


Cytotoxic Cyclolignans Obtained by the Enlargement of the Cyclolignan Skeleton of Podophyllic Aldehyde, a Selective Podophyllotoxin-Derived Cyclolignan

 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.1.1. Addition of Carbon Nucleophiles
2.1.2. Addition of Nitrogen Nucleophiles
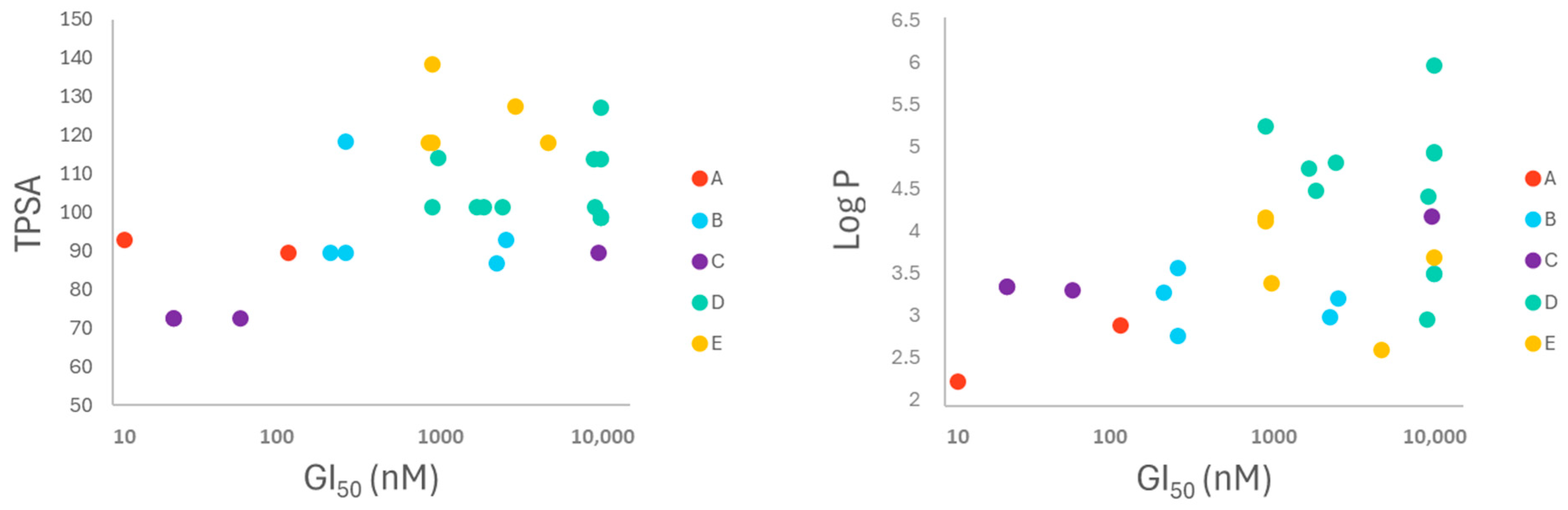
2.2. Bioactivity
3. Materials and Methods
3.1. Chemistry
- Compound 3. A mixture of aldehyde 2 (100 mg, 0.24 mmol), ammonium acetate (23 mg, 0.3 mmol), and nitromethane (5 mL) was heated at reflux under an inert atmosphere for 4 h, chilled, and concentrated under vacuum. The residue was redissolved in CH2Cl2 and washed with brine, dried over Na2SO4, filtered, and evaporated. Purification by silica gel CC of the crude provided 3 (CH2Cl2/EtOAc 97:3, 8 mg, 8%) and unreacted 2 (CH2Cl2/EtOAc 9:1, 39 mg, 39%). Data for 3: 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.79 (d, 1H, H9, J = 13 Hz), 7.14 (d, 1H, H1″, J = 13 Hz), 7.08 (s, 1H, H7), 6.81 (s, 1H, H6), 6.69 (s, 1H, H3), 6.22 (s, 2H, H2′ and H6′), 6.01 (d, 1H, H10a, J = 1.3 Hz), 6.00 (d, 1H, H10b, J = 1.3 Hz), 4.58 (d, 1H, H7′, J = 1.5 Hz), 3.78 (s, 3H, H11′), 3.75 (s, 6H, H10′ and H12′), 3.69 (s, 3H, CH3O-9′), 3.55 (d, 1H, H8′, J = 1.5 Hz); 13C NMR (CDCl3) δ (ppm) 171.3 (C9′), 153.4 (C3′ and C5′), 150.1 (C4), 147.4 (C5), 141.1 (C7),140.3 (C9), 137.6 (C1′), 137.2 (C4′), 136.2 (1″), 132.3 (C2), 125.8 (C1), 124.5 (C8), 109.9 (C3), 108.6 (C6), 104.4 (C2′ and C6′), 101.8 (C10), 60.8 (C11′), 56.2 (C10′ and C12′), 52.9 (CH3O-9′), 48.0 (C8′), 46.3 (C7′). IR νmax/cm−1 (film): 2923, 1731, 1604, 1586, 1504, 1460, 1376, 1242, 1126, 974.
- Compound 4. From 2 (140 mg, 0.33 mmol) and (thiphenylphosphoranylidene)acetaldehyde (155 mg, 0.49 mmol) in 60 mL of toluene for 7 d. CC on silica gel (CH2Cl2/EtOAc 96:4) of the crude yielded a 6:4 mixture of 2 and 4 (150 mg). Data for 4: 1H NMR (CDCl3, 200 MHz) δ (ppm) 9.58 (d, 1H, H2″, J = 7.7 Hz), 7.26 (d, 1H, H9, J = 16 Hz), 6.98 (s, 1H, H7), 6.80 (s, 1H, H6), 6.70 (s, 1H, H3), 6.24 (s, 2H, H2′ and H6′), 6.16 (dd, 1H, H1″, J = 16 and 7.7 Hz), 6.00 (d, 1H, H10a, J = 1.1 Hz), 5.98 (d, 1H, H10b, J = 1.1 Hz), 4.57 (d, 1H, H7′, J = 1.5 Hz), 3.88 (s, 3H, H11′), 3.71 (d, 1H, H8′, J = 1.5 Hz), 3.74 (s, 6H, H10′ and H12′), 3.66 (s, 3H, CH3O-9′); 13C NMR (CDCl3) δ (ppm) 193.6 (C2″) 171.7 (C9′), 153.2 (C3′ and C5′), 152.9 (C9), 149.4 (C4), 147.3 (C5), 145.6 (C1′), 137.8 (C7), 137.0 (C4′), 131.3 (C2), 127.5 (C1″), 128.7 (C8), 126.1 (C1), 109.9 (C3), 108.3 (C6), 104.3 (C2′ and C6′), 101.6 (C10), 60.8 (C11′), 56.1 (C10′ and C12′), 52.7 (CH3O-9′), 47.4 (C7′), 46.4 (C8′). EM: 452 m/z.
- Compound 5. From 2 (100 mg, 0.23 mmol) and (triphenylphosphoranylidene)propan-2-one (76 mg, 0.24 mmol) in 20 mL of toluene for 2 d. CC of the crude on silica gel (CH2Cl2/EtOAc 9:1) yielded 5 (90 mg, 82%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.30 (d, 1H, H9, J = 16 Hz), 6.92 (s, 1H, H7), 6.78 (s, 1H, H6), 6.68 (s, 1H, H3), 6.25 (s, 2H, H2′ and H6′), 6.13 (d, 1H, H1″, J = 16 Hz), 5.98 (d, 1H, H10a, J = 1.1 Hz), 5.97 (d, 1H, H10b, J = 1.1 Hz), 4.54 (d, 1H, H7′, J = 1.5 Hz), 3.78 (s, 3H, H11′), 3.74 (s, 6H, H10′ and H12′), 3.71 (d, 1H, H8′, J = 1.5 Hz), 3.66 (s, 3H, CH3O-9′), 2.29 (s, 3H, H3″); 13C NMR (CDCl3) δ (ppm) 198.4 (C2″) 171.9 (C9′), 153.2 (C3′ and C5′), 148.9 (C4), 147.2 (C5), 144.2 (C9), 138.2 (C1′), 137.0 (C7 and C4′), 131.1 (C2), 128.8 (C8), 126.4 (C1), 126.0 (C1″),109.7 (C3), 108.1 (C6), 104.3 (C2′ and C6′), 101.5 (C10), 60.8 (C11′), 56.0 (C10′ and C12′), 52.6 (CH3O-9′), 47.3 (C8′), 46.5 (C7′), 27.4 (C3″). EM: 466 m/z. IR νmax/cm−1 (film) 2921, 1731, 1667, 1651, 1578, 1505, 1458, 1224, 1126, 1005, 752.
