HCl (628 mg, 9 mmol) in EtOH (20 mL) and 2% aq. NaOH (2 mL) to give a pure product of 885 mg (83% yield), after filtration as a brick-yellow solid: mp 196–197 °C (EtOH/H2O); 1H NMR (400 MHz, DMSO-d6) δ 12.35 (s, 1H), 9.28 (s, 1H), 8.55 (d, 1H, J = 8.8 Hz), 8.31–8.30 (m, 1H), 8.19 (d, 1H, J = 8.6 Hz), 8.12 (d, 1H, J = 8.9 Hz), 7.89 (ddd, 1H, J = 8.4, 6.5, 1.4 Hz), 7.77 (dd, 1H, J = 8.9, 1.9 Hz), 7.68 (ddd, 1H, J = 8.2, 6.6, 1.3 Hz), 2.56 (s, 1H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 146.3 (br. s, C), 145.8 (br. s, C), 145.1 (CH), 136.8 (C), 135.1 (br. s, C), 133.9 (CH), 130.7 (CH), 128.1 (br. s, CH), 127.9 (br. s, CH), 126.9 (CH), 125.8 (CH), 123.64 (CH), 123.61 (C), 123.56 (C), 21.7 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C15H13N2O+ 237.1022, found 237.1017.
- 2-Nitroacridine-9-carbaldehyde oxime (1c). Compound 1c was prepared according to the published procedure [29] from acridinecarbaldehyde 5c (200 mg, 0.8 mmol) and H2NOH·HCl (138 mg, 2 mmol) in EtOH (5 mL) and 2% aq. NaOH (0.5 mL) to give a pure product of 162 mg (76% yield), after filtration as a yellow-green solid: mp 202–214 °C (dec., EtOH/H2O); 1H NMR (400 MHz, DMSO-d6) δ 12.67 (s, 1H), 9.65 (d, 1H, J = 2.5 Hz), 9.44 (s, 1H), 8.56 (d, 1H, J = 8.8 Hz), 8.49 (dd, 1H, J = 9.5, 2.7 Hz), 8.34 (d, 1H, J = 9.5 Hz), 8.24 (d, 1H, J = 8.6 Hz), 8.01 (t, 1H, J = 7.7 Hz), 7.77 (t, 1H, J = 7.7 Hz); 13C{1H} NMR (100 MHz, DMSO-d6) δ 150.0 (C), 148.7 (C), 145.0 (C), 144.5 (CH), 141.7 (CH), 137.7 (C), 132.0 (CH), 131.4 (CH), 129.5 (CH), 127.5 (CH), 124.9 (CH), 124.0 (CH), 122.3 (CH), 120.9 (C); HRMS (ESI) m/z [M + H]+ calcd for C14H10N3O3+ 268.0717, found 268.0719.
- 9-Methylbenzo[c]acridine-7-carbaldehyde oxime (1d). Compound 1d was prepared according to the published procedure [29] from acridinecarbaldehyde 5d (935 mg, 3.5mmol) and H2NOH·HCl (600 mg, 8.6 mmol) in EtOH (20 mL) and 2% aq. NaOH (2 mL) to give a pure product of 984 mg (99% yield), after filtration as a brick-orange solid: mp 237–238 °C (EtOH/H2O); 1H NMR (400 MHz, DMSO-d6) δ 12.02 (br. s, 1H), 9.42–9.40 (m, 1H), 9.23 (s, 1H), 8.31–8.27 (m, 3H), 8.02–8.00 (m, 1H), 7.90 (d, 1H, J = 9.5 Hz), 7.83–7.78 (m, 3H), 2.61 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 145.5 (C), 145.2 (CH), 145.0 (C), 136.9 (C), 133.9 (C), 133.0 (C), 132.9 (CH), 130.3 (C), 129.5 (CH), 129.0 (CH), 128.3 (CH), 128.0 (CH), 127.6 (CH), 124.8 (CH), 124.2 (C), 123.8 (CH), 122.9 (CH), 122.3 (C), 21.7 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C19H15N2O+ 287.1179, found 287.1170.
- 9-Phenylacridine-2-carbaldehyde oxime (1e). Compound 1e was prepared according to the published procedure [29] from acridinecarbaldehyde 5e (693 mg, 2.5 mmol) and H2NOH·HCl (425 mg, 6.1 mmol) in EtOH (20 mL) and 2% aq. NaOH (2 mL) to give a pure product of 639 mg (88% yield), after filtration as a bright yellow solid: mp 212–215 °C (dec., EtOH/H2O); 1H NMR (400 MHz, DMSO-d6) δ 11.71 (br. s, 1H), 8.62–8.42 (m, 3H0, 8.34 (s, 1H), 8.24–8.16 (m, 1H), 7.92–7.90 (m,1H), 7.81–7.73 (m, 5H), 7.61–7.57 (m, 2H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 155.8 (br. s, C), 147.2 (CH), 142.1 (br. s, C), 141.4 (br. s, C), 135.2 (br. s, CH), 133.3 (C), 132.4 (br. s, C), 132.3 (CH), 130.0 (CH), 129.8 (CH), 128.9 (CH), 128.0 (CH), 127.6 (CH), 125.5 (CH), 125.2 (C), 124.9 (C), 123.1 (br. s, CH), 122.3 (br. s, CH); HRMS (ESI) m/z [M + H]+ calcd for C20H15N2O+ 299.1179, found 299.1183.
- N-Hydroxyacridine-9-carbimidoyl chloride hydrochloride (2a-HCl). Compound 2a-HCl was prepared according to the general procedure GP-A from oxime 1a (2.0 g, 9 mmol) in DCM (30 mL) and DMF (3 mL) to give a pure product of 1.84 g (70% yield), after filtration as a bright yellow solid: mp 226–230 °C (dec., DCM); 1H NMR (400 MHz, DMSO-d6) δ 13.29 (s, 1H), 8.42 (d, 2H, J = 8.7 Hz), 8.14–8.07 (m, 4H), 7.89–7.85 (m, 2H), 4.46 (br. s); 13C{1H} NMR (100 MHz, DMSO-d6) δ 144.9 (C), 140.4 (C), 133.6 (CH), 128.7 (CH), 127.4 (C), 125.8 (CH), 125.2 (CH), 123.6 (C); HRMS (ESI) m/z [M − Cl]+ calcd for C14H10ClN2O+ 257.0476, found 257.0482.
- N-Hydroxy-2-methylacridine-9-carbimidoyl chloride hydrochloride (2b-HCl). Compound 2b-HCl was prepared according to the general procedure GP-A from oxime 1b (842 mg, 2.3 mmol) in DCM (20 mL) and DMF (2 mL) to give a pure product of 838 mg (83% yield), after filtration as a bright yellow-orange solid: mp 231–232 °C (dec., DCM); 1H NMR (400 MHz, DMSO-d6) δ 13.33 (s, 1H), 8.46–8.44 (m, 2H), 8.39 (d, 1H, J = 9.0 Hz), 8.12–8.08 (m, 2H), 7.99 (dd, 1H, J = 8.9, 1.9 Hz), 7.90–7.86 (m, 2H), 7.61 (s, 3H), 5.81 (br. s); 13C{1H} NMR (100 MHz, DMSO-d6) δ 143.2 (C), 142.8 (C), 140.0 (C), 138.8 (C), 136.6 (CH), 133.3 (CH), 128.4 (CH), 127.0 (C), 124.9 (CH), 124.6 (CH), 124.4 (CH), 123.6 (C), 123.6 (C), 122.8 (CH), 21.4 (CH3); HRMS (ESI) m/z [M − Cl]+ calcd for C14H20ClN2O+ 257.0476, found 257.0482.
- N-Hydroxy-2-nitroacridine-9-carbimidoyl chloride hydrochloride (2c-HCl). Compound 2c-HCl was prepared according to the general procedure GP-A from oxime 1c (155 mg, 0.6 mmol) in DCM (3 mL) and DMF (0.3 mL) to give a pure product of 136 mg (69% yield), after filtration as an orange-brown solid: mp 229–230 °C (dec., DCM); 1H NMR (400 MHz, DMSO-d6) δ 13.47 (s, 1H), 8.86 (t, 1H, J = 2.0 Hz), 8.55 (dt, 1H, J = 9.4, 2.4 Hz), 8.45 (dd, 1H, J = 9.4, 2.2 Hz), 8.32 (d, 1H, J = 8.7 Hz), 8.12 (d, 1H, J = 8.8 Hz), 8.10–8.06 (m, 1H), 7.88 (dd, 1H, J = 8.7, 6.7 Hz), 7.51–6.61 (br. s); 13C{1H} NMR (100 MHz, DMSO-d6) δ 150.2 (C), 148.4 (C), 145.6 (C), 139.9 (C), 133.3 (CH), 131.7 (CH), 129.6 (CH), 129.4 (CH), 127.4 (C), 125.1 (CH), 123.9 (C), 123.7 (CH), 122.0 (CH), 121.5 (C); HRMS (ESI) m/z [M − Cl]+ calcd for C14H9ClN3O3+ 302.0327, found 302.0320.
- N-Hydroxy-9-methylbenzo[c]acridine-7-carbimidoyl chloride hydrochloride (2d-HCl). Compound 2d-HCl was prepared according to the general procedure GP-A from oxime 1d (855 mg, 3 mmol) in DCM (30 mL) and DMF (3 mL) to give a pure product of 908 mg (85% yield), after filtration as a bright yellow solid: mp 267–272 °C (dec., DCM); 1H NMR (400 MHz, DMSO-d6) δ 13.07 (s, 1H), 9.39–9.37 (m, 1H), 8.31 (d, 1H, J = 8.7 Hz), 8.07–8.04 (m, 1H), 8.01 (d, 1H, J = 9.4 Hz), 7.86–7.76 (m, 5H), 5.61–5.16 (br. s), 2.61 (s, 3H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 146.0 (C), 145.6 (C), 138.0 (C), 135.1 (C), 133.3 (CH), 133.0 (C), 130.5 (C), 129.78 (CH), 129.75 (CH), 129.5 (CH), 128.8 (C), 128.4 (CH), 128.1 (CH), 124.6 (CH), 123.8 (C), 122.6 (CH), 122.2 (C), 121.8 (CH), 21.7 (CH3); HRMS (ESI) m/z [M − Cl]+ calcd for C19H14ClN2O+ 321.0789, found 321.0801.
- N-Hydroxy-9-phenylacridine-2-carbimidoyl chloride hydrochloride (2e-HCl). Compound 2e-HCl was prepared according to the general procedure GP-A from oxime 1e (623 mg, 2.1 mmol) in DCM (20 mL) and DMF (2 mL) to give a pure product of 721 mg (94% yield), after filtration as a bright yellow solid: mp 228–229 °C (dec., DCM); 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.63–8.56 (m, 3H), 8.27–8.23 (m, 1H), 8.16 (d, 1H, J = 1.9 Hz), 7.83 (d, 1H, J = 3.9 Hz), 7.79–7.75 (m, 3H), 7.64–7.62 (m, 2H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 156.7 (C), 142.0 (C), 141.8 (C), 135.9 (CH), 134.4 (C), 133.1 (C), 132.1 (CH), 131.1 (C), 130.0 (CH), 130.0 (CH), 128.9 (CH), 128.3 (CH), 127.7 (CH), 125.8 (CH), 125.3 (C), 124.4 (C), 123.0 (CH), 122.3 (CH); HRMS (ESI) m/z [M + H]+ calcd for C20H14ClN2O+ 333.0789, found 333.0777.
- 3-(Acridin-9-yl)-5-phenylisoxazole (7a). Compound 7a was prepared according to the general procedure GP-B from chlorooxime 2a-HCl (171 mg, 0.58 mmol) and phenylacetylene 6a (298 mg, 2.92 mmol, 5 eq.) in DCM (10 mL) and sat. aq. NaHCO3 (5 mL) to give a pure product of 178 mg (95% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a bright yellow solid: mp 258–260 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3): δ 8.32 (d, 2H, J = 8.7 Hz), 8.01 (d, 2H, J = 8.7 Hz), 7.96–7.94 (m, 2H), 7.83 (ddd, 2H, J = 8.5, 6.6, 1.4 Hz), 7.58–7.53 (m, 5H), 6.85 (s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 170.8 (C), 159.4 (C), 148.7 (C), 133.7 (C), 130.8 (CH), 130.3 (CH), 129.9 (CH), 129.2 (CH), 127.0 (C), 126.8 (CH), 126.1 (CH), 125.8 (CH), 125.0 (C), 102.6 (CH); HRMS (ESI) m/z [M + H]+ calcd for C29H23N2O+ 415.1805, found 415.1799.
