
Developing an Improved Cycle Architecture for AI-Based Generation of New Structures Aimed at Drug Discovery


Abstract
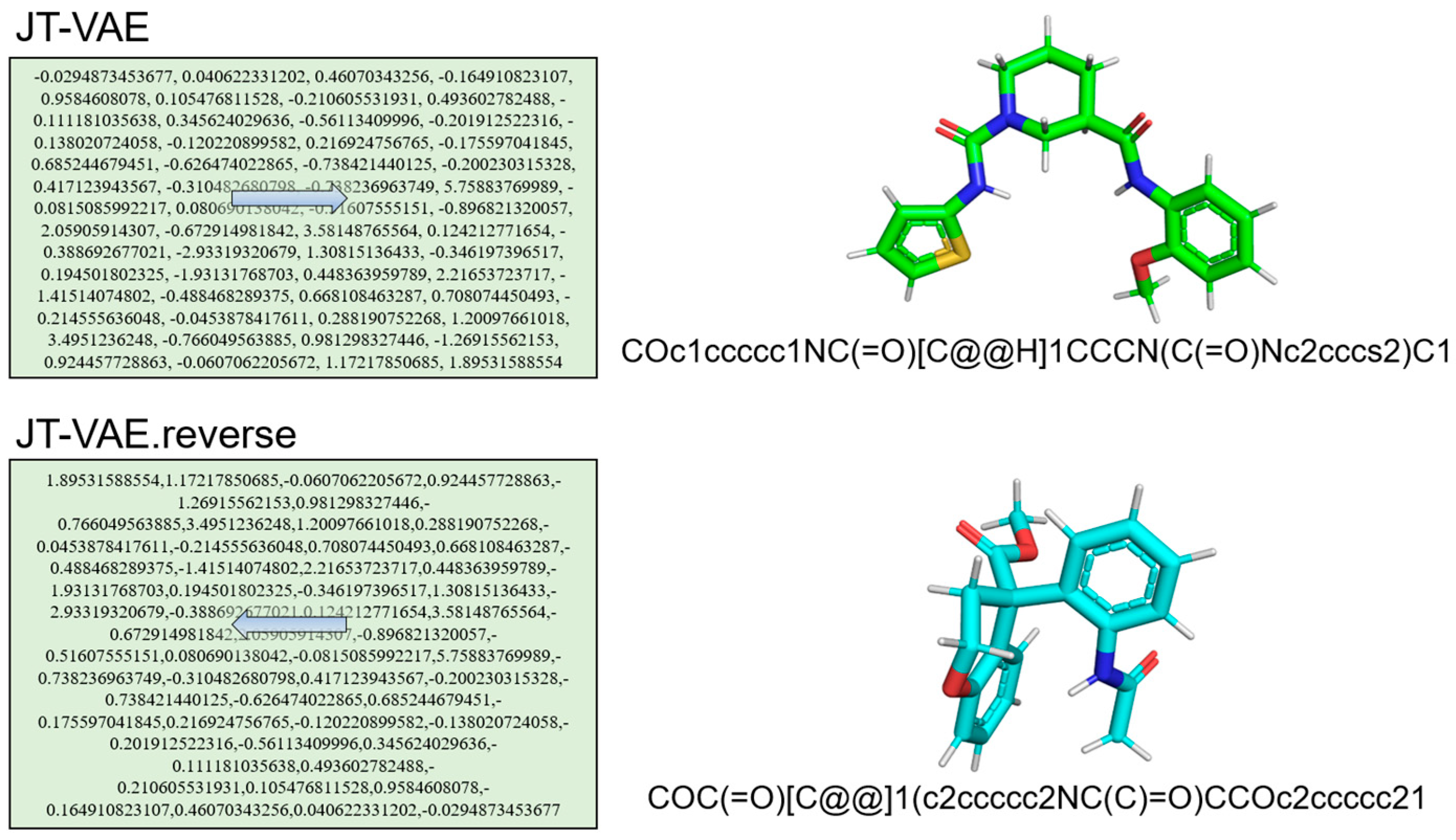
1. Introduction
2. Results
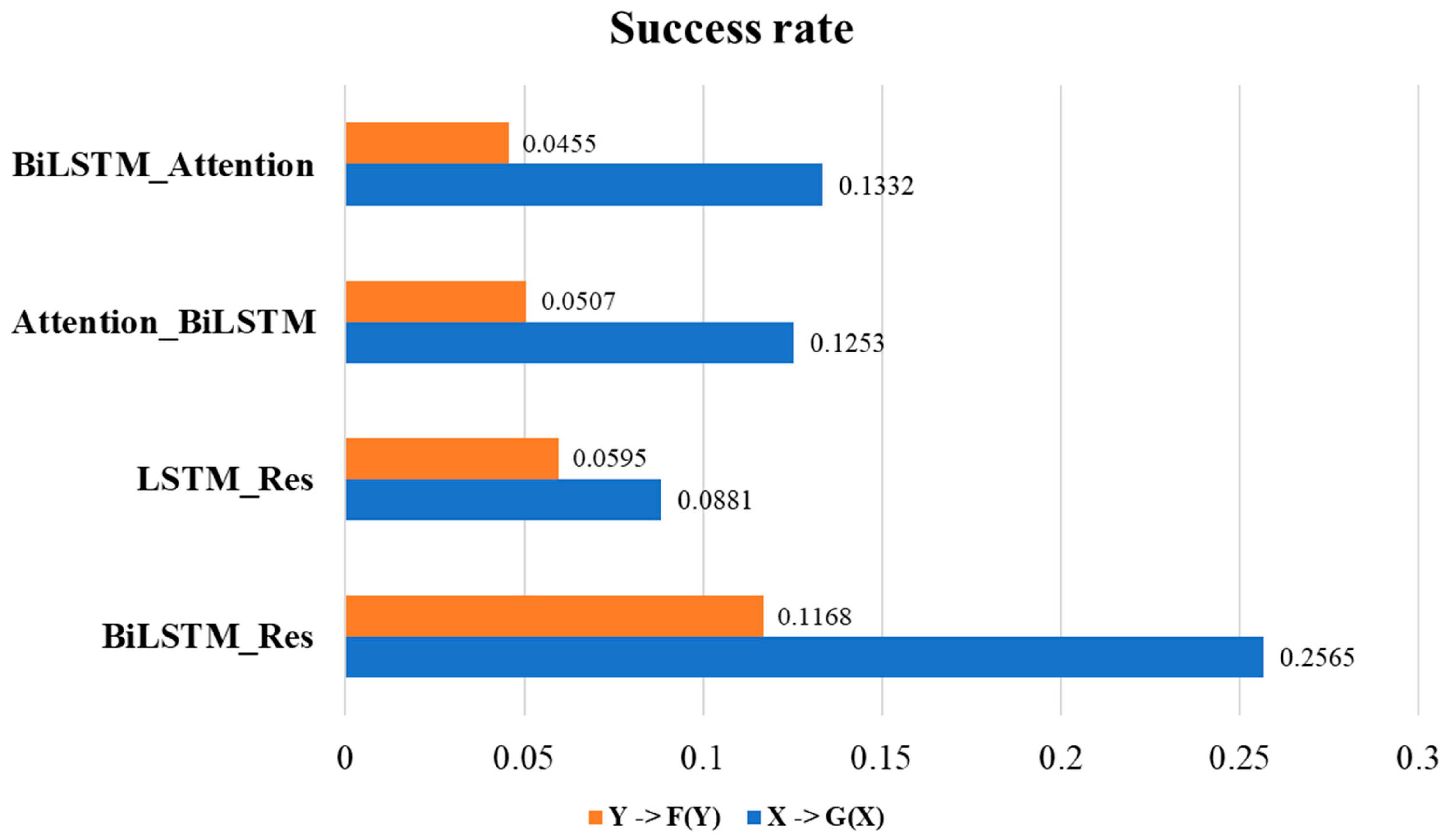
2.1. Ablation Experiment to Identify Architecture of Model
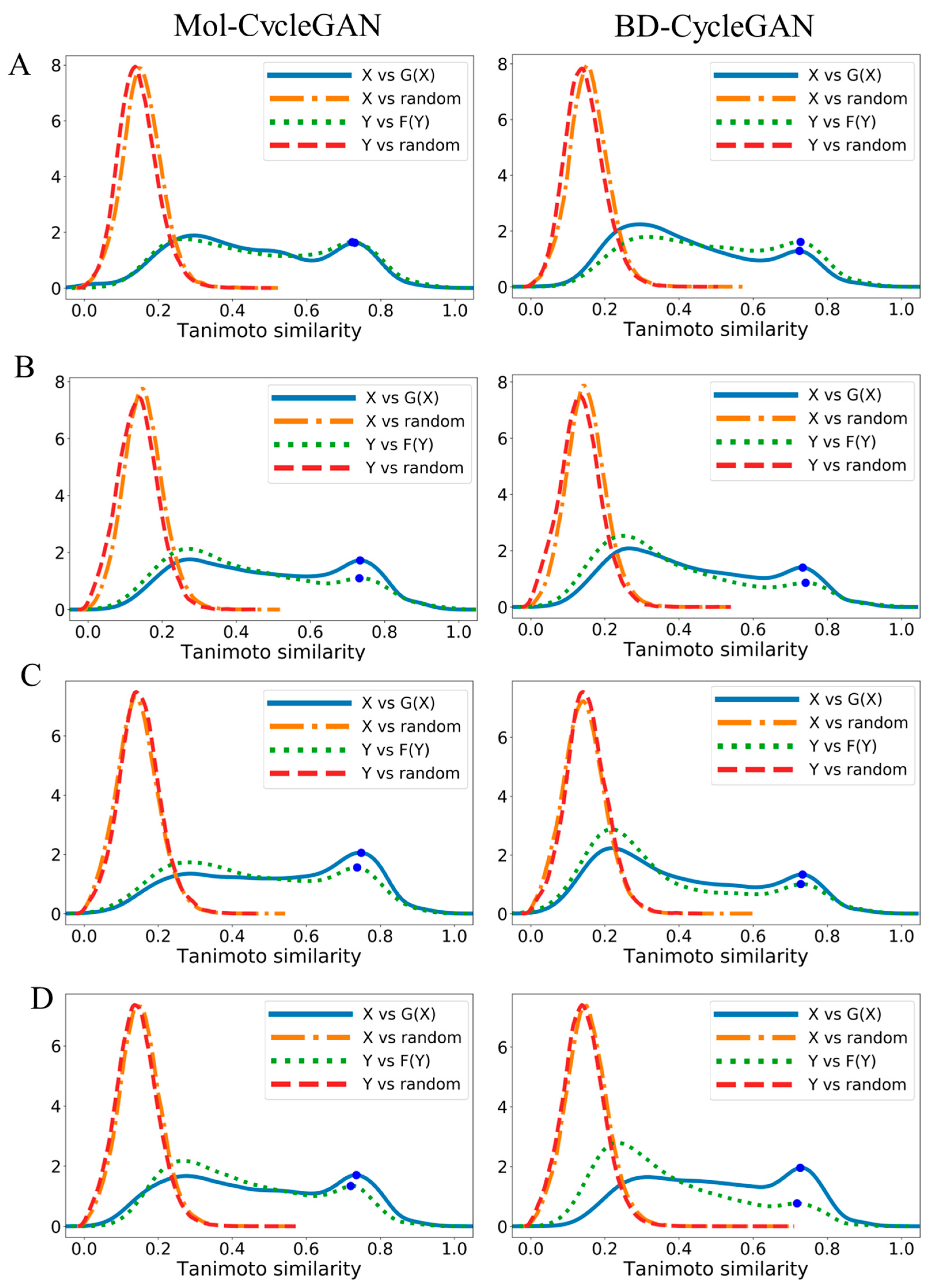
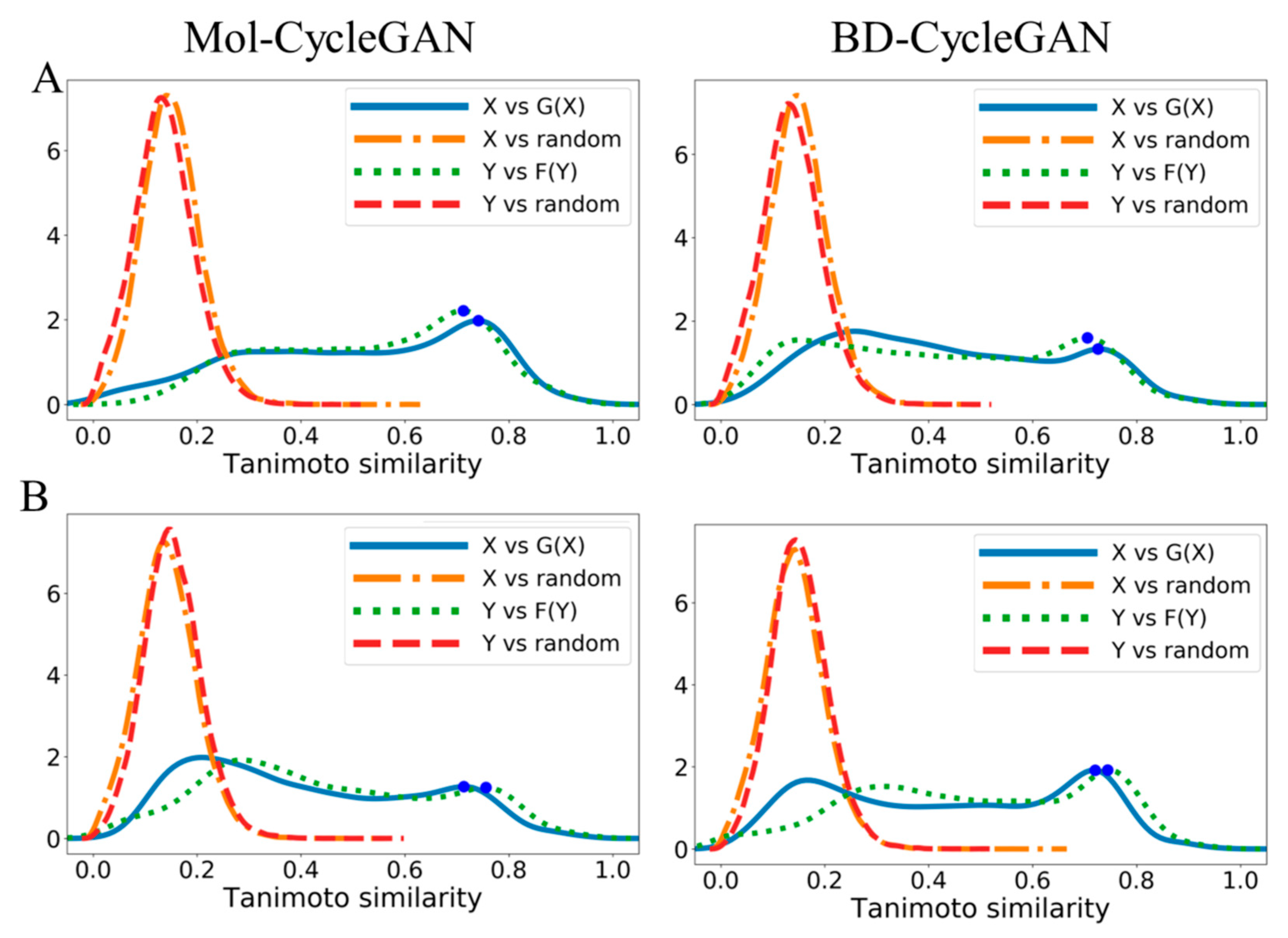
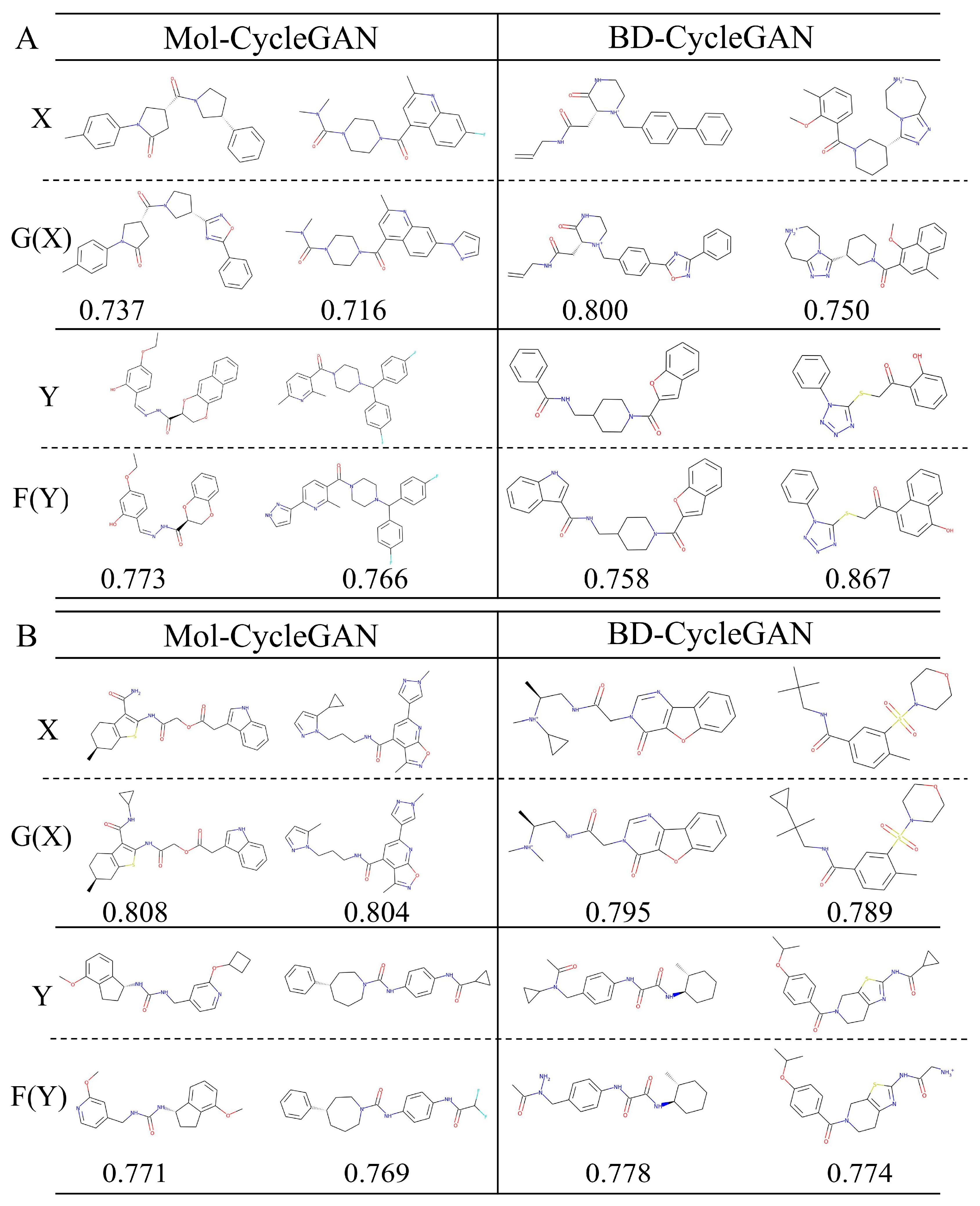
2.2. Molecular Generation with Specific Structural Group
2.3. Performance Evaluation on the Chemical Structure
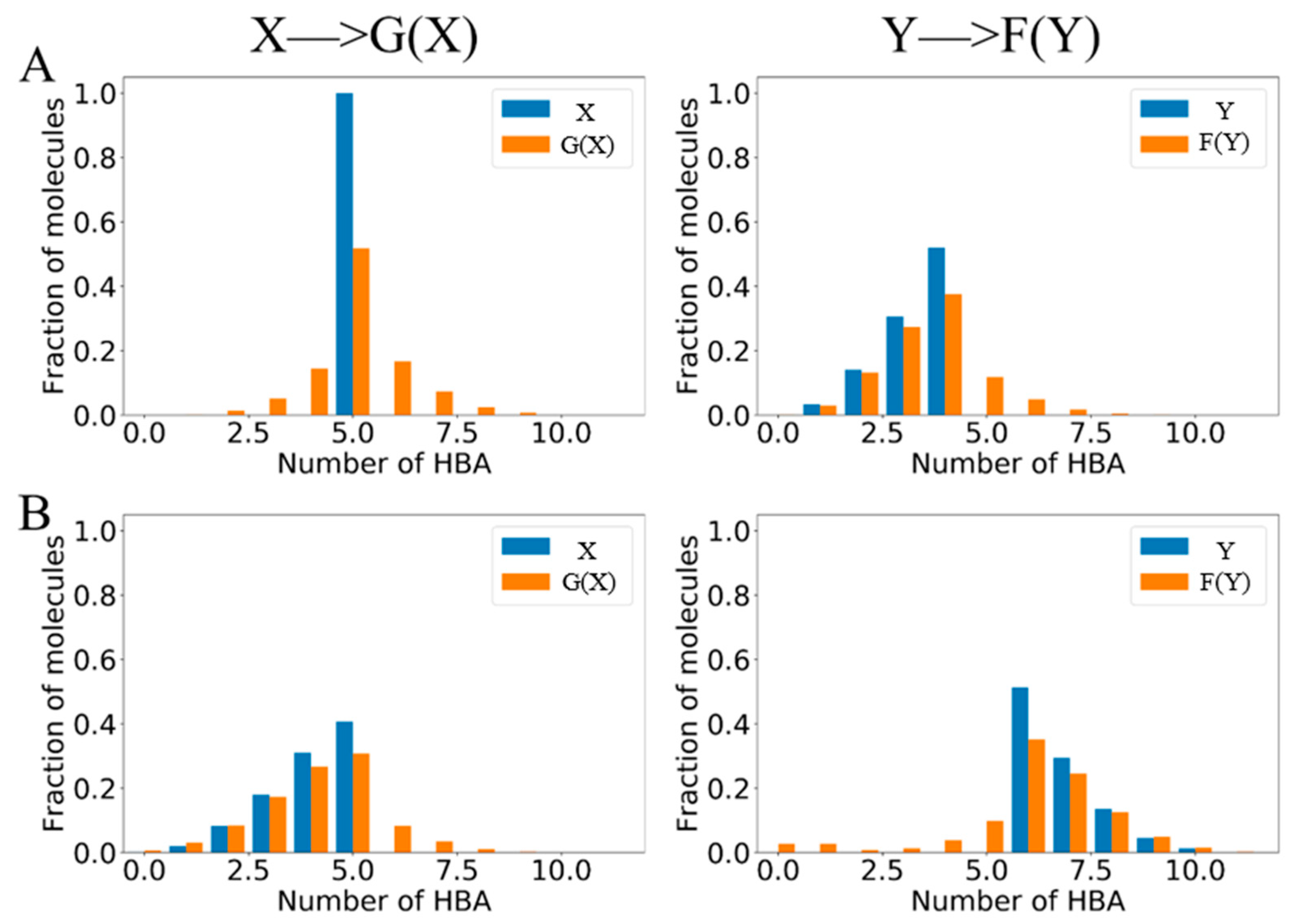
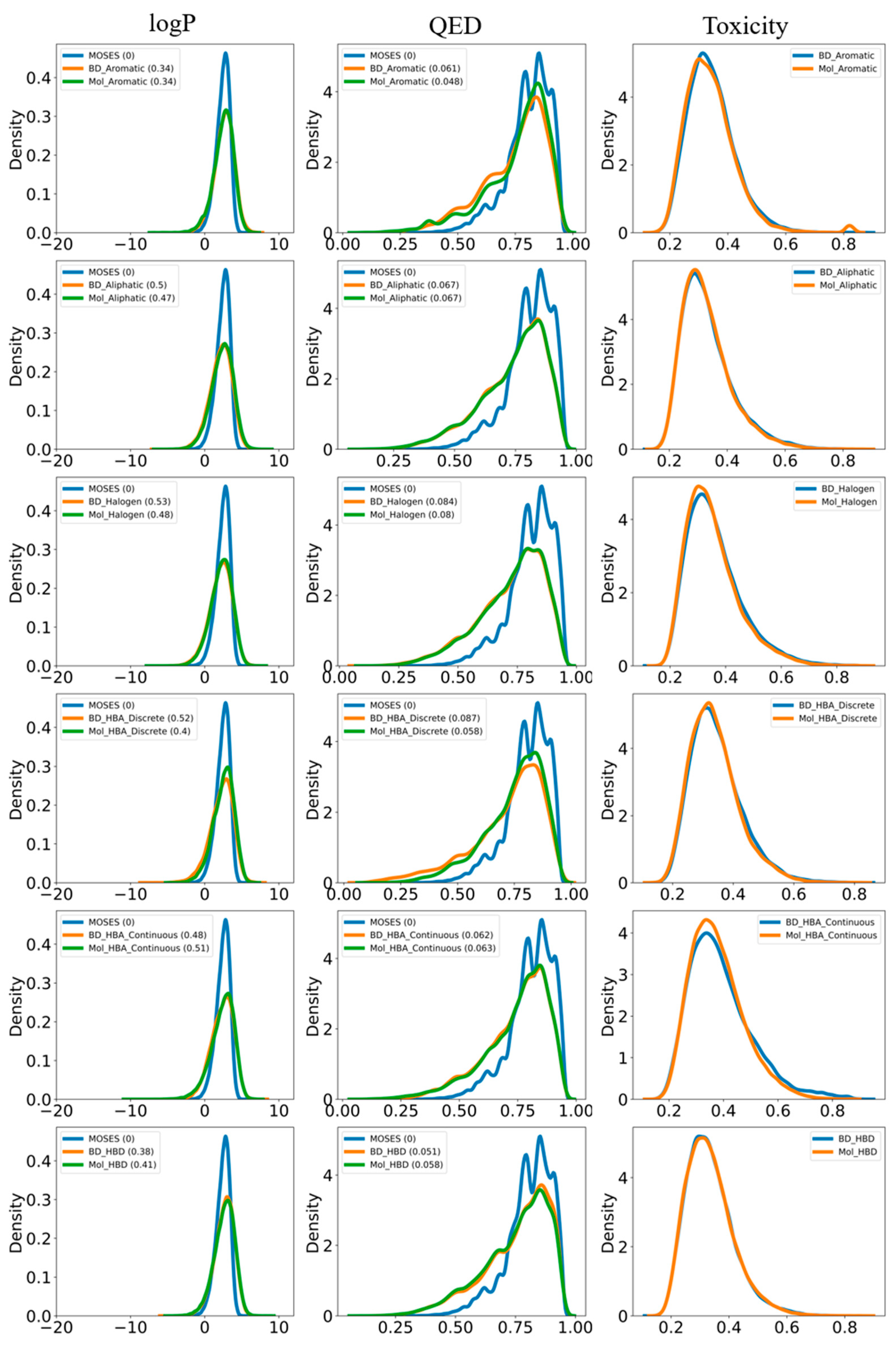
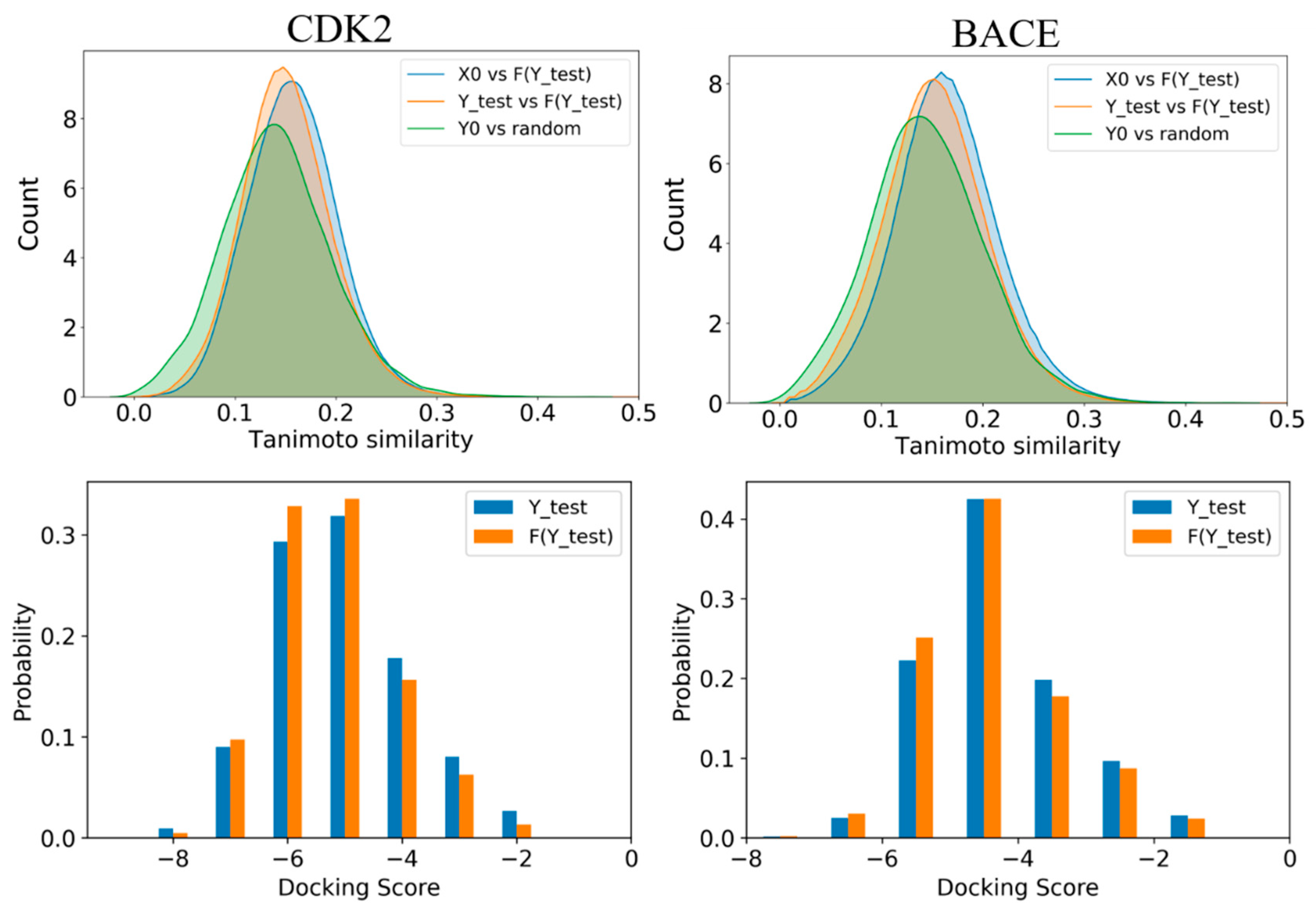
2.4. Structure and Property Analysis
2.5. Applications in Active and Decoy Generation
3. Discussion
4. Materials and Methods
4.1. BiLSTM for Processing of Bidirectional Molecular Representation
4.2. Attention Mechanism for Focusing Molecular Information
4.3. Residual Connection for Keeping Molecular Information
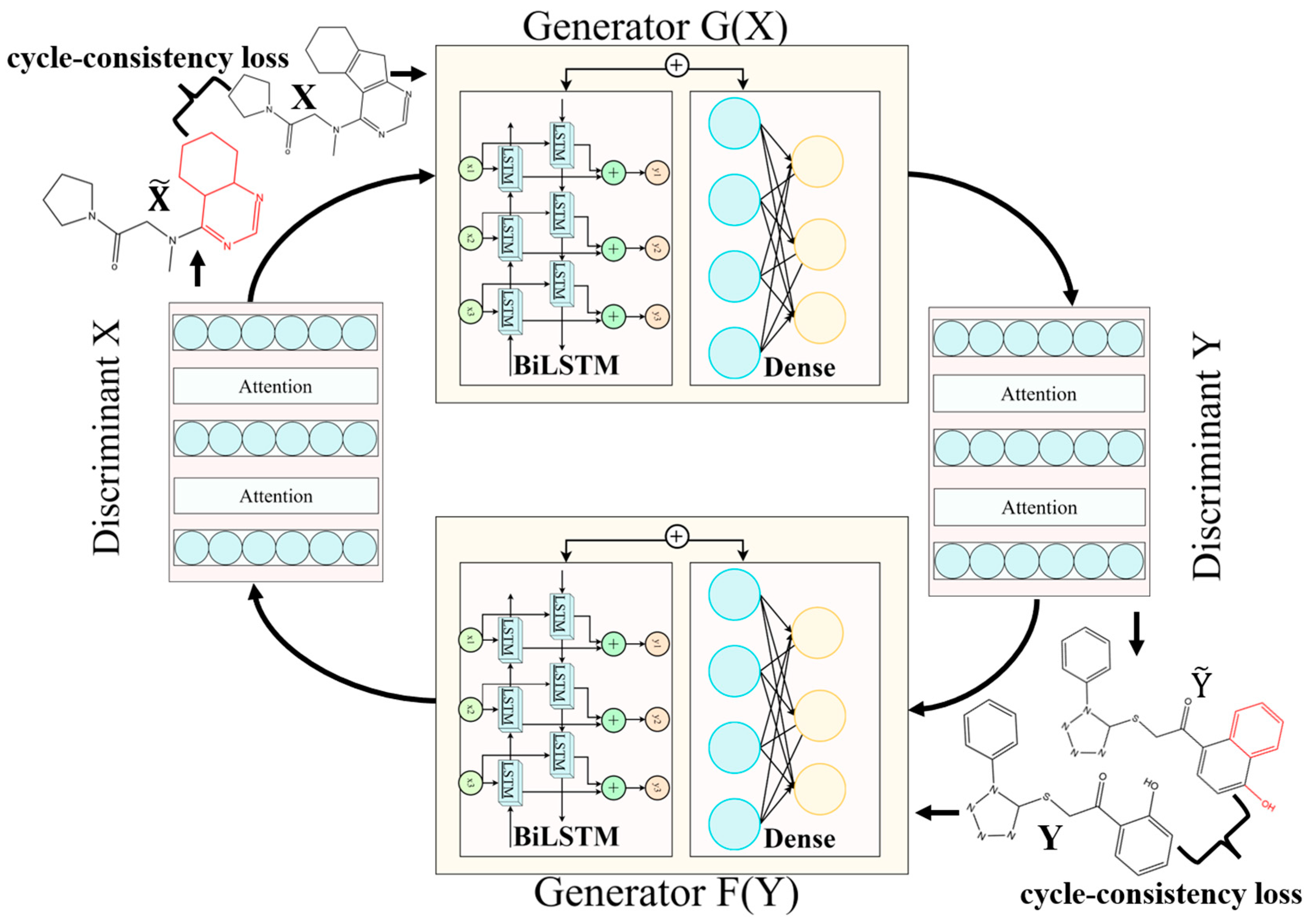
4.4. CycleGAN for Molecular Generation in a Cycle Way
4.5. Model Selection
4.6. Workflow
4.7. Evaluation Metrics
4.8. Data Set