- Compound 6. From 2 (200 mg, 0.47 mmol) and methyl (thiphenylphosphoranylidene)acetate (320 mg, 0.94 mmol) in 15 mL of toluene for 34 h. CC of the crude on silica gel (CH2Cl2/EtOAc 95:5) yielded 6 (190 mg, 84%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.46 (d, 1H, H9, J = 16 Hz), 6.85 (s, 1H, H7), 6.75 (s, 1H, H6), 6.66 (s, 1H, H3), 6.25 (s, 2H, H2′ and H6′), 5.95 (d, 1H, H10a, J = 1.4 Hz), 5.94 (d, 1H, H10b, J = 1.4 Hz), 5.87 (d, 1H, H1″, J = 16 Hz), 4.52 (d, 1H, H7′, J = 1.6 Hz), 3.77 (s, 3H, H11′), 3.73 (s, 6H, H10′ and H12′), 3.69 (d, 1H, H8′, J = 1.6 Hz), 3.73 (s, 3H, CH3O-3″), 3.64 (s, 3H, CH3O-9′); 13C NMR (CDCl3) δ (ppm) 171.9 (C9′), 167.5 (C2″), 153.1 (C3′ and C5′), 148.7 (C4), 147.1 (C5), 145.4 (C9), 138.2 (C1′), 136.9 (C4′), 136.2 (C7), 131.2 (C2), 128.5 (C8), 126.2 (C1), 116.6 (C1″),109.6 (C3), 108.1 (C6), 104.3 (C2′ and C6′), 101.4 (C10), 60.7 (C11′), 56.0 (C10′ and C12′), 52.5 (CH3O-9′), 51.5 (C3″), 47.2 (C8′), 46.5 (C7′). HRMS calcd for C26H26O9 [M + H]+ 482.1577 u, found 482.1572 m/z; IR νmax/cm−1 (film) 2940, 1730, 1715, 1600, 1510, 1495, 1325, 1135, 1050, 1015. UV (EtOH) λmax 213 (lg ε 4.4), 267 (lg ε 4.2), 368 (lg ε 4.3). [α]22D −197° (c 0.97%).
- Compounds 7 and 8. To a solution of 6 (88 mg, 0.18 mmol) in dry ethyl ether (10 mL), a suspension of lithium aluminum hydride (14 mg, 0.37 mmol) in dry ether (5 mL) was added. It was stirred under argon at −15 °C for 3.5 h. The excess hydride was decomposed with wet EtOAc. The organic layer was dried over Na2SO4, filtered, and evaporated. CC of the crude afforded 7 (CH2Cl2/EtOAc 8:2, 14 mg, 14%) and 8 (EtOAc, 29 mg, 37%).
- Compound 7: 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.41 (d, 1H, H9, J = 16 Hz), 6.77 (s, 1H, H7), 6.74 (s, 1H, H6), 6.71 (s, 1H, H3), 6.26 (s, 2H, H2′ and H6′), 5.98 (s, 2H, H10), 5.97 (d, 1H, H1″, J = 16 Hz), 4.30 (s, 1H, H7′), 3.78 (s, 3H, H11′), 3.75 (s, 3H, CH3O-3″), 3.74 (s, 6H, H10′ and H12′), 3.68 (m, 1H, H9a’), 3.39 (t, 1H, H9b’, J = 10 Hz), 3.01 (dd, 1H, H8′, J = 10 and 4.9 Hz); 13C NMR (CDCl3) δ (ppm) 167.9 (C2″), 152.9 (C3′ and C5′), 148.7 (C4), 147.0 (C5), 145.4 (C9), 139.6 (C1′), -(C4′), 135.8 (C7), 131.5 (C2 and C8), 126.4 (C1), 115.9 (C1″), 110.4 (C3), 107.8 (C6), 104.2 (C2′ and C6′), 101.3 (C10), 62.7 (C9′), 60.7 (C11′), 55.9 (C10′ and C12′), 51.6 (C3″), 44.7 (C7′ and 8′). IR νmax/cm−1 (film) 3468, 2924, 1717, 1588, 1504, 1463, 1370, 1237, 1195, 1038, 934.
- Compound 8: 1H NMR (CDCl3, 200 MHz) δ (ppm) 6.67 (s, 1H, H3), 6.65 (s, 1H, H6), 6.40 (s, 1H, H7), 6.30 (s, 2H, H2′ and H6′), 6.28 (d, 1H, H9, J = 16 Hz), 5.93 (d, 1H, H10a, J = 1.5 Hz), 5.92 (d, 1H, H10b, J = 1.5 Hz), 5.89 (m, 1H, H1″), 4.23 (m, 1H, H2″), 3.79 (d, 1H, H7′, J = 1.6 Hz), 3.77 (s, 3H, H11′), 3.73 (s, 6H, H10′ and H12′), 3.69 (dd, 1H, H9a′, J = 11 and 4.6 Hz), 3.37 (dd, 1H, H9b’, J = 11 and 9.5 Hz), 3.01 (ddd, 1H, H8′, J = 9.5, 4.6 and 1.6 Hz); 13C NMR (CDCl3) δ (ppm) 152.9 (C3′ and C5′), 147.3 (C4), 146.7 (C5), 140.2 (C1′), 136.5 (C4′), 132.8 (C2), 132.1 (C7), -(C8), 129.9 (C1), 128.5 (C9), 127.2 (C1″), 110.2 (C3), 107.1 (C6), 104.5 (C2′ and C6′), 101.0 (C10), 63.6 (C2″), 63.1 (C9′), 60.7 (C11′), 56.0 (C10′ and C12′), 44.9 (C7′ and 8′). IR νmax/cm−1 (film) 3430, 2934, 1590, 1505, 1484, 1236, 1126, 1039, 934. UV (EtOH) λmax 209 (lg ε 4.5), 295 (lg ε 3.7), 316 (lg ε 3.7). [α]22D −37.1° (c 0.41%).
- Compound 9. A 3.0 M solution of methylmagnesium iodide in diethyl ether (0.17 mL) was added dropwise to a stirred solution of 2 (209 mg, 0.49 mmol) in dry THF at −78 °C under an argon atmosphere. Then, it was stirred and left to reach rt for 90 min and stirred for an additional 2 h. Then, a solution of saturated ammonium chloride was added and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and evaporated. Insolubilization in EtOAc gave a mixture of epimers, 9 (23 mg, 11%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 6.65/6.63 (s, 1H, H6), 6.34 (m, 1H, H7), 6.21 (s, 1H, H3), 6.53 (bs, 2H, H2′ and H6′), 6.28 (d, 1H, H9, J = 16 Hz), 5.92 (d, 1H, H10a, J = 1.3 Hz), 5.90 (d, 1H, H10b, J = 1.3 Hz), 5.20 (m, 1H, H9), 4.02/4.10 (s, 1H, H7′), 3.89 (s, 3H, H11′), 3.85 (s, 6H, H10′ and H12′), 3.80 (m, 1H, H8′), 1.63/1.54 (d, 3H, CH3-9, J = 6.0/6.8 Hz); 13C NMR (CDCl3) δ (ppm) 174.1 (C9′), 153.4 (C3′ and C5′), 147.5 (C4), 146.5 (C5), 136.9/136.1 (C1′), 136.5/136.6 (C4′), 130.9 (C2), 128.8 (C1), 126.8/127.1 (C8), 119.3/119.8 (C7), 109.3 (C3), 107.1 (C6), 103.6 (C2′ and C6′), 101.3 (C10), 76.8/77.3 (C9), 60.9 (C11′), 56.1 (C10′ and C12′), 46.9 (C7′), 45.7/43.4 (C8′), 18.8/21.5 (Me). IR νmax/cm−1 (film) 2924, 1774, 1592, 1506, 1483, 1234, 1132, 1034, 929. Purification by preparative thin-layer chromatography (Hexane/EtOAc) afforded compounds 9a (3 mg), 9b (3 mg), and 9c (4 mg).
- Compound 9a: 1H NMR (CDCl3, 200 MHz) δ (ppm) 6.71 (s, 1H, H6), 6.64 (s, 1H, H3), 6.35 (s, 2H, H2′ and H6′), 5.95 (d, 1H, H10a, J = 1.3 Hz), 5.93 (d, 1H, H10b, J = 1.3 Hz), 5.04 (qd, 1H, J = 6.8 and 1.8, H9), 4.78 (m, 1H, H7′), 3.81 (dd, 1H, J = 23 and 4.2, H7a), 3.79 (s, 3H, H11′), 3.78 (s, 6H, H10′ and H12′), 3.59 (dd, 1H, J = 23 and 4.2, H7b), 1.50 (d, 3H, CH3-9, J = 6.8 Hz); 13C NMR (CDCl3) δ (ppm) 171.4 (C9′), 161.1 (C8), 153.2 (C3′ and C5′), 147.1 (C4), 146.9 (C5), 138.4 (C1′), 136.8 (C4′), 129.8 (C1), 127.7 (C2), 123.4 (C8′), 109.4 (C3), 107.8 (C6), 105.2 (C2′ and C6′), 101.2 (C10), 78.3 (C9), 60.7 (C11′), 56.0 (C10′ and C12′), 42.7 (C7′), 28.4(C7), 18.5 (Me). IR νmax/cm−1 (film) 2927, 1753, 1591, 1505, 1485, 1233, 1127, 1037, 934. EM: 410 m/z. UV (EtOH) λmax 207 (lg ε 4.4), 259 (lg ε 4.2), 290 (lg ε 3.6), 310 (lg ε 3.6), 349 (lg ε 3.3). [α]22D +26.9° (c 0.23%).