- 3-(Acridin-9-yl)-5-(trimethylsilyl)isoxazole (7b). Compound 7b was prepared according to the general procedure GP-B from chlorooxime 2a-HCl (255 mg, 0.87 mmol) and trimethylsilylacetylene 6b (1.94 g, 17.4 mmol, 20 eq.) in DCM (15 mL) and sat. NaHCO3 aq. (8 mL) to give a pure product of 256 mg (92% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a bright yellow solid: mp 151–152 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.29 (dt, 2H, J = 8.8, 1.0 Hz), 7.87 (ddd, 2H, J = 8.7, 1.4, 0.7 Hz), 7.80 (ddd, 2H, J = 8.9, 6.6, 1.4 Hz), 7.52 (ddd, 2H, J = 8.7, 6.6, 1.2 Hz), 6.74 (s, 1H), 0.49 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 179.1 (C), 156.9 (C), 148.6 (C), 134.1 (C), 130.2 (CH), 129.8 (CH), 126.5 (CH), 125.9 (CH), 125.1 (C), 115.4 (CH), −1.8 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C19H19N2OSi+ 319.1261, found 319.1258.
- 3-(Acridin-9-yl)-5-((4-isopropylphenoxy)methyl)isoxazole (7c). Compound 7c was prepared according to the general procedure GP-B from chlorooxime 2a-HCl (155 mg, 0.51 mmol) and 1-isopropyl-4-(prop-2-yn-1-yloxy)benzene 6c (446 mg, 2.56 mmol, 5 eq.) in DCM (10 mL) and sat. NaHCO3 aq. (5 mL) to give a pure product of 198 mg (98% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a bright yellow solid: mp 94–95 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.30 (d, 2H, J = 8.8 Hz), 7.91 (d, 2H, J = 8.7 Hz), 7.83–7.79 (m, 2H), 7.56–7.52 (m, 2H), 7.21 (d, 2H, J = 8.2 Hz), 6.99 (d, 2H, J = 8.2 Hz), 6.67 (s, 1H), 5.37 (s, 2H), 2.90 (hept, 1H, J = 7.0 Hz), 1.25 (d, 6H, J = 7.0 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ 169.2 (C), 158.9 (C), 155.8 (C), 148.6 (C), 142.7 (C), 133.3 (C), 130.3 (CH), 129.9 (CH), 127.6 (CH), 126.8 (CH), 125.7 (CH), 124.9 (C), 114.8 (CH), 106.4 (CH), 61.8 (CH2), 33.3 (CH), 24.1 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C26H23N2O2+ 395.1754, found 395.1752.
- Methyl 3-(2-methylacridin-9-yl)isoxazole-5-carboxylate (7d). Compound 7d was prepared according to the general procedure GP-B from chlorooxime 2b-HCl (330 mg, 1.1 mmol) and methyl propiolate 6d (452 mg, 5.4 mmol, 5 eq.) in DCM (20 mL) and sat. NaHCO3 aq. (10 mL) to give a pure product of 179 mg (52% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a yellow solid: mp 169–170 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.28 (dd, 1H, J = 9.1, 1.2 Hz), 8.20 (d, 1H, J = 8.9 Hz), 7.80–776 (m, 2H), 7.65 (dd, 1H, J = 8.9, 1.6 Hz), 7.55–7.51 (m, 2H), 7.25 (s, 1H), 4.08 (s, 3H), 2.52 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.9 (C), 159.6 (C), 157.0 (C), 148.0 (C), 147.6 (C), 137.3 (C), 133.3 (CH), 130.5 (C), 130.0 (CH), 129.8 (CH), 129.7 (CH), 127.0 (CH), 125.0 (CH), 124.8 (C), 124.8 (C), 123.1 (CH), 112.3 (CH), 53.1 (CH3), 22.1 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C19H15N2O3+ 319.1077, found 319.1076.
- 5-Methoxy-4-((3-(2-methylacridin-9-yl)isoxazol-5-yl)methyl)-3-(naphthalen-2-yl)isoxazole (7e). Compound 7e was prepared according to the general procedure GP-B from chlorooxime 2b-HCl (150 mg, 0.49 mmol) and 5-methoxy-3-(naphthalen-2-yl)-4-(prop-2-yn-1-yl)isoxazole 6e (296 mg, 1.12 mmol, 2.3 eq.) in DCM (20 mL) and sat. NaHCO3 aq. (10 mL) to give a pure product of 227 mg (93% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1–6:1, (v/v)) as a bright yellow solid: mp 157–158 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.21 (d, 1H, J = 8.7 Hz), 8.14 (d, 1H, J = 8.7 Hz), 8.13 (s, 1H), 7.96 (d, 1H, J = 8.5 Hz), 7.92–7.89 (m, 2H), 7.78 (dd, 1H, J = 8.5, 1.8 Hz), 7.70 (ddd, 1H, J = 8.1, 6.7, 1.3 Hz), 7.61–7.52 (m, 5H), 7.24–7.20 (m, 1H), 6.23 (s, 1H), 4.25 (s, 3H), 4.18 (s, 2H), 2.40 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.3 (C), 170.4 (C), 164.3 (C), 159.0 (C), 147.8 (C), 147.4 (C), 136.7 (C), 133.8 (C), 133.1 (CH), 133.0 (C), 132.2 (C), 129.6 (CH), 129.6 (CH), 129.4 (CH), 128.8 (CH), 128.5 (CH), 127.8 (CH), 127.6 (CH), 127.2 (CH), 126.8 (CH), 126.7 (C), 126.4 (CH), 125.3 (CH), 124.9 (C), 124.8 (C), 124.6 (CH), 123.5 (CH), 105.2 (CH), 85.9 (CH), 58.1 (CH3), 22.0 (CH3), 20.1 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C32H24N3O3+ 498.1812, found 498.1807.
- 5-(4-Chlorophenyl)-3-(2-nitroacridin-9-yl)isoxazole (7f). Compound 7f was prepared according to the general procedure GP-B from chlorooxime 2c-HCl (150 mg, 0.44 mmol) and 1-chloro-4-ethynylbenzene 6f (305 mg, 2.2 mmol, 5 eq.) in DCM (20 mL) and sat. aq. NaHCO3 (10 mL) to give a pure product of 159 mg (89% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1–0:1, (v/v)) as a bright yellow solid: mp 232–233 °C (ethyl acetate); 1H NMR (400 MHz, DMSO-d6) δ 8.90 (d, 1H, J = 2.5 Hz), 8.55 (dd, 1H, J = 9.5, 2.5 Hz), 8.48 (d, 1H, J = 9.5 Hz), 8.36 (d, 1H, J = 8.8 Hz), 8.11–8.05 (m, 5H), 7.79 (dd, 1H, J = 8.7, 6.6 Hz), 7.71–7.69 (m, 2H); 13C{1H} NMR (CDCl3, 100 MHz) δ 169.3 (C), 158.0 (C), 149.9 (C), 148.4 (C), 145.1 (C), 136.4 (C), 135.5 (C), 132.4 (CH), 131.5 (CH), 129.4 (CH), 129.1 (CH), 128.2 (CH), 127.5 (CH), 125.6 (CH), 124.9 (C), 124.4 (C), 122.9 (CH), 122.8 (CH), 122.0 (C), 103.9 (CH); HRMS (ESI) m/z [M + H]+ calcd for C22H13ClN3O3+ 402.0640, found 402.0635.
- 5-(2-Fluorophenyl)-3-(9-methylbenzo[c]acridin-7-yl)isoxazole (7g). Compound 7g was prepared according to the general procedure GP-B from chlorooxime 2d-HCl (170 mg, 0.48 mmol) and 1-ethynyl-2-fluorobenzene 6g (230 mg, 1.9 mmol, 4 eq.) in DCM (10 mL) and sat. NaHCO3 aq. (5 mL) to give a pure product of 174 mg (90% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a brick-yellow solid: mp 234–235 °C (ethyl acetate); 1H NMR (CDCl3, 400 MHz) δ 9.56 (d, 1H, J = 7.9 Hz), 8.36–8.34 (m, 1H), 8.21 (td, 1H, J = 7.6, 1.8 Hz), 7.86 (dd, 1H, J = 7.6, 1.5 Hz), 7.82–7.78 (m, 1H), 7.76–7.68 (m, 5H), 7.52 (tdd, 1H, J = 7.5, 5.1, 1.8 Hz), 7.40 (td, 1H, J = 7.5, 1.2 Hz), 7.29–7.25 (m, 1H), 7.06 (d, 1H, J = 3.8 Hz), 2.55 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz) δ 164.5 (d, C, J = 2.5 Hz), 160.1 (C), 159.3 (d, C, J = 253.6 Hz), 146.6 (C), 146.0 (C), 136.9 (C), 133.3 (C), 132.4 (CH), 132.1 (d, CH, J = 8.6 Hz), 131.55 (C), 131.48 (C), 129.9 (CH), 129.0 (CH), 128.6 (CH), 127.84 (CH), 127.82 (CH), 127.5 (CH), 125.4 (C), 125.3 (CH), 124.9 (d, CH, J = 3.6 Hz), 123.8 (CH), 123.6 (C), 122.9 (CH), 116.4 (d, CH, J = 21.0 Hz), 115.6 (d, C, J = 12.3 Hz), 106.7 (d, CH, J = 11.3 Hz), 22.3 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C27H18FN2O+ 405.1398, found 405.1392.
- 5-((4-Isopropylphenoxy)methyl)-3-(9-methylbenzo[c]acridin-7-yl)isoxazole (7h). Compound 7h was prepared according to the general procedure GP-B from chlorooxime 2d-HCl (150 mg, 0.58 mmol) and 1-isopropyl-4-(prop-2-yn-1-yloxy)benzene 6c (370 mg, 2.1 mmol, 5 eq.) in DCM (10 mL) and sat. NaHCO3 aq. (5 mL) to give a pure product of 157 mg (95% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a bright yellow solid: mp 169–170 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 9.58 (br. s, 1H), 8.38 (br. s, 1H), 7.87–7.85 (m, 1H), 7.82–7.72 (m, 2H), 7.69 (d, 2H, J = 9.2 Hz), 7.63–7.61 (m, 2H), 7.24–7.20 (m, 2H), 7.02–6.98 (m, 2H), 6.66 (s, 1H), 5.39 (s, 2H), 2.91 (hept, 1H, J = 7.0 Hz), 2.55 (s, 3H), 1.25 (d, 6H, J = 7.0 Hz); 13C{1H} NMR (100 MHz, CDCl3) δ 169.1 (C), 159.3 (C), 155.9 (C), 146.6 (C), 146.0 (C), 142.6 (C), 136.9 (C), 133.3 (C), 132.4 (CH), 131.6 (C), 131.3 (C), 129.9 (CH), 129.1 (CH), 128.6 (CH), 127.8 (CH), 127.6 (CH), 127.5 (CH), 125.4 (C), 125.3 (CH), 123.7 (CH), 123.5 (C), 122.9 (CH), 114.8 (CH), 106.4 (CH), 61.9 (CH2), 33.3 (CH), 24.2 (CH3), 22.1 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C31H27N2O2+ 459.2068, found 459.2061.