4.9. Applications in Active and Decoy Generation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walters, W.P.; Barzilay, R. Applications of Deep Learning in Molecule Generation and Molecular Property Prediction. Acc. Chem. Res. 2021, 54, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef]
- McNair, D. Artificial Intelligence and Machine Learning for Lead-to-Candidate Decision-Making and Beyond. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 77–97. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80. [Google Scholar] [CrossRef] [PubMed]
- Zhavoronkov, A.; Vanhaelen, Q.; Oprea, T.I. Will artificial intelligence for drug discovery impact clinical pharmacology? Clin. Pharmacol. Ther. 2020, 107, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Urbina, F.; Lentzos, F.; Invernizzi, C.; Ekins, S. Dual use of artificial-intelligence-powered drug discovery. Nat. Mach. Intell. 2022, 4, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; MacKerell, A.D., Jr. Computer-Aided Drug Design Methods. Methods Mol. Biol. 2017, 1520, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Xing, J.; Li, X.; Liu, X.; Fu, Z.; Xiong, Z.; Lu, D.; Wu, X.; Zhao, J.; Tan, X. Artificial intelligence in drug design. Sci. China Life Sci. 2018, 61, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yang, Z.; Ojima, I.; Samaras, D.; Wang, F. Artificial intelligence in drug discovery: Applications and techniques. Brief. Bioinform. 