- Compound 9b: 1H NMR (CDCl3, 200 MHz) δ (ppm) 6.72 (s, 1H, H6), 6.62 (s, 1H, H3), 6.36 (s, 2H, H2′ and H6′), 5.95 (d, 1H, H10a, J = 1.5 Hz), 5.94 (d, 1H, H10b, J = 1.5 Hz), 5.07 (q, 1H, J = 6.8, H9), 4.79 (t, 1H, J = 4.2 Hz, H7′), 3.73 (d, 1H, J = 3.8 Hz, H7a), 3.78 (s, 3H, H11′), 3.77 (s, 6H, H10′ and H12′), 3.63 (d, 1H, J = 4.1, H7b), 1.51 (d, 3H, CH3-9, J = 6.8 Hz); 13C NMR (CDCl3) δ (ppm) 171.3 (C9′), 160.9 (C8), 153.1 (C3′ and C5′), 147.1 (C4), 146.9 (C5), 138.3 (C1′), 136.9 (C4′), 129.6 (C1), 127.6 (C2), 123.8 (C8′), 109.4 (C3), 107.8 (C6), 105.5 (C2′ and C6′), 101.2 (C10), 78.1 (C9), 60.7 (C11′), 56.1 (C10′ and C12′), 42.7 (C7′), 28.3 (C7), 18.1 (Me). IR νmax/cm−1 (film) 2980, 1757, 1584, 1505, 1485, 1237, 1126, 1037, 938. EM: 410 m/z. UV (EtOH) λmax 210 (lg ε 4.5), 258 (lg ε 4.5), 310 (lg ε 3.8), 350 (lg ε 3.6). [α]22D +23.0° (c 0.58%).
- Compound 9c: 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.65 (s, 1H, H7), 7.21 (s, 1H, H6), 7.10 (m, 1H, H3), 6.56 (d, 1H, J = 2.8 Hz, H2′), 6.54 (d, 1H, J = 2.8 Hz, H6′), 6.09 (s, 2H, H10), 5.62 (q, 1H, J = 6.6 Hz, H9), 3.97 (s, 3H, H11′), 3.85 (s, 3H, H10′), 3.84 (s, 3H, H12′), 1.73 (d, 3H, CH3-9, J = 6.6 Hz); 13C NMR (CDCl3) δ (ppm) 169.0 (C9′), 152.9 (C3′ and C5′), 150.0 (C4), 148.7 (C5), 144.8 (C8), 140.3 (C7′), 137.7 (C4′), 134.6 (C1′), 130.4, 130.3 (C2 and C1), 118.8 (C8′), 118.7 (C7), 107.3 (C3), 107.1 (C6), 103.8 (C2′ and C6′), 101.8 (C10), 75.8 (C9), 61.8 (C11′), 56.1 (C10′ and C12′), 21.1 (Me). EM: 408 m/z.
- Compound 10. A stirred suspension of zinc (62 mg, 0.52 mmol) in dry THF was refluxed in a two-necked flask under an inert atmosphere. Then, a solution of ethyl bromodifluoroacetate (0.12 mL, 0.94 mmol) and aldehyde 2 (200 mg, 0.47 mmol) in a 1:1 mixture of dry CH2Cl2:THF was added through a septum. The reaction mixture was refluxed for 24 h, and the solvent was removed under vacuum. The residue was dissolved in chloroform, washed with a 5% tetrasodium ethylenediaminetretraacetate (EDTA) solution (pH = 10) and brine, dried over Na2SO4, filtered, and evaporated. Purification by silica gel CC (CH2Cl2/EtOAc 92:8) yielded compound 10 (50 mg, 21%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 6.67 (s, 1H, H6), 6.60 (d, 1H J = 3.0 Hz, H7), 6.21 (s, 1H, H3), 6.50 (bs, 2H, H2′ and H6′), 5.93 (d, 1H, H10a, J = 1.3 Hz), 5.91 (d, 1H, H10b, J = 1.3 Hz), 5.47 (dd, 1H, J = 16 and 7.2 Hz, H9), 4.41 (q, 2H, J = 7.1 Hz, O-CH2-CH3), 4.04 (d, 1H, J = 16 Hz, H7′), 3.89 (s, 3H, H11′), 3.84 (s, 6H, H10′ and H12′), 3.76 (m, 1H, H8′), 1.39 (t, 3H, J = 7.1 Hz, O-CH2-CH3); 13C NMR (CDCl3) δ (ppm) 172.6 (C9′), 161.9 (t, C2″), 153.6 (C3′ and C5′), 148.2 (C4), 146.7 (C5), 137.4 (C4′), 135.5 (C1′), 131.5 (C2), 126.2 (C1 and C8), 125.9 (C7), 111.2 (t, C1″), 109.7 (C2′ and C6′), 109.3 (C3), 107.6 (C6), 101.4 (C10), 77.2 (t, C9), 63.7 (O-CH2-CH3), 60.8 (C11′), 56.1 (C10′ and C12′), 47.0 (C7′), 43.9 (C8′), 13.9 (O-CH2-CH3).); HRMS calcd for C26H24O9F [M + H]+ 518.1388 u, found 518.1397 m/z; IR νmax/cm−1 (film) 2938, 1789, 1752, 1592, 1509, 1485, 1259, 1128, 1033, 928. UV (EtOH) λmax 210 (lg ε 4.5), 224 (lg ε 4.4), 301 (lg ε 3.9), 317 (lg ε 3.9). [α]22D −37.3° (c 0.51%).
- The general procedure for the synthesis of benzimidazoles 11–15. To a solution of 2 (0.10–0.16 mmol) in EtOH (5 mL), p-benzoquinone (p-BQ) (0.11–0.16 mmol) and the corresponding phenylenediamine (0.10–0.16 mmol) were added. The mixture was stirred at reflux for a specified time and then concentrated under vacuum. The reaction product was purified by CC on silica gel (CH2Cl2/EtOAc 8:2) to yield the corresponding benzimidazole.
- Benzimidazole 11. From 2 (48 mg, 0.11 mmol) in EtOH (5 mL), p-BQ (15 mg, 0.14 mmol), and 1,2-phenylenediamine (16 mg, 0.14 mmol) following the general procedure for 4.5 h. CC of the crude on silica gel (CH2Cl2/EtOAc 8:2) afforded 11 (33 mg, 57%). 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.38 (bs, 1H, H7), 7.38 (m, 2H, H3″ and H6″), 7.15 (m, 2H, H4″ and H5″), 6.70 (s, 1H, H3), 6.61 (bs, 1H, H6), 6.35 (s, 2H, H2′ and H6′), 5.95 (d, 1H, H10a, J = 1.3 Hz), 5.93 (d, 1H, H10b, J = 1.3 Hz), 4.65 (d, 1H, H7′, J = 1.6 Hz), 4.50 (bs, 1H, H8′), 3.74 (s, 3H, H11′), 3.69 (s, 6H, H10′ and H12′), 3.65 (s, 3H, CH3O-9′); 13C NMR (CDCl3, 200 MHz) δ (ppm) 173.2 (C9′), 153.0 (C3′ and C5′), 151.5 (C9), 148.3 (C4), 147.0 (C5), 143.0 (C1″), 138.0 (C1′), 136.7 (C4′), 134.2 (C2″), 130.1 (C2), 129.2 (C7), 126.0 (C1), 122.7 (C4″ and C5″), 122.0 (C8), 119.0 (C6″), 110.5 (C3″), 109.6 (C3), 107.9 (C6), 104.8 (C2′ and C6′), 101.3 (C10), 60.7 (C11′), 56.0 (C10′ and C12′), 52.7 (CH3O-9′), 47.7 (C8′), 46.1 (C7′); HRMS calcd for C29H27N2O7 [M+H]+ 515.1818 u, found 515.1829 m/z; IR νmax/cm−1 (film) 2953, 2924, 1731, 1590, 1504, 1484, 1459, 1278, 1234, 1126, 1037, 744.