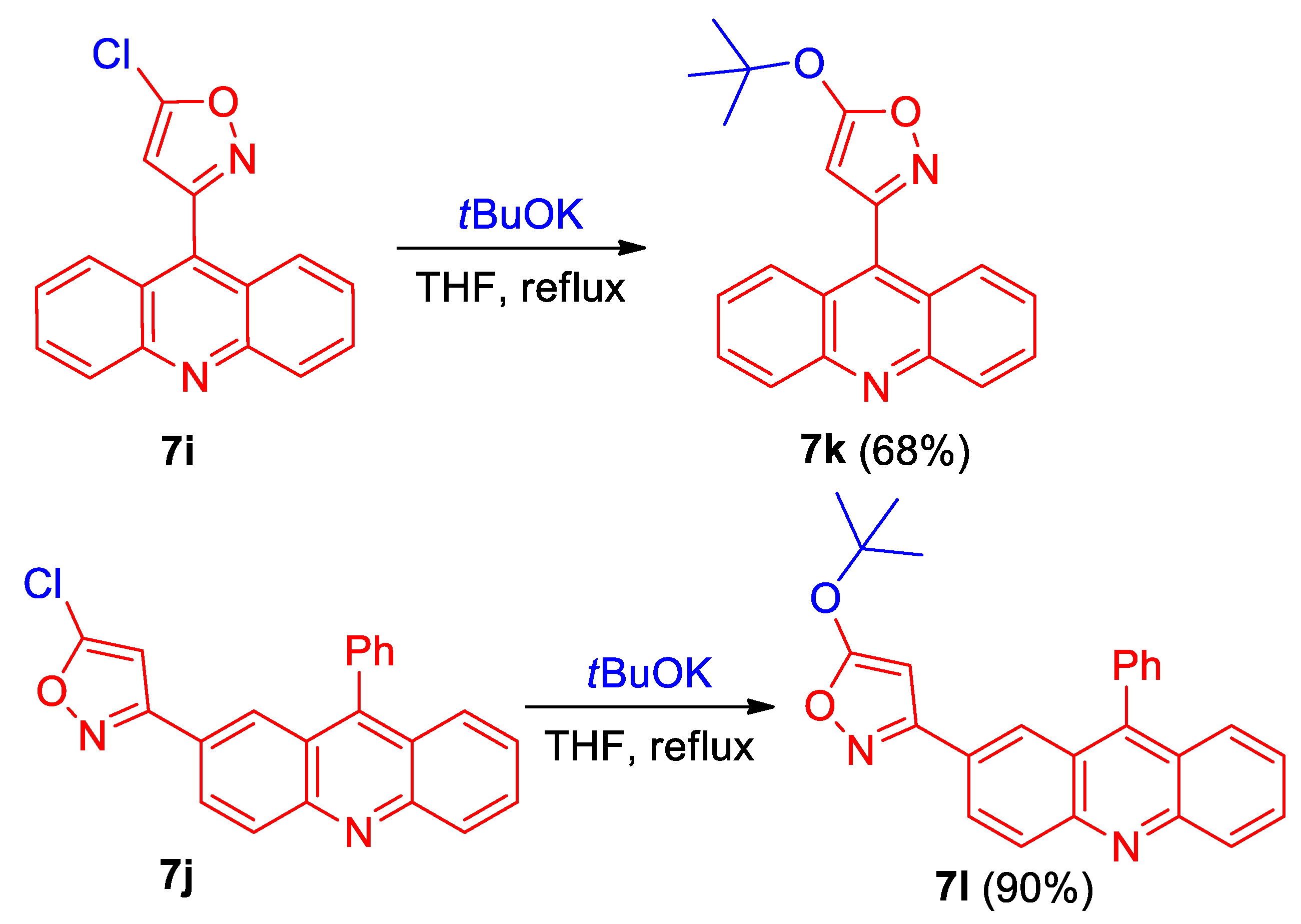
- 3-(Acridin-9-yl)-5-chloroisoxazole (7i). Compound 7i was prepared according to the general procedure GP-B from chlorooxime 2a-HCl (365 mg, 0.58 mmol) and 1,1-dichloroethylene 8 (2 mL, 24.9 mmol, 20 eq.) in DCM (20 mL) and sat. NaHCO3 aq. (10 mL) to give a pure product of 218 mg (62% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a bright yellow solid: mp 181–182 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.31 (dt, 2H, J = 8.8, 1.0 Hz), 7.91 (dt, 2H, J = 8.8, 1.0 Hz), 7.83 (ddd, 2H, J = 8.8, 6.6, 1.4 Hz), 7.52 (ddd, 2H, J = 8.7, 6.6, 1.2 Hz), 6.52 (s, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 160.9 (C), 155.9 (C), 148.6 (C), 132.1 (C), 130.3 (CH), 130.0 (CH), 127.1 (CH), 125.3 (CH), 124.7 (C), 104.5 (CH); HRMS (ESI) m/z [M + H]+ calcd for C16H10ClN2O+ 291.0477, found 291.0470.
- 5-Chloro-3-(9-phenylacridin-2-yl)isoxazole (7j). Compound 7j was prepared according to the general procedure GP-B from chlorooxime 2d-HCl (620 mg, 1.7 mmol) and 1,1-dichloroethylene 8 (2.7 mL, 33.6 mmol, 20 eq.) in DCM (30 mL) and sat. aq. NaHCO3 (15 mL) to give a pure product of 381 mg (64% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1, (v/v)) as a bright yellow solid: mp 159–160 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.36 (d, 1H, J = 9.0 Hz), 8.29 (d, 1H, J = 8.8 Hz), 8.23–8.20 (m, 1H), 8.01 (d, 1H, J = 1.9 Hz), 7.82 (ddd, 1H, J = 8.6, 6.6, 1.4 Hz), 7.72 (dd, 1H, J = 8.8, 1.3 Hz), 7.68–7.61 (m, 3H), 7.49–7.45 (m, 3H), 6.41 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz) δ 163.7 (C), 155.3 (C), 149.5 (C), 149.0 (C), 148.2 (C), 135.2 (C), 130.8 (CH), 130.7 (CH), 130.4 (CH), 129.7 (CH), 128.8 (CH), 128.7 (CH), 127.3 (CH), 127.0 (CH), 126.2 (CH), 125.8 (CH), 125.5 (C), 125.4 (C), 124.6 (C), 99.6 (CH); HRMS (ESI) m/z [M + H]+ calcd for C22H14ClN2O+ 357.0789, found 357.0785.
- 3-(Acridin-9-yl)-5-(tert-butoxy)isoxazole (7k). Compound 7k was prepared according to the published procedure [56] from chloroisoxazole 7i (165 mg, 0.59 mmol) and tBuOK (100 mg, 0.88 mmol) in THF (6 mL) to give a pure product of 128 mg (68% yield), after column chromatography on silica (light petroleum/ethyl acetate, 8:1, (v/v)) as a colorless solid: mp 166–167 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.28 (dt, 2H, J = 8.9, 0.9 Hz), 8.02 (dt, 2H, J = 8.7, 1.1 Hz), 7.80 (ddd, 2H, J = 8.7, 6.6, 1.4 Hz), 7.55 (ddd, 2H, J = 8.7, 6.6, 1.2 Hz), 1.64 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz) δ 172.2 (C), 160.6 (C), 148.7 (C), 134.3 (C), 130.2 (CH), 129.8 (CH), 126.6 (CH), 125.8 (CH), 124.8 (C), 87.3 (CH), 85.7 (C), 28.4 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C20H19N2O2+ 319.1441, found 319.1438.
- 5-(tert-Butoxy)-3-(9-phenylacridin-2-yl)isoxazole (7l). Compound 7l was prepared according to the published procedure [56] from chloroisoxazole 7j (320 mg, 0.9 mmol) and tBuOK (152 mg, 1.35 mmol) in THF (10 mL) to give a pure product of 318 mg (90% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a light yellow solid: mp 114–115 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 9.1 Hz, 1H), 8.29 (d, 1H, J = 8.6 Hz), 8.21 (dd, 1H, J = 9.1, 1.9 Hz), 8.04 (dd, 1H, J = 1.9, 0.7 Hz), 7.79 (ddd, 1H, J = 8.7, 6.5, 1.4 Hz), 7.71 (ddd, 1H, J = 8.8, 1.4, 0.7 Hz), 7.66–7.60 (m, 3H), 7.48–7.43 (m, 3H), 5.58 (s, 1H), 1.52 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz) δ 172.2 (C), 163.4 (C), 149.2 (C), 149.0 (C), 148.0 (C), 135.4 (C), 130.5 (CH), 130.4 (CH), 129.7 (CH), 128.63 (CH), 128.57 (CH), 127.7 (CH), 127.0 (C), 126.9 (CH), 126.0 (CH), 125.5 (C), 125.0 (CH), 124.7 (C), 85.3 (C), 82.6 (CH), 28.4 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C26H23N2O2+ 395.1754, found 395.1753.
- 3-(2-Methylacridin-9-yl)-4,5-dihydroisoxazole-5-carbonitrile (10). Compound 10 was prepared according to the general procedure GP-B from chlorooxime 2b-HCl (150 mg, 0.49 mmol) and acrylonitrile 9 (518 mg, 9.8 mmol, 20 eq.) in DCM (20 mL) and sat. NaHCO3 aq. (10 mL) to give a pure product of 116 mg (83% yield), after column chromatography on silica (light petroleum/ethyl acetate, 6:1–0:1, (v/v)) as a bright yellow solid: mp 217–218 °C (ethyl acetate); 1H NMR (CDCl3, 400 MHz) δ 8.28 (d, 1H, J = 8.8 Hz), 8.21–8.19 (m, 1H), 7.92 (d, 1H, J = 8.8 Hz), 7.82 (ddd, 1H, J = 8.4, 6.7, 1.4 Hz), 7.70–7.67 (m, 2H), 7.64 (dd, 1H, J = 8.2, 6.7, 1.2 Hz), 5.65 (dd, 1H, J = 10.7, 4.8 Hz), 3.93 (dd, 1H, J = 17.6, 10.7 Hz), 3.81 (dd, 1H, J = 17.6, 4.8 Hz), 2.61 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz) δ 154.6 (C), 147.8 (C), 147.5 (C), 138.0 (C), 133.6 (CH), 130.2 (CH), 130.1 (CH), 129.9 (CH), 129.5 (C), 127.6 (CH), 124.2 (C), 124.2 (C), 124.0 (CH), 122.1 (CH), 117.0 (C), 66.8 (CH), 46.2 (CH2), 22.2 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C18H14N3O+ 288.1131, found 288.1125.
- tert-Butyl 3-(9-phenylacridin-2-yl)-2H-azirine-2-carboxylate (11a). A mixture of isoxazole 7l (123 mg, 0.31 mmol) and FeCl2·4H2O (6.2 mg, 0.03 mmol) was stirred in MeCN (10 mL) at rt overnight. After the evaporation of the solvent, the product was filtered through a pad of silica (light petroleum/ethyl acetate, 10:1, (v/v)) to give pure product 11a at 107 mg (87% yield) as a yellow solid: mp 155–156 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.42 (d, 1H, J = 8.9 Hz), 8.31 (d, 1H, J = 8.7 Hz), 8.21–8.17 (m, 2H), 7.89–7.85 (m,1H), 7.77–7.75 (m, 1H), 7.67–7.58 (m, 3H), 7.57–7.46 (m, 2H), 7.44–7.41 (m, 1H), 2.77 (s, 1H), 1.43 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz) δ 170.7 (C), 159.0 (C), 150.4 (C), 149.7 (C), 149.6 (C), 134.8 (C), 132.6 (CH), 131.6 (CH), 131.4 (CH), 130.4 (CH), 130.3 (CH), 129.9 (CH), 129.0 (CH), 128.8 (CH), 128.6 (CH), 128.1 (CH), 127.2 (CH), 126.6 (CH), 125.6 (C), 124.5 (C), 119.8 (C), 81.7 (C), 30.9 (CH), 28.0 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C26H23N2O2+ 395.1754, found 395.1753.