2022, 23, bbab430. [Google Scholar] [CrossRef] [PubMed]
- Delijewski, M.; Haneczok, J. AI drug discovery screening for COVID-19 reveals zafirlukast as a repurposing candidate. Med. Drug Discov. 2021, 9, 100077. [Google Scholar] [CrossRef]
- Mokaya, M.; Imrie, F.; van Hoorn, W.P.; Kalisz, A.; Bradley, A.R.; Deane, C.M. Testing the limits of SMILES-based de novo molecular generation with curriculum and deep reinforcement learning. Nat. Mach. Intell. 2023, 5, 386–394. [Google Scholar] [CrossRef]
- Arnold, C. Inside the nascent industry of AI-designed drugs. Nat. Med. 2023, 29, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Mamoshina, P.; Vieira, A.; Putin, E.; Zhavoronkov, A. Applications of Deep Learning in Biomedicine. Mol. Pharm. 2016, 13, 1445–1454. [Google Scholar] [CrossRef]
- Lavecchia, A. Deep learning in drug discovery: Opportunities, challenges and future prospects. Drug Discov. Today 2019, 24, 2017–2032. [Google Scholar] [CrossRef] [PubMed]
- Askr, H.; Elgeldawi, E.; Aboul Ella, H.; Elshaier, Y.A.M.M.; Gomaa, M.M.; Hassanien, A.E. Deep learning in drug discovery: An integrative review and future challenges. Artif. Intell. Rev. 2023, 56, 5975–6037. [Google Scholar] [CrossRef] [PubMed]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef] [PubMed]
- Özçelik, R.; van Tilborg, D.; Jiménez-Luna, J.; Grisoni, F. Structure-Based Drug Discovery with Deep Learning. ChemBioChem 2023, 24, e202200776. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Sercu, T.; Wadhawan, K.; Padhi, I.; Gehrmann, S.; Cipcigan, F.; Chenthamarakshan, V.; Strobelt, H.; dos Santos, C.; Chen, P.-Y.; et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat. Biomed. Eng. 2021, 5, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Coley, C.W. The Synthesizability of Molecules Proposed by Generative Models. J. Chem. Inf. Model. 2020, 60, 5714–5723. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.-H.; Xie, L.; Zhang, P. FAME: Fragment-based Conditional Molecular Generation for Phenotypic Drug Discovery. In Proceedings of the 2022 SIAM International Conference on Data Mining (SDM), Virtually, 28–30 April 2022; pp. 720–728. [Google Scholar]