- Benzimidazole 12. To a solution of 2 (49 mg, 0.12 mmol) in abs EtOH, aq HCl 2 N (0.5 mL) and 1,2-phenylenediamine (31 mg, 0.28 mmol) were added and continuously stirred at 135 °C under an argon atmosphere for 24 h. The reaction was diluted with water and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, and filtered, and the solvent was evaporated. CC of the residue (CH2Cl2/EtOAc 8:2) yielded aldehyde 2 (41%) and compound 12 (28%). 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.52 (bs, 1H, H7), 7.48 (m, 2H, H3″ and H6″), 7.16 (m, 2H, H4″ and H5″), 6.76 (s, 1H, H6), 6.70 (bs, 1H, H3), 6.36 (s, 2H, H2′ and H6′), 5.98 (s, 1H, H10a), 5.97 (s, 1H, H10b), 4.64 (bs, 1H, H7′), 4.39 (bs, 1H, H8′), 4.13 (dq, 2H, J = 7.1 and 2.3, CH3-CH2-O-9′), 3.73 (s, 3H, H11′), 3.71 (s, 6H, H10′ and H12′), 1.14 (t, 3H, J = 7.1, CH3-CH2-O-9′); 13C NMR (CDCl3, 200 MHz) δ (ppm) 172.4 (C9′), 153.1 (C3′ and C5′), 151.2 (C9), 148.6 (C4), 147.0 (C5), 137.7 (C1′), 136.9 (C4′), 130.6 (C2), 123.1 (C7), 125.8 (C1), 123.1 (C4″ and C5″), 109.6 (C3), 108.2 (C6), 104.7 (C2′ and C6′), 101.4 (C10), 61.7 (CH3-CH2-O-9′), 60.7 (C11′), 56.1 (C10′ and C12′), 48.2 (C8′), 46.0 (C7′), 14.0 (CH3-CH2-O-9′); HRMS calcd for C30H28N2O7 [M+H]+ 529.1974 u, found 529.1921 m/z; IR νmax/cm−1 (film) 2954, 2923, 1728, 1589, 1504, 1485, 1460, 1235, 1126, 1036, 746. [α]22D −72.4° (c 0.17%)
- Benzimidazole 13. From 2 (70 mg, 0.16 mmol) in EtOH (5 mL), p-BQ (18 mg, 0.16 mmol), and 4,5-dimethyl-1,2-phenylenediamine (27 mg, 0.20 mmol) following the general procedure for 4.5 h. CC of the crude on silica gel (CH2Cl2/EtOAc 9:1) afforded 13 (14 mg, 16%). 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.41 (m, 1H, H6″), 7.31 (s, 1H, H7), 7.04 (m, 1H, H3″), 6.70 (s, 1H, H3), 6.65 (s, 1H, H6), 6.36 (s, 2H, H2′ and H6′), 5.96 (d, 1H, H10a, J = 1.4 Hz), 5.95 (d, 1H, H10b, J = 1.4 Hz), 4.63 (d, 1H, H7′, J = 2.1 Hz), 4.48 (d, 1H, H8′, J = 2.1 Hz), 3.73 (s, 1H, H11′), 3.69 (s, 2H, H10′ and H12′), 3.67 (s, 3H, CH3O-9′), 2.30 (s, 6H, CH3-4″ and CH3-5″); 13C NMR (CDCl3, 200 MHz) δ (ppm) 173.3 (C9′), 153.0 (C3′ and C5′), 150.5 (C9), 148.2 (C4), 146.9 (C5), 142.1 (C1″), 137.9 (C1′), 136.8 (C4′), 132.3 (C2″ and C5″), 131.1 (C4″), 130.3 (C2), 128.3 (C7), 126.1 (C1), 122.2 (C8), 119.2 (C6″), 110.7 (C3″), 109.6 (C3), 108.0 (C6), 104.7 (C2′ and C6′), 101.3 (C10), 60.7 (C11′), 56.1 (C10′ and C12′), 52.7 (CH3O-9′), 47.7 (C8′), 46.2 (C7′), 20.3 (CH3-4″ and CH3-5″); HRMS calcd for C31H31N2O7 [M + H]+ 543.2131 u, found 543.2147 m/z; IR νmax/cm−1 (film) 3310, 2931, 2873, 2852, 1695, 1465, 1454, 1236, 1128, 1036, 890, 737.
- Compound 13 was also obtained from the following procedure: A mixture of aldehyde 2 (52 mg, 0.12 mmol) and 4,5-dimethyl-1,2-phenylenediamine (18 mg, 0.13 mmol) in acetonitrile (2 mL) was stirred at 90 °C for 1 h. FeCl3·6H2O (1 mg, 0.004 mmol) was then added, and the mixture was stirred with heating at 90 °C, under continuous O2 bubbling, for 7 h. The solvent was evaporated, and the residue was purified by silica gel CC (CH2Cl2/EtOAc 9:1) to give 13 (37 mg, 56%).
- Benzimidazole 14. From 2 (46 mg, 0.11 mmol) in EtOH (4 mL), p-BQ (14 mg, 0.13 mmol), and 4-methoxy-1,2-phenylenediamine (23 mg, 0.11 mmol) following the general procedure for 6.5 h. CC of the crude on silica gel (CH2Cl2/EtOAc 7:3) afforded 14 (38 mg, 65%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.37 (s, 1H, H7), 7.35 (m, 1H, H5″), 6.83 (d, 1H, H6″, J = 2.5 Hz), 6.78 (d, 1H, H3″, J = 2.5 Hz), 6.70 (s, 1H, H3), 6.66 (s, 1H, H6), 6.34 (s, 2H, H2′ and H6′), 5.97 (d, 1H, H10a, J = 1.5 Hz), 5.96 (d, 1H, H10b, J = 1.5 Hz), 4.65 (d, 1H, H7′, J = 2.0 Hz), 4.46 (d, 1H, H8′, J = 2.0 Hz), 3.81 (s, 3H, CH3O-4″), 3.73 (s, 1H, H11′), 3.71 (s, 2H, H10′ and H12′), 3.69 (s, 3H, CH3O-9′); 13C NMR (CDCl3, 200 MHz) δ (ppm) 173.4 (C9′), 156.7 (C4″), 153.0 (C3′ and C5′), 150.8 (C9), 148.4 (C4), 147.1 (C5), 137.9 (C1′), 136.9 (C4′), 130.2 (C2), 129.0 (C7), 126.0 (C1), 121.4 (C8), 112.6 (C3″ and C6″), 109.6 (C3), 108.1 (C6), 104.8 (C2′ and C6′), 101.4 (C10), 60.7 (C11′), 56.1 (C10′ and C12′), 55.6 (CH3O-4″), 52.8 (CH3O-9′), 47.7 (C8′), 46.1 (C7′); HRMS calcd for C30H29N2O8 [M + H]+ 545.1924 u, found 545.1974 m/z; IR νmax/cm−1 (film) 2926, 1737, 1732, 1590, 1505, 1485, 1463, 1456, 1417, 1373, 1327, 1274, 1237, 1126, 1036, 825, 730.
- Benzimidazole 15. From 2 (45 mg, 0.11 mmol) in EtOH (5 mL), p-BQ (14 mg, 0.13 mmol), and 4,5-dichloro-1,2-phenylenediamine (20 mg, 0.11 mmol) following the general procedure for 6 h. CC of the crude on silica gel (CH2Cl2/EtOAc 85:15) afforded 15 (62 mg, 99%). 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.57 (bs, 1H, H6″), 7.15 (s, 1H, H7), 7.01 (bs, 1H, H3″), 6.69 (s, 1H, H3), 6.33 (s, 2H, H2′ and H6′), 6.24 (s, 1H, H6), 5.97 (d, 1H, H10a, J = 1.3 Hz), 5.94 (d, 1H, H10b, J = 1.3 Hz), 4.68 (d, 1H, H7′, J = 2.8 Hz), 4.66 (d, 1H, H8′, J = 2.8 Hz), 3.82 (s, 3H, CH3O-9′), 3.74 (s, 1H, H11′), 3.71 (s, 2H, H10′ and H12′); 13C NMR (CDCl3, 200 MHz) δ (ppm) 174.7 (C9′), 153.1 (C3′ and C5′), 152.9 (C9), 148.6 (C4), 147.0 (C5), 142.6 (C1″), 137.7 (C1′), 137.1 (C4′), 133.0 (C2″), 130.2 (C7), 130.1 (C2), 126.8 (C5″), 126.0 (C4″), 125.6 (C1), 121.3 (C8), 119.9 (C3″), 111.5 (C6″), 109.6 (C3), 108.2 (C6), 105.1 (C2′ and C6′), 101.5 (C10), 60.7 (C11′), 56.2 (C10′ and C12′), 53.2 (CH3O-9′), 47.6 (C8′), 46.3 (C7′); HRMS calcd for C29H25N2O7 [M + H]+ 583.0960 u, found 583.1016 m/z; Anal. calcd for C29H24N2O7Cl2: C, 59.70; H, 4.15; N, 4.80; found: C, 57.53; H, 4.25; N, 4.83; IR νmax/cm−1 (film) 3290, 2952, 2920, 2850, 1731, 1592, 1504, 1485, 1462, 1454, 1417, 1374, 1295, 1235, 1126, 1098, 1038, 1005, 828.
- The general procedure for the synthesis of benzoxazoles 16–18. A solution of compound 2 (0.12–0.14 mmol) and the corresponding 2-aminophenol (0.12–0.20 mmol) in dry EtOH (2 mL) was stirred at reflux for the specified time. The solvent was evaporated, and the residue was redissolved in glacial acetic acid (1 mL). Pb(AcO)4 (0.12–0.34 mmol) was added to the solution, and it was then stirred at room temperature for a time. After dilution with water, the mixture was extracted with EtOAc, and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and evaporated. Purification by silica gel column chromatography (Hexane/EtOAc) of the crude provided the corresponding benzoxazoles.