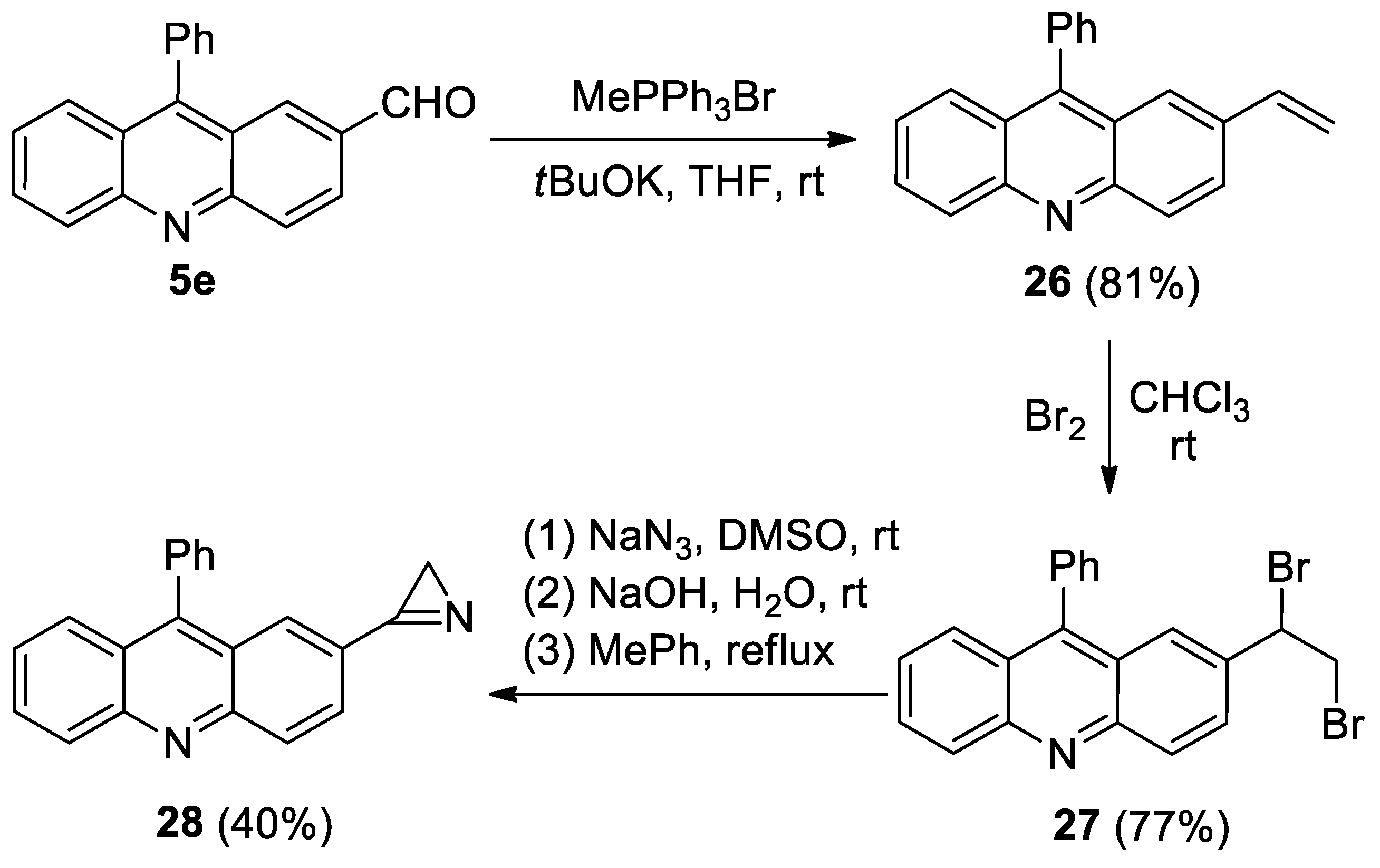
- 9-Phenyl-2-vinylacridine (26). Compound 26 was prepared according to the published procedure [57] from aldehyde 5e (400 mg, 1.4 mmol), methyltriphenylphosphonium bromide (1.51 g, 4.2 mmol) and tBuOK (475 mg, 4.2 mmol) in THF (25 mL) at rt for 48 h to give a pure product pf 397 mg (81% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a bright yellow solid: mp 108–109 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.24 (dd, 2H, J = 12.2, 8.9 Hz), 7.98 (dd, 1H, J = 9.2, 2.0 Hz), 7.75 (ddd, 1H, J = 8.5, 6.6, 11.5 Hz), 7.67 (dd, 1H, J = 8.8, 1.3 Hz), 7.65–7.58 (m, 3H), 7.52 (d, 1H, J = 2.0 Hz), 7.46–7.40 (m, 3H), 6.78 (dd, 1H, J = 17.6, 10.9 Hz), 5.83 (d, 1H, J = 17.6 Hz), 5.32 (d, 1H, J = 10.9 Hz); 13C{1H} NMR (CDCl3, 100 MHz) δ 148.8 (C), 148.6 (C), 147.0 (C), 136.5 (CH), 135.8 (C), 134.6 (C), 130.5 (CH), 129.94 (CH), 129.89 (CH), 129.6 (CH), 128.5 (CH), 128.4 (CH), 127.1 (CH), 126.8 (CH), 125.7 (CH), 125.5 (C), 125.3 (CH), 125.2 (C), 115.1 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C21H16N+ 282.1277, found 282.1290.
- 2-(1,2-Dibromoethyl)-9-phenylacridine (27). Compound 27 was prepared according to the published procedure [58] from styrene 26 (320 mg, 1.14 mmol) and Br2 (0.7 mL, 1.4 mmol0) in CHCl3 (5 mL) at 0 °C for 30 min to give a pure product of 387 mg (77% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a bright yellow solid: mp 154–155 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.34 (d, 1H, J = 9.1 Hz), 8.29 (d, 1H, J = 8.8 Hz), 7.84–7.79 (m, 2H), 7.71 (d, 1H, J = 8.8 Hz), 7.72–7.60 (m, 4H), 7.47–7.43 (m, 3H), 5.22 (dd, 1H, J = 10.6, 5.5 Hz), 4.11–4.01 (m, 2H); 13C{1H} NMR (CDCl3, 100 MHz) δ 149.0 (br. s, C), 148.3 (br. s, C), 135.3 (C), 130.9 (br. s, C), 130.6 (br. s, CH), 130.5 (CH), 130.4 (CH), 129.4 (br. s, CH), 128.7 (CH), 128.6 (C), 128.6 (CH), 128.3 (br. s, CH), 126.9 (CH), 126.3 (CH), 126.1 (CH), 125.5 (C), 124.3 (C), 51.1 (CH), 34.3 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C21H16Br2N+439.9644, found 439.9648.
- 2-(2H-Azirin-3-yl)-9-phenylacridine (28). Compound 28 was prepared according to the published procedure [59] from dibromide 27 (380 mg, 0.86 mmol), NaN3 (84 mg, 1.3 mmol), and NaOH (40 mg, 1 mmol) in DMSO (2 mL) and toluene (5 mL) to give a pure product of 102 mg (40% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a yellow-brown solid: mp 192–193 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.41 (d, 1H, J = 9.0 Hz), 8.32 (d, 1H, J = 8.8 Hz), 8.26 (dd, 1H, J = 8.9, 1.8 Hz), 8.20 (d, 1H, J = 1.6 Hz), 7.86 (ddd, 1H, J = 8.5, 6.5, 1.4 Hz), 7.75 (d, 1H, J = 8.6 Hz), 7.68–7.62 (m, 3H), 7.52–7.47 (m, 3H), 1.82 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz) δ 165.7 (C), 150.0 (C), 149.7 (C), 149.4 (C), 135.0 (C), 131.6 (CH), 131.3 (CH), 131.0 (CH), 130.4 (CH), 129.7 (CH), 128.9 (CH), 128.7 (CH), 127.9 (CH), 127.2 (CH), 126.5 (CH), 125.6 (C), 124.6 (C), 122.8 (C), 20.5 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C21H15N2+ 295.1230, found 295.1238.
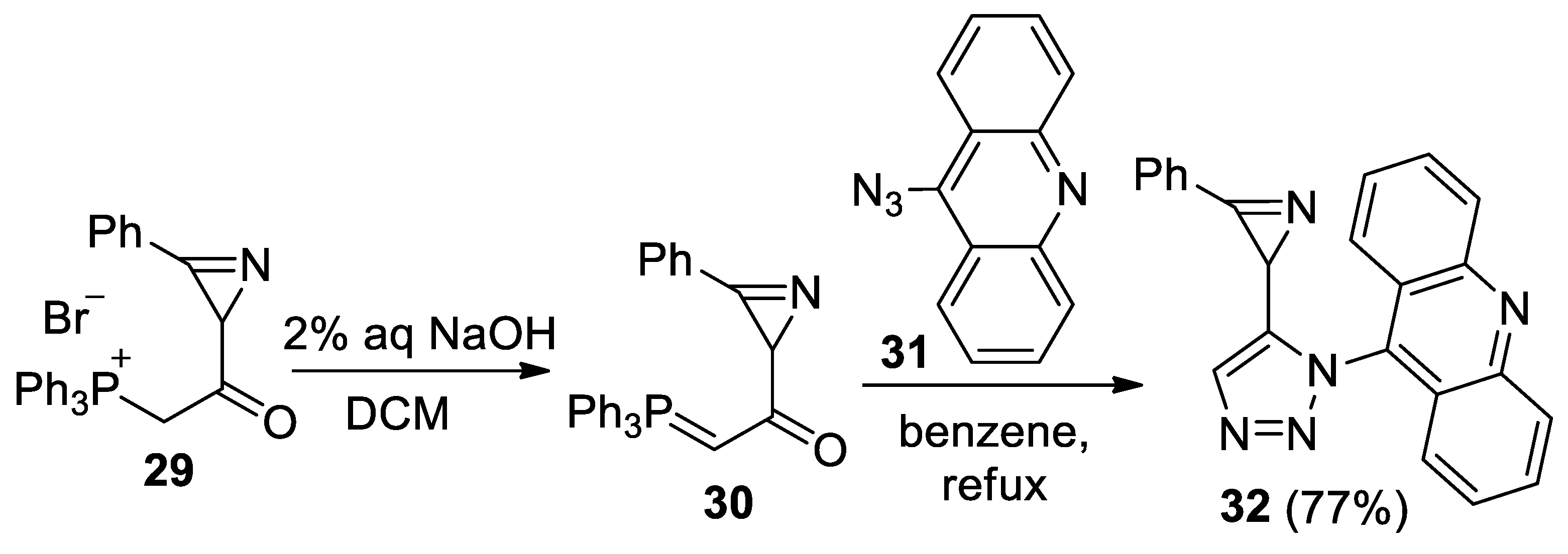
- 9-(5-(3-Phenyl-2H-azirin-2-yl)-1H-1,2,3-triazol-1-yl)acridine (32). Compound 32 was prepared according to the published procedure [55] from phosphonium salt 29 (250 mg, 0.5 mmol) and azide 31 (165 mg, 0.75 mmol) in benzene (10 mL) for 4 h to give a pure product of 139 mg (77% yield), after column chromatography on silica (light petroleum/ethyl acetate, 8:1–3:1, (v/v)) as a beige solid: mp 194–196 °C (dec., light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 8.30 (d, 1H, J = 8.8 Hz), 8.23 (d, 1H, J = 8.8 Hz), 7.89–7.85 (m, 1H), 7.84–7.80 (m, 1H), 7.82 (s, 1H), 7.66–7.58 (m, 2H), 7.51–7.47 (m, 1H), 7.38 (d, 2H, J = 9.2 Hz), 7.32–7.28 (m, 4H), 2.86 (s, 1H); 13C{1H} NMR (CDCl3, 100 MHz) δ 161.4 (C), 149.2 (C), 149.1 (C), 141.4 (C), 136.1 (C), 133.7 (CH), 132.3 (CH), 130.8 (CH), 130.7 (CH), 129.76 (CH), 129.74 (CH), 129.1 (CH), 129.0 (CH), 128.4 (CH), 128.2 (CH), 123.2 (C), 123.2 (C), 122.6 (CH), 122.4 (CH), 122.0 (C), 23.1 (CH); HRMS (ESI) m/z [M + H]+ calcd for C23H16N5+ 362.1400, found 362.1399.