- Wang, X.; Gao, C.; Han, P.; Li, X.; Chen, W.; Rodríguez Patón, A.; Wang, S.; Zheng, P. PETrans: De Novo Drug Design with Protein-Specific Encoding Based on Transfer Learning. Int. J. Mol. Sci. 2023, 24, 1146. [Google Scholar] [CrossRef] [PubMed]
- Creswell, A.; White, T.; Dumoulin, V.; Arulkumaran, K.; Sengupta, B.; Bharath, A.A. Generative adversarial networks: An overview. IEEE Signal Process. Mag. 2018, 35, 53–65. [Google Scholar] [CrossRef]
- Metz, L.; Poole, B.; Pfau, D.; Sohl-Dickstein, J. Unrolled generative adversarial networks. arXiv 2016, arXiv:1611.02163. [Google Scholar]
- Guimaraes, G.L.; Sanchez-Lengeling, B.; Outeiral, C.; Farias, P.L.C.; Aspuru-Guzik, A. Objective-reinforced generative adversarial networks (organ) for sequence generation models. arXiv 2017, arXiv:1705.10843. [Google Scholar]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.-C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminformatics 2019, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-Y.; Park, T.; Isola, P.; Efros, A.A. Unpaired image-to-image translation using cycle-consistent adversarial networks. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017; pp. 2223–2232. [Google Scholar]