- Benzoxazole 16. From 2 (50 mg, 0.12 mmol) and 2-aminophenol (13 mg, 0.12 mmol) in dry EtOH (2 mL) for 46 h. Then, in glacial acetic acid (1 mL) with Pb(AcO)4 (55 mg, 0.12 mmol) for 24 h. CC (Hexane/ EtOAc 7:3) of the crude provided 16 (36 mg, 59%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.73 (s, 1H, H7), 7.67 (m, 1H, H3″), 7.49 (m, 1H, H6″), 7.31 (m, 1H, H4″), 7.29 (m, 1H, H5″), 6.90 (s, 1H, H6), 6.72 (s, 1H, H3), 6.30 (s, 2H, H2′ and H6′), 6.01 (d, 1H, H10a, J = 1.3 Hz), 5.99 (d, 1H, H10b, J = 1.3 Hz), 4.72 (d, 1H, H7′, J = 1.8 Hz), 4.40 (d, 1H, H8′, J = 1.8 Hz), 3.75 (s, 1H, H11′), 3.72 (s, 2H, H10′ and H12′), 3.65 (s, 3H, CH3O-9′); 13C NMR (CDCl3, 400 MHz) δ (ppm) 172.2 (C9′), 163.0 (C9), 153.1 (C3′ and C5′), 150.5 (C1″), 149.0 (C4), 147.2 (C5), 142.1 (C2″), 137.6 (C1′), 137.0 (C4′), 132.7 (C7), 131.0 (C2), 126.0 (C1), 125.1 (C5″), 124.4 (C4″), 119.8 (C3″), 119.4 (C8), 110.2 (C6″), 110.0 (C3), 108.4 (C6), 104.8 (C2′ and C6′), 101.5 (C10), 60.7 (C11′), 56.0 (C10′ and C12′), 52.7 (CH3O-9′), 47.4 (C8′), 46.2 (C7′); HRMS calcd for C29H26NO8 [M + H)]+ 516.1658 u, found 516.1691 m/z; IR νmax/cm−1 (film) 2952, 2937, 1732, 1590, 1504, 1485, 1455, 1418, 1371, 1328, 1242, 1128, 1037, 1007, 933, 808, 765, 747.
- Benzoxazole 17. From 2 (58 mg, 0.14 mmol) and 2-amino-4-methylphenol (17 mg, 0.14 mmol) in dry EtOH (2 mL) for 23 h. Then, in glacial acetic acid (1 mL) with Pb(AcO)4 (159 mg, 0.34 mmol) for 44 h. CC (Hexane/EtOAc 8:2) of the crude provided 17 (41 mg, 57%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.70 (s, 1H, H7), 7.44 (bs, 1H, H3″), 7.35 (d, 1H, H6″, J = 8.4 Hz), 7.10 (dd, 1H, H5″, J1 = 8.4 Hz, J2 = 1.5 Hz), 6.88 (s, 1H, H6), 6.71 (s, 1H, H3), 6.30 (s, 2H, H2′ and H6′), 5.99 (d, 1H, H10a, J = 1.5 Hz), 5.97 (d, 1H, H10b, J = 1.5 Hz), 4.71 (d, 1H, H7′, J = 2.0 Hz), 4.39 (d, 1H, H8′, J = 2.0 Hz), 3.74 (s, 1H, H11′), 3.71 (s, 2H, H10′ and H12′), 3.63 (s, 3H, CH3O-9′), 2.43 (s, 3H, CH3-4″); 13C NMR (CDCl3, 200 MHz) δ (ppm) 172.3 (C9′), 163.1 (C9), 153.1 (C3′ and C5′), 149.0 (C4), 148.7 (C1″), 147.3 (C5), 142.2 (C2″), 137.7 (C1′), 136.9 (C4′), 134.3 (C4″), 132.5 (C7), 131.0 (C2), 126.2 (C5″), 126.0 (C1), 119.7 (C3″), 119.5 (C8), 110.0 (C3), 109.6 (C6″), 108.4 (C6), 104.7 (C2′ and C6′), 101.5 (C10), 60.7 (C11′), 56.0 (C10′ and C12′), 52.7 (CH3O-9′), 47.4 (C8′), 46.2 (C7′), 21.5 (CH3-4″); HRMS calcd for C30H28NO8 [M + H]+ 530.1815 u, found 530.1863 m/z; IR νmax/cm−1 (film) 2952, 2937, 1732, 1591, 1504, 1485, 1462, 1418, 1371, 1329, 1239, 1127, 1037, 1010, 933, 819, 735.
- Benzoxazole 18. From 2 (54 mg, 0.13 mmol) and 2-amino-4-chlorophenol (29 mg, 0.20 mmol) in dry EtOH (2 mL) for 47 h. Then, in glacial acetic acid (1 mL) with Pb(AcO)4 (60 mg, 0.13 mmol) for 26 h. CC of the crude on silica gel (CH2Cl2/EtOAc 98:2) provided 18 (27 mg, 39%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.73 (s, 1H, H7), 7.62 (d, 1H, H3″, J = 2.2 Hz), 7.39 (d, 1H, H6″, J = 8.4 Hz), 7.26 (dd, 1H, H5″, J1 = 8.4 Hz, J2 = 2.2 Hz), 6.89 (s, 1H, H6), 6.71 (s, 1H, H3), 6.29 (s, 2H, H2′ and H6′), 6.01 (d, 1H, H10a, J = 1.3 Hz), 5.99 (d, 1H, H10b, J = 1.3 Hz), 4.72 (d, 1H, H7′, J = 2.0 Hz), 4.36 (d, 1H, H8′, J = 2.0 Hz), 3.75 (s, 1H, H11′), 3.72 (s, 2H, H10′ and H12′), 3.65 (s, 3H, CH3O-9′); 13C NMR (CDCl3, 200 MHz) δ (ppm) 172.1 (C9′), 164.3 (C9), 153.1 (C3′ and C5′), 149.2 (C1″), 149.0 (C4), 147.3 (C5), 143.2 (C2″), 137.5 (C1′), 137.0 (C4′), 133.5 (C7), 131.1 (C4″), 129.8 (C2), 125.8 (C1), 125.3 (C5″), 119.7 (C3″), 118.9 (C8), 110.9 (C6″), 110.0 (C3), 108.5 (C6), 104.6 (C2′ and C6′), 101.6 (C10), 60.7 (C11′), 56.0 (C10′ and C12′), 52.7 (CH3O-9′), 47.4 (C8′), 46.2 (C7′); Anal. calcd for C29H24NO8Cl: C, 63.33; H, 4.40; N, 2.55; found: C, 63.37; H, 4.69; N, 2.54; IR νmax/cm−1 (film) 2934, 2838, 1732, 1589, 1537, 1505, 1485, 1455, 1255, 1238, 1127, 1037, 1010, 934, 816, 735, 703.
- Synthesis of benzothiazoles 19 and 20. To a solution of aldehyde 2 (50 mg, 0.12 mmol), 2-aminothiophenol (15 μL, 0.14 mmol) or 2-amino-4-(trifluoromethyl)thiophenol (28 mg, 0.12 mmol) and p-toluensulfonic acid (2 mg, 0.01 mmol) in dry toluene (5 mL) MgSO4 (34 mg, 0.28 mmol) were added, and the mixture was stirred at reflux for 24 h. The mixture was then filtered, and the solvent was evaporated under vacuum. The residue was redissolved in EtOAc and neutralized with aq saturated NaHCO3 and washed with brine, dried over Na2SO4, filtered, and evaporated off. CC of the crude on silica gel (CH2Cl2/EtOAc 96:4 or 98:2) afforded 19 (32 mg, 52%) or 20 (55 mg, 77%).
- Compound 19. 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.92 (m, 1H, H3″), 7.82 (m, 1H, H6″), 7.42 (m, 1H, H4″), 7.40 (s, 1H, H7), 7.34 (m, 1H, H5″), 6.86 (s, 1H, H6), 6.73 (s, 1H, H3), 6.38 (s, 2H, H2′ and H6′), 5.99 (s, 1H, H10a), 5.98 (s, 1H, H10b), 4.67 (d, 1H, H7′, J = 2.4 Hz), 4.61 (d, 1H, H8′, J = 2.4 Hz), 3.75 (s, 1H, H11′), 3.71 (s, 2H, H10′ and H12′), 3.62 (s, 3H, CH3O-9′); 13C NMR (CDCl3, 200 MHz) δ (ppm) 172.5 (C9′), 167.8 (C9), 153.7 (C1″), 153.0 (C3′ and C5′), 148.7 (C4), 147.1 (C5), 137.8 (C1′), 136.8 (C4′), 134.4 (C2″), 131.8 (C7), 131.3 (C2), 127.4 (C8), 126.1 (C1), 126.0 (C4″), 125.2 (C5″), 123.0 (C3″), 121.3 (C6″), 109.8 (C3), 108.2 (C6), 104.6 (C2′ and C6′), 101.4 (C10), 60.7 (C11′), 56.0 (C10′ and C12′), 52.5 (CH3O-9′), 48.3 (C8′), 46.5 (C7′); HRMS calcd for C29H26NO7S [M + H]+ 532.1430 u, found 532.1381 m/z; Anal. calcd for C29H25NO7S: C, 65.52; H, 4.74; N, 2.63; S, 6.03; found: C, 65.05; H, 4.96; N, 2.54; S, 5.94; IR νmax/cm−1 (film) 2952, 2927, 1732, 1588, 1504, 1486, 1456, 1238, 1126, 1036, 1009, 731, 686.