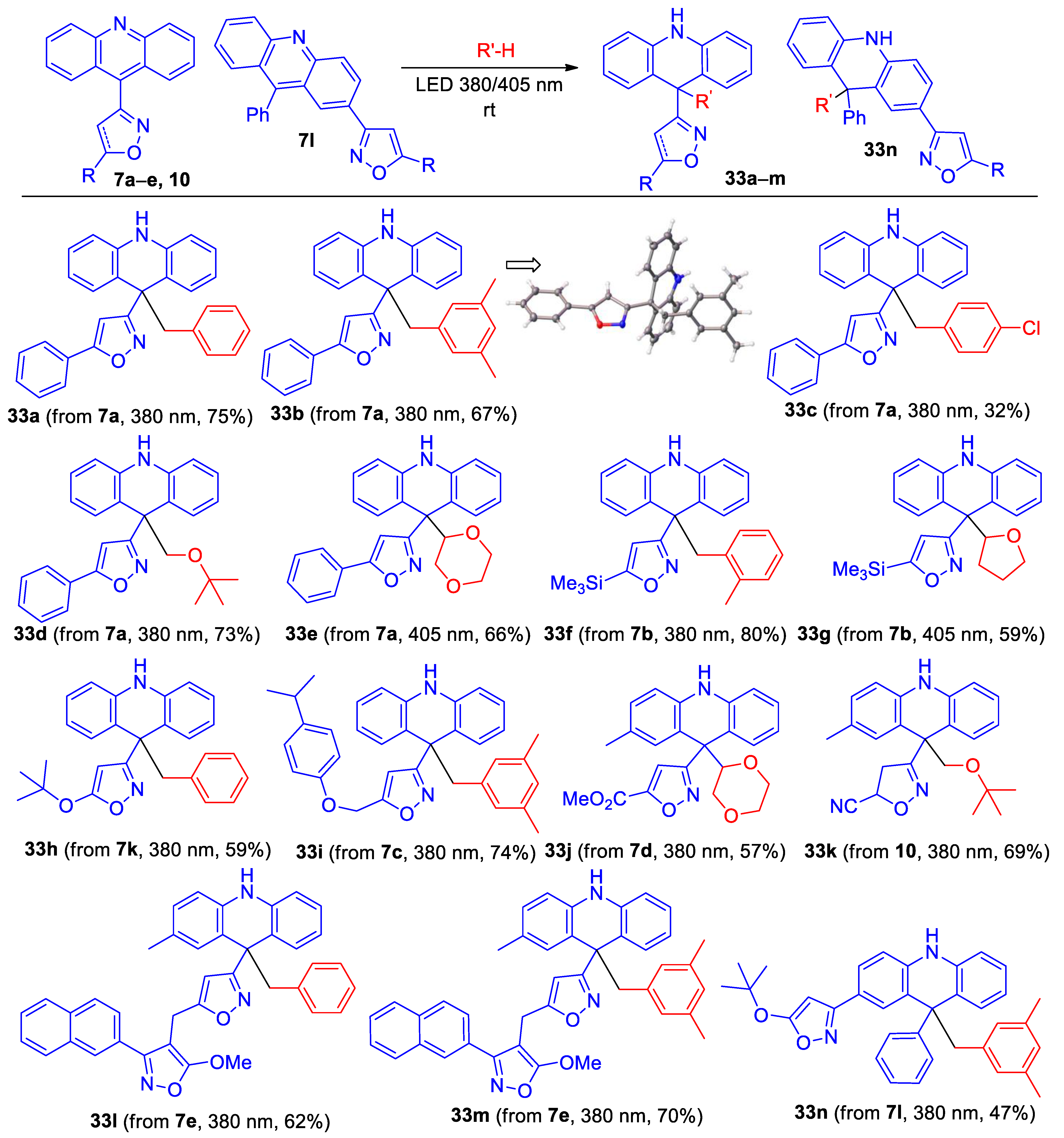
- 3-(9-Benzyl-9,10-dihydroacridin-9-yl)-5-phenylisoxazole (33a). Compound 33a was prepared according to the general procedure GP-C from isoxazole 7a (34 mg, 0.105 mmol) in toluene (4 mL) at 380 nm for 50 min to give a pure product of 33 mg (75% yield), after column chromatography on silica (light petroleum/ethyl acetate, 15:1, (v/v)) as a colorles solid: mp 143–144 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.76–7.73 (m, 2H), 7.467.40 (m, 3H), 7.12–7.04 (m, 5H), 6.94–6.90 (m, 1H), 6.88–6.84 (m, 1H), 6.45–6.43 (m, 2H), 6.32–6.30 (m, 2H), 6.23 (s, 1H), 5.65 (s, 1H), 3.60 (s, 2H); 13C{1H} NMR (CDCl3, 100 MHz) δ 170.7 (C), 169.4 (C), 138.5 (C), 137.0 (C), 130.7 (CH), 129.9 (CH), 129.3 (CH), 128.8 (CH), 128.0 (CH), 127.5 (C), 126.9 (CH), 125.83 (CH), 125.77 (CH), 122.0 (C), 120.4 (CH), 113.2 (CH), 101.0 (CH), 50.6 (CH2), 48.2 (C); HRMS (ESI) m/z [M + H]+ calcd for C9H23N2O+ 415.1805, found 415.1810.
- 3-(9-(3,5-Dimethylbenzyl)-9,10-dihydroacridin-9-yl)-5-phenylisoxazole (33b). Compound 33b was prepared according to the general procedure GP-C from isoxazole 7a (52 mg, 0.16 mmol) in mesitylene (10 mL) at 380 nm for 90 min to give a pure product of 48 mg (67% yield), after column chromatography on silica (light petroleum/ethyl acetate, 15:1, (v/v)) as a light yellow solid: mp 187–188 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.73 (dd, 2H, J = 7.7, 2.0 Hz), 7.44–7.38 (m, 3H), 7.10–7.06 (m, 4H), 6.84 (t, 2H, J = 7.5 Hz), 6.68 (br. s, 1H), 6.41 (dd, 2H, J = 8.3, 1.2 Hz), 6.23 (s, 1H), 5.86 (s, 2H), 5.64 (br. s, 1H), 3.47 (s, 2H), 1.98 (s, 6H); 13C{1H} NMR (CDCl3, 100 MHz) δ 170.6 (C), 169.3 (C), 138.7 (C), 136.7 (C), 136.0 (C), 129.9 (CH), 129.3 (CH), 128.8 (CH), 128.6 (CH), 127.8 (CH), 127.6 (C), 127.2 (CH), 125.8 (CH), 122.2 (C), 120.3 (CH), 112.9 (CH), 101.1 (CH), 50.7 (CH2), 48.3 (C), 21.0 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C31H27N2O+ 443.2118, found 443.2124.
- 3-(9-(4-Chlorobenzyl)-9,10-dihydroacridin-9-yl)-5-phenylisoxazole (33c). Compound 33c was prepared according to the general procedure GP-C from isoxazole 7a (60 mg, 0.19 mmol) and 4-chlorotoluene (2.36 g, 18.6 mmol, 100 eq.) in α,α,α-trifluorotoluene (12 mL) at 380 nm for 40 h to give a pure product of 27 mg (32% yield), after column chromatography on silica (light petroleum/ethyl acetate, 25:1, (v/v)) as a light yellow solid: mp 183–184 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, C6D6) δ 7.36–7.34 (m, 2H), 7.10–7.09 (m, 2H), 6.97–6.92 (m, 5H), 6.84–6.81 (m, 2H), 6.770–6.66 (m, 2H), 6.21–6.19 (m, 2H), 6.03–6.00 (m, 2H), 5.97 (s, 1H), 4.92 (br. s, 1H), 3.71 (s, 2H); 13C{1H} NMR (C6D6, 100 MHz) δ 170.8 (C), 169.9 (C), 138.9 (C), 136.3 (C), 132.5 (CH), 132.4 (C), 129.9 (CH), 129.7 (CH), 128.9 (CH), 128.4 (CH), 128.2 (CH), 127.5 (CH), 126.1 (CH), 122.4 (C), 120.9 (CH), 113.6 (CH), 101.4 (CH), 50.6 (CH2), 48.6 (C); HRMS (ESI) m/z [M + Na]+ calcd for C29H21ClN2NaO+ 471.1235, found 471.1236.
- 3-(9-(tert-Butoxymethyl)-9,10-dihydroacridin-9-yl)-5-phenylisoxazole (33d). Compound 33d was prepared according to the general procedure GP-C from isoxazole 7a (60 mg, 0.19 mmol) in MTBE (7 mL) at 380 nm for 2.5 h to give a pure product of 56 mg (73% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a colorless solid: mp 180–181 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, C6D6): δ 7.36–7.33 (m, 2H), 7.24–7.22 (m, 2H), 7.05–7.01 (m, 2H), 6.96–6.93 (m, 3H), 6.77–6.73 (m, 2H), 6.37–6.34 (m, 2H), 6.03 (s,1H), 5.62–5.60 (br. s, 1H), 4.32 (s, 2H), 0.87 (s, 9H); 13C NMR (100 MHz, C6D6): δ 169.5 (C), 169.2 (C), 139.4 (C), 130.0 (CH), 129.7 (CH), 128.9 (CH), 128.4 (CH), 128.0 (CH), 126.1 (CH), 122.7 (C), 120.6 (CH), 113.6 (CH), 101.4 (CH), 73.0 (C), 71.2 (CH2), 48.4 (C), 27.3 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C27H27N2O2+ 411.2064, found 411.2067.
- 3-(9-(1,4-Dioxan-2-yl)-9,10-dihydroacridin-9-yl)-5-phenylisoxazole (33e). Compound 33e was prepared according to the general procedure C from isoxazole 7a (50 mg, 0.16 mmol) in 1,4-dioxane (5 mL) at 405 nm for 90 min to give a pure product pf 52 mg (66% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a light brown semi-solid; NMR spectra indicate that the product is a mixture of two diastereomers in a ~1:1 ratio; 1H NMR (400 MHz, CDCl3): δ 7.72–7.68 (m, 2H), 7.42–7.36 (m, 3H), 7.22–7.18 (m, 1H), 7.14–7.11 (m, 2H), 7.04–7.02 (m, 1H), 6.87–6.71 (m, 4H), 6.27 (br. S, 1H), 6.12 (s, 1H), 4.46–4.43 (m, 1H), 3.84–3.79 (m, 2H), 3.64–3.54 (m, 2H), 3.37–3.31 (m, 1H), 3.24–3.19 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 168.9 ©, 168.3 (br. s, C), 138.4 (br. s, C), 138.3 (C), 132.2 (br. s, CH), 129.9 (CH), 129.6 (br. s, CH), 128.8 (CH), 128.5 (br. s, CH), 128.3 (br. s, CH), 127.5 (C), 125.7 (CH), 120.7 (br. s, CH), 120.1 (br. s, CH), 119.6 (br. s, C), 119.1 (br. s, C), 113.7 (br. s, CH), 113.3 (br. s, CH), 101.2 (CH), 81.2 (CH), 67.7 (CH2), 67.3 (CH2), 66.2 (CH2), 48.7 (br. s, C); HRMS (ESI) m/z [M + H]+ calcd for C26H23N2O3+ 411.1703, found 411.1700.
- 3-(9-(2-Methylbenzyl)-9,10-dihydroacridin-9-yl)-5-(trimethylsilyl)isoxazole (33f). Compound 33f was prepared according to the general procedure GP-C from isoxazole 7b (57 mg, 0.18 mmol) in o-xylene (8 mL) at 380 nm for 60 min to give a pure product of 61 mg (80% yield), after column chromatography on silica (light petroleum/ethyl acetate, 10:1, (v/v)) as a light yellow solid: mp 187–188 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.09 (td, 2H, J = 7.7, 1.5 Hz), 6.96–6.92 (m, 3H), 6.85–6.81 (m, 3H), 6.67 (td, 1H, J = 7.5, 1.5 Hz), 6.42 (d, 2H, J = 7.9 Hz), 6.12 (s, 1H), 6.08 (dd, 1H, J = 7.7, 1.4 Hz), 5.65 (br. s, 1H), 3.62 (s, 2H), 1.48 (s, 3H), 0.32 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 177.5 (C), 168.3 (C), 139.0 (C), 138.0 (C), 135.3 (C), 131.7 (CH), 129.6 (CH), 129.4 (CH), 127.8 (CH), 125.9 (CH), 124.2 (CH), 122.6 (C), 120.4 (CH), 114.1 (CH), 113.2 (CH), 48.1 (C), 47.2 (CH2), 18.8 (CH3), −1.9 (CH3); HRMS (ESI) m/z [M + Na]+ calcd for C27H28N2NaOSi+ 447.1863, found 447.1868.
- 3-(9-(Tetrahydrofuran-2-yl)-9,10-dihydroacridin-9-yl)-5-(trimethylsilyl)isoxazole (33g). Compound 33g was prepared according to the general procedure GP-C from isoxazole 7b (100 mg, 0.31 mmol) in THF (10 mL) at 405 nm for 3 h to give a pure product of 72 mg (59% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a colorless solid: mp 79–80 °C (light petroleum/ethyl acetate); NMR spectra indicate that the product is a mixture of two diastereomers in a ~1:1 ratio; 1H NMR (400 MHz, CDCl3): δ 7.16–7.05 (m, 3H), 6.95–6.92 (m, 1H), 6.82–6.75 (m, 2H), 6.2–6.68 (m, 2H), 6.23 (br. s, 1H), 6.05 (s, 1H), 4.86 (t, 1H, J = 7.2 Hz), 3.75 (td, 1H, J = 7.4, 4.8 Hz), 3.36 (q, 1H, 7.4 Hz), 1.73–1.69 (m, 2H), 1.33–1.23 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 176.9 (C), 167.0 (C), 138.8 (C), 138.2 (C), 132.1 (CH), 129.7 (CH), 128.1 (CH), 127.9 (CH), 125.1 (C), 121.2 (C), 120.2 (CH), 120.0 (CH), 119.9 (C), 115.4 (C), 114.1 (CH), 113.4 (CH), 112.9 (CH), 86.2 (CH), 69.2 (CH2), 49.8 (C), 27.7 (CH2), 25.7 (CH2), −1.9 (CH3); HRMS (ESI) m/z [M + Na]+ calcd for C23H26N2NaO2Si+ 413.1656, found 413.1658.