- Maziarka, Ł.; Pocha, A.; Kaczmarczyk, J.; Rataj, K.; Danel, T.; Warchoł, M. Mol-CycleGAN: A generative model for molecular optimization. J. Cheminformatics 2020, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. Artif. Intell. Drug Discov. 2020, 75, 228. [Google Scholar]
- Wang, F.; Feng, X.; Guo, X.; Xu, L.; Xie, L.; Chang, S. Improving de novo Molecule Generation by Embedding LSTM and Attention Mechanism in CycleGAN. Front. Genet. 2021, 12, 709500. [Google Scholar] [CrossRef] [PubMed]
- Yulita, I.N.; Fanany, M.I.; Arymuthy, A.M. Bi-directional Long Short-Term Memory using Quantized data of Deep Belief Networks for Sleep Stage Classification. Procedia Comput. Sci. 2017, 116, 530–538. [Google Scholar] [CrossRef]
- Niu, Z.; Zhong, G.; Yu, H. A review on the attention mechanism of deep learning. Neurocomputing 2021, 452, 48–62. [Google Scholar] [CrossRef]
- Pu, L.; Naderi, M.; Liu, T.; Wu, H.C.; Mukhopadhyay, S.; Brylinski, M. eToxPred: A machine learning-based approach to estimate the toxicity of drug candidates. BMC Pharmacol. Toxicol. 2019, 20, 2. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Enamine. Targeted Libraries-Enamine. Available online: https://enamine.net/compound-libraries/targeted-libraries (accessed on 8 May 2023).