- Compound 20. 1H NMR (CDCl3, 400 MHz) δ (ppm) 8.19 (bs, 1H, H3″), 7.92 (d, 1H, H6″, J = 8.4 Hz), 7.56 (dd, 1H, H5″, J1 = 8.4 Hz, J2 = 1.5 Hz), 7.43 (s, 1H, H7), 6.87 (s, 1H, H6), 6.73 (s, 1H, H3), 6.36 (s, 2H, H2′ and H6′), 6.01 (d, 1H, H10a, J = 1.3 Hz), 5.99 (d, 1H, H10b, J = 1.3 Hz), 4.69 (d, 1H, H7′, J = 2.2 Hz), 4.58 (d, 1H, H8′, J = 2.2 Hz), 3.75 (s, 1H, H11′), 3.72 (s, 2H, H10′ and H12′), 3.64 (s, 3H, CH3O-9′); 13C NMR (CDCl3, 200 MHz) δ (ppm) 172.3 (C9′), 169.8 (C9), 153.3 (C4″), 153.1 (C3′ and C5′), 149.0 (C4), 147.2 (C5), 137.8 (C1″), 137.7 (C1′), 137.0 (C4′), 133.1 (C7), 131.5 (C2), 129.1 (C2″), 127.0 (C8), 125.8 (C1), 125.5 (CF3-4″), 121.8 (C6″), 121.5 (C5″), 120.1 (C3″), 109.9 (C3), 108.3 (C6), 104.6 (C2′ and C6′), 101.5 (C10), 60.7 (C11′), 56.1 (C10′ and C12′), 52.6 (CH3O-9′), 48.4 (C8′), 46.5 (C7′); HRMS calcd for C30H25NO7SF3 [M + H]+ 600.1304 u, found 600.1337 m/z; IR νmax/cm−1 (film) 2953, 2839, 1732, 1589, 1505, 1487, 1463, 1418, 1330, 1232, 1126, 1037, 1008, 933, 736.
- Compound 21. To a solution of compound 2 (50 mg, 0.12 mmol) in nitrobenzene (5 mL), 2,3-diaminopyridine (26 mg, 0.24 mmol) was added. The mixture was stirred at 145 °C under a N2 atmosphere for 3 d. Purification by silica gel CC (CH2Cl2/MeOH 97:3) provided compound 21 (48 mg, 79%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 8.35 (d, 1H, H6″, J = 5.1 Hz), 8.03 (m, 1H, H4″), 7.65 (s, 1H, H7), 7.26 (m, 1H, H5″), 6.84 (s, 1H, H6), 6.75 (s, 1H, H3), 6.36 (s, 2H, H2′ and H6′), 6.02 (s, 1H, H10a), 6.00 (s, 1H, H10b), 4.75 (d, 1H, H7′, J = 1.9 Hz), 4.61 (d, 1H, H8′, J = 1.9 Hz), 3.72 (s, 1H, H11′), 3.67 (s, 2H, H10′ and H12′), 3.63 (s, 3H, CH3O-9′); 13C NMR (CDCl3, 200 MHz) δ (ppm) 172.6 (C9′), 153.8 (C9), 153.0 (C3′ and C5′), 149.5 (C2″), 148.7 (C4), 147.2 (C5), 142.4 (C6″), 137.7 (C1′), 136.7 (C4′), 130.8 (C7), 130.6 (C2), 126.9 (C3″), 126.0 (C1), 122.4 (C8), 118.2 (C5″), 109.9 (C3), 107.9 (C6), 104.6 (C2′and C6′), 101.5 (C10), 60.6 (C11′), 55.9 (C10′ and C12′), 52.6 (CH3O-9′), 47.5 (C8′), 46.2 (C7′); HRMS calcd for C28H26N3O7 [M + H]+ 516.1770 u, found 516.1799 m/z; IR νmax/cm−1 (film) 2953, 2921, 2850, 1738, 1732, 1589, 1504, 1485, 1463, 1456, 1414, 1279, 1259, 1235, 1125, 1037, 1007, 935, 900, 785.
- Compound 22. To a solution of 4,5-diaminopyrimidine (10 mg, 0.09 mmol) in N,N-dimethylacetamide (2 mL), Na2S2O5 (23 mg, 0.12 mmol) was first added, and then aldehyde 2 (38 mg, 0.09 mmol). The mixture was stirred at 100 °C for 97 h. The solvent was evaporated under reduced pressure, and the crude product was purified by CC on silica gel (EtOAc), providing 22 (26 mg, 56%). 1H NMR (CDCl3, 200 MHz) δ (ppm) 9.04 (s, 1H, H6″), 8.95 (s, 1H, H2″), 7.58 (s, 1H, H7), 6.75 (s, 1H, H6), 6.73 (s, 1H, H3), 6.31 (s, 2H, H2′ and H6′), 6.01 (s, 1H, H10a), 6.00 (s, 1H, H10b), 4.74 (d, 1H, H7′, J = 1.8 Hz), 4.55 (d, 1H, H8′, J = 1.8 Hz), 3.73 (s, 1H, H11′), 3.70 (s, 2H, H10′ and H12′), 3.69 (s, 3H, CH3O-9′); 13C NMR (CDCl3, 200 MHz) δ (ppm) 172.5 (C9′), 154.4 (C9), 153.2 (C4″), 153.1 (C3′ and C5′), 151.5 (C2″), 149.3 (C4), 147.3 (C5), 147.2 (C6″), 137.5 (C1′), 137.1 (C4′), 135.2 (C5″), 132.3 (C7), 131.0 (C2), 125.4 (C1), 121.4 (C8), 110.0 (C3), 108.2 (C6), 104.8 (C2′ and C6′), 101.6 (C10), 60.7 (C11′), 56.1 (C10′ and C12′), 52.8 (CH3O-9′), 47.6 (C8′), 46.1 (C7′); HRMS calcd for C27H25N4O7 [M + H]+ 517.1723, found 517.1669; Anal. calcd for C27H24N4O7: C, 62.79; H, 4.68; N, 10.85; found: C, 62.18; H, 5.02; N, 10.34; IR νmax/cm−1 (film) 2930, 2851, 1732, 1606, 1590, 1505, 1485, 1463, 1417, 1377, 1260, 1238, 1126, 1037, 1007, 910, 730.
- Compound 22 was also obtained through the following procedure: To a solution of compound 2 (52 mg, 0.12 mmol) in dry EtOH (2 mL), MgSO4 (30 mg, 0.25 mmol) and 4,5-diaminopyrimidine (17 mg, 0.15 mmol) were added, and the mixture was stirred at reflux for 98 h. The solvent was then evaporated, and the residue was redissolved in glacial acetic acid (2 mL). The reaction mixture was stirred at reflux for 48 h. After dilution with water, the mixture was extracted with EtOAc, and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and evaporated. Purification by silica gel CC (CH2Cl2/MeOH 96:4) of the crude provided 22 (16 mg, 25%).
- General procedures for 1,3-thiazolidin-4-ones 28–32.
- Method A from imines 23–27. To a solution of the corresponding imine in dry benzene, MgSO4 and thioglycolic acid were added, and the reaction was refluxed for the specified time in a flask equipped with a Dean–Stark trap under a nitrogen atmosphere. After cooling, the mixture was filtered, concentrated under reduced pressure, diluted with an aq sat NaHCO3 solution, and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, and evaporated. Purification of the crude by silica gel CC (CH2Cl2/EtOAc) provided the corresponding thiazolidinones.
- Method B from aldehyde 2. To a suspension of aldehyde 2 in dry benzene was added the appropriate amine, and the mixture was refluxed for the specified time in a flask equipped with a Dean–Stark trap under a nitrogen atmosphere. After cooling to room temperature, thioglycolic acid was added dropwise to the solution, and the resulting mixture was refluxed for additional time. It was then cooled and concentrated under reduced pressure. The obtained residue was diluted with an aq sat NaHCO3 solution and extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, and evaporated. Purification of the crude by silica gel CC (CH2Cl2/EtOAc) provided the corresponding thiazolidinones.