- 3-(9-Benzyl-9,10-dihydroacridin-9-yl)-5-(tert-butoxy)isoxazole (33h). Compound 33h was prepared according to the general procedure GP-C from isoxazole 7k (41 mg, 0.13 mmol) in toluene (5 mL) at 380 nm for 90 min to give a pure product of 31 mg (59% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a colorless solid: mp 161–162 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.11–7.00 (m, 5H), 6.90–6.82 (m, 4H), 6.38 (dd, 2H, J = 7.9, 1.2 Hz), 6.26–6.23 (m, 2H), 5.58 (s, 1H), 5.00 (s, 1H), 3.45 (s, 2H), 1.43 (s, 9H); 13C NMR (100 MHz, C6D6) δ 172.0 (C), 171.8 (C), 138.9 (C), 137.8 (C), 131.3 (CH), 129.8 (CH), 128.4 (CH), 128.4 (CH), 127.9 (CH), 127.3 (CH), 126.2 (CH), 122.9 (C), 120.7 (CH), 113.5 (CH), 85.7 (CH), 83.7 (C), 50.7 (CH2), 49.2 (C), 27.9 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C27H27N2O2+ 411.2067, found 411.2070.
- 3-(9-(3,5-Dimethylbenzyl)-9,10-dihydroacridin-9-yl)-5-((4-isopropylphenoxy)methyl)isoxazole (33i). Compound 33i was prepared according to the general procedure GP-C from isoxazole 7k (55 mg, 0.14 mmol) in mesitylene (10 mL) at 380 nm for 90 min to give a pure product of 53 mg (74% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a colorless solid: mp 167–168 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.15–7.12 (m, 2H), 7.08–7.04 (m, 2H), 6.98 (d, 2H, J = 7.6 Hz), 6.87–6.81 (m, 4H), 6.67 (s, 1H), 6.40–6.38 (m, 2H), 6.04 (s, 1H), 5.83 (s, 2H), 5.60 (s, 1H), 5.08 (s, 2H), 3.42 (s, 2H), 2.85 (hept, 1H, J = 6.9 Hz), 1.97 (s, 6H), 1.22 (d, 6H, J = 6.9 Hz); 13C NMR (100 MHz, CDCl3) δ 169.9 (C), 167.6 (C), 156.1 (C), 142.3 (C), 138.7 (C), 136.7 (C), 136.0 (C), 129.2 (CH), 128.7 (CH), 127.8 (CH), 127.4 (CH), 127.2 (CH), 122.2 (C), 120.3 (CH), 114.9 (CH), 113.0 (CH), 104.8 (CH), 61.9 (CH2), 50.7 (CH2), 48.3 (C), 33.3 (CH), 24.1 (CH3), 20.9 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C35H35N2O2+ 515.2693, found 515.2692.
- Methyl 3-(9-(1,4-dioxan-2-yl)-2-methyl-9,10-dihydroacridin-9-yl)isoxazole-5-carboxylate (33j). Compound 33j was prepared according to the general procedure GP-C from isoxazole 7d (90 mg, 0.28 mmol) in 1,4-dioxane (20 mL) at 380 nm for 2 h to givea pure product of 65 mg (57% yield), after column chromatography on silica (light petroleum/MTBE, 3:1, (v/v)) as a light yellow solid: mp 79–81 °C (light petroleum/MTBE); NMR spectra indicate that the product is a mixture of two diastereomers in a ~1:1 ratio; 1H NMR (400 MHz, C6D6) δ 7.31–7.29 (m, 0.5 H), 7.05–7.00 (m, 0.5 H), 6.99–6.96 (m, 1H), 6.89–6.86 (m, 0.5 H), 6.78–6.74 (m, 1H), 6.64–6.59 (m, 0.5 H), 6.49 (s, 0.5 H), 6.48 (s, 0.5 H), 6.30–6.24 (m, 1H), 6.21–6.14 (m, 1H), 5.44 (s, 0.5 H), 5.43 (s, 0.5 H), 4.78 (dd, 0.5 H, J = 10.2, 2.3 Hz), 4.72 (dd, 0.5 H, J = 10.2, 2.2 Hz), 3.91–3.86 (m, 1H), 3.70–3.60 (m, 1H), 3.44–3.37 (m, 2H), 3.14 (s, 1.5 H), 3.14 (s, 1.5 H), 3.16–3.10 (m, 1H), 3.06–3.00 (m, 1H), 2.04 (s, 1.5 H), 1.92 (s, 1.5 H); 13C NMR (100 MHz, C6D6) δ 168.99 (C), 168.96 (C), 160.1 (C), 157.04 (C), 157.03 (C), 139.1 (C), 136.7 (C), 136.5 (C), 132.8 (CH), 132.5 (CH), 130.3 (C), 129.78 (CH), 129.75 (CH), 129.6 (CH), 129.5 (CH), 128.66 (CH), 128.62 (CH), 128.4 (CH), 120.8 (CH), 120.3 (CH), 119.6 (C), 119.5 (C), 119.3 (C), 119.1 (C), 114.3 (CH), 114.1 (CH), 113.9 (CH), 113.7 (CH), 111.48 (CH), 111.46 (CH), 82.1 (CH), 81.9 (CH), 67.9 (CH2), 67.72 (CH2), 67.66 (CH2), 66.16 (CH2), 66.12 (CH2), 51.8 (CH3), 49.33 (C), 49.25 (C), 30.2 (C), 20.9 (CH3), 20.7 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C23H25N2O5+ 407.1601, found 407.1601.
- 3-(9-(tert-Butoxymethyl)-2-methyl-9,10-dihydroacridin-9-yl)-4,5-dihydroisoxazole-5-carbonitrile (33k). Compound 33k was prepared according to the general procedure GP-C from isoxazole 10 (60 mg, 0.18 mmol) in MTBE (20 mL) at 380 nm for 2 h to give a pure product of 47 mg (69% yield), after column chromatography on silica (light petroleum/MTBE, 1:1, (v/v)) as a light brow oil; NMR spectra indicate that the product is a mixture of two diastereomers in a ~1:1 ratio; 1H NMR (400 MHz, C6D6) δ 7.23–7.19 (m, 1 H), 7.03–6.91 (m, 2 H), 6.84–6.78 (m, 1.5 H), 6.73–6.69 (m, 0.5 H), 6.26–6.19 (m, 2H), 5.38 (s, 0.5 H), 5.37 (s, 0.5 H), 4.07–3.94 (m, 3H), 2.51–2.27 (m, 2H), 2.21 (s, 1.5 H), 2.08 (s, 1.5 H), 0.77 (s, 4.5 H), 0.77 (s, 4.5 H); 13C NMR (100 MHz, C6D6) δ 161.1 (C), 160.9 (C), 139.9 (C), 139.4 (C), 137.3 (C), 136.9 (C), 129.6 (C), 129.4 (CH), 129.3 (CH), 128.63 (CH), 128.56 (CH), 128.45 (CH), 128.42 (CH), 128.39 (CH), 121.1 (CH), 120.3 (CH), 120.0 (C), 119.8 (C), 119.2 (C), 119.1 (C), 117.9 (C), 117.7 (C), 114.1 (CH), 114.0 (CH), 113.94 (CH), 113.90 (CH), 73.0, 71.68 (CH2), 71.63 (CH2), 65.9 (CH), 65.8 (CH), 48.6, 48.5, 42.0 (CH2), 27.1 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C23H26N3O2+ 376.2020, found 376.2013.
- 4-((3-(9-Benzyl-2-methyl-9,10-dihydroacridin-9-yl)isoxazol-5-yl)methyl)-5-methoxy-3-(naphthalen-2-yl)isoxazole (33l). Compound 33l was prepared according to the general procedure GP-C from isoxazole 7e (50 mg, 0.10 mmol) in toluene (10 mL) at 380 nm for 5h to give a pure product of 37 mg (62% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a colorless solid: mp 157–158 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.91 (s, 1H), 7.86–7.84 (m, 2H), 7.71 (d, 1H, J = 8.0 Hz), 7.61 (d, 1H, J = 8.6 Hz), 7.54 (t, 1H, J = 7.3 Hz), 7.49 (t, 1H, J = 7.4 Hz), 7.02 (d, 1H, J = 7.8 Hz), 6.98 (d, 1H, J = 8.4 Hz), 6.89–6.78 (m, 5H), 6.622 (t, 1H, J = 7.7 Hz), 6.34 (d, 1H, J = 7.9 Hz), 6.29 (d, 1H, J = 8.0 Hz), 6.24 (d, 2H, J = 7.6 Hz), 5.65 (s, 1H), 5.50 (s, 1H), 4.12 (s, 3H), 3.85 (s, 2H), 3.51–3.43 (m, 2H), 2.13 (s, 3H); 1H NMR (400 MHz, C6D6) δ 8.03 (s, 1H), 7.83–7.81 (m, 1H), 7.58–7.56 (m, 2H), 7.53–7.51 (m, 1H), 7.28–7.20 (m, 2H), 7.03–6.98 (m, 2H), 6.92–6.84 (m, 4H), 6.75–6.73 (m, 1H), 6.57–6.53 (m, 1H), 6.50–6.47 (m, 2H), 5.98–5.94 (m, 2H), 5.55 (s, 1H), 4.87 (s, 1H), 4.87 (s, 1H), 3.89–3.79 (m, 2H), 3.48 (s, 2H), 3.33 (s, 3H), 1.93 (s, 3H); 13C NMR (100 MHz, C6D6) δ 170.7 ©, 170.6 (C), 170.4 (C), 164.5 (C), 139.0 (C), 137.8 (C), 136.6 (C), 134.2 (C), 133.6 (C), 131.2 (CH), 129.8 (CH), 129.68 (CH), 129.66 (C), 129.02 (CH), 128.9 (CH), 128.8 (CH), 128.4 (CH), 128.2 (CH), 128.0 (CH), 127.9 (CH), 127.8 (C), 127.3 (CH), 127.1 (CH), 126.7 (CH), 126.3 (CH), 125.5 (CH), 122.5 (C), 120.3 (CH), 113.6 (CH), 113.4 (C), 103.6 (CH), 86.8 (C), 57.4 (CH3), 51.2 (CH2), 48.7 (C), 20.7 (CH3), 20.1 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C39H32N3O3+ 590.2438, found 590.2424.
- 4-((3-(9-(3,5-Dimethylbenzyl)-2-methyl-9,10-dihydroacridin-9-yl)isoxazol-5-yl)methyl)-5-methoxy-3-(naphthalen-2-yl)isoxazole (33m). Compound 33m was prepared according to the general procedure GP-C from isoxazole 7e (50 mg, 0.10 mmol) in mesytilene (10 mL) at 380 nm for 5h to give a pure product of 43 mg (70% yield), after column chromatography on silica (light petroleum/ethyl acetate, 20:1, (v/v)) as a beige solid: mp 162–163 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, C6D6): δ 8.03 (s, 1H), 7.84–7.81 (m, 1H), 7.58–7.56 (m, 2H), 7.54–7.51 (m, 1H), 7.27–7.20 (m, 2H), 7.03–7.00 (m, 2H), 6.92–6.88 (m, 1H), 6.77–6.74 (m, 1H), 6.61 (s, 1H), 6.57–6.53 (m, 1H), 6.10 (s, 2H), 6.01–5.97 (m, 2H), 5.58 (s, 1H), 4.91 (s, 1H), 3.84–3.75 (m, 2H), 3.49 (s, 2H), 3.33 (s, 3H), 1.99 (s, 6H), 1.94 (s, 3H); 13C NMR (100 MHz, C6D6) δ 170.7 (C), 170.5 (C), 170.4 (C), 164.5 (C), 139.2 (C), 137.4 (C), 136.8 (C), 136.0 (C), 134.2 (C), 133.6 (C), 129.8 (CH), 129.7 (CH), 129.6 (C), 129.4 (CH), 129.0 (CH), 128.9 (CH), 128.6 (CH), 128.4 (CH), 128.4 (CH), 128.2 (CH), 128.0 (CH), 127.9 (CH), 127.7 (CH), 127.1 (CH), 126.7 (CH), 125.5 (CH), 122.8 (C), 120.3 (CH), 113.3 (CH), 113.2 (CH), 103.7 (CH), 86.8 (C), 57.4 (CH3), 51.2 (CH2), 48.8 (C), 21.3 (CH3), 20.7 (CH3), 20.1 (CH2); HRMS (ESI) m/z [M + H]+ calcd for C41H36N3O3+ 618.2751, found 618.2738.