- Graves, A. (Ed.) Long short-term memory. In Supervised Sequence Labelling with Recurrent Neural Networks; Springer: Berlin/Heidelberg, Germany, 2012; Volume 385, pp. 37–45. [Google Scholar]
- Wang, S.; Wang, X.; Wang, S.; Wang, D. Bi-directional long short-term memory method based on attention mechanism and rolling update for short-term load forecasting. Int. J. Electr. Power Energy Syst. 2019, 109, 470–479. [Google Scholar] [CrossRef]
- Guo, M.-H.; Xu, T.-X.; Liu, J.-J.; Liu, Z.-N.; Jiang, P.-T.; Mu, T.-J.; Zhang, S.-H.; Martin, R.R.; Cheng, M.-M.; Hu, S.-M. Attention mechanisms in computer vision: A survey. Comput. Vis. Media 2022, 8, 331–368. [Google Scholar] [CrossRef]
- Zheng, S.; Yan, X.; Yang, Y.; Xu, J. Identifying structure–property relationships through SMILES syntax analysis with self-attention mechanism. J. Chem. Inf. Model. 2019, 59, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Rolnick, D.; Tegmark, M. The power of deeper networks for expressing natural functions. arXiv 2017, arXiv:1705.05502. [Google Scholar]
- Lin, X.; Quan, Z.; Wang, Z.-J.; Huang, H.; Zeng, X. A novel molecular representation with BiGRU neural networks for learning atom. Brief. Bioinform. 2020, 21, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 2019, 63, 8749–8760. [Google Scholar] [CrossRef]
- Winter, R.; Montanari, F.; Steffen, A.; Briem, H.; Noé, F.; Clevert, D.-A. Efficient multi-objective molecular optimization in a continuous latent space. Chem. Sci. 2019, 10, 8016–8024. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kang, S.; Yoo, J.; Kwon, Y.; Nam, Y.; Lee, D.; Kim, I.; Choi, Y.-S.; Jung, Y.; Kim, S. Deep-learning-based inverse design model for intelligent discovery of organic molecules. Comput. Mater. 2018, 4, 67. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Liu, Z. Multi-objective de novo drug design with conditional graph generative model. J. Cheminform. 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M. Molecular sets (MOSES): A benchmarking platform for molecular generation models. Front. Pharmacol. 2020, 11, 565644. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | X—>G(X) | Y—>F(Y) | |||||
|---|---|---|---|---|---|---|---|
| Data | Model | Success Rate | Diversity | Non-Identity | Success Rate | Diversity | Non-Identity |
| Aromatic rings | Mol-CycleGAN | 0.231 | 0.981 | 0.762 | 0.102 | 0.997 | 0.582 |
| BD-CycleGAN | 0.257 | 0.995 | 0.804 | 0.117 | 0.997 | 0.680 | |
| Aliphatic rings | Mol-CycleGAN | 0.183 | 0.996 | 0.713 | 0.154 | 0.994 | 0.769 |
| BD-CycleGAN | 0.222 | 0.996 | 0.814 | 0.196 | 0.996 | 0.861 | |
| Halogen | Mol-CycleGAN | 0.032 | 0.997 | 0.417 | 0.145 | 0.993 | 0.717 |
| BD-CycleGAN | 0.121 | 0.994 | 0.714 | 0.257 | 0.991 | 0.793 | |
| HBD | Mol-CycleGAN | 0.226 | 0.994 | 0.718 | 0.147 | 0.991 | 0.822 |
| BD-CycleGAN | 0.193 | 0.996 | 0.782 | 0.178 | 0.996 | 0.923 | |
| HBA_Discrete | Mol-CycleGAN | 0.154 | 0.986 | 0.389 | 0.030 | 0.999 | 0.328 |
| BD-CycleGAN | 0.376 | 0.995 | 0.782 | 0.078 | 0.996 | 0.662 | |
| HBA_Continuous | Mol-CycleGAN | 0.106 | 0.995 | 0.518 | 0.102 | 0.994 | 0.475 |
| BD-CycleGAN | 0.085 | 0.974 | 0.662 | 0.142 | 0.966 | 0.662 | |
| Structure | Model | X—>G(X) | Y—>F(Y) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Filters | Valid | IntDiv | IntDiv2 | Novelty | Filters | Valid | IntDiv | IntDiv2 | Novelty | ||
| Aromatic | Mol-Cycle | 0.621 | 0.989 | 0.865 | 0.859 | 0.954 | 0.598 | 0.998 | 0.869 | 0.863 | 0.947 |
| BD-Cycle | 0.639 | 0.995 | 0.866 | 0.860 | 0.961 | 0.599 | 0.998 | 0.868 | 0.863 | 0.957 | |
| Aliphatic | Mol-Cycle | 0.576 | 0.998 | 0.862 | 0.857 | 0.960 | 0.518 | 0.994 | 0.865 | 0.859 | 0.974 |
| BD-Cycle | 0.585 | 0.997 | 0.863 | 0.857 | 0.970 | 0.519 | 0.996 | 0.867 | 0.861 | 0.980 | |
| Halogen | Mol-Cycle | 0.587 | 0.998 | 0.869 | 0.863 | 0.927 | 0.618 | 0.994 | 0.865 | 0.859 | 0.963 |
| BD-Cycle | 0.576 | 0.998 | 0.873 | 0.867 | 0.960 | 0.534 | 0.994 | 0.870 | 0.864 | 0.975 | |
| HBD | Mol-Cycle | 0.691 | 0.995 | 0.865 | 0.859 | 0.950 | 0.549 | 0.993 | 0.871 | 0.866 | 0.975 |