- 1,3-Thiazolidin-4-one 28: Following method A from imine 23 (78 mg, 0.17 mmol) in dry benzene (10 mL), MgSO4 (43 mg, 0.35 mmol), and thioglycolic acid (26 μL, 0.37 mmol). The mixture was refluxed for 20 h. Purification by silica gel CC (CH2Cl2/EtOAc 8:2) provided compound 28 (33 mg, 31%) as a 7:3 mixture of epimers. 1H NMR (CDCl3, 200 MHz) δ (ppm) 6.73/6.71 (s, 1H, H6), 6.60/6.56 (s, 1H, H3), 6.47/6.55 (s, 1H, H7), 6.18/6.28 (s, 2H, H2′ and H6′), 5.97/5.94 (d, 1H, J = 1.4 Hz, H10a), 5.96/5.93 (d, 1H, J = 1.4 Hz, H10b), 5.27/5.25 (d, 1H, J = 1.4/1.9 Hz, H9), 4.41/4.32 (d, 1H, J = 1.8/2.8 Hz, H7′), 3.77/3.80 (s, 1H, H11′), 3.73/3.76 (s, 6H, H10′ and H12′), 3.66/3.61 (s, 3H, CH3O-9′), 3.45 (m, 2H, H2″), 3.23/3.63 (d, 1H, J = 1.8/2.8Hz, H8′), 2.08/2.77 and 3.40/3.60 (m, 2H, N-CH2-CH3), 0.95/1.5 (t, 3H, J = 7.2 Hz, N-CH2-CH3); 13C NMR (CDCl3, 200 MHz) Table S4. HRMS calcd for C27H29NO8S [M + H]+ 528.1692 u, found 528.1723 m/z; IR νmax/cm−1 (film) 2935, 1732, 1678, 1589, 1505, 1485, 1461, 1417, 1270, 1237, 1126, 1036, 1008, 734.
- 1,3-Thiazolidin-4-one 29: Following method A from imine 24 (49 mg, 0.10 mmol) in dry benzene (10 mL), MgSO4 (43 mg, 0.35 mmol), and thioglycolic acid (8 μL, 0.12 mmol). The mixture was refluxed for 2 h. Purification by silica gel CC (CH2Cl2/EtOAc 8:2) provided aldehyde 2 (35%) and compound 29 (37 mg, 59%) as a 7:3 mixture of epimers. 1H NMR (CDCl3, 200 MHz) δ (ppm) 6.72/6.70 (s, 1H, H6), 6.58/6.54 (s, 1H, H3), 6.46/6.54 (s, 1H, H7), 6.15/6.27 (s, 2H, H2′ and H6′), 5.96/5.93 (s, 1H, H10a), 5.94/5.92 (s, 1H, H10b), 5.24 bs/5.21 d (1H, J = 1.8 Hz, H9), 4.39 bs/4.30 d (1H, J = 3.3 Hz, H7′), 3.75/3.78 (s, 1H, H11′), 3.71/3.76 (s, 6H, H10′ and H12′), 3.64/3.60 (s, 3H, CH3O-9′), 3.45 (m, 2H, H2″), 3.20 d/3.46 bs (1H, J = 1.8 Hz, H8′), 2.08/2.70 and 3.45/3.60 (m, 2H, N-CH2-CH2-CH3), 1.40 (m, 2H, N-CH2-CH2-CH3), 0.73/0.84 (t, 3H, J = 7.5 Hz, N-CH2-CH2-CH3); 13C NMR (CDCl3, 200 MHz) Table S4. HRMS calcd for C28H31NO8S [M + H]+ 542.1848 u, found 542.1896 m/z; IR νmax/cm−1 (film) 2958, 1733, 1681-1674, 1589, 1505, 1485, 1463, 1417, 1267, 1238, 1126, 1037, 1009, 735.
- 1,3-Thiazolidin-4-one 30: Following method B from aldehyde 2 (50 mg, 0.12 mmol) and ethanolamine (14 μL, 0.23 mmol) in dry benzene (10 mL) for 24 h and thioglycolic acid (10 μL, 0.14 mmol) for an additional 48 h. Purification by silica gel CC (acetone) provided compound 30 (33 mg, 52%) as a 7:3 mixture of epimers. 1H NMR (C DCl3, 200 MHz) δ (ppm) 6.73/6.71 (s, 1H, H6), 6.59/6.60 (s, 1H, H3), 6.51/6.55 (s, 1H, H7), 6.17/6.26 (s, 2H, H2′ and H6′), 5.97/5.94 (d, 1H, J = 1.5 Hz, H10a), 5.95/5.93 (d, 1H, J = 1.5 Hz, H10b), 5.41 bs/5.45 d (1H, J = 1.8 Hz, H9), 4.39 bs/4.32 d (1H, J = 3.3 Hz, H7′), 3.75/3.79 (s, 1H, H11′), 3.72/3.76 (s, 6H, H10′ and H12′), 3.65/3.61 (s, 3H, CH3O-9′), 3.50 (m, 2H, H2″), 3.22 d/3.60 bs (1H, J = 1.8 Hz, H8′), 2.30 and 3.50 (m, 2H, N-CH2-CH2-OH), 3.50–3.80 (m, 2H, N-CH2-CH2-OH); 13C NMR (CDCl3, 200 MHz) Table S4. HRMS calcd for C27H29NO9S [M + H]+ 544.1641 u, found 544.1698 m/z; IR νmax/cm−1 (film) 3450, 2934, 1732, 1678, 1589, 1504, 1485, 1462, 1418, 1269, 1238, 1126, 1036, 1007, 734.
- 1,3-Thiazolidin-4-one 31: Following method A from imine 26 (87 mg, 0.17 mmol) in dry dichloromethane (10 mL), MgSO4 (50 mg, 0.41 mmol), and thioglycolic acid (30 μL, 0.43 mmol). The mixture was kept at rt for 15 d. Evaporation of the solvent and CC on silica gel (CH2Cl2/EtOAc 1:1) provided compound 31 (31 mg, 31%) as a 7:3 mixture of epimers. 1H NMR (CDCl3, 200 MHz) δ (ppm) 7.20 (m, 5H, phenyl), 6.63/6.67 (s, 1H, H6), 6.58/6.53 (s, 1H, H3), 6.38/6.51 (s, 1H, H7), 6.10/6.17 (s, 2H, H2′ and H6′), 5.94 (d, 1H, J = 1.5 Hz, H10a), 5.92 (d, 1H, J = 1.5 Hz, H10b), 5.68/5.53 (s, 1H, H9), 4.50/4.31 (d, 1H, J = 2.2/2.8 Hz, H7′), 3.71/3.82 (s, 1H, H11′), 3.69/3.80 (s, 6H, H10′ and H12′), 3.68/3.51 (s, 3H, CH3O-9′), 3.51/3.48 (s, 2H, H2″), 3.46 d/3.52 bs (1H, J = 2.2 Hz, H8′); 13C NMR (CDCl3, 200 MHz) Table S4. HRMS calcd for C31H29NO8S [M + H]+ 576.1692 u, found 576.1653 m/z; IR νmax/cm−1 (film) 2935, 1732, 1693–1684, 1590, 1504, 1485, 1462, 1270, 1238, 1126, 1036, 1008, 733.
- 1,3-Thiazolidin-4-one 32: Following method A from imine 27 (252 mg, 0.47 mmol) in dry benzene (15 mL), MgSO4 (100 mg, 0.82 mmol), and thioglycolic acid (68 μL, 0.98 mmol). The mixture was refluxed for 72 h. Purification by silica gel CC (CHCl3/MeOH 98:2) provided compound 32 (90 mg, 31%) as a 7:3 mixture of epimers. 1H NMR (CDCl3, 200 MHz) δ (ppm) 6.99/7.12 and 6.75/6.86 (d, J = 9.0 Hz, AB system, phenyl), 6.64/6.66 (s, 1H, H6), 6.55/6.51 (s, 1H, H3), 6.40/6.52 (s, 1H, H7), 6.07/6.18 (s, 2H, H2′ and H6′), 5.93 (d, 1H, J = 1.2 Hz, H10a), 5.92 (d, 1H, J = 1.2 Hz, H10b), 5.65/5.53 (s, 1H, H9), 4.42/4.31 (d, 1H, J = 1.6/2.6 Hz, H7′), 3.80/3.81 (s, 1H, H11′), 3.66/3.71 (s, 6H, H10′ and H12′), 3.65/3.53 (s, 3H, CH3O-9′), 3.60/3.48 (s, 2H, H2″), 3.43 d/3.52 bs (1H, J = 2.0 Hz, H8′), 3.79/3.80 (s, 3H, CH3O-Ph); 13C NMR (CDCl3, 200 MHz) Table S4. HRMS calcd for C32H31NO8S [M + H]+ 606.1797 u, found 606.1827 m/z; IR νmax/cm−1 (film) 2930, 1732, 1688–1682, 1590, 1512, 1485, 1463, 1270, 1127, 1036, 1009, 735.