- 5-(tert-Butoxy)-3-(9-(3,5-dimethylbenzyl)-9-phenyl-9,10-dihydroacridin-2-yl)isoxazole (33n). Compound 33n was prepared according to the general procedure GP-C from isoxazole 7l (56 mg, 0.14 mmol) in mesytilene (15 mL) at 380 nm for 8h to give a pure product of 34 mg (47% yield), after column chromatography on silica (light petroleum/MTBE, 20:1, (v/v)) as a colorless solid: mp 138–139 °C (light petroleum/ethyl acetate); 1H NMR (400 MHz, C6D6) δ 7.68–7.66 (m, 1H), 7.53–7.47 (m, 3H), 7.12–7.10 (m, 2H), 7.04–7.01 (m, 1H), 6.92–6.88 (m, 1H), 6.82–6.80 (m, 1H), 6.68–6.64 (m, 2H), 6.02 (s, 2H), 5.99–5.94 (m, 2H), 5.37 (s, 1H), 4.92 (s, 1H), 3.40–3.30 (m, 2H), 2.00 (s, 6H), 1.11 (s, 9H); 13C NMR (100 MHz, C6D6) δ 172.3 (C), 163.9 (C), 151.3 (C), 140.4 (C), 138.6 (C), 137.6 (C), 136.1 (C), 130.8 (CH), 130.0 (CH), 129.1 (CH), 129.0 (CH), 128.4 (CH), 128.3 (CH), 127.8 (CH), 127.3 (C), 127.1 (C), 127.0 (CH), 126.4 (CH), 125.5 (CH), 122.4 (C), 120.8 (CH), 113.3 (CH), 113.0 (CH), 84.1 (C), 82.6 (CH), 52.5 (CH2), 51.7 (C), 28.2 (CH3), 21.3 (CH3); HRMS (ESI) m/z [M + H]+ calcd for C35H35N2O2+ 515.2693, found 515.2690.
3.2.5. Cell Culture
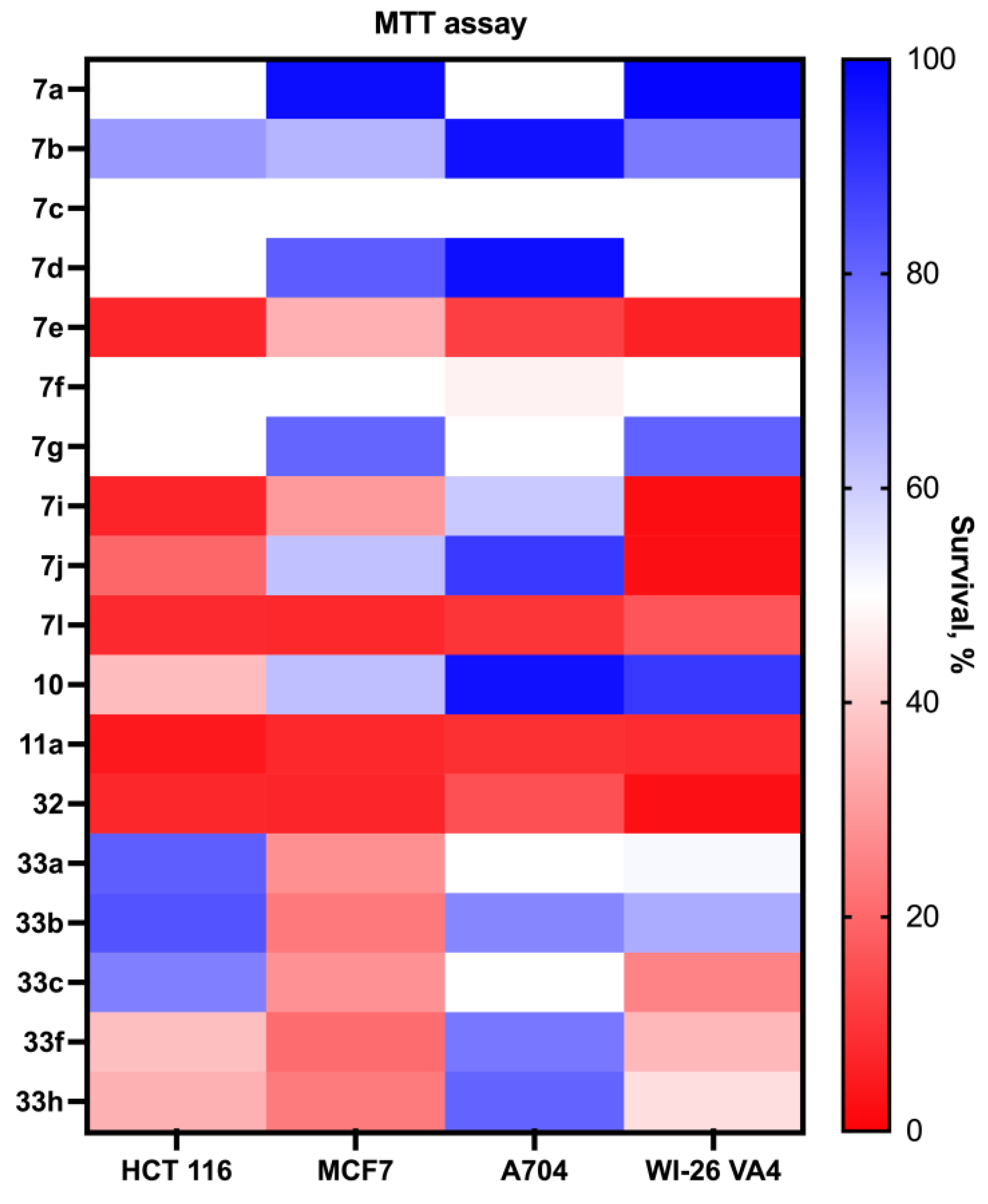
3.2.6. Antiproliferative Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gensicka-Kowalewska, M.; Cholewiński, G.; Dzierzbicka, K. Recent developments in the synthesis and biological activity of acridine/acridone analogues. RSC Adv. 2017, 7, 15776–15804. [Google Scholar] [CrossRef]
- Ježek, J.; Hlaváček, J.; Šebestík, J. Biomedical Applications of Acridines; Progress in Drug Research; Rainsford, R.D., Ed.; Springer: Cham, Switzerland, 2017; Volume 72, pp. 1–243. [Google Scholar]
- Varakumar, P.; Rajagopal, K.; Aparna, B.; Raman, K.; Byran, G.; Gonçalves Lima, C.M.; Rashid, S.; Nafady, M.H.; Emran, T.B.; Wybraniec, S. Acridine as an Anti-Tumour Agent: A Critical Review. Molecules 2023, 28, 193. [Google Scholar] [CrossRef]
- Piorecka, K.; Kurjata, J.; Stanczyk, W.A. Acriflavine, an acridine derivative for biomedical application: Current state of the art. J. Med. Chem. 2022, 65, 11415–11432. [Google Scholar] [CrossRef] [PubMed]
- Prasher, P.; Sharma, M. Medicinal chemistry of acridine and its analogues. MedChemComm 2018, 9, 1589–1618. [Google Scholar] [CrossRef] [PubMed]
- Viegas, C.J.; Danuello, A.; Bolzani, V.S.; Barreiro, E.J.; Fraga, C.M. Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar]
- Zhang, Q.; Yu, X. Current Scenario of Acridine Hybrids with Anticancer Potential. Curr. Top. Med. Chem. 2021, 21, 1773–1786. [Google Scholar] [CrossRef] [PubMed]
- Chufarova, N.; Czarnecka, K.; Skibiński, R.; Cuchra, M.; Majsterek, I.; Szymański, P. New tacrine–acridine hybrids as promising multifunctional drugs for potential treatment of Alzheimer’s disease. Arch. Pharm. 2018, 351, e1800050. [Google Scholar] [CrossRef]
- Maciejewska, K.; Czarnecka, K.; Kręcisz, P.; Niedziałek, D.; Wieczorek, G.; Skibiński, R.; Szymański, P. Novel Cyclopentaquinoline and Acridine Analogs as Multifunctional, Potent Drug Candidates in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 5876. [Google Scholar] [CrossRef]
- Remya, R.S.; Ramalakshmi, N.; Nalini, C.N.; Amuthalakshmi, S. Design, synthesis, and in vitro evaluation of novel acridine derivatives as monoamine oxidase inhibitors. Rasayan J. Chem. 2022, 15, 2318–2325. [Google Scholar] [CrossRef]
- Fonte, M.; Tassi, N.; Gomes, P.; Teixeira, C. Acridine-Based Antimalarials—From the Very First Synthetic Antimalarial to Recent Developments. Molecules 2021, 26, 600. [Google Scholar] [CrossRef]
- Kumar, A.; Srivastava, K.; Kumar, S.R.; Puri, S.K.; Chauhan, P.M. Synthesis of new 4-aminoquinolines and quinoline–acridine hybrids as antimalarial agents. Bioorganic Med. Chem. Lett. 2010, 20, 7059–7063. [Google Scholar] [CrossRef]
- Pandey, S.K.; Biswas, S.; Gunjan, S.; Chauhan, B.S.; Singh, S.; Srivastava, K.; Singh, S.; Batra, S.; Tripathi, R. Pyrrolidine-Acridine hybrid in Artemisinin-based combination: A pharmacodynamic study. Parasitology 2016, 143, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Awad, H.M.; El-Sayed, I.E.T.; El Gokha, A.A. Synthesis and antiproliferative activity of new hybrids bearing neocryptolepine, acridine and α-aminophosphonate scaffolds. J. Iran. Chem. Soc. 2020, 17, 1211–1221. [Google Scholar] [CrossRef]
- Lisboa, T.; Silva, D.; Duarte, S.; Ferreira, R.; Andrade, C.; Lopes, A.L.; Ribeiro, J.; Farias, D.; Moura, R.; Reis, M.; et al. Toxicity and antitumor activity of a thiophene–acridine hybrid. Molecules 2019, 25, 64. [Google Scholar] [CrossRef] [PubMed]
- Pandhurnekar, C.P.; Pandhurnekar, H.C.; Mungole, A.J.; Butoliya, S.S.; Yadao, B.G. A review of recent synthetic strategies and biological activities of isoxazole. J. Heterocycl. Chem. 2022, 60, 537–565. [Google Scholar] [CrossRef]
- Agrawal, N.; Mishra, P. The synthetic and therapeutic expedition of isoxazole and its Analogs. Med. Chem. Res. 2018, 27, 1309–1344. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mo, J.; Lin, H.Z.; Chen, Y.; Sun, H.P. The Recent Progress of Isoxazole in Medicinal Chemistry. Bioorg. Med. Chem. 2018, 26, 3065–3075. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, Q.; Tang, H.; Pan, X. Isoxazole/Isoxazoline Skeleton in the Structural Modification of Natural Products: A Review. Pharmaceuticals 2023, 16, 228. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, M.; Mohtadi-Haghighi, D.; Mirfazli, S.S.; Mahdavi, M.; Hariri, R.; Lotfian, H.; Edraki, N.; Iraji, A.; Firuzi, O.; Akbarzadeh, T. Design and synthesis of selective acetylcholinesterase inhibitors: Arylisoxazole-phenylpiperazine derivatives. Chem. Biodivers. 2019, 16, e180043. [Google Scholar] [CrossRef]
- Rastegari, A.; Safavi, M.; Vafadarnejad, F.; Najafi, Z.; Hariri, R.; Bukhari, S.N.A.; Iraji, A.; Edraki, N.; Firuzi, O.; Saeedi, M.; et al. Synthesis and evaluation of novel arylisoxazoles linked to tacrine moiety: In vitro and in vivo biological activities against Alzheimer’s disease. Mol. Divers. 2022, 26, 409–428. [Google Scholar] [CrossRef]
- Wang, M.; Li, L.; Yang, S.; Guo, F.; Zhu, G.; Zhu, B.; Chang, J. Synthesis of novel oxazol-5-one derivatives containing chiral trifluoromethyl and isoxazole moieties as potent antitumor agents and the mechanism investigation. Bioorg. Chem. 2023, 135, 106505. [Google Scholar] [CrossRef] [PubMed]
- Lingala, A.K.; Murahari, K.K.; Desireddi, J.R.; Mothe, T.; Mait, B.; Manchal, R. Design, synthesis and biological evaluation of isoxazole bearing 1,3-oxazole-1,3,4-oxadiazole derivatives as anticancer agents. Chem. Data Collect. 2023, 43, 100959. [Google Scholar] [CrossRef]
- Hawash, M.; Kahraman, D.C.; Ergun, S.G.; Cetin-Atalay, R.; Baytas, S.N. Synthesis of novel indole-isoxazole hybrids and evaluation of their cytotoxic activities on hepatocellular carcinoma cell lines. BMC Chem. 2021, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Güiza, F.M.; Duarte, Y.B.; Mendez-Sanchez, S.C.; Bohórquez, A.R.R. Synthesis and in vitro evaluation of substituted tetrahydroquinoline-isoxazole hybrids as anticancer agents. Med. Chem. Res. 2019, 28, 1182–1196. [Google Scholar] [CrossRef]
- Xie, X.; Xiong, S.S.; Li, X.; Huang, H.; Wu, F.B.; Shen, P.F.; Peng, C.; He, G.; Han, B. Design and organocatalytic synthesis of spirooxindole–cyclopentene–isoxazole hybrids as novel MDM2–p53 inhibitors. Org. Chem. Front. 2021, 8, 1836–1843. [Google Scholar] [CrossRef]
- Kushwaha, P.K.; Srivastava, K.S.; Kumari, N.; Kumar, R.; Mitra, D.; Sharon, A. Synthesis and anti-HIV activity of a new isoxazole containing disubstituted 1,2,4-oxadiazoles analogs. Bioorg. Med. Chem. 2022, 56, 116612. [Google Scholar] [CrossRef] [PubMed]
- Kalirajan, R.; Rafick, M.H.; Sankar, S.; Gowramma, B. Green synthesis of some novel chalcone and isoxazole substituted 9-anilinoacridine derivatives and evaluation of their antimicrobial and larvicidal activities. Indian J. Chem. 2018, 57B, 583–590. [Google Scholar]