| BD-Cycle | 0.699 | 0.997 | 0.863 | 0.857 | 0.956 | 0.554 | 0.997 | 0.874 | 0.868 | 0.986 | |
| HBA_Discete | Mol-Cycle | 0.636 | 0.988 | 0.865 | 0.859 | 0.905 | 0.519 | 0.999 | 0.882 | 0.874 | 0.943 |
| BD-Cycle | 0.611 | 0.996 | 0.868 | 0.862 | 0.956 | 0.454 | 0.999 | 0.887 | 0.879 | 0.965 | |
| HBA_Continuous | Mol-Cycle | 0.533 | 0.997 | 0.880 | 0.873 | 0.939 | 0.647 | 0.997 | 0.863 | 0.857 | 0.940 |
| BD-Cycle | 0.589 | 0.998 | 0.882 | 0.874 | 0.953 | 0.649 | 0.997 | 0.866 | 0.858 | 0.952 | |
| X—>F(X) | FCD | SNN | Scaff | Frag | |||||
|---|---|---|---|---|---|---|---|---|---|
| Data | Model | Test | TestSF | Test | TestSF | Test | TestSF | Test | TestSF |
| Aromatic Rings | Mol-CycleGAN | 0.627 | 4.487 | 0.609 | 0.467 | 0.901 | 0.143 | 0.998 | 0.909 |
| BD-CycleGAN | 0.831 | 4.460 | 0.578 | 0.466 | 0.887 | 0.149 | 0.997 | 0.989 | |
| Aliphatic Rings | Mol-CycleGAN | 0.278 | 5.971 | 0.669 | 0.466 | 0.922 | 0.125 | 0.999 | 0.990 |
| BD-CycleGAN | 0.464 | 6.275 | 0.603 | 0.456 | 0.895 | 0.098 | 0.999 | 0.990 | |
| Halogen | Mol-CycleGAN | 0.082 | 5.397 | 0.825 | 0.480 | 0.942 | 0.200 | 0.999 | 0.987 |
| BD-CycleGAN | 0.591 | 5.700 | 0.640 | 0.448 | 0.847 | 0.170 | 0.997 | 0.986 | |
| HBD | Mol-CycleGAN | 0.419 | 4.08 | 0.653 | 0.476 | 0.907 | 0.138 | 0.998 | 0.993 |
| BD-CycleGAN | 0.368 | 4.126 | 0.637 | 0.476 | 0.904 | 0.143 | 0.999 | 0.993 | |
| HBA_Discrete | Mol-CycleGAN | 0.160 | 4.016 | 0.882 | 0.498 | 0.901 | 0.143 | 0.999 | 0.993 |
| BD-CycleGAN | 0.864 | 4.833 | 0.596 | 0.461 | 0.717 | 0.145 | 0.998 | 0.991 | |
| HBA_Continuous | Mol-CycleGAN | 0.212 | 6.288 | 0.734 | 0.450 | 0.957 | 0.196 | 0.997 | 0.968 |
| BD-CycleGAN | 0.562 | 5.944 | 0.692 | 0.450 | 0.883 | 0.191 | 0.997 | 0.982 | |
| Y—>F(Y) | FCD | SNN | Scaff | Frag | |||||
|---|---|---|---|---|---|---|---|---|---|
| Data | Model | Test | TestSF | Test | Test | Test | TestSF | Test | TestSF |
| Aromatic Rings | Mol-CycleGAN | 0.135 | 4.741 | 0.727 | 0.469 | 0.908 | 0.104 | 0.999 | 0.990 |
| BD-CycleGAN | 0.175 | 4.915 | 0.675 | 0.465 | 0.887 | 0.101 | 0.999 | 0.991 | |
| Aliphatic Rings | Mol-CycleGAN | 0.494 | 10.810 | 0.607 | 0.438 | 0.433 | 0.011 | 0.998 | 0.971 |
| BD-CycleGAN | 0.633 | 10.403 | 0.538 | 0.428 | 0.371 | 0.018 | 0.997 | 0.974 | |
| Halogen | Mol-CycleGAN | 0.558 | 5.778 | 0.638 | 0.462 | 0.841 | 0.135 | 0.998 | 0.984 |
| BD-CycleGAN | 2.153 | 7.560 | 0.551 | 0.433 | 0.605 | 0.148 | 0.982 | 0.975 | |
| HBD | Mol-CycleGAN | 0.358 | 5.553 | 0.594 | 0.447 | 0.904 | 0.204 | 0.997 | 0.988 |
| BD-CycleGAN | 0.758 | 6.229 | 0.513 | 0.429 | 0.840 | 0.172 | 0.994 | 0.986 | |
| HBA_Discrete | Mol-CycleGAN | 0.034 | 8.755 | 0.862 | 0.452 | 0.982 | 0.179 | 0.999 | 0.941 |
| BD-CycleGAN | 0.470 | 10.012 | 0.674 | 0.422 | 0.927 | 0.182 | 0.977 | 0.880 | |
| HBA_Continuous | Mol-CycleGAN | 0.272 | 4.975 | 0.755 | 0.483 | 0.824 | 0.106 | 0.999 | 0.994 |
| BD-CycleGAN | 0.543 | 4.721 | 0.673 | 0.476 | 0.666 | 0.109 | 0.999 | 0.993 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Xie, L.; Lu, X.; Mao, R.; Xu, L.; Xu, X. Developing an Improved Cycle Architecture for AI-Based Generation of New Structures Aimed at Drug Discovery. Molecules 2024, 29, 1499. https://doi.org/10.3390/molecules29071499
Zhang C, Xie L, Lu X, Mao R, Xu L, Xu X. Developing an Improved Cycle Architecture for AI-Based Generation of New Structures Aimed at Drug Discovery. Molecules. 2024; 29(7):1499. https://doi.org/10.3390/molecules29071499
Chicago/Turabian StyleZhang, Chun, Liangxu Xie, Xiaohua Lu, Rongzhi Mao, Lei Xu, and Xiaojun Xu. 2024. "Developing an Improved Cycle Architecture for AI-Based Generation of New Structures Aimed at Drug Discovery" Molecules 29, no. 7: 1499. https://doi.org/10.3390/molecules29071499
APA StyleZhang, C., Xie, L., Lu, X., Mao, R., Xu, L., & Xu, X. (2024). Developing an Improved Cycle Architecture for AI-Based Generation of New Structures Aimed at Drug Discovery. Molecules, 29(7), 1499. https://doi.org/10.3390/molecules29071499