3.2. Biological Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J. Natural products and drug discovery. Natl. Sci. Rev. 2022, 9, nwac206. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Davison, E.K.; Brimble, M.A. Natural product derived privileged scaffolds in drug discovery. Curr. Opin. Chem. Biol. 2019, 52, 1–8. [Google Scholar] [CrossRef]
- San Feliciano, A.; Castro, M.Á.; López-Pérez, J.L.; del Olmo, E. The importance of structural manipulation of natural compounds in drug discovery and development. In Plant Bioactives and Drug Discovery: Principles, Practice, and Perspectives; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 127–160. [Google Scholar]
- Zalesak, F.; Bon, D.J.-Y.D.; Pospisil, J. Lignans and Neolignans: Plant secondary metabolites as a reservoir of biologically active substances. Pharmacol. Res. 2019, 146, 104284. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. Int. J. Kuwait Univ. Health Sci. Cent. 2016, 25 (Suppl. S2), 41–59. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Cong, Y.; Li, H.-M.; Li, S.; Shen, Y.; Qi, Q.; Zhang, Y.; Li, Y.-Z.; Tang, Y.-J. Challenges and potential for improving the druggability of podophyllotoxin-derived drugs in cancer chemotherapy. Nat. Prod. Rep. 2021, 38, 470–488. [Google Scholar] [CrossRef]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anti-Cancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef]
- Fan, H.Y.; Zhu, Z.L.; Xian, H.C.; Wang, H.F.; Chen, B.J.; Tang, Y.J.; Tang, Y.L.; Liang, X.H. Insight Into the Molecular Mechanism of Podophyllotoxin Derivatives as Anticancer Drugs. Front. Cell Dev. Biol. 2021, 9, 709075. [Google Scholar] [CrossRef]
- Miranda-Vera, C.; Hernández, A.P.; García-García, P.; Díez, D.; García, P.A.; Castro, M.A. Podophyllotoxin: Recent Advances in the Development of Hybridization Strategies to Enhance Its Antitumoral Profile. Pharmaceutics 2023, 15, 2728. [Google Scholar] [CrossRef]
- Moss, P.G. Nomenclature of Lignans and Neolignans. Pure Appl. Chem. 2000, 72, 1493–1523. [Google Scholar] [CrossRef]
- Castro, M.Á.; Miguel del Corral, J.M.; García, P.A.; Rojo, M.V.; Bento, A.C.; Mollinedo, F.; Francesch, A.M.; San Feliciano, A. Lignopurines: A new family of hybrids between cyclolignans and purines. Synthesis and biological evaluation. Eur. J. Med. Chem. 2012, 58, 377–389. [Google Scholar] [CrossRef]
- Hernandez, A.P.; Diez, P.; Garcia, P.A.; Perez-Andres, M.; Veselinova, A.; Jambrina, P.G.; San Feliciano, A.; Diez, D.; Fuentes, M.; Castro, M.A. Improving Properties of Podophyllic Aldehyde-Derived Cyclolignans: Design, Synthesis and Evaluation of Novel Lignohydroquinones, Dual-Selective Hybrids against Colorectal Cancer Cells. Pharmaceutics 2023, 15, 886. [Google Scholar] [CrossRef]
- Gordaliza, M.; Castro, M.A.; San Feliciano, A.; Del Corral, J.M.M.; Lopez, M.L.; Faircloth, G.T. Immunosuppressive Cyclolignan Derivatives. EP0711765A1, 27 August 2003. [Google Scholar]
- Castro, M.A.; Miguel del Corral, J.M.; García, P.A.; Rojo, M.V.; de la Iglesia-Vicente, J.; Mollinedo, F.; Cuevas, C.; San Feliciano, A. Synthesis and biological evaluation of new podophyllic aldehyde derivatives with cytotoxic and apoptosis-inducing activities. J. Med. Chem. 2010, 53, 983–993. [Google Scholar] [CrossRef]
- GarciaRocha, M.; Garcia-Grávalos, M.; Avila, J. Characterisation of antimitotic products from marine organisms that disorganise the microtubule network: Ecteinascidin 743, isohomohalichondrin-B and LL-15. Br. J. Cancer 1996, 73, 875–883. [Google Scholar] [CrossRef][Green Version]
- Castro, M.Á.; Miguel del Corral, J.M.; Gordaliza, M.; Grande, C.; Gómez-Zurita, A.; García-Grávalos, D.; San Feliciano, A. Synthesis and cytotoxicity of podophyllotoxin analogues modified in the A ring. Eur. J. Med. Chem. 2003, 38, 65–74. [Google Scholar] [CrossRef]
- Zhao, H.; Neamati, N.; Mazumder, A.; Sunder, S.; Pommier, Y.; Burke, T.R. Arylamide inhibitors of HIV-1 integrase. J. Med. Chem. 1997, 40, 1186–1194. [Google Scholar] [CrossRef]
- Markgraf, J.H.; Finkelstein, M.; Cort, J.R. Canthine analogs via intramolecular Diels-Alder reactions. Tetrahedron 1996, 52, 461–470. [Google Scholar] [CrossRef]
- Kunz, T.; Reissig, H.U. A new route to trans-substituted gamma-lactones. Angew. Chem.-Int. Ed. Engl. 1988, 27, 268–270. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Arimoto, M.; Nakajima, S.; Tanoguchi, M.; Fukada, Y. Syntheses of epipodophyllotoxin and podophyllotoxin from deoxypodophyllotoxin. Chem. Pharm. Bull. 1986, 34, 2056–2060. [Google Scholar] [CrossRef]
- Gordaliza, M.; Castro, M.; Miguel del Corral, J.; López-Vázquez, M.; García, P.A.; San Feliciano, A.; García-Grávalos, M.; Broughton, H. Preparation and cytotoxicity of podophyllotoxin derivatives lacking the lactone ring. Tetrahedron 1997, 53, 15743–15760. [Google Scholar] [CrossRef]
- Castro, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; Garcia, P.A.; Gomez-Zurita, M.A.; Garcia-Grávalos, M.D.; de la Iglesia-Vicente, J.; Gajate, C.; An, F.Y.; Mollinedo, F.; et al. Synthesis and biological evaluation of new selective cytotoxic cyclolignans derived from podophyllotoxin. J. Med. Chem. 2004, 47, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. Chemmedchem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.Y.; Tong, Y.R.; Luo, Y.F.; Huang, L.Q.; Gao, W. Biosynthesis, total synthesis, and pharmacological activities of aryltetralin-type lignan podophyllotoxin and its derivatives. Nat. Prod. Rep. 2022, 39, 1856–1875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Rakesh, K.P.; Shantharam, C.S.; Manukumar, H.M.; Asiri, A.M.; Marwani, H.M.; Qin, H.L. Podophyllotoxin derivatives as an excellent anticancer aspirant for future chemotherapy: A key current imminent needs. Bioorganic Med. Chem. 2018, 26, 340–355. [Google Scholar] [CrossRef]
- Yu, X.; Che, Z.; Xu, H. Recent Advances in the Chemistry and Biology of Podophyllotoxins. Chem.-A Eur. J. 2017, 23, 4467–4526. [Google Scholar] [CrossRef]
- Castro, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; Garcia, P.A.; Gomez-Zurita, M.A.; San Feliciano, A. Synthesis and cytotoxic evaluation of C-9 oxidized podophyllotoxin derivatives. Bioorganic Med. Chem. 2007, 15, 1670–1678. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay foranticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García, P.A.; Hernández, Á.-P.; Gómez-Zurita, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; Francesch, A.; San Feliciano, A.; Castro, M.Á. Cytotoxic Cyclolignans Obtained by the Enlargement of the Cyclolignan Skeleton of Podophyllic Aldehyde, a Selective Podophyllotoxin-Derived Cyclolignan. Molecules 2024, 29, 1442. https://doi.org/10.3390/molecules29071442
García PA, Hernández Á-P, Gómez-Zurita MA, Miguel del Corral JM, Gordaliza M, Francesch A, San Feliciano A, Castro MÁ. Cytotoxic Cyclolignans Obtained by the Enlargement of the Cyclolignan Skeleton of Podophyllic Aldehyde, a Selective Podophyllotoxin-Derived Cyclolignan. Molecules. 2024; 29(7):1442. https://doi.org/10.3390/molecules29071442
Chicago/Turabian StyleGarcía, Pablo A., Ángela-Patricia Hernández, Mª Antonia Gómez-Zurita, José M. Miguel del Corral, Marina Gordaliza, Andrés Francesch, Arturo San Feliciano, and Mª Ángeles Castro. 2024. "Cytotoxic Cyclolignans Obtained by the Enlargement of the Cyclolignan Skeleton of Podophyllic Aldehyde, a Selective Podophyllotoxin-Derived Cyclolignan" Molecules 29, no. 7: 1442. https://doi.org/10.3390/molecules29071442
APA StyleGarcía, P. A., Hernández, Á.-P., Gómez-Zurita, M. A., Miguel del Corral, J. M., Gordaliza, M., Francesch, A., San Feliciano, A., & Castro, M. Á. (2024). Cytotoxic Cyclolignans Obtained by the Enlargement of the Cyclolignan Skeleton of Podophyllic Aldehyde, a Selective Podophyllotoxin-Derived Cyclolignan. Molecules, 29(7), 1442. https://doi.org/10.3390/molecules29071442