- Mosher, M.D.; Natale, N.R. The preparation of intercalating isoxazoles via a nitrile oxide cycloaddition. J. Heterocycl. Chem. 1995, 32, 779–781. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Hussain, F.H.; Quadrelli, P. 9-Anthraldehyde oxime: A synthetic tool for variable applications. Monatshefte Für Chem.-Chem. Mon. 2020, 151, 1643–1658. [Google Scholar] [CrossRef]
- Galenko, E.E.; Khlebnikov, A.F.; Novikov, M.S. Isoxazole-azirine isomerization as a reactivity switch in the synthesis of heterocycles. Chem. Heterocycl. Compd. 2016, 52, 637–650. [Google Scholar] [CrossRef]
- Lim, R.K.; Lin, Q. Azirine ligation: Fast and selective protein conjugation via photoinduced azirine–alkene cycloaddition. Chem. Commun. 2010, 46, 7993–7995. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, Z.; Ma, W.; Bao, G.; Li, Y.; Yu, C.; Li, J.; E, R.; Xu, Z.; Wang, R.; et al. Copper (I)-Catalyzed Late-Stage Introduction of Oxime Ethers into Peptides at the Carboxylic Acid Site. Org. Lett. 2022, 24, 9248–9253. [Google Scholar] [CrossRef]
- Sakharov, P.A.; Novikov, M.S.; Rostovskii, N.V. 2H-Azirines in medicinal chemistry. Chem. Heterocycl. Compd. 2021, 57, 512–521. [Google Scholar] [CrossRef]
- Mueller, J.O.; Schmidt, F.G.; Blinco, J.P.; Barner-Kowollik, C. Visible-Light-Induced Click Chemistry. Angew. Chem. Int. Ed. 2015, 54, 10284–10288. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.S.; Lin, Q. Light-Triggered Click Chemistry. Chem. Rev. 2021, 121, 6991–7031. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, B.D.; Macdougall, L.J.; Mavila, S.; Sinha, J.; Kirkpatrick, B.E.; Anseth, K.S.; Bowman, C.N. Photoclick Chemistry: A Bright Idea. Chem. Rev. 2021, 121, 6915–6990. [Google Scholar] [CrossRef] [PubMed]
- Luque, A.; Paternoga, J.; Opatz, T. Strain Release Chemistry of Photogenerated Small-Ring Intermediates. Chem. Eur. J. 2021, 27, 4500–4516. [Google Scholar] [CrossRef]
- 9-Phenyl Acridine. Available online: https://www.hampfordresearch.com/wp-content/uploads/2022/11/FP_5400_9_PA.pdf (accessed on 11 July 2014).
- Hansda, S.; Ghosh, G.; Ghosh, R. 9-phenyl acridine photosensitizes A375 cells to UVA radiation. Helion 2020, 6, e04733. [Google Scholar] [CrossRef]
- Padwa, A. Cycloaddition and cyclization chemistry of 2H-azirines. In Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 2010; Volume 99, pp. 1–31. [Google Scholar]
- Khlebnikov, A.F.; Novikov, M.S. Ring Expansions of Azirines and Azetines. Top. Heterocycl. Chem. 2015, 41, 143–232. [Google Scholar]
- Krivolapova, Y.V.; Tomashenko, O.A.; Funt, L.D.; Spiridonova, D.V.; Novikov, M.S.; Khlebnikov, A.F. Azirine-triazole hybrids: Selective synthesis of 5-(2H-azirin-2-yl)-, 5-(1H-pyrrol-2-yl)-1H-1,2,3-triazoles and 2-(5-(2H-azirin-2-yl)-1H-1,2,3-triazol-1-yl)pyridines. Org. Biomol. Chem. 2022, 20, 5434–5443. [Google Scholar] [CrossRef]
- Periasamy, N. Nanosecond laser flash photolysis of acridine in organic solvents. Proc. Indian Acad. Sci. (Chem. Sci.) 1984, 93, 1361–1375. [Google Scholar] [CrossRef]
- Braun, D.; Sfudenroth, R. Synthese von 9-Phenyl-9-benzylacridan. Liebigs Ann. Chem. 1981, 1981, 999–1002. [Google Scholar] [CrossRef]
- Castellano, A.; Catteau, J.-P.; Lablache-Combier, A.; Allan, G. Photochemical Studies. XVI. Second Moment Analysis of the Electron Spin Resonance Spectra of Radicals Formed by Ultraviolet Irradiation of 9-Phenylacridine and of Acridine. Can. J. Chem. 1973, 51, 3508–3513. [Google Scholar] [CrossRef]
- Niizuma, S.; Koizumi, M. Radicals Produced by UV Irradiation of Acridane and Acridine in Glassy and Crystalline Medium at 77°K. Bull. Chem. Soc. Jpn. 1968, 41, 1090–1096. [Google Scholar] [CrossRef]
- Agafonova, A.V.; Sakharov, P.A.; Smetanin, I.A.; Rostovskii, N.V.; Khlebnikov, A.F.; Novikov, M.S. Stannyl radical-mediated synthesis of 6H-1,3-oxazin-6-ones from 2-acyloxyazirines or whether free radicals can open the azirine ring? Org. Chem. Front. 2022, 9, 4118–4127. [Google Scholar] [CrossRef]
- Ye, Y.; Holder, G.M.; Duke, C.C. The preparation of 2H and 3H-labelled 7, 9-, and 7, 10-dimethylbenz[c]acridine by catalytic dehalogenation. J. Label. Compd. Radiopharm. 1993, 33, 1–10. [Google Scholar] [CrossRef]
- Rosewear, J.; Wilshire, J.F. The preparation of some 2-nitroacridines and related compounds. Aust. J. Chem. 1981, 34, 839–853. [Google Scholar]
- Tsuge, O.; Torii, A. Compounds Related to Acridine. X. The Reaction of 9-Ethynylacridine with Active Methylene Compounds. Bull. Chem. Soc. Jpn. 1973, 46, 283–285. [Google Scholar] [CrossRef]
- Sakharov, P.A.; Novikov, M.S.; Khlebnikov, A.F. 2-Diazoacetyl-2H-azirines: Source of a Variety of 2H-Azirine Building Blocks with Orthogonal and Domino Reactivity. J. Org. Chem. 2018, 83, 8304–8314. [Google Scholar] [CrossRef]
- Singer, B.; Maas, G. Dikationether, 3 [1]. Substitutionen an Bis (acridinium) ethern und 9-Trifloxyacridinium-Salzen mit Halogeniden, Pseudohalogeniden und Schwefel-Nucleophilen. Z. Für Naturforschung B 1984, 39, 1399–1408. [Google Scholar] [CrossRef]
- Suzuki, H.; Tanaka, Y. An unusually acidic methyl group directly bound to acridinium cation. J. Org. Chem. 2001, 66, 2227–2231. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, J.; Peuntinger, K.; Fröhlich, R.; Guldi, D.M.; Mattay, J. Synthesis and properties of acridine and acridinium dye functionalized bis (terpyridine) ruthenium (II) complexes. Eur. J. Org. Chem. 2018, 2018, 2682–2700. [Google Scholar] [CrossRef]
- Rostovskii, N.V.; Ruvinskaya, J.O.; Novikov, M.S.; Khlebnikov, A.F.; Smetanin, I.A.; Agafonova, A.V. Switchable Synthesis of Pyrroles and Pyrazines via Rh(II)-Catalyzed Reaction of 1,2,3-Triazoles with Isoxazoles: Experimental and DFT Evidence for the 1,4-Diazahexatriene Intermediate. J. Org. Chem. 2017, 82, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Baś, S.; Yamashita, Y.; Kobayashi, S. Development of Brønsted Base–Photocatalyst Hybrid Systems for Highly Efficient C–C Bond Formation Reactions of Malonates with Styrenes. ACS Catal. 2020, 10, 10546–10550. [Google Scholar] [CrossRef]
- Molloy, J.J.; Seath, C.P.; West, M.J.; McLaughlin, C.; Fazakerley, N.J.; Kennedy, A.R.; Nelson, D.J.; Watson, A.J. Interrogating Pd (II) anion metathesis using a bifunctional chemical probe: A transmetalation switch. J. Am. Chem. Soc. 2018, 140, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Mosiagin, I.P.; Tomashenko, O.A.; Spiridonova, D.V.; Novikov, M.S.; Tunik, S.P.; Khlebnikov, A.F. Free-radical cyclization approach to polyheterocycles containing pyrrole and pyridine rings. Beilstein J. Org. Chem. 2021, 17, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 1054104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 11, 6378–6396. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galenko, E.E.; Novikov, M.S.; Bunev, A.S.; Khlebnikov, A.F. Acridine–Isoxazole and Acridine–Azirine Hybrids: Synthesis, Photochemical Transformations in the UV/Visible Radiation Boundary Region, and Anticancer Activity. Molecules 2024, 29, 1538. https://doi.org/10.3390/molecules29071538
Galenko EE, Novikov MS, Bunev AS, Khlebnikov AF. Acridine–Isoxazole and Acridine–Azirine Hybrids: Synthesis, Photochemical Transformations in the UV/Visible Radiation Boundary Region, and Anticancer Activity. Molecules. 2024; 29(7):1538. https://doi.org/10.3390/molecules29071538
Chicago/Turabian StyleGalenko, Ekaterina E., Mikhail S. Novikov, Alexander S. Bunev, and Alexander F. Khlebnikov. 2024. "Acridine–Isoxazole and Acridine–Azirine Hybrids: Synthesis, Photochemical Transformations in the UV/Visible Radiation Boundary Region, and Anticancer Activity" Molecules 29, no. 7: 1538. https://doi.org/10.3390/molecules29071538
APA StyleGalenko, E. E., Novikov, M. S., Bunev, A. S., & Khlebnikov, A. F. (2024). Acridine–Isoxazole and Acridine–Azirine Hybrids: Synthesis, Photochemical Transformations in the UV/Visible Radiation Boundary Region, and Anticancer Activity. Molecules, 29(7), 1538. https://doi.org/10.3390/molecules